DAYVIGO- lemborexant tablet, film coated
DAYVIGO by
Drug Labeling and Warnings
DAYVIGO by is a Prescription medication manufactured, distributed, or labeled by Eisai Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DAYVIGO safely and effectively. See full prescribing information for DAYVIGO.
DAYVIGOTM (lemborexant) tablets, for oral use, [controlled substance schedule pending]
Initial U.S. Approval: [pending controlled substance scheduling]
INDICATIONS AND USAGE
DAYVIGO is an orexin receptor antagonist indicated for the treatment of adult patients with insomnia, characterized by difficulties with sleep onset and/or sleep maintenance. (1)
DOSAGE AND ADMINISTRATION
- Recommended dose is 5 mg taken no more than once per night, immediately before going to bed, with at least 7 hours remaining before the planned time of awakening. Dosage may be increased to 10 mg based on clinical response and tolerability. (2.1)
- The maximum recommended dose is 10 mg once daily. (2.1)
- Time to sleep onset may be delayed if taken with or soon after a meal. (2.1)
- Hepatic Impairment: (2.3)
○ Moderate hepatic impairment: Initial and maximum recommended dosage is 5 mg no more than once per night.
○ Severe hepatic impairment: Not recommended.
DOSAGE FORMS AND STRENGTHS
Tablets: 5 mg, 10 mg (3)
CONTRAINDICATIONS
DAYVIGO is contraindicated in patients with narcolepsy. (4)
WARNINGS AND PRECAUTIONS
- CNS Depressant Effects and Daytime Impairment: Impairs alertness and motor coordination including morning impairment. Risk increases with dose and use with other central nervous system (CNS) depressants. For patients taking DAYVIGO 10 mg, caution against next-day driving and other activities requiring complete mental alertness. (5.1)
- Sleep Paralysis, Hypnagogic/Hypnopompic Hallucinations, and Cataplexy-like Symptoms: May occur with use of DAYVIGO. (5.2)
- Complex Sleep Behaviors: Behaviors including sleep-walking, sleep-driving, and engaging in other activities while not fully awake may occur. Discontinue immediately if a complex sleep behavior occurs. (5.3)
- Compromised Respiratory Function: Effect on respiratory function should be considered. (5.4, 8.8)
- Worsening of Depression/Suicidal Ideation: Worsening of depression or suicidal thinking may occur. Prescribe the lowest number of tablets feasible to avoid intentional overdosage. (5.5)
- Need to Evaluate for Co-morbid Diagnoses: Reevaluate if insomnia persists after 7 to 10 days of treatment. (5.6)
ADVERSE REACTIONS
The most common adverse reaction (reported in ≥5% of patients treated with DAYVIGO and at least twice the rate of placebo) was somnolence. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Eisai Inc. at 1-888-274-2378 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2019
- Recommended dose is 5 mg taken no more than once per night, immediately before going to bed, with at least 7 hours remaining before the planned time of awakening. Dosage may be increased to 10 mg based on clinical response and tolerability. (2.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
2.2 Dosage Recommendations for Concomitant Use with CYP3A Inhibitors or CYP3A Inducers
2.3 Dosage Recommendations for Patients with Hepatic Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 CNS Depressant Effects and Daytime Impairment
5.2 Sleep Paralysis, Hypnagogic/Hypnopompic Hallucinations, and Cataplexy-like Symptoms
5.3 Complex Sleep Behaviors
5.4 Patients with Compromised Respiratory Function
5.5 Worsening of Depression/Suicidal Ideation
5.6 Need to Evaluate for Co-morbid Diagnoses
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Drugs Having Clinically Important Interactions with DAYVIGO
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
8.8 Patients with Compromised Respiratory Function
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
9.2 Abuse
9.3 Dependence
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Controlled Clinical Studies
14.2 Special Safety Studies
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
DAYVIGO is indicated for the treatment of adult patients with insomnia, characterized by difficulties with sleep onset and/or sleep maintenance [see Clinical Studies (14.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
The recommended dosage of DAYVIGO is 5 mg taken no more than once per night, immediately before going to bed, with at least 7 hours remaining before the planned time of awakening. The dose may be increased to the maximum recommended dose of 10 mg based on clinical response and tolerability. Time to sleep onset may be delayed if taken with or soon after a meal [see Clinical Pharmacology (12.3)].
2.2 Dosage Recommendations for Concomitant Use with CYP3A Inhibitors or CYP3A Inducers
Co-administration with Strong or Moderate CYP3A Inhibitors
Avoid concomitant use of DAYVIGO with strong or moderate CYP3A inhibitors [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Co-administration with Weak CYP3A Inhibitors
The maximum recommended dosage of DAYVIGO is 5 mg no more than once per night when co-administered with weak CYP3A inhibitors [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Co-administration with Strong or Moderate CYP3A Inducers
Avoid concomitant use of DAYVIGO with strong or moderate CYP3A inducers [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
2.3 Dosage Recommendations for Patients with Hepatic Impairment
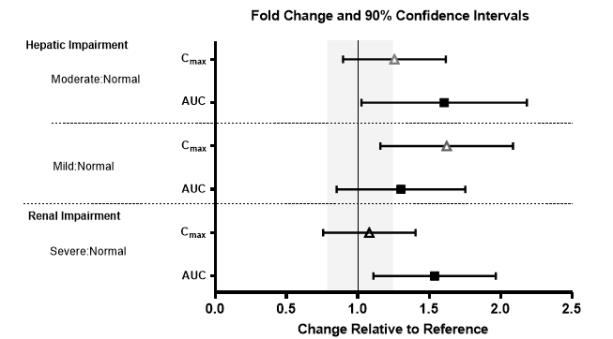
The maximum recommended dose of DAYVIGO is 5 mg no more than once per night in patients with moderate hepatic impairment [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
DAYVIGO is not recommended in patients with severe hepatic impairment [see Use in Specific Populations (8.7)].
-
3 DOSAGE FORMS AND STRENGTHS
DAYVIGO (lemborexant) tablets are available as:
- 5 mg tablets: pale yellow, round, biconvex, film-coated tablets, and debossed with "5" on one side and "LЄM" on the other side.
- 10 mg tablets: orange, round, biconvex, film-coated tablets, and debossed with "10" on one side and "LЄM" on the other side.
- 5 mg tablets: pale yellow, round, biconvex, film-coated tablets, and debossed with "5" on one side and "LЄM" on the other side.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 CNS Depressant Effects and Daytime Impairment
DAYVIGO is a central nervous system (CNS) depressant that can impair daytime wakefulness even when used as prescribed. CNS depressant effects may persist in some patients for up to several days after discontinuing DAYVIGO. Prescribers should advise patients about the potential for next-day somnolence.
Driving ability was impaired in some subjects taking DAYVIGO 10 mg [see Clinical Studies (14.2)]. The risk of daytime impairment is increased if DAYVIGO is taken with less than a full night of sleep remaining or if a higher than recommended dose is taken [see Dosage and Administration (2.1)]. If DAYVIGO is taken in these circumstances, patients should be cautioned against driving and other activities requiring complete mental alertness.
Co-administration with other CNS depressants (e.g., benzodiazepines, opioids, tricyclic antidepressants, alcohol) increases the risk of CNS depression, which can cause daytime impairment. Dosage adjustments of DAYVIGO and of concomitant CNS depressants may be necessary when administered together because of potentially additive effects. The use of DAYVIGO with other drugs to treat insomnia is not recommended. Patients should be advised not to consume alcohol in combination with DAYVIGO because of additive effects [see Drug Interactions (7.1)].
Because DAYVIGO can cause drowsiness, patients, particularly the elderly, are at a higher risk of falls.
5.2 Sleep Paralysis, Hypnagogic/Hypnopompic Hallucinations, and Cataplexy-like Symptoms
Sleep paralysis, an inability to move or speak for up to several minutes during sleep-wake transitions, and hypnagogic/hypnopompic hallucinations, including vivid and disturbing perceptions, can occur with the use of DAYVIGO. Prescribers should explain the nature of these events to patients when prescribing DAYVIGO.
Symptoms similar to mild cataplexy can occur with DAYVIGO. Such symptoms can include periods of leg weakness lasting from seconds to a few minutes, can occur either at night or during the day, and may not be associated with an identified triggering event (e.g., laughter or surprise).
5.3 Complex Sleep Behaviors
Complex sleep behaviors, including sleep-walking, sleep-driving, and engaging in other activities while not fully awake (e.g., preparing and eating food, making phone calls, having sex), have been reported to occur with the use of hypnotics such as DAYVIGO. These events can occur in hypnotic-naïve as well as in hypnotic-experienced persons. Patients usually do not remember these events. Complex sleep behaviors may occur following the first or any subsequent use of DAYVIGO, with or without the concomitant use of alcohol and other CNS depressants [see Drug Interactions (7.1)]. Discontinue DAYVIGO immediately if a patient experiences a complex sleep behavior.
5.4 Patients with Compromised Respiratory Function
The effect of DAYVIGO on respiratory function should be considered if prescribed to patients with compromised respiratory function. DAYVIGO has not been studied in patients with moderate to severe obstructive sleep apnea (OSA) or in patients with chronic obstructive pulmonary disease (COPD) [see Use in Special Populations (8.8)].
5.5 Worsening of Depression/Suicidal Ideation
In clinical studies of DAYVIGO in patients with insomnia, the incidence of suicidal ideation or any suicidal behavior, as assessed by questionnaire, was higher in patients receiving DAYVIGO than in those receiving placebo (0.3% for DAYVIGO 10 mg, 0.4% for DAYVIGO 5 mg, and 0.2% for placebo).
In primarily depressed patients treated with hypnotics, worsening of depression and suicidal thoughts and actions (including completed suicides) have been reported. Suicidal tendencies may be present in such patients and protective measures may be required. Intentional overdose is more common in this group of patients; therefore, the lowest number of tablets that is feasible should be prescribed at any one time.
The emergence of any new behavioral sign or symptom of concern requires careful and immediate evaluation.
5.6 Need to Evaluate for Co-morbid Diagnoses
Because sleep disturbances may be the presenting manifestation of a medical and/or psychiatric disorder, treatment of insomnia should be initiated only after careful evaluation of the patient. The failure of insomnia to remit after 7 to 10 days of treatment may indicate the presence of a primary psychiatric and/or medical illness that should be evaluated. Worsening of insomnia or the emergence of new cognitive or behavioral abnormalities may be the result of an unrecognized underlying psychiatric or medical disorder and can emerge during the course of treatment with sleep-promoting drugs such as DAYVIGO.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in detail in other sections of the labeling:
- CNS Depressant Effects and Daytime Impairment [see Warnings and Precautions (5.1)]
- Sleep Paralysis, Hypnagogic/Hypnopompic Hallucinations, and Cataplexy-like Symptoms [see Warnings and Precautions (5.2)]
- Complex Sleep Behaviors [see Warnings and Precautions (5.3)]
- Patients with Compromised Respiratory Function [see Warnings and Precautions (5.4)]
- Worsening of Depression/Suicidal Ideation [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of DAYVIGO was evaluated in 1418 adult patients with insomnia disorder (age 18 to 88 years) from two controlled efficacy trials (Study 1 and Study 2). Study 1 was a 6-month placebo-controlled trial assessing DAYVIGO 5 or 10 mg once nightly, followed by a 6-month parallel-group extension period in which patients initially treated with DAYVIGO continued on the same dose, and patients who received placebo were re-randomized to receive DAYVIGO 5 or 10 mg once nightly. In Study 1, 434 patients were treated with DAYVIGO for one year. Study 2 was a 30-day placebo- and active-controlled trial assessing DAYVIGO 5 or 10 mg once nightly.
Adverse Reactions Resulting in Discontinuation of Treatment
The frequencies of discontinuation due to adverse reactions in Study 1 (the first 30 days) and Study 2 were 2.6% and 1.4% for patients treated with 10 mg and 5 mg DAYVIGO, respectively, compared to 1.5% for patients in the placebo group. The most common adverse reactions leading to discontinuation of DAYVIGO were somnolence (1.0% for 10 mg, 0.7% for 5 mg, and 0.4% for placebo) and nightmares (0.3% for 10 mg, 0.3% for 5 mg, and 0% for placebo).
The frequencies of discontinuation due to adverse reactions in the 6-month placebo-controlled period of Study 1 were 8.3% and 4.1% for patients treated with DAYVIGO 10 mg and 5 mg, respectively, compared to 3.8% for patients in the placebo group. The most common reasons for discontinuation of DAYVIGO and occurring in more than one patient within a treatment arm were somnolence (2.9% for 10 mg, 1.0% for 5 mg, and 0.6% for placebo), nightmares (1.3% for 10 mg, 0.3% for 5 mg, and 0% for placebo), and palpitations (0.6% for 10 mg, 0% for 5 mg, and 0% for placebo).
Most Common Adverse Reactions
The most common adverse reaction (reported in 5% or more of patients treated with DAYVIGO and at least twice the rate of placebo) in Study 1 (the first 30 days) and Study 2 was somnolence (10% for DAYVIGO 10 mg, 7% for DAYVIGO 5 mg, and 1% for placebo).
Table 1 presents the adverse reactions based on the pooled data from the first 30 days of Study 1 (6-month controlled efficacy trial) and Study 2 (1-month controlled efficacy trial) where the incidence was ≥2% in DAYVIGO-treated patients and greater than in placebo-treated patients.
Table 1: Adverse Reactions Reported in ≥2% of DAYVIGO-Treated Patients and at a Greater Frequency than Placebo-Treated Patients During the First 30 Days of Study 1 and Study 2 DAYVIGO Placebo 5 mg 10 mg n=528 n=580 n=582 (%) (%) (%) Somnolence or fatigue* 1.3 6.9 9.6 Headache 3.4 5.9 4.5 Nightmare or abnormal dreams 0.9 0.9 2.2 *Combines preferred terms somnolence, lethargy, fatigue, sluggishness
Other Adverse Reactions Observed During Clinical Trials (Studies 1 and 2)
Other adverse reactions of <2% incidence but greater than placebo are shown below. The following list does not include adverse reactions 1) for which a drug cause was remote, 2) that were so general to be uninformative, or 3) that were not considered to have clinically significant implications.
- Sleep paralysis was reported in 1.6% and 1.3% of patients receiving DAYVIGO 10 mg and 5 mg, respectively, compared to no reports for placebo. Hypnagogic hallucinations were reported in 0.7% and 0.1% of patients receiving DAYVIGO 10 mg and 5 mg, respectively, compared to no reports for placebo [see Warnings and Precautions (5.2)].
- Two events of complex sleep behavior were reported, both in patients receiving DAYVIGO 10 mg [see Warnings and Precautions (5.3)].
- CNS Depressant Effects and Daytime Impairment [see Warnings and Precautions (5.1)]
-
7 DRUG INTERACTIONS
7.1 Drugs Having Clinically Important Interactions with DAYVIGO
Table 2: Clinically Important Drug Interactions with DAYVIGO Effect of Other Drugs on DAYVIGO Strong, Moderate, and Weak CYP3A Inhibitors Clinical Impact: Concomitant use with a strong, moderate, or weak CYP3A inhibitor increases lemborexant AUC and Cmax which may increase the risk of DAYVIGO adverse reactions [see Clinical Pharmacology (12.3)]. Intervention: Avoid concomitant use of DAYVIGO with strong or moderate CYP3A inhibitors [see Dosage and Administration (2.2)].
The maximum recommended dose of DAYVIGO with weak CYP3A inhibitors is 5 mg [see Dosage and Administration (2.2)].Examples: Strong CYP3A inhibitors: itraconazole, clarithromycin
Moderate CYP3A inhibitors: fluconazole, verapamil
Weak CYP3A inhibitors: chlorzoxazone, ranitidineStrong and Moderate CYP3A Inducers Clinical Impact: Concomitant use with a strong or moderate CYP3A inducer decreases lemborexant exposure, which may reduce DAYVIGO efficacy [see Clinical Pharmacology (12.3)]. Intervention: Avoid concomitant use of DAYVIGO with strong or moderate CYP3A inducers [see Dosage and Administration (2.2)]. Examples: Strong CYP3A inducers: rifampin, carbamazepine, St. John’s wort
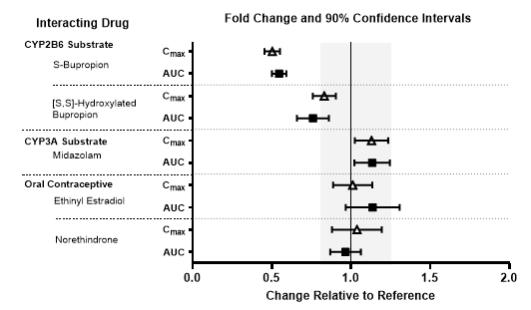
Moderate CYP3A inducers: bosentan, efavirenz, etravirine, modafinilAlcohol Clinical Impact: Concomitant use of alcohol increases lemborexant Cmax and AUC. Co-administration of DAYVIGO with alcohol produced a numerically greater negative impact on postural stability and memory as compared with alcohol alone when assessed near the tmax of DAYVIGO (2 hours post-dose) [see Clinical Pharmacology (12.2)]. Intervention: Avoid alcohol consumption with DAYVIGO [see Warnings and Precautions (5.1)]. Effect of DAYVIGO on Other Drugs CYP2B6 Substrates Clinical Impact: Concomitant use of DAYVIGO decreases the AUC of drugs that are CYP2B6 substrates, which may result in reduced efficacy for these concomitant medications [see Clinical Pharmacology (12.3)]. Intervention: Patients receiving DAYVIGO and CYP2B6 substrates concurrently should be monitored for adequate clinical response. Increasing the doses of CY2B6 substrates may be considered as needed. Examples: Bupropion, methadone -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women who are exposed to DAYVIGO during pregnancy. Healthcare providers are encouraged to register patients in the DAYVIGO pregnancy registry by calling 1-888-274-2378.
Risk Summary
There are no available data on DAYVIGO use in pregnant women to evaluate for drug-associated risks of major birth defects, miscarriage or adverse maternal or fetal outcomes.
In animal reproduction studies, oral administration of lemborexant to pregnant rats and rabbits during the period of organogenesis caused toxicities only at high multiples of the human exposure at the maximum recommended human dose (MRHD) based on AUC. The no observed adverse effect levels (NOAEL) are approximately >100 and 23 times the MRHD based on AUC in rats and rabbits, respectively. Similarly, oral administration of lemborexant to pregnant and lactating rats caused toxicities only at high multiples of the human exposure at the MRHD based on AUC. The NOAEL is 93 times the MRHD based on AUC [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies are 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Lemborexant was administered orally to pregnant rats during the period of organogenesis in 2 studies at doses of 60, 200, and 600 mg/kg/day or 20, 60, and 200 mg/kg/day, which are approximately 6 to >300 times the MRHD based on AUC. Lemborexant caused maternal toxicity, manifested by decreased body weight and food consumption, decreased mean fetal body weight, an increased number of dead fetuses, and skeletal, external and visceral malformations (omphalocele, cleft palate, and membranous ventricular septal defect) at >300 times the MRHD based on AUC. The NOAEL of 200 mg/kg/day is approximately 143 times the MRHD based on AUC.
Lemborexant was administered orally to pregnant rabbits during the period of organogenesis at doses of 10, 30, and 100 mg/kg/day, which are approximately 7 to 139 times the MRHD based on AUC. Lemborexant caused maternal toxicity that consisted of decreased body weight and food consumption and a higher incidence of skeletal variations (presence of cervical ribs and supernumerary lung lobes) at approximately 139 times the MRHD based on AUC. The NOAEL of 30 mg/kg/day is approximately 23 times the MRHD based on AUC.
Lemborexant was administered orally to pregnant rats during pregnancy and lactation at doses of 30, 100, and 300 mg/kg/day, which are approximately 15 to 206 times the MRHD based on AUC. Lemborexant caused maternal toxicity that consisted of decreased body weight and food consumption and toxicity to offspring consisting of decreased pup body weights, decreased femur length, and decreased acoustic startle responses at 206 times the MRHD based on AUC. The NOAEL of 100 mg/kg/day is approximately 93 times the MRHD based on AUC.
8.2 Lactation
Risk Summary
There are no data on the presence of lemborexant in human milk, the effects on the breastfed infant, or the effects on milk production. Lemborexant and its metabolites are present in the milk of lactating rats. When a drug is present in animal milk, it is likely that the drug will be present in human milk. Infants exposed to DAYVIGO through breastmilk should be monitored for excessive sedation. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for DAYVIGO and any potential adverse effects on the breastfed infant from DAYVIGO or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of DAYVIGO have not been established in pediatric patients.
8.5 Geriatric Use
Of the total number of patients treated with DAYVIGO (n=1418) in controlled Phase 3 studies, 491 patients were 65 years and over, and 87 patients were 75 years and over. Overall, efficacy results for patients <65 years of age were similar compared to patients ≥65 years.
In a pooled analysis of Study 1 (the first 30 days) and Study 2, the incidence of somnolence in patients ≥65 years with DAYVIGO 10 mg was higher (9.8%) compared to 7.7% in patients <65 years. The incidence of somnolence with DAYVIGO 5 mg was similar in patients ≥65 years (4.9%) and <65 years (5.1%). The incidence of somnolence in patients treated with placebo was 2% or less regardless of age [see Clinical Studies (14.2)]. Because DAYVIGO can increase somnolence and drowsiness, patients, particularly the elderly, are at a higher risk of falls [see Warning and Precautions (5.1)]. Exercise caution when using doses higher than 5 mg in patients ≥65 years old.
8.6 Renal Impairment
No dose adjustment is required in patients with mild, moderate, or severe renal impairment.
DAYVIGO exposure (AUC) was increased in patients with severe renal impairment. Patients with severe renal impairment may experience an increased risk of somnolence [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
DAYVIGO has not been studied in patients with severe hepatic impairment. Use in this population is not recommended [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)].
DAYVIGO exposure (AUC and Cmax) and terminal half-life were increased in patients with moderate hepatic impairment (Child-Pugh class B). Dosage adjustment is recommended in patients with moderate hepatic impairment (Child-Pugh class B) [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)].
DAYVIGO exposure (AUC) was increased in patients with mild hepatic impairment (Child-Pugh class A), but the terminal half-life was not changed. Patients with mild hepatic impairment may experience an increased risk of somnolence [see Clinical Pharmacology (12.3)].
8.8 Patients with Compromised Respiratory Function
In a study of patients with mild OSA (apnea-hypopnea index <15 events per hour of sleep), DAYVIGO did not increase the frequency of apneic events or cause oxygen desaturation.
DAYVIGO has not been studied in patients with COPD or moderate to severe OSA. Clinically meaningful respiratory effects of DAYVIGO in COPD or moderate to severe OSA cannot be excluded [see Warnings and Precautions (5.4)].
-
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
DAYVIGO contains lemborexant. (Controlled substance schedule to be determined after review by the Drug Enforcement Administration).
9.2 Abuse
Abuse is the intentional, non-therapeutic use of a drug, even once, for its desirable psychological or physiological effects. In a human abuse potential study conducted in recreational sedative abusers (n=29), lemborexant 10 mg, 20 mg (two times the maximum recommended dose), and 30 mg (three times the maximum recommended dose) produced responses on positive subjective measures such as “Drug Liking,” “Overall Drug Liking,” “Take Drug Again,” and “Good Drug Effects” that were statistically similar to those produced by the sedatives zolpidem (30 mg) and suvorexant (40 mg), and statistically greater than the responses on these measures that were produced by placebo. Because individuals with a history of abuse or addiction to alcohol or other drugs may be at increased risk for abuse and addiction to DAYVIGO, follow such patients carefully.
9.3 Dependence
Physical dependence is a state that develops as a result of physiological adaptation in response to repeated drug use, manifested by withdrawal signs and symptoms after abrupt discontinuation or a significant dose reduction of a drug. In animal studies and clinical trials evaluating physical dependence, chronic administration of lemborexant did not produce withdrawal signs or symptoms upon drug discontinuation. This suggests that lemborexant does not produce physical dependence.
-
10 OVERDOSAGE
There is limited clinical experience with DAYVIGO overdose. In clinical pharmacology studies, healthy patients who were administered multiple doses of up to 75 mg (7.5 times the maximum recommended dose) of DAYVIGO showed dose-dependent increases in the frequency of somnolence.
There is no available specific antidote to an overdose of DAYVIGO. In the event of overdose, standard medical practice for the management of any overdose should be used. In managing overdose, provide supportive care, including close medical supervision and monitoring and consider the possibility of multiple drug involvement. Consult a Certified Poison Control Center for the most up to date information on the management of overdosage (1-800-222-1222 or www.poison.org).
The value of dialysis in the treatment of overdosage has not been determined with lemborexant. As lemborexant is highly protein-bound, hemodialysis is not expected to contribute to elimination of lemborexant.
-
11 DESCRIPTION
DAYVIGO contains lemborexant, an orexin receptor antagonist. The chemical name of lemborexant is (1R,2S)-2-{[(2,4-dimethylpyrimidin-5-yl)oxy]methyl}-2-(3-fluorophenyl)-N-(5-fluoropyridin-2-yl) cyclopropanecarboxamide. The molecular formula is C22H20F2N4O2. The molecular weight is 410.42.
The structural formula is:
![The structural formula for DAYVIGO contains lemborexant, an orexin receptor antagonist. The chemical name of lemborexant is (1R,2S)-2-{[(2,4-dimethylpyrimidin-5-yl)oxy]methyl}-2-(3-fluorophenyl)-N-(5-fluoropyridin-2-yl) cyclopropanecarboxamide. The molecular formula is C22H20F2N4O2. The molecular weight is 410.42.](https://fda.report/DailyMed/7074cb65-77b3-45d2-8e8d-da8dc0f70bfd/dayvigo-01.jpg)
Lemborexant is a white to off-white powder that is practically insoluble in water.
DAYVIGO tablets are intended for oral administration. Each film coated tablet contains 5 mg or 10 mg of lemborexant. The inactive ingredients are: hydroxypropyl cellulose, lactose monohydrate, low-substituted hydroxypropyl cellulose, and magnesium stearate.
In addition, the film coating contains the following inactive ingredients: hypromellose 2910, polyethylene glycol 8000, talc, titanium dioxide, and either (a) ferric oxide yellow for the 5 mg tablet; or, (b) both ferric oxide yellow and ferric oxide red for the 10 mg tablet.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism of action of lemborexant in the treatment of insomnia is presumed to be through antagonism of orexin receptors. The orexin neuropeptide signaling system plays a role in wakefulness. Blocking the binding of wake-promoting neuropeptides orexin A and orexin B to receptors OX1R and OX2R is thought to suppress wake drive.
12.2 Pharmacodynamics
Lemborexant binds to orexin receptors OX1R and OX2R and acts as a competitive antagonist (IC50 values of 6.1 nM and 2.6 nM, respectively). A major metabolite of lemborexant, M10, binds with comparable affinity as the parent drug to orexin receptors OX1R and OX2R (IC50 values of 4.2 nM and 2.9 nM), respectively.
Cardiac Electrophysiology
In a concentration-QTcF analysis using the data from two randomized, double-blind, placebo-controlled, multiple ascending dose studies in healthy subjects, lemborexant does not prolong the QTcF interval to any clinically relevant extent at a dose 5 times the maximum recommended dose.
Drug Interactions
Lemborexant co-administered with alcohol produced a numerically greater negative impact on postural stability and memory as compared with alcohol alone at approximately 2 hours post-dose [see Drug Interactions 7.1].
12.3 Pharmacokinetics
Following single doses of lemborexant 2.5 to 75 mg, geometric mean Cmax and AUC0-24h increased slightly less than in proportion to dose. The extent of accumulation of lemborexant at steady-state is 1.5- to 3-fold across this dose range.
Absorption
The time to peak concentration (tmax) of lemborexant is approximately 1 to 3 hours.
Effect of Food
Lemborexant Cmax decreased by 23%, AUC0-inf increased by 18%, and tmax was delayed by 2 hours following administration of a high-fat and high-calorie meal (containing approximately 150, 250, and 500 to 600 calories from protein, carbohydrate, and fat, respectively).
Distribution
The volume of distribution of lemborexant is 1970 L. Protein binding of lemborexant is approximately 94% in vitro. The blood to plasma concentration ratio of lemborexant is 0.65.
Elimination
Metabolism
Lemborexant is primarily metabolized by CYP3A4, and to a lesser extent by CYP3A5. The major circulating metabolite is M10.
Excretion
Following administration of an oral dose, 57.4% of the dose was recovered in the feces and 29.1% in the urine (<1% as unchanged). The effective half-life for lemborexant 5 mg and 10 mg is 17 and 19 hours, respectively.
Specific Populations
No clinically significant differences in the pharmacokinetics of lemborexant were observed based on age, sex, race/ethnicity, or body mass index. No studies have been conducted to investigate the pharmacokinetics of lemborexant in pediatric patients. Exposures of lemborexant in patients with hepatic and renal impairment are summarized in Figure 1.
Figure 1. Effects of Hepatic and Renal Impairment on Lemborexant Pharmacokinetics
Drug Interaction Studies
The effects of other drugs on the exposures of lemborexant are summarized in Figure 2. The effects of lemborexant on the exposures of other drugs are summarized in Figure 3.
Physiologically-based pharmacokinetic (PBPK) modeling predicted that concomitant use of weak CYP3A inhibitors increased lemborexant exposure by less than 2-fold. Based on these results, drug interactions between lemborexant and strong CYP3A inducers, strong CYP3A inhibitors, moderate CYP3A inhibitors, and CYP2B6 substrates are clinically significant.
Figure 2. Effects of Co-administered Drugs on the Pharmacokinetics of Lemborexant 10 mg
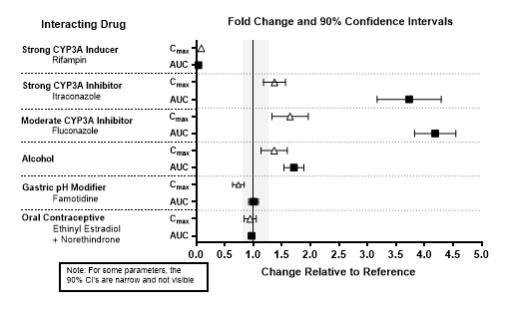
Figure 3. Effects of Lemborexant 10 mg on the Pharmacokinetics of Co-Administered Drugs
In Vitro Studies
In vitro metabolism studies demonstrated that lemborexant and M10 have the potential to induce CYP3A and the weak potential to inhibit CYP3A and induce CYP2B6. Lemborexant and M10 do not inhibit other CYP isoforms (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2D6, CYP2A6, CYP2C19, and CYP2E1) or transporters (P-gp, BCRP, BSEP, OAT1, OAT3, OATP1B1, OATP1B3, OCT1, OCT2, MATE1, and MATE2-K). Lemborexant is a potential poor substrate of P-gp, but M10 is a substrate of P-gp. Lemborexant and M10 are not substrates of BCRP, OATP1B1, or OATP1B3.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Lemborexant did not increase the incidence of tumors in rats treated for 2 years at oral doses of 30, 100, and 300 mg/kg/day (males) and 10, 30, and 100 mg/kg/day (females), which are >80 times the MRHD based on AUC. Lemborexant did not increase the incidence of tumors in Tg ras H2 mice treated for 26 weeks at oral doses of 50, 150, and 500 mg/kg/day.
Mutagenesis
Lemborexant was not mutagenic in the in vitro bacterial reverse mutation (Ames) assay or in the in vitro mouse lymphoma thymidine kinase assay, and was not clastogenic in the in vivo rat micronucleus assay.
Impairment of Fertility
Lemborexant was orally administered to female rats at doses of 30, 100, or 1000 mg/kg/day prior to and throughout mating and continuing to gestation Day 6. These doses are approximately 12 to >500 times the MRHD based on AUC. Irregular estrous cycles and decreased pregnancy rate were observed at 60 times the MRHD based on AUC, and decreased numbers of corpora lutea, implantations, and live embryos were observed at >500 times the MRHD based on AUC. The exposure at the NOAEL of 30 mg/kg/day is approximately 12 times the MRHD based on AUC. Lemborexant did not affect fertility when orally administered to male rats at doses of 30, 100, or 1000 mg/kg/day prior to and throughout mating; the highest dose is approximately 138 times the MRHD based on AUC.
-
14 CLINICAL STUDIES
14.1 Controlled Clinical Studies
DAYVIGO was evaluated in two clinical trials in patients with insomnia characterized by difficulties with sleep onset and/or sleep maintenance (Study 1, NCT02952820 and Study 2, NCT02783729).
Study 1 was a 6-month, randomized, double-blind, placebo-controlled, multi-center trial in adult patients age 18 or older who met DSM-5 criteria for insomnia disorder. Patients were randomized to placebo (n=325), DAYVIGO 5 mg (n=323), or DAYVIGO 10 mg (n=323) once nightly. The primary efficacy endpoint was the mean change from baseline to end of treatment at 6 months for log-transformed patient-reported (subjective) sleep onset latency (sSOL), defined as the estimated minutes from the time that the patient attempted to sleep until sleep onset. Pre-specified secondary efficacy endpoints for sleep maintenance were change from baseline to end of treatment at 6 months for patient-reported sleep efficiency (sSEF) and wake after sleep onset (sWASO). sSEF is defined as the proportion of time spent asleep per time in bed. sWASO is defined as the minutes of wake from the onset of sleep until wake time. The primary and pre-specified secondary efficacy endpoints were measured by sleep diary.
The demographic characteristics of patients in Study 1 were similar across the treatment arms. Patients had a median age of 55 years (range 18 to 88) and were 68% female, 72% White, 8% Black or African American, 17% Japanese, and 3.5% other; 28% were elderly (≥65 years).
Examination of subgroups by age, race, and sex did not suggest differences in response to DAYVIGO. In Study 1, DAYVIGO 5 mg and 10 mg demonstrated statistically significant superiority on the primary efficacy measure, sSOL, compared to placebo (Table 3). DAYVIGO 5 mg and 10 mg also showed statistically significant superiority in sSEF and sWASO.
Table 3: Primary and Secondary Efficacy Results for Change from Baseline in Sleep Onset and Sleep Maintenance at 6 Months in Patients with Insomnia (Study 1)
Endpoint Treatment Group Number of Patients
ITTBaseline Meana
(SD)Month 6
LS Meana
(SE)Treatment Effect
(95% CI)Sleep Onset
sSOL
(minutes)DAYVIGO 5 mg* 316 43.0 (31.5) 20.0 (1.1) 0.7 (0.6, 0.8) DAYVIGO 10 mg* 315 45.0 (33.4) 19.2 (1.1) 0.7 (0.6, 0.8) Placebo 318 45.0 (31.8) 27.3 (1.4) (ratio vs placebo)b Sleep Maintenance
sSEF
(%)DAYVIGO 5 mg* 316 63.1 (18.2) 75.9 (0.9) 4.5 (2.2, 6.9) DAYVIGO 10 mg* 315 62.0 (17.2) 75.9 (0.9) 4.7 (2.4, 7.0) Placebo 318 61.3 (17.8) 71.4 (0.8) (%)c Sleep Maintenance
sWASO
(minutes)DAYVIGO 5 mg* 316 132.8 (82.5) 87.9 (3.7) -17.5 (-27.3, -7.6) DAYVIGO 10 mg* 315 136.8 (87.4) 92.7 (3.7) -12.7 (-22.4, -3.0) Placebo 318 132.5 (80.2) 105.3 (3.6) (minutes)c ITT (intention to treat); sSOL (subjective sleep onset latency); SD (standard deviation); LS (least squares); SE (standard error); CI (unadjusted confidence interval); sSEF (subjective sleep efficiency); sWASO (subjective wake after sleep onset)
a For the sleep onset sSOL endpoint, the mean refers to geometric mean, which was used due to the approximately log normal distribution of the outcomes; SD for the geometric mean is calculated as GM*SD (log transformed sSOL); SE for the least squares geometric mean is calculated in the same way as the SD.
b For the sleep onset sSOL endpoint, treatment effect refers to the ratio of [Month 6 sSOL / Baseline sSOL] for DAYVIGO versus placebo, such that a smaller ratio corresponds to a greater improvement.
c Treatment effect refers to the treatment difference between DAYVIGO versus placebo, such that a larger value for sSEF and smaller value for sWASO correspond to greater improvement.
* Doses that were statistically significantly superior (p<0.05) to placebo after multiplicity adjustment.Study 2 was a 1-month, randomized, double-blind, placebo- and active-controlled, multi-center, parallel-group clinical trial in adult female patients age 55 and older and male patients 65 years and older who met DSM-5 criteria for insomnia disorder. Patients were randomized to placebo (n=208), DAYVIGO 5 mg (n=266) or 10 mg (n=269), or active comparator (n=263) once nightly.
The primary efficacy endpoint was the mean change in log-transformed latency to persistent sleep (LPS) from baseline to end of treatment (Days 29/30), as measured by overnight polysomnography (PSG) monitoring. LPS was defined as the number of minutes from lights off to the first 10 consecutive minutes of non-wakefulness. The pre-specified secondary efficacy endpoints in Study 2 were the mean change from baseline to end of treatment (Days 29/30) in sleep efficiency (SEF) and wake after sleep onset (WASO) measured by PSG.
The demographic and baseline characteristics of patients in Study 2 were similar across the treatment arms. Patients had a median age of 63 years (range 55 to 88) and were 86% female, 72% White, 25% Black or African American, and 2% other; 45% were elderly (≥65 years).
In Study 2, DAYVIGO 5 mg and 10 mg demonstrated statistically significant superiority on the primary efficacy measure, LPS, compared to placebo (Table 4). DAYVIGO 5 mg and 10 mg demonstrated statistically significant improvement in SEF and WASO compared to placebo.
Table 4: Primary and Secondary Efficacy Results for Change from Baseline in Sleep Onset and Sleep Maintenance at 1 Month in Patients with Insomnia (Study 2)
Endpoint Treatment Group Number of Patients
ITTBaseline Meana
(SD)Day 29/30
LS Meana
(SE)Treatment Effect
(95% CI)
Sleep Onset
LPS
(minutes)DAYVIGO 5 mg* 266 33.0 (27.2) 15.5 (0.8) 0.8 (0.7, 0.9) DAYVIGO 10 mg* 269 33.3 (27.2) 14.5 (0.7) 0.7 (0.6, 0.8) Placebo 208 33.6 (25.9) 20.0 (1.1) (ratio vs. placebo)b Sleep Maintenance
SEF
(%)DAYVIGO 5 mg* 266 68.4 (11.3) 80.7 (0.5) 7.1 (5.6, 8.5) DAYVIGO 10 mg* 269 67.8 (10.8) 82.7 (0.5) 8.0 (6.6, 9.5) Placebo 208 68.9 (9.6) 74.6 (0.6) (%)c Sleep Maintenance
WASO
(minutes)DAYVIGO 5 mg* 266 113.4 (39.0) 68.3 (2.2) -24.0 (-30.0, -18.0) DAYVIGO 10 mg* 269 114.8 (40.0) 66.9 (2.2) -25.3 (-31.4, -19.3) Placebo 208 111.7 (37.2) 92.2 (2.5) (minutes)c ITT (intention to treat); LPS (latency to persistent sleep); SD (standard deviation); LS (least squares); SE (standard error); CI (unadjusted confidence interval); SEF (sleep efficiency); WASO (wake after sleep onset)
a For the sleep onset LPS endpoint, the mean refers to geometric mean, which was used due to the approximately log normal distribution of the outcomes; SD for the geometric mean is calculated as GM*SD (log transformed LPS); SE for the least squares geometric mean is calculated in the same way as the SD.
b For the LPS endpoint, treatment effect refers to the ratio of [Day 29/30 LPS / Baseline LPS] for DAYVIGO versus placebo, such that a smaller ratio corresponds to a greater improvement.
c Treatment effect refers to the treatment difference between DAYVIGO versus placebo, such that a larger value for SEF and smaller value for WASO correspond to greater improvement.
* Doses that were statistically significantly superior (p<0.05) to placebo after multiplicity adjustment.The effects of DAYVIGO at the beginning of treatment were generally consistent with later timepoints.
14.2 Special Safety Studies
Middle of the Night Safety
The effect of DAYVIGO on middle of the night safety was evaluated in a randomized, placebo- and active-controlled trial in healthy female subjects ≥55 years or male subjects ≥65 years. Postural stability, the ability to awaken in response to a sound stimulus, and attention and memory were assessed following a scheduled awakening 4 hours after the start of the 8-hour time in bed. Postural stability was measured by assessing body sway using an ataxia meter. Nighttime dosing of DAYVIGO 5 mg and 10 mg resulted in impairment of balance (measured by body sway area) at 4 hours as compared to placebo.
The ability to awaken to sound in the middle of the night was assessed using an audiometer that delivered 1000 Hz tones up to 105 dB. There were no meaningful differences between DAYVIGO (5 mg or 10 mg) and placebo on ability to awaken to sound.
A computerized performance assessment battery was administered to assess attention and memory after middle of the night awakening (4 hours postdose) in subjects receiving DAYVIGO 5 mg or 10 mg. DAYVIGO was associated with dose-dependent worsening on measures of attention and memory as compared to placebo.
Patients should be cautioned about the potential for middle of the night postural instability, as well as attention and memory impairment.
Effects on Next-day Postural Stability and Memory
The effects of DAYVIGO on next day postural stability and memory were evaluated in two randomized, placebo- and active-controlled trials in healthy subjects and insomnia patients age 55 and older.
There were no meaningful differences between DAYVIGO (5 mg or 10 mg) and placebo on next-day postural stability or memory compared to placebo.
Effects on Driving
A randomized, double-blind, placebo- and active-controlled, four-period crossover study evaluated the effects of nighttime administration of DAYVIGO on next-morning driving performance approximately 9 hours after dosing in 24 healthy elderly subjects (≥65 years, median age 67 years; 14 men, 10 women) and 24 adult subjects (median age 49 years; 12 men, 12 women). The primary driving performance outcome measure was change in Standard Deviation of Lateral Position (SDLP). Testing was conducted after one night (a single dose) and after eight consecutive nights of treatment with DAYVIGO. Although DAYVIGO at doses of 5 mg and 10 mg did not cause statistically significant impairment in next-morning driving performance in adult or elderly subjects (compared with placebo), driving ability was impaired in some subjects taking 10 mg DAYVIGO.
Patients using the 10 mg dose should be cautioned about the potential for next-morning driving impairment because there is individual variation in sensitivity to DAYVIGO.
Rebound Insomnia
Rebound insomnia was assessed by comparing sleep diary-recorded sSOL and sWASO from the screening period to the two weeks following treatment discontinuation in both Studies 1 and 2. Analyses of group means and the proportion of patients with rebound insomnia suggest that DAYVIGO was not associated with rebound insomnia following treatment discontinuation.
Withdrawal Effects
In 12-month and 1-month controlled safety and efficacy trials (Studies 1 and 2, respectively), withdrawal effects were assessed by the Tyrer Benzodiazepine Withdrawal Symptom Questionnaire following discontinuation from study drug in patients who received DAYVIGO 5 mg or 10 mg. There was no evidence of withdrawal effects following DAYVIGO discontinuation at either dose.
-
16
HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
DAYVIGO tablets are available as:
- 5 mg, pale yellow, round, biconvex, film-coated tablets, and debossed with "5" on one side and "LЄM" on the other side.
NDC: 62856-405-30, bottle of 30 with child-resistant closure
NDC: 62856-405-90, bottle of 90 with child-resistant closure
- 10 mg, orange, round, biconvex, film-coated tablets, and debossed with "10" on one side and "LЄM" on the other side.
NDC: 62856-410-30, bottle of 30 with child-resistant closure
NDC: 62856-410-90, bottle of 90 with child-resistant closure
-
17
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Administration Instructions
Advise patients to take DAYVIGO only when preparing for or getting into bed and only if they can stay in bed for a full night (at least 7 hours) before being active again [see Dosage and Administration (2.1)].
Advise patients that the effect of DAYVIGO may be delayed if taken with or soon after a meal [see Dosage and Administration (2.1), Clinical Pharmacology (12.3)].
CNS Depressant Effects and Daytime Impairment
Advise patients that DAYVIGO can impair daytime wakefulness even when used as prescribed. The risk of daytime impairment is increased if DAYVIGO is taken with less than a full night of sleep remaining or if a higher than recommended dose is taken. If DAYVIGO is taken in these circumstances, caution patients against driving and other activities requiring complete mental alertness. Advise patients that increased drowsiness may increase the risk of falls in some patients [see Warnings and Precautions (5.1)].
Sleep Paralysis, Hypnagogic/Hypnopompic Hallucinations, and Cataplexy-Like Symptoms
Advise patients and their families that DAYVIGO may cause sleep paralysis, which is an inability to move or speak for several minutes during sleep-wake transitions, despite being aware of surroundings; hypnagogic/hypnopompic hallucinations, including vivid and disturbing perceptions; and symptoms similar to mild cataplexy [see Warnings and Precautions (5.2)].
Complex Sleep Behaviors
Instruct patients and their families that DAYVIGO may cause complex sleep behaviors, including sleep-walking, sleep-driving, preparing and eating food, making phone calls, or having sex while not being fully awake. Tell patients to discontinue DAYVIGO and notify their healthcare provider immediately if they develop any of these symptoms [see Warnings and Precautions (5.3)].
Worsening of Depression/Suicidal Ideation
Tell patients to report any worsening of depression or suicidal thoughts immediately [see Warnings and Precautions (5.4)].
Pregnancy
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to DAYVIGO during pregnancy [see Use in Specific Populations (8.1)].
Concomitant Medications
Ask patients about alcohol consumption, medicines they are taking, and drugs they may be taking without a prescription. Advise patients not to consume alcohol in combination with DAYVIGO [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Tolerance, Abuse, and Dependence
Tell patients not to increase the dose of DAYVIGO on their own, and to inform you if they believe the drug “does not work” [see Drug Abuse and Dependence (9)].
Distributed by:
Eisai Inc.
Woodcliff Lake, NJ 07677DAYVIGOTM is a trademark of Eisai R&D Management Co., Ltd. and is licensed to Eisai Inc.
© 2019 Eisai Inc.
-
MEDICATION GUIDE
MEDICATION GUIDE
DAYVIGOTM (daye-vi'-goe)
(lemborexant)
tablets, for oral use [pending controlled substance schedule]What is the most important information I should know about DAYVIGO?
DAYVIGO may cause serious side effects including:
-
Decreased awareness and alertness. The morning after you take DAYVIGO, your ability to drive safely and think clearly may be decreased. You may also have sleepiness during the day.
○ Do not take more DAYVIGO than prescribed.
○ Do not take DAYVIGO unless you are able to stay in bed for a full night (at least 7 hours) before you must be active again.
○ Take DAYVIGO right before going to bed.
What is DAYVIGO?
- DAYVIGO is a prescription medicine for adults who have trouble falling or staying asleep (insomnia).
- It is not known if DAYVIGO is safe and effective in children under the age of 18 years.
DAYVIGO is a federally controlled substance because it can be abused or cause dependence. Keep DAYVIGO in a safe place to prevent misuse and abuse. Selling or giving away DAYVIGO may harm others and is against the law. Tell your doctor if you have ever abused or have been dependent on alcohol, prescription medicines or street drugs. Who should not take DAYVIGO?
Do not take DAYVIGO if you fall asleep often at unexpected times (narcolepsy).Before taking DAYVIGO, tell your healthcare provider about all of your medical conditions, including if you:
- have a history of depression, mental illness, or suicidal thoughts.
- have a history of drug or alcohol abuse or addiction.
- have a history of a sudden onset of muscle weakness (cataplexy).
- have a history of daytime sleepiness.
- have lung problems or breathing problems, including sleep apnea.
- have liver problems.
- are pregnant or plan to become pregnant. It is not known if DAYVIGO can harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if DAYVIGO passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby during treatment with DAYVIGO.
- Taking DAYVIGO with certain other medicines can cause serious side effects. DAYVIGO may affect the way other medicines work and other medicines may affect the way DAYVIGO works.
-
Do not take DAYVIGO with other medicines that can make you sleepy unless your healthcare provider tells you to.
- Know the medicines you take. Keep a list of your medicines with you to show your healthcare provider and pharmacist each time you get a new medicine.
How should I take DAYVIGO? - Take DAYVIGO exactly as your healthcare provider tells you to take it.
- Only take DAYVIGO one time each night, right before going to bed.
- Only take DAYVIGO when you can stay in bed for a full night (at least 7 hours).
- DAYVIGO may take longer to work if you take it with or soon after a meal.
- Do not increase your dose of DAYVIGO without talking to your healthcare provider first. Call your healthcare provider if your insomnia (sleep problem) worsens or is not improved within 7 to 10 days. This may mean that there is another condition causing your sleep problem.
- If you take too much DAYVIGO, call your healthcare provider or go to the nearest hospital emergency room right away.
What should I avoid while taking DAYVIGO? -
Do not drink alcohol while taking DAYVIGO. It can increase your chances of getting serious side effects.
-
Do not drive, operate heavy machinery, do anything dangerous, or do other activities that require clear thinking if you take DAYVIGO and have had less than a full night of sleep (at least 7 hours) or if you have taken more DAYVIGO than prescribed by your healthcare provider.
- You may still feel drowsy the next day after taking DAYVIGO. Do not drive or do other dangerous activities until you feel fully awake.
What are the possible side effects of DAYVIGO? See “What is the most important information I should know about DAYVIGO?”
DAYVIGO may cause serious side effects, including:
-
temporary inability to move or talk (sleep paralysis) for up to several minutes while you are going to sleep or waking up.
-
temporary weakness in your legs that can happen during the day or at night.
-
complex sleep behaviors such as sleep-walking, sleep-driving, preparing and eating food, making phone calls, having sex or doing other activities while not fully awake that you may not remember the next morning. Call your healthcare provider right away if you experience a complex sleep behavior.
- worsening depression and suicidal thoughts have happened during treatment with DAYVIGO. Call your healthcare provider right away if you have any worsening depression or thoughts of suicide or dying.
These are not all of the possible side effects of DAYVIGO. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store DAYVIGO? - Store DAYVIGO at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep DAYVIGO and all medicines out of the reach of children.
General information about the safe and effective use of DAYVIGO.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use DAYVIGO for a condition for which it was not prescribed. Do not give DAYVIGO to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about DAYVIGO that is written for healthcare professionals.What are the ingredients in DAYVIGO?
Active ingredient: lemborexant
Inactive ingredients: hydroxypropyl cellulose, lactose monohydrate, low-substituted hydroxypropyl cellulose, and magnesium stearate. The tablet film coating contains: hypromellose 2910, polyethylene glycol 8000, talc, titanium dioxide, and either ferric oxide yellow for the 5 mg tablet; or both ferric oxide yellow and ferric oxide red for the 10 mg tablet.Distributed by:
Eisai Inc.
Woodcliff Lake, NJ 07677
DAYVIGOTM is a trademark of Eisai R&D Management Co., Ltd. and is licensed to Eisai Inc.
© 2019 Eisai Inc.
For more information, go to www.DAYVIGO.com or call 1-888-274-2378This Medication Guide has been approved by the U.S. Food and Drug Administration. Issued: 12/2019
-
Decreased awareness and alertness. The morning after you take DAYVIGO, your ability to drive safely and think clearly may be decreased. You may also have sleepiness during the day.
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
DAYVIGO
lemborexant tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 62856-405 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LEMBOREXANT (UNII: 0K5743G68X) (LEMBOREXANT - UNII:0K5743G68X) LEMBOREXANT 5 mg Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) LOW-SUBSTITUTED HYDROXYPROPYL CELLULOSE (11% HYDROXYPROPYL; 120000 MW) (UNII: NZ94SDL6WR) HYDROXYPROPYL CELLULOSE (45000 WAMW) (UNII: 8VAB711C5E) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TALC (UNII: 7SEV7J4R1U) POLYETHYLENE GLYCOL 8000 (UNII: Q662QK8M3B) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color YELLOW (pale yellow) Score no score Shape ROUND Size 7mm Flavor Imprint Code 5;LEM Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 62856-405-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 04/10/2020 2 NDC: 62856-405-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 04/10/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212028 04/10/2020 DAYVIGO
lemborexant tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 62856-410 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LEMBOREXANT (UNII: 0K5743G68X) (LEMBOREXANT - UNII:0K5743G68X) LEMBOREXANT 10 mg Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) LOW-SUBSTITUTED HYDROXYPROPYL CELLULOSE (11% HYDROXYPROPYL; 120000 MW) (UNII: NZ94SDL6WR) HYDROXYPROPYL CELLULOSE (45000 WAMW) (UNII: 8VAB711C5E) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TALC (UNII: 7SEV7J4R1U) POLYETHYLENE GLYCOL 8000 (UNII: Q662QK8M3B) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color ORANGE Score no score Shape ROUND Size 7mm Flavor Imprint Code 10;LEM Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 62856-410-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 04/10/2020 2 NDC: 62856-410-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 04/10/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212028 04/10/2020 Labeler - Eisai Inc. (831600833)




Trademark Results [DAYVIGO]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 DAYVIGO 88655847 not registered Live/Pending |
Eisai R&D Management Co., Ltd. 2019-10-15 |
 DAYVIGO 88058574 not registered Live/Pending |
Eisai R&D Management Co., Ltd. 2018-07-30 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.