ELFABRIO- pegunigalsidase alfa injection, solution, concentrate
ELFABRIO by
Drug Labeling and Warnings
ELFABRIO by is a Prescription medication manufactured, distributed, or labeled by Chiesi USA, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ELFABRIO® safely and effectively. See full prescribing information for ELFABRIO.
ELFABRIO (pegunigalsidase alfa-iwxj) injection, for intravenous use
Initial U.S. Approval: 2023
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS
See full prescribing information for complete boxed warning.
Appropriate medical support measures, including cardiopulmonary resuscitation equipment, should be readily available. If a severe hypersensitivity reaction occurs, discontinue ELFABRIO immediately and initiate appropriate medical treatment. (5.1)
INDICATIONS AND USAGE
ELFABRIO is a hydrolytic lysosomal neutral glycosphingolipid-specific enzyme indicated for the treatment of adults with confirmed Fabry disease. (1)
DOSAGE AND ADMINISTRATION
- For pretreatment recommendations, see Full Prescribing Information. (2.1)
- Recommended dosage is 1 mg/kg every 2 weeks administered as an intravenous infusion. (2.2)
- For dosage and administration modifications due to hypersensitivity reactions or infusion-associated reactions (IARs), see Full Prescribing Information. (2.3)
- For instructions on preparation (including dilution), storage, and administration (including rates for the initial 4-6 infusions for ERT-experienced and ERT-naïve patients), see Full Prescribing Information. (2.4, 2.5, 2.6)
DOSAGE FORMS AND STRENGTHS
Injection: 20 mg/10 mL or 5 mg/2.5 mL (2 mg/mL) solution in a single-dose vial. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
-
Infusion-Associated Reactions: If severe IARs occur, discontinue ELFABRIO and initiate appropriate medical treatment. (5.2)
- Membranoproliferative Glomerulonephritis: Monitor serum creatinine and urinary protein to creatinine ratio. Discontinue ELFABRIO if glomerulonephritis is suspected, until a diagnostic evaluation can be conducted. (5.3)
ADVERSE REACTIONS
Most common adverse reactions (≥15%) are: infusion-associated reactions, nasopharyngitis, headache, diarrhea, fatigue, nausea, back pain, pain in extremity, and sinusitis. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Chiesi USA, Inc. at 1-888-661-9260 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.See 17 for PATIENT COUNSELING INFORMATION.
Revised: 5/2024
- For pretreatment recommendations, see Full Prescribing Information. (2.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommendations Prior to ELFABRIO Treatment
2.2 Recommended Dosage and Administration
2.3 Administration Modifications Due to Hypersensitivity Reactions and/or Infusion-Associated Reactions
2.4 Preparation Instructions
2.5 Storage of the Diluted Solution
2.6 Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions Including Anaphylaxis
5.2 Infusion-Associated Reactions
5.3 Membranoproliferative Glomerulonephritis
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Patients with Prior Enzyme Replacement Therapy
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS
Patients treated with ELFABRIO have experienced hypersensitivity reactions, including anaphylaxis. Appropriate medical support measures, including cardiopulmonary resuscitation equipment, should be readily available during ELFABRIO administration. If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue ELFABRIO immediately and initiate appropriate medical treatment. In patients with severe hypersensitivity reaction, a desensitization procedure to ELFABRIO may be considered [see Warnings and Precautions (5.1)].
- 1 INDICATIONS AND USAGE
-
2
DOSAGE AND ADMINISTRATION
2.1 Recommendations Prior to ELFABRIO Treatment
Pretreatment
- In enzyme replacement therapy (ERT)-experienced patients, if pretreatment with antihistamines, antipyretics, and/or corticosteroids was used prior to ERT administration, consider similar pretreatment with these medications before the first several ELFABRIO infusions. After 4 to 6 ELFABRIO infusions, a stepwise decrease in the pretreatment medication dose(s) and/or discontinuation of the pretreatment medication(s) may be considered if treatment with ELFABRIO was tolerated.
- In ERT-naïve patients, prior to ELFABRIO administration, pre-treating with antihistamines, antipyretics, and/or corticosteroids may be considered [see Warnings and Precautions (5.1, 5.2)].
Medical Support
Appropriate medical support measures including cardiopulmonary resuscitation equipment should be readily available during ELFABRIO administration.
2.2 Recommended Dosage and Administration
The recommended dosage of ELFABRIO, based on actual body weight, is 1 mg/kg administered by intravenous infusion every 2 weeks.
The initial recommended ELFABRIO infusion rates for ERT-experienced or ERT-naïve patients are based on actual body weight [see Tables 1 and 2].
If one or more doses are missed, restart ELFABRIO treatment as soon as possible, maintaining the 2 week interval between infusions thereafter. Do not double a dose to compensate for a missed dose.
2.3 Administration Modifications Due to Hypersensitivity Reactions and/or Infusion-Associated Reactions
In the event of a severe hypersensitivity reaction (e.g., anaphylaxis) or severe infusion-associated reaction (IAR), immediately discontinue ELFABRIO administration and initiate appropriate medical treatment. For additional recommendations in the event of a severe hypersensitivity reaction or IAR, see Warnings and Precautions (5.1, 5.2).
In the event of a mild to moderate hypersensitivity reaction or a mild to moderate IAR, consider temporarily holding the infusion for 15 to 30 minutes or slowing the infusion rate by 25% to 50% [see Dosage and Administration (2.6)], and initiating appropriate medical treatment [see Warnings and Precautions (5.1, 5.2)].
- If symptoms persist despite holding or slowing the infusion, stop the infusion and monitor the patient. Consider re-initiating the infusion within 7 to 14 days at 25% to 50% of the rate at which the reaction occurred with appropriate pretreatment.
- If symptoms subside after holding the infusion, resume infusion at a 25% to 50% reduced rate as tolerated. Alternatively, if symptoms subside after slowing the infusion, complete infusion at the reduced rate as tolerated.
- Starting with the next infusion, increase the infusion rate by increments of 25% every third infusion as tolerated until the infusion rate at which the reaction occurred is reached. Closely monitor the patient.
2.4 Preparation Instructions
Use aseptic technique during preparation. Dilute ELFABRIO in the following manner:
- Determine the number of ELFABRIO vials to be diluted based on actual body weight in kg and the recommended dose [see Dosage and Administration (2.2) and Dosage Forms and Strengths (3)]. Round the number of vials up to the next whole number.
- Remove the appropriate number of ELFABRIO vials from the refrigerator and allow the vials to sit for 15-30 minutes at room temperature 20°C to 25°C (68°F to 77°F) before use. Do not use an external heat source to heat the product because heat may damage the product.
- Visually inspect the solution in the vials for particulate matter and discoloration. The solution should be clear and colorless. Discard if the solution is discolored or if visible particulate matter is present.
- Dilute the supplied ELFABRIO solution required for a dose in 0.9% Sodium Chloride Injection to a total volume based on actual body weight specified in Tables 1 and 2 below.
- Prior to adding the volume of ELFABRIO required for the dose, remove the equal volume of 0.9% Sodium Chloride Injection from the infusion bag.
- Withdraw the volume of ELFABRIO required for the dose from the vials (discard any unused solution remaining in the vial).
- Inject the ELFABRIO solution directly into the 0.9% Sodium Chloride Injection solution through the port of the infusion bag. Do not inject in the airspace within the infusion bag.
- Gently invert infusion bag to mix the solution. Avoid vigorous shaking or agitation.
2.5 Storage of the Diluted Solution
- If the diluted ELFABRIO solution is not used immediately:
○ Refrigerate the diluted solution at 2°C to 8°C (36°F to 46°F) for up to 24 hours. The solution must be infused within 8 hours after removal from the refrigerator, inclusive of the total infusion time, or discarded.
○ Store the diluted solution at room temperature at 20°C to 25°C (68°F to 77°F) for up to 8 hours. The solution must be used within 8 hours, inclusive of infusion time, or discarded.
- Do not freeze or shake.
2.6 Administration Instructions
Administer ELFABRIO as follows:
- Use an in-line low protein-binding, 0.2 micron, in-line filter during administration.
- For the initial 4-6 infusions, infuse ELFABRIO using the infusion rates described in Table 1 for ERT-experienced patients and Table 2 for ERT-naïve patients.
- If a patient tolerates the initial 4-6 ELFABRIO infusions, the duration of every third infusion may be decreased in decrements of 30 minutes as tolerated. The minimum recommended infusion duration is 1.5 hours.
- At the end of the infusion, flush the line with 0.9% Sodium Chloride Injection using the same infusion rate as the one used for the last part of the ELFABRIO infusion.
- Do not infuse ELFABRIO in the same intravenous line with other products.
Table 1 presents the recommended infusion rates for the initial 4-6 ELFABRIO infusions based on actual body weight for ERT-experienced patients.
Table 1: Recommended Infusion Rate1 for ERT-Experienced Patients for the Initial 4-6 ELFABRIO Intravenous Infusions Based on Actual Body Weight Actual Body Weight Total Infusion Volume Infusion Rate1 ˂ 70 kg 150 mL 0.83 mL/min (50 mL/h) 70 -100 kg 250 mL 1.39 mL/min (83 mL/h) > 100 kg 500 mL 2.78 mL/min (167 mL/h) 1 Infusion rate may be increased if the patient tolerates the initial 4-6 infusions (see above). Infusion rate may be slowed in case of a hypersensitivity reaction or an IAR [see Dosage and Administration (2.3)].
Table 2 presents the recommended infusion rates for the initial 4-6 ELFABRIO infusions based on actual body weight for ERT-naïve patients.
Table 2: Recommended Infusion Rate1 for ERT-Naïve Patients for the Initial 4-6 ELFABRIO Intravenous Infusions Based on Actual Body Weight Actual Body Weight Total Infusion Volume Infusion Rate1 ˂ 70 kg 150 mL 0.63 mL/min (37.5 mL/h) 70 -100 kg 250 mL 1 mL/min (60 mL/h) > 100 kg 500 mL 1.38 mL/min (83 mL/h) 1 Infusion rate may be increased if the patient tolerates the initial 4-6 infusions (see above). Infusion rate may be slowed in case of a hypersensitivity reaction or an IAR [see Dosage and Administration (2.3)].
Administration of ELFABRIO to Patients Previously Treated with ERT with an Infusion Duration Over 3 Hours
In patients previously treated with an ERT with an infusion duration over 3 hours:
- Use the same infusion rate for the ELFABRIO infusion.
- May decrease the duration of every third infusion after the initial 4-6 ELFABRIO infusions in decrements of 30 minutes as tolerated. The recommended minimum infusion duration of the maintenance infusion is 1.5 hours.
Home Infusion
Home administration under the supervision of a healthcare provider may be considered for patients who have reached an infusion duration that is tolerated well [see Dosage and Administration (2.1, 2.3)]. The decision to have a patient move to home infusion should be made after evaluation and recommendation by a healthcare provider.
The infusion duration should remain constant for home administration and the duration should only be decreased in a healthcare facility. In case of a missed dose or delayed infusion, a healthcare provider should be contacted.
- In enzyme replacement therapy (ERT)-experienced patients, if pretreatment with antihistamines, antipyretics, and/or corticosteroids was used prior to ERT administration, consider similar pretreatment with these medications before the first several ELFABRIO infusions. After 4 to 6 ELFABRIO infusions, a stepwise decrease in the pretreatment medication dose(s) and/or discontinuation of the pretreatment medication(s) may be considered if treatment with ELFABRIO was tolerated.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5
WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions Including Anaphylaxis
Hypersensitivity reactions including anaphylaxis have been reported in ELFABRIO-treated patients. In clinical trials, 20 (14%) of ELFABRIO-treated patients experienced hypersensitivity reactions. In these trials, 4 ELFABRIO-treated patients (3%; 1 naïve to enzyme replacement therapy (ERT) and 3 ERT-experienced patients) experienced anaphylaxis during the initial infusion and were positive for anti-pegunigalsidase alfa-iwxj IgE antibodies (referred to as IgE ADA) [see Adverse Reactions (6.16.1) and Clinical Pharmacology (12.6)]. The risk of pegunigalsidase alfa-iwxj-related hypersensitivity may be increased in certain patients with pre-existing ADA from prior ERT [see Use In Specific Populations (8.6)].
Anaphylaxis (reported as Type I hypersensitivity reaction, hypersensitivity reaction, or bronchospasm) occurred within 5 to 40 minutes of the start of the initial infusion. Signs and symptoms included headache, nausea, vomiting, throat tightness, facial and oral edema, truncal rash, tachycardia, hypotension, rigors, urticaria, intense pruritus, moderate upper airway obstructions, macroglossia, and mild lip edema. Patients received treatment that included epinephrine, antihistamines and/or systemic corticosteroids.
Prior to ELFABRIO administration, consider pretreating with antihistamines, antipyretics, and/or corticosteroids. Appropriate medical support measures, including cardiopulmonary resuscitation equipment, should be readily available during ELFABRIO administration.
- If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue ELFABRIO immediately and initiate appropriate medical treatment. Consider the risks and benefits of re-administering ELFABRIO following severe hypersensitivity reactions (including anaphylaxis). Patients may be rechallenged using slower infusion rates. In patients with severe hypersensitivity reaction, desensitization measures to ELFABRIO may be considered. If the decision is made to readminister ELFABRIO, ensure the patient tolerates the infusion. If the patient tolerates the infusion, the rate may be increased to reach the recommended rate.
- If a mild or moderate hypersensitivity reaction occurs, consider temporarily holding the infusion or slowing the infusion rate [see Dosage and Administration (2.3)].
Consider monitoring patients who demonstrate hypersensitivity reactions during ELFABRIO treatment for the presence of IgG and IgE ADA [see Clinical Pharmacology (12.6)].
5.2 Infusion-Associated Reactions
Infusion-associated reactions (IARs) have been reported in ELFABRIO-treated patients. In clinical trials, 41 (29%) of ELFABRIO-treated patients experienced one or more IARs, defined as any adverse reaction with onset after start of the infusion and up to 24 hours after the end of infusion. The risk of pegunigalsidase alfa-iwxj-related IARs may be increased in certain patients with pre-existing ADA from prior ERT [see Use In Specific Populations (8.6)].
IARs included anaphylaxis reactions during the initial ELFABRIO administration [see Warnings and Precautions (5.1)].
In addition to the hypersensitivity reactions described above [see Warnings and Precautions (5.1)], other IARs included nausea, chills, pruritus, rash, chest pain, dizziness, vomiting, asthenia, pain, sneezing, dyspnea, nasal congestion, throat irritation, abdominal pain, erythema, diarrhea, burning sensation, neuralgia, headache, paresthesia, tremor, agitation, increased body temperature, flushing, bradycardia, myalgia, hypertension, and hypotension [see Adverse Reactions (6.1)].
Up to 40% of patients were pretreated with diphenhydramine, prednisone and/or acetaminophen at least once during the clinical trials. Severe reactions in the trials were generally managed with administration of antipyretics, antihistamines, corticosteroids, intravenous fluids, and/or oxygen.
IARs were more frequently observed in ELFABRIO-treated patients who developed IgG anti-drug antibodies (ADA) including patients who had pre-existing IgG ADA [see Adverse Reactions (6.16.1) and Clinical Pharmacology (12.6)]. Consider monitoring patients who demonstrate IARs during ELFABRIO treatment for the presence of IgG and IgE ADA.
Patients with advanced Fabry disease may have compromised cardiac function which may predispose them to a higher risk of severe complications from IARs. Closely monitor patients with compromised cardiac function if ELFABRIO is administered to these patients.
Prior to ELFABRIO administration, consider pre-treating with antihistamines, antipyretics, and/or corticosteroids to reduce the risk of IARs. However, IARs may still occur in patients after receiving pre-treatment.
- If a severe IAR occurs, discontinue ELFABRIO immediately and initiate appropriate medical treatment. Consider the risks and benefits of re-administering ELFABRIO following a severe IAR. Patients may be rechallenged using slower infusion rates. Once a patient tolerates the infusion, the infusion rate may be increased to reach the recommended infusion rate.
- If a mild or moderate IAR occurs, consider temporarily holding the infusion or slowing the infusion rate [see Dosage and Administration (2.3)].
5.3 Membranoproliferative Glomerulonephritis
A case of membranoproliferative glomerulonephritis with immune depositions in the kidney was reported during clinical trials [see Adverse Reactions (6.1)]. This event led to a decline in renal function that slowly improved upon discontinuation of ELFABRIO but did not return to baseline by the end of the trial. Monitor serum creatinine and urinary protein to creatinine ratio. If glomerulonephritis is suspected, discontinue ELFABRIO until a diagnostic evaluation can be conducted.
- If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue ELFABRIO immediately and initiate appropriate medical treatment. Consider the risks and benefits of re-administering ELFABRIO following severe hypersensitivity reactions (including anaphylaxis). Patients may be rechallenged using slower infusion rates. In patients with severe hypersensitivity reaction, desensitization measures to ELFABRIO may be considered. If the decision is made to readminister ELFABRIO, ensure the patient tolerates the infusion. If the patient tolerates the infusion, the rate may be increased to reach the recommended rate.
-
6
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in labeling:
- Hypersensitivity Reactions Including Anaphylaxis [see Warnings and Precautions (5.1)]
- Infusion-Associated Reactions (IARs) [see Warnings and Precautions (5.2)]
- Membranoproliferative Glomerulonephritis [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trial of another drug and may not reflect the rates observed in clinical practice.
Adverse Reactions From Trial 2
The safety of ELFABRIO in adults with confirmed Fabry disease who had been previously treated with agalsidase beta was evaluated in Trial 2 which included a total of 52 ELFABRIO-treated patients (29 male, 23 female aged 20 to 60 years old) with Fabry disease [see Clinical Studies (14)]. Patients received 1 mg/kg of ELFABRIO given intravenously every 2 weeks for at least 104 weeks.
The most common adverse reactions (≥15%) reported with ELFABRIO were infusion-associated reactions which occurred in 17 patients (32%); followed by, nasopharyngitis and headache each in 11 patients (21%); diarrhea in 10 patients (19%); fatigue and nausea each in 9 patients (17%); and back pain, pain in extremity, and sinusitis each in 8 patients (15%).
One ELFABRIO-treated patient experienced a severe hypersensitivity reaction during the first infusion. The patient withdrew from the trial following a moderate hypersensitivity reaction during the second infusion.
Table 3 lists adverse reactions reported in at least 5% of ELFABRIO-treated patients in Trial 2.
Table 3: Adverse Reactions in Adults With Fabry Disease (Trial 2)1 Adverse Reaction ELFABRIO
N=52
n (%)Agalsidase beta
N=25
n (%)Infusion-Associated Reaction2,4 17 (32) 8 (32) Nasopharyngitis 11 (21) 4 (16) Headache 11 (21) 5 (20) Diarrhea 10 (19) 6 (24) Fatigue 9 (17) 4 (16) Nausea 9 (17) 3 (12) Back pain 8 (15) 5 (20) Pain in Extremity 8 (15) 4 (16) Sinusitis 8 (15) 3 (12) Abdominal Pain 6 (12) 0 (0) Proteinuria 6 (12) 0 (0) Hypersensitivity3,4 5 (9) 4 (16) Upper Respiratory Tract Congestion 4 (8) 0 (0) Neuralgia 4 (8) 0 (0) Peripheral Neuropathy 3 (6) 0 (0) Sciatica 3 (6) 0 (0) Infusion Site Extravasation 3 (6) 0 (0) Hematuria 3 (6) 0 (0) 1 Adverse reactions were those that occurred in ≥ 5% of ELFABRIO-treated patients.
2 “Infusion-associated reaction” includes nausea, vomiting, abdominal pain, diarrhea, fatigue, chills, malaise, non-cardiac chest pain, hypersensitivity, body temperature increased, burning sensation, neuralgia, agitation, throat irritation, pruritic rash, and flushing. Events occurring within 24 hours.
3 “Hypersensitivity” includes macular rash, pruritic rash, and face swelling. Events occurring within 24 hours.
4 The events of hypersensitivity and pruritic rash fall in both hypersensitivity and IAR categories.Membranoproliferative Glomerulonephritis
A case of membranoproliferative glomerulonephritis with immune depositions in the kidney was reported in an ELFABRIO-treated patient.
Immunogenicity: Anti-Drug Antibody-Associated Adverse Reactions
Of the patients who experienced serious hypersensitivity reactions during the first ELFABRIO infusion and had pre-infusion samples and samples available for testing at the time of the event, all but one had pre-existing IgE ADAs and all tested positive for IgE ADAs at the time of the reaction.
In the overall clinical program, IARs occurred in 51% (19/37) of patients who were IgG ADA positive at baseline compared to 16% (13/84) in IgG ADA negative patients [see Clinical Pharmacology (12.6)].
- Hypersensitivity Reactions Including Anaphylaxis [see Warnings and Precautions (5.1)]
-
8
USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on ELFABRIO use in pregnant females to evaluate a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes; however, as an enzyme replacement, ELFABRIO is not expected to cause adverse outcomes. Animal reproduction studies have been conducted with pegunigalsidase alfa-iwxj in pregnant rats and rabbits. No adverse effects on embryofetal development were observed in pregnant rats intravenously administered pegunigalsidase alfa-iwxj twice per week at exposures up to 3.6 times that of the maximum recommended human dose (MRHD) (based on area under the concentration-time curve (AUC)). Maternal toxicity was observed in pregnant rabbits intravenously administered pegunigalsidase alfa-iwxj twice per week at doses that were ≥ 3.2 times the MRHD (based on human equivalent dose) [see Data].
The estimated background risk of major birth defects and miscarriage in the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
There is a pregnancy safety study for ELFABRIO. If a patient becomes pregnant while receiving ELFABRIO, healthcare providers should report ELFABRIO exposure by calling 1-888-661-9260 or visiting https://chiesirarediseases.com/contact-us/medical-information-form.
Data
Animal Data
In an embryofetal development study in the rat, pegunigalsidase alfa-iwxj was administered during the period of organogenesis on gestation day 6, 9, 12, and 15. No maternal or fetal adverse effects were noted at exposures that were up to 3.6-fold greater than the recommended dose of 1 mg/kg every two weeks.
In an embryofetal development study in the rabbit, administration of pegunigalsidase alfa-iwxj during the period of organogenesis on gestation day 6, 9, 12, 15, and 18, resulted in maternal toxicity, including maternal mortality, decreased body weight, and decreased feed consumption. These effects were observed at exposures that were ≥ 3.2-fold greater than the recommended dose of 1 mg/kg every two weeks. Adverse embryofetal effects included abortion, increased late resorptions, number of does with resorptions, and increased post-implantation loss at exposures that were 6.5 fold greater than the recommended dose of 1 mg/kg every two weeks. Decreased fetal body weight was observed at exposures that were ≥ 3.2 times greater than the recommended dose of 1 mg/kg every two weeks. There was no increase in fetal external, skeletal, or visceral malformations.
8.2 Lactation
Risk Summary
There are no data on the presence of pegunigalsidase alfa-iwxj in either human or animal milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ELFABRIO and any potential adverse effects on the breastfed infant from pegunigalsidase alfa-iwxj or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of ELFABRIO have not been established in pediatric patients.
8.5 Geriatric Use
Clinical trials of ELFABRIO did not include patients 65 years of age and older to determine if they respond differently from younger adult patients.
8.6 Patients with Prior Enzyme Replacement Therapy
Patients that received prior ERT are more likely to have pre-existing anti-drug antibodies (ADA) to pegunigalsidase alfa-iwxj which could be due to the ADA cross-reactivity to pegunigalsidase alfa-iwxj by prior ERT. When switching from other ERT to ELFABRIO:
- Pre-existing ADA may reduce the plasma pegunigalsidase alfa-iwxj concentrations, which may reduce ELFABRIO efficacy [see Clinical Pharmacology (12.2, 12.6)].
- The risk of ELFABRIO-related hypersensitivity and infusion-associated reactions may be increased in certain patients with pre-existing ADA from prior ERT [see Warnings and Precautions (5.1, 5.2) and Adverse Reactions (6.1)].
Consider monitoring clinical or pharmacodynamic responses (e.g., plasma lyso-Gb3 levels) when switching from agalsidase beta to ELFABRIO, in patients with pre-existing ADA.
- Pre-existing ADA may reduce the plasma pegunigalsidase alfa-iwxj concentrations, which may reduce ELFABRIO efficacy [see Clinical Pharmacology (12.2, 12.6)].
-
11
DESCRIPTION
Pegunigalsidase alfa-iwxj, a hydrolytic lysosomal neutral glycosphingolipid-specific enzyme, is a PEGylated and crosslinked, chemically modified, recombinant human alpha-galactosidase A enzyme that is produced by genetically modified Bright Yellow 2 (Nicotiana tabacum) plant cells. The amino acid sequence of one subunit of pegunigalsidase alfa-iwxj consists of 405 amino acids, of which 398 amino acids are identical to human alpha-GAL-A with an additional 6 amino acids (SEKDEL) included at the C-terminal to encode an endoplasmic retrieval signal, and an additional glycine at the N-terminus derived from the signal peptide.
Pegunigalsidase alfa-iwxj is a homodimeric glycoprotein covalently crosslinked with an average of nine 2.3 kDa PEG per dimer. The total molecular weight of the cross-linked dimer is approximately 116 kDa. Pegunigalsidase alfa-iwxj has specific activity of approximately 35-62 U/mg (one enzyme unit is defined as the amount of enzyme which catalyzes the hydrolysis of one micromole of synthetic substrate, p-nitrophenyl-α-D-galactopyranoside per minute at 37°C).
ELFABRIO (pegunigalsidase alfa-iwxj) injection is a sterile, preservative-free, 20 mg/10 mL or 5 mg/2.5 mL (2 mg/mL) solution in a single-dose vial for intravenous infusion after dilution. Each mL contains 2 mg of pegunigalsidase alfa-iwxj, anhydrous citric acid (0.2 mg), sodium chloride (7.06 mg), sodium citrate (6.73 mg), and Water for Injection, USP. The pH is approximately 5.9 to 6.4.
-
12
CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Fabry disease is caused by deficiency of the lysosomal enzyme alpha-galactosidase A. ELFABRIO provides an exogenous source of alpha-galactosidase A. ELFABRIO is internalized and transported into lysosomes where it is thought to exert enzymatic activity and reduce accumulated globotriaosylceramide (Gb3).
12.2 Pharmacodynamics
Plasma globotriaosylsphingosine (lyso-Gb3, a metabolite of Gb3) concentrations are elevated in patients with Fabry disease. In patients with Fabry disease who were [see Clinical Studies (14)]:
- ERT-naïve or had not received ERT treatment for at least 26 weeks and had a negative test for anti-pegunigalsidase alfa-iwxj antibodies (Trial 1), ELFABRIO treatment resulted in significant reductions in median plasma lyso-Gb3 concentrations compared to baseline of approximately:
○ -43% (Week 4), -57% (Week 26), -68% (Week 52), and -84% (Week 104) in male patients, and
○ -3% (Week 4), -19% (Week 26), -32% (Week 52), and -75% (Week 104) in female patients.
- ERT-experienced for at least 104 weeks (Trial 2), switching to ELFABRIO treatment resulted in increases in median plasma lyso-Gb3 concentrations compared to baseline of:
○ Approximately 11% (Week 6), 15% (Week 26), and 18% (Week 104) in male patients, and
○ No significant changes in median plasma lyso-Gb3 concentrations in female patients.
Pegunigalsidase alfa-iwxj exposure-response relationship is unknown.
12.3 Pharmacokinetics
The pharmacokinetics (PK) of pegunigalsidase alfa-iwxj were evaluated in adult patients with Fabry disease and are presented as mean (standard deviation, SD) unless otherwise specified. The pharmacokinetics of pegunigalsidase alfa-iwxj in plasma following intravenous infusion of ELFABRIO 1 mg/kg every 2 weeks in adult treatment-naïve patients with Fabry disease are summarized in Table 4. The maximum plasma concentration (Cmax) and area under the concentration-time curve (AUC) of pegunigalsidase alfa-iwxj increased with longer duration of treatment. In adult ERT-experienced patients with Fabry disease, mean Cmax ranged from 21.2 to 23.3 μg/mL and mean AUCtau ranged from 958 to 1074 μg·h/mL following intravenous infusion of ELFABRIO 1 mg/kg every 2 weeks.
Table 4: Pharmacokinetics of Pegunigalsidase Alfa-iwxj in Adult ERT-Naïve1 Patients With Fabry Disease Following Intravenous Infusion of ELFABRIO 1 mg/kg Every 2 Weeks PK Parameters Pegunigalsidase Alfa-iwxj Day 1 Week 13 Week 26 Week 52 Mean Infusion Duration (h) 5.5 4.4 3.9 3.3 General Information Cmax (μg/mL) 11.1±2.4 11.9±2.4 13.3±3.0 17.3±6.1 AUCinf (μg·h/mL) 391±136 510±174 748±200 1428±875 Distribution V (mL/kg) 321±71 271±89 226±116 186±91 Elimination t1/2 (h) 78.9±10.3 85.7±28.4 96.5±31.4 121±22 CL (mL/h/kg) 2.9±0.7 2.3±0.8 1.6±0.6 1.1±0.7 Metabolism Expected metabolism into small peptides by catabolic pathways Cmax=maximum plasma concentration; AUC=area under the plasma concentration-time curve; V= terminal volume of distribution; t1/2=elimination half-life; CL=clearance
1 Includes patients who had not received ERT for at least 26 Weeks and who tested negative for anti-pegunigalsidase alfa-iwxj antibodies at screening.12.6 Immunogenicity
The observed incidence of IgG anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADA in other studies, including those of pegunigalsidase alfa-iwxj ELFABRIO or of other pegunigalsidase alfa products.
The incidences of IgG ADA in ELFABRIO-treated patients with Fabry disease in Trials 1 and 2 are shown in Table 5.
Immunogenicity in ERT-Naïve Patients or Those Who Did Not Receive ERT for 26 Weeks
In Trial 1 (ERT-naïve patients with Fabry disease or those who did not receive ERT for 26 weeks) [see Clinical Studies (14)]:
- Four of the 14 ELFABRIO-treated who were IgG anti-pegunigalsidase alfa-iwxj antibodies (anti-drug antibodies or ADA) negative at baseline became ADA positive during ELFABRIO treatment. The onset of ADA positivity in 3 of these patients occurred within 26 weeks after starting ELFABRIO treatment.
- Two patients had ADA prior to receiving their first ELFABRIO dose. One of these patients remained ADA positive after receiving ELFABRIO treatment (this patient had boosted antibody titers during ELFABRIO treatment).
Immunogenicity in ERT-Experienced Patients
In Trial 2 (ERT-experienced patients with Fabry disease who switched from agalsidase beta [see Clinical Studies (14)]:
- Three of the 34 ELFABRIO-treated patients who were ADA negative at baseline became ADA positive during ELFABRIO treatment. The onset of ADA positivity occurred within 26 weeks after starting ELFABRIO treatment in 1 patient and more than 52 weeks in the remaining 2 patients.
- Eighteen ELFABRIO-treated patients had ADA prior to receiving their first ELFABRIO dose:
○ 17 (94%) patients remained ADA positive after receiving ELFABRIO treatment at 1 or more timepoints.
○ 3 (17%) patients had boosted ADA titers during ELFABRIO treatment.
Table 5: ADA Incidence in ELFABRIO-Treated Patients with Fabry Disease (Trials 1 and 2) Trial 1
ERT-Naïve1 PatientsTrial 2
ERT-Experienced PatientsBaseline Male
(N=11)Female
(N=7)Male
(N=29)Female
(N=23)ADA 1 (9%) 1 (14%) 18 (62%) 0 Neutralizing antibody2 0 0 17/18 0 Anti-enzyme antibody3 1/1 1/1 18/18 0 Anti-PEG antibody4 0 0 2/18 0 Anti-plant glycan antibody5 1/1 1/1 0 0 At any time during ELFABRIO treatment* Male
(N=9)Female
(N=7)Male
(N=29)Female
(N=23)ADA 5 (56%) 0 17 (59%) 3 (13%) Neutralizing antibody2 3/5 0 14/17 1/3 Anti-enzyme antibody3 4/5 0 17/17 1/3 Anti-PEG antibody4 1/5 0 2/17 1/3 Anti-plant glycan antibody5 2/5 0 0 0 ADA, anti-drug antibodies
*ADA incidence after ELFABRIO treatment was determined if positive at any timepoint during ELFABRIO treatment.
In Trial 1, of those that were positive at baseline (N=2) or any timepoint during ELFABRIO treatment (N=5), 4 became negative for ADA during Trial 1 through their last timepoint on study. In Trial 2, of those that were positive at baseline (N=18) or any timepoint during ELFABRIO treatment (N=20), 8 became negative for ADA during Trial 2 through their last timepoint on study.
1 Includes patients who had not received ERT for at least 26 Weeks and who tested negative for anti-pegunigalsidase alfa-iwxj antibodies at screening.
2Neutralizing antibodies that inhibit enzyme activity, tested for IgG ADA positive samples only. Assessments for neutralizing antibodies that inhibit cellular uptake of enzyme have not been performed.
3 Anti-pegunigalsidase alfa-iwxj antibodies specific to the enzyme moiety on pegunigalsidase alfa-iwxj, tested for IgG ADA positive samples only
4 Anti-pegunigalsidase alfa-iwxj antibodies specific to the PEG moieties on pegunigalsidase alfa-iwxj, tested for IgG ADA positive samples only
5 Anti-pegunigalsidase alfa-iwxj antibodies specific to the plant glycan motifs on pegunigalsidase alfa-iwxj, tested for IgG ADA positive samples onlyADA Effects on Safety and Efficacy
IARs occurred more frequently in ELFABRIO-treated patients who were IgG ADA positive compared to those that were IgG ADA negative [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)]. The effect of IgE ADA on hypersensitivity reactions of ELFABRIO treatment has not been fully characterized [see Warnings and Precautions (5.1)].
The effect of IgG ADA on the effectiveness of ELFABRIO has not been fully characterized [see Use in Specific Populations (8.6)].
ADA Effects on Pharmacodynamics
In Trial 2, baseline and post-treatment plasma lyso-Gb3 levels were higher in ELFABRIO-treated patients who were IgG ADA positive at baseline or at any time during the treatment compared with patients who were IgG ADA negative at baseline and at any time during the treatment [see Clinical Pharmacology (12.2)]. The IgG ADA positive patient who had plasma pegunigalsidase alfa-iwxj concentrations below the limit of quantification of the assay had the highest plasma lyso-Gb3 levels among ELFABRIO-treated patients.
ADA Effects on Pharmacokinetics
IgG ADA including pre-existing IgG ADA reduced plasma pegunigalsidase alfa-iwxj concentrations in ELFABRIO-treated patients with Fabry disease. Patients with higher ADA titers had lower pegunigalsidase alfa-iwxj concentrations compared to those with lower ADA titers.
In Trial 1 in patients who were negative for ADA at screening, 3 patients developed ADA following ELFABRIO treatment and had plasma pegunigalsidase alfa-iwxj concentration measures. Of these three patients:
- The plasma pegunigalsidase alfa-iwxj concentrations were transiently decreased in two of the patients who received 0.2 mg/kg of ELFABRIO intravenously every other week (0.2 times the approved recommended dosage), and
- No ADA effect on pegunigalsidase alfa-iwxj concentrations was observed in the third patient who received 1 mg/kg of ELFABRIO intravenously every other week.
In Trial 2, 3 of 17 patients who had plasma pegunigalsidase alfa-iwxj concentration measures had pre-existing ADA at baseline and maintained ADA positive status following ELFABRIO treatment [see Use in Specific Populations (8.6)]. Of these three patients:
- 1 patient with the highest ADA titer had plasma pegunigalsidase alfa-iwxj concentrations that were below the limit of quantification of the assay at all trial visits with pharmacokinetic assessments, and
- 2 patients had low plasma pegunigalsidase alfa-iwxj AUC, approximately 5% of the expected AUC for ADA-negative ELFABRIO-treated patients.
- ERT-naïve or had not received ERT treatment for at least 26 weeks and had a negative test for anti-pegunigalsidase alfa-iwxj antibodies (Trial 1), ELFABRIO treatment resulted in significant reductions in median plasma lyso-Gb3 concentrations compared to baseline of approximately:
-
13
NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Animal studies to evaluate the carcinogenic potential of pegunigalsidase alfa-iwxj have not been conducted. No effect on fertility was observed when pegunigalsidase alfa-iwxj was administered to adult rats at exposures ≤ 3.6-fold greater than the recommended dose of 1 mg/kg every two weeks.
-
14
CLINICAL STUDIES
Trial 1 was an open-label dose-ranging trial in adults diagnosed with Fabry disease (NCT01678898). Patients received ELFABRIO at 0.2 mg/kg, 1 mg/kg, or 2 mg/kg given intravenously every other week for 52 weeks. The 0.2 mg/kg and 2 mg/kg dosage regimens are not approved and are not recommended [see Dosage and Administration (2.2)].
Trial 1 enrolled 18 patients who were ERT-naïve or who had not received ERT for more than 26 weeks and had a negative test for anti-pegunigalsidase alfa-iwxj IgG antibodies prior to enrollment. Two patients in the 1 mg/kg treatment group discontinued the trial after their first infusion; one of them discontinued due to severe hypersensitivity reaction. Among the remaining 16 patients who completed Trial 1, 9 (56%) were males and 7 (44%) were females ranging in age from 17 to 54 years with a median age of 30 years. Twelve patients were White (75%) and 3 were Black or African American (19%). Three patients were Hispanic/Latino and 13 patients were not Hispanic/Latino. Of the 9 males, 7 (78%) had the classic phenotype. The median baseline eGFR and proteinuria was 115 mL/min/1.73 m2 and 0.11 g/g respectively. Among the male patients, the median value of residual alpha-galactosidase A activity was 2.4% (range: 0.0%-9.3%) in plasma and 1.3% (range: 0.0%-3.4%) in leukocytes.
The average number of globotriaosylceramide (Gb3) inclusions per renal peritubular capillary (PTC) in renal biopsy specimens of patients was assessed by light microscopy using the quantitative Barisoni Lipid Inclusion Scoring System (BLISS). Evaluable renal biopsies were obtained at baseline and at 26 weeks of treatment in 14 of the 16 patients who completed Trial 1.
Table 6 shows the changes from baseline to 26 weeks in the BLISS score (average number of Gb3 inclusions per renal PTC) for these 14 ELFABRIO-treated patients.
Table 6: Summary of the Renal Biopsy BLISS Score1 of Gb3 Inclusions at Baseline and after 26 Weeks of ELFABRIO Treatment in Adults with Fabry Disease (Trial 1) All Patients Males Females (N = 14) (N = 8) (N = 6) Median (range) Baseline 3.2 (0.4, 9.0) 6.8 (0.4, 9.0) 1.2 (0.8, 3.3) Week 26 0.7 (0.3, 2.5) 0.7 (0.3, 2.5) 0.7 (0.3, 1.4) Change at Week 26 -2.5 (-8.5, 0.5) -5.3 (-8.5, 0.5) -0.7 (-2.5, 0.1) Mean Change at Week 26 (95% CI) -3.1 (-4.8, -1.4) -4.7 (-7.1, -2.3) -1.0 (-2.1, 0.1) 1 The BLISS methodology counts the number of Gb3 inclusions in each renal PTC contained in a biopsy specimen. For each biopsy specimen (slide), approximately 300 renal PTCs were scored, and the final biopsy score for each patient was determined as the average number of Gb3 inclusions per PTC.
Trial 2 was a randomized, double-blind, and active-controlled trial (NCT02795676) in ERT-experienced adults diagnosed with Fabry disease. Eligible patients were treated with agalsidase beta for at least one year prior to trial entry (the mean duration of agalsidase beta treatment prior to enrollment was 5.7 years). Patients were randomized 2:1 to receive ELFABRIO (1 mg/kg intravenous infusion) or agalsidase beta (1 mg/kg intravenous infusion) every 2 weeks for 104 weeks.
A total of 77 patients were randomized and received at least one dose of ELFABRIO (N = 52, 68%) or agalsidase beta (N = 25, 32%). Of these patients, 47 (61%) were males and 30 (39%) were females. Patients were 18 to 60 years of age with a median age of 46 years at baseline; 72 (94%) were White, 3 (4%) were Black or African American and 2 (3%) were Asian. Two patients were Hispanic/Latino and 75 patients were not Hispanic/Latino. Forty-one (53%) patients had the classic phenotype. The median baseline eGFR and proteinuria was 75 mL/min/1.73 m2 and 0.11 g/g, respectively.
The primary efficacy endpoint was the annualized rate of change in eGFR (eGFR slope) assessed over 104 weeks. The estimated mean eGFR slope was -2.4 and -2.3 mL/min/1.73 m2/year on ELFABRIO and agalsidase beta respectively. The estimated treatment difference was -0.1 (95% CI: -2.3, 2.1) mL/min/1.73 m2/year.
-
16
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
ELFABRIO (pegunigalsidase alfa-iwxj) injection is a sterile, preservative-free, clear and colorless solution supplied in a single-dose vial. Each vial contains 20 mg/10 mL or 5 mg/2.5 mL (2 mg/mL) of pegunigalsidase alfa-iwxj. ELFABRIO is available as:
- One single-dose 20 mg/10 mLvial in a carton (NDC: 10122-160-02)
- One single-dose 5 mg/2.5 mL vial in a carton (NDC: 10122-165-02)
- Five single-dose 20 mg/10 mL vials in a carton (NDC: 10122-160-05)
- Ten single-dose 20 mg/10 mL vials in a carton (NDC: 10122-160-10)
Storage and Handling
Store refrigerated at 2°C to 8°C (36°F to 46°F). Do not freeze. Do not shake.
-
17
PATIENT COUNSELING INFORMATION
Hypersensitivity Reactions Including Anaphylaxis and Infusion-Associated Reactions (IARs)
Advise the patient and/or caregiver that reactions related to the infusion may occur during and after ELFABRIO treatment, including anaphylactic reactions, other serious or severe hypersensitivity reactions, and IARs. Inform the patient and/or caregiver of the signs and symptoms of hypersensitivity reactions and IARs and to seek immediate medical care should these signs and symptoms occur [see Warnings and Precautions (5.1, 5.2)].
Pregnancy Safety Study
Advise a patient who is exposed to ELFABRIO during pregnancy that there is a pregnancy safety study that monitors pregnancy outcomes. Encourage the patient to report the pregnancy to Chiesi USA, Inc. at 1-888-661-9260 and https://chiesirarediseases.com/contact-us/medical-information-form [see Use in Specific Populations (8.1)].
Manufactured by:
Chiesi Farmaceutici S.p.A.
Via Palermo 26/A, 43122 Parma, Italy
U.S. License No. 2245Manufactured at:
Chiesi Farmaceutici S.p.A.
43122 Parma, ItalyManufactured for:
Chiesi USA, Inc.
Cary NC, 27518, USA
Product of Israel.ELFABRIO is a registered trademark of Chiesi Farmaceutici S.p.A.
Licensed from Protalix Ltd.
CTPA-001-0324-01-W -
PRINCIPAL DISPLAY PANEL
NDC: 10122-160-02
ELFABRIO
(pegunigalsidase alfa-iwxj)
Injection
20 mg/ 10 mL (2 mg/ml)
For Intravenous Infusion After Dilution
Rx Only
1 x 10 mL single-dose vial
Discard unused portion.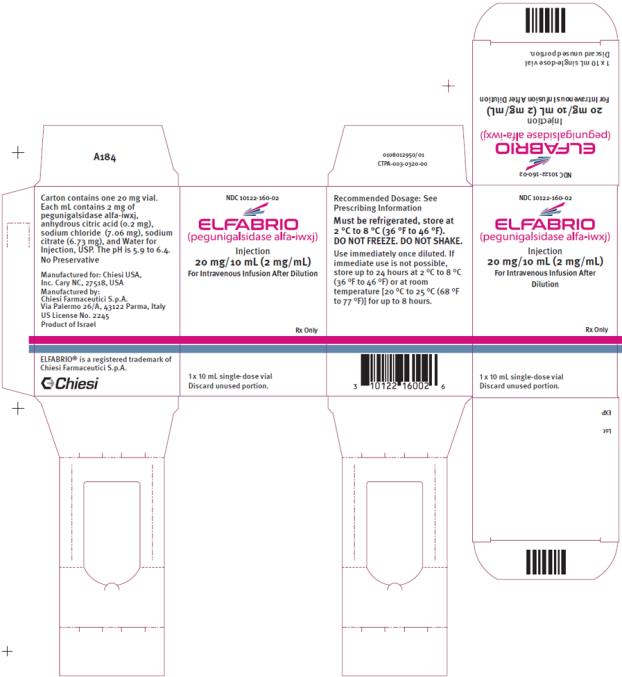
-
PRINCIPAL DISPLAY PANEL
NDC: 10122-165-02
ELFABRIO
(pegunigalsidase alfa-iwxj)
Injection
5 mg/ 2.5 mL (2 mg/ml)
For Intravenous Infusion After Dilution
Rx Only
1 x 2.5 mL single-dose vial
Discard unused portion.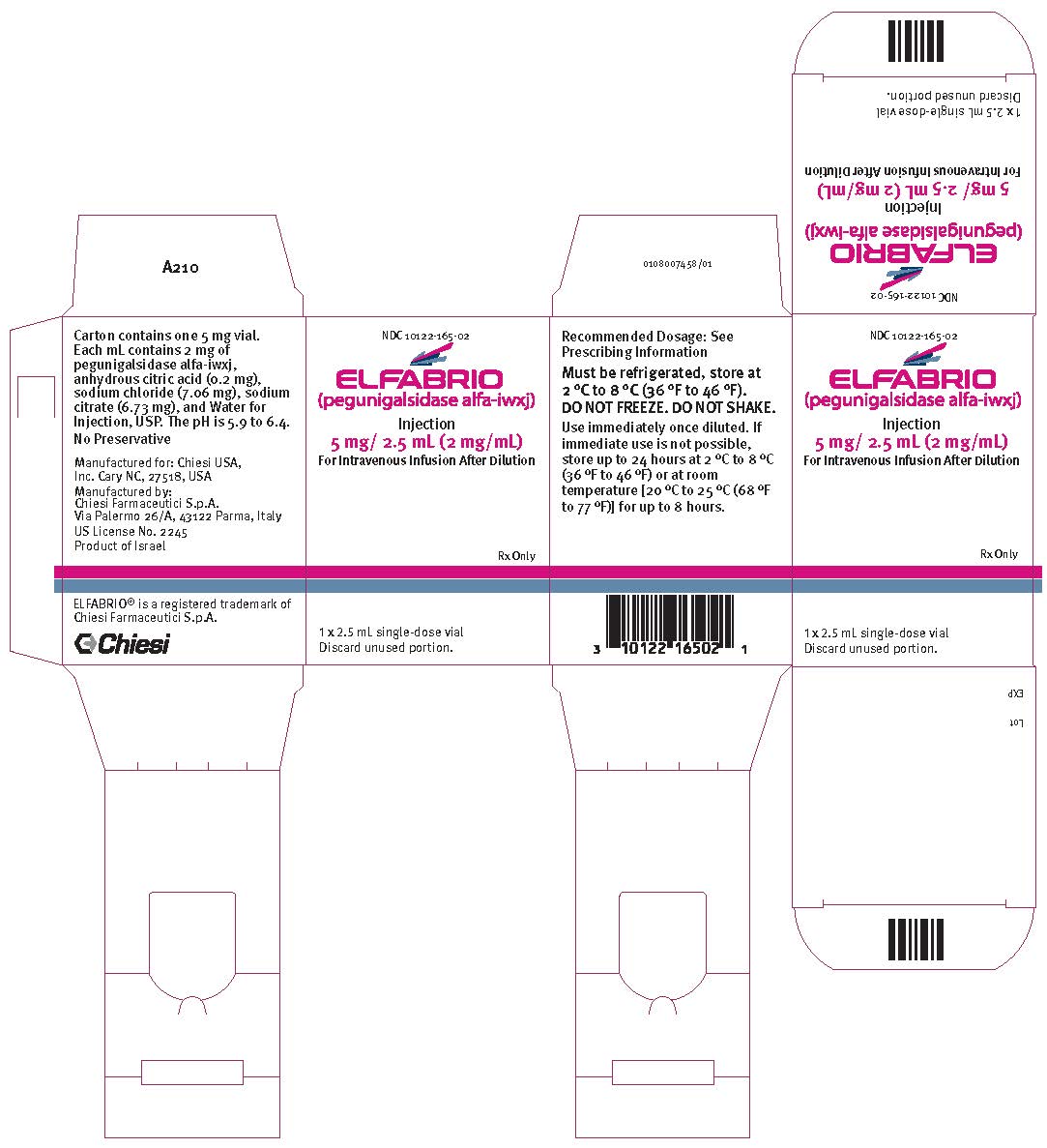
-
INGREDIENTS AND APPEARANCE
ELFABRIO
pegunigalsidase alfa injection, solution, concentrateProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 10122-160 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PEGUNIGALSIDASE ALFA (UNII: 8M7V7Q6537) (PEGUNIGALSIDASE ALFA - UNII:8M7V7Q6537) PEGUNIGALSIDASE ALFA 20 mg in 10 mL Inactive Ingredients Ingredient Name Strength ANHYDROUS CITRIC ACID (UNII: XF417D3PSL) 2.0 mg in 10 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 70.6 mg in 10 mL TRISODIUM CITRATE DIHYDRATE (UNII: B22547B95K) 76.7 mg in 10 mL WATER (UNII: 059QF0KO0R) 9916.7 mg in 10 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 10122-160-02 1 in 1 CARTON 05/23/2023 1 NDC: 10122-160-01 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product 2 NDC: 10122-160-05 5 in 1 CARTON 05/23/2023 2 NDC: 10122-160-01 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product 3 NDC: 10122-160-10 10 in 1 CARTON 05/23/2023 3 NDC: 10122-160-01 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761161 05/23/2023 ELFABRIO
pegunigalsidase alfa injection, solution, concentrateProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 10122-165 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PEGUNIGALSIDASE ALFA (UNII: 8M7V7Q6537) (PEGUNIGALSIDASE ALFA - UNII:8M7V7Q6537) PEGUNIGALSIDASE ALFA 5 mg in 2.5 mL Inactive Ingredients Ingredient Name Strength ANHYDROUS CITRIC ACID (UNII: XF417D3PSL) SODIUM CHLORIDE (UNII: 451W47IQ8X) TRISODIUM CITRATE DIHYDRATE (UNII: B22547B95K) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 10122-165-02 1 in 1 CARTON 07/08/2024 1 2.5 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761161 05/23/2023 Labeler - Chiesi USA, Inc. (088084228)
Trademark Results [ELFABRIO]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ELFABRIO 88226849 not registered Live/Pending |
CHIESI FARMACEUTICI S.P.A. 2018-12-12 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.