ZERBAXA- ceftolozane and tazobactam injection, powder, lyophilized, for solution
ZERBAXA by
Drug Labeling and Warnings
ZERBAXA by is a Prescription medication manufactured, distributed, or labeled by Merck Sharp & Dohme Corp., Cubist Pharmaceuticals LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ZERBAXA safely and effectively. See full prescribing information for ZERBAXA.
ZERBAXA® (ceftolozane and tazobactam) for injection, for intravenous use
Initial U.S. Approval: 2014RECENT MAJOR CHANGES
INDICATIONS AND USAGE
ZERBAXA (ceftolozane and tazobactam) is a combination of ceftolozane, a cephalosporin antibacterial, and tazobactam, a beta-lactamase inhibitor, indicated in patients 18 years or older for the treatment of the following infections caused by designated susceptible microorganisms:
- Complicated Intra-abdominal Infections (cIAI), used in combination with metronidazole (1.1)
- Complicated Urinary Tract Infections (cUTI), Including Pyelonephritis (1.2)
- Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP) (1.3)
To reduce the development of drug-resistant bacteria and maintain the effectiveness of ZERBAXA and other antibacterial drugs, ZERBAXA should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria. (1.4)
DOSAGE AND ADMINISTRATION
- Administer all doses of ZERBAXA every 8 hours by intravenous infusion over 1 hour in patients 18 years or older. (2.1, 2.2)
- See Full Prescribing Information for instructions on the preparation of solutions. (2.3)
- For doses above 1.5 g, reconstitute a second vial in the same manner as the first one, withdraw an appropriate volume (per Table 3 in the Full Prescribing Information), and add to the same infusion bag. (2.3)
Recommended Dosage of ZERBAXA by Infection in Patients 18 years or older with Creatinine Clearance (CrCl) Greater than 50 mL/min (2.1) Infection Dose Duration of Treatment - * Used in conjunction with metronidazole 500 mg intravenously every 8 hours
Complicated Intra-abdominal Infections (cIAI)* 1.5 g 4-14 days Complicated Urinary Tract Infections (cUTI), Including Pyelonephritis 1.5 g 7 days Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP) 3 g 8-14 days Recommended Dosage of ZERBAXA in Patients 18 years or older with CrCl 50 mL/min or less (2.2) Estimated CrCl
(mL/min)*cIAI and cUTI, including pyelonephritis HABP/VABP - * CrCl estimated using Cockcroft-Gault formula
30 to 50 ZERBAXA 750 mg (500 mg and 250 mg) intravenously every 8 hours ZERBAXA 1.5 g (1 g and 0.5 g) intravenously every 8 hours 15 to 29 ZERBAXA 375 mg (250 mg and 125 mg) intravenously every 8 hours ZERBAXA 750 mg (500 mg and 250 mg) intravenously every 8 hours End-stage renal disease (ESRD) on hemodialysis (HD) A single loading dose of ZERBAXA 750 mg (500 mg and 250 mg) followed by a ZERBAXA 150 mg (100 mg and 50 mg) maintenance dose administered intravenously every 8 hours for the remainder of the treatment period (on hemodialysis days, administer the dose at the earliest possible time following completion of dialysis) A single loading dose of ZERBAXA 2.25 g (1.5 g and 0.75 g) followed by a ZERBAXA 450 mg (300 mg and 150 mg) maintenance dose administered every 8 hours for the remainder of the treatment period (on hemodialysis days, administer the dose at the earliest possible time following completion of dialysis) DOSAGE FORMS AND STRENGTHS
- ZERBAXA 1.5 g (ceftolozane and tazobactam) for injection supplied as a sterile powder for reconstitution in single-dose vials containing ceftolozane 1 g (equivalent to 1.147 g ceftolozane sulfate) and tazobactam 0.5 g (equivalent to 0.537 g tazobactam sodium). (3)
CONTRAINDICATIONS
- ZERBAXA is contraindicated in patients with known serious hypersensitivity to the components of ZERBAXA (ceftolozane and tazobactam), piperacillin/tazobactam, or other members of the beta-lactam class. (4)
WARNINGS AND PRECAUTIONS
- Decreased efficacy was observed in a Phase 3 cIAI trial in a subgroup of patients with baseline CrCl of 30 to 50 mL/min. Monitor CrCl at least daily in patients with changing renal function and adjust the dose of ZERBAXA accordingly. (5.1)
- Serious hypersensitivity (anaphylactic) reactions have been reported with beta-lactam antibacterial drugs. Exercise caution in patients with known hypersensitivity to beta-lactam antibacterial drugs. If an anaphylactic reaction to ZERBAXA occurs, discontinue the drug and institute appropriate therapy. (5.2)
- Clostridium difficile-associated diarrhea (CDAD) has been reported with nearly all systemic antibacterial agents, including ZERBAXA. Evaluate if diarrhea occurs. (5.3)
ADVERSE REACTIONS
The most common adverse reactions (≥5% in either cIAI or cUTI indication) are nausea, diarrhea, headache and pyrexia. The most common adverse reactions (≥5% in the HABP/VABP indication) are increase in hepatic transaminases, renal impairment/renal failure, and diarrhea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., at 1-877-888-4231 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
- Geriatrics: Higher incidence of adverse reactions was observed in patients aged 65 years and older. In a Phase 3 cIAI trial, cure rates were lower in patients 65 years and older. (8.5)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Complicated Intra-abdominal Infections
1.2 Complicated Urinary Tract Infections, Including Pyelonephritis
1.3 Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP)
1.4 Usage
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dosage Adjustments in Patients with Renal Impairment
2.3 Preparation of Solutions
2.4 Compatibility
2.5 Storage of Constituted Solutions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Decreased Efficacy in Patients with Baseline Creatinine Clearance of 30 to 50 mL/min
5.2 Hypersensitivity Reactions
5.3 Clostridium difficile-associated Diarrhea
5.4 Development of Drug-Resistant Bacteria
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Patients with Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Complicated Intra-abdominal Infections
14.2 Complicated Urinary Tract Infections, Including Pyelonephritis
14.3 Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP)
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Complicated Intra-abdominal Infections
ZERBAXA used in combination with metronidazole is indicated for the treatment of patients 18 years and older with complicated intra-abdominal infections (cIAI) caused by the following susceptible Gram-negative and Gram-positive microorganisms: Enterobacter cloacae, Escherichia coli, Klebsiella oxytoca, Klebsiella pneumoniae, Proteus mirabilis, Pseudomonas aeruginosa, Bacteroides fragilis, Streptococcus anginosus, Streptococcus constellatus, and Streptococcus salivarius.
1.2 Complicated Urinary Tract Infections, Including Pyelonephritis
ZERBAXA is indicated for the treatment of patients 18 years and older with complicated urinary tract infections (cUTI), including pyelonephritis, caused by the following susceptible Gram-negative microorganisms: Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, and Pseudomonas aeruginosa.
1.3 Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP)
ZERBAXA is indicated for the treatment of patients 18 years and older with hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia, caused by the following susceptible Gram-negative microorganisms: Enterobacter cloacae, Escherichia coli, Haemophilus influenzae, Klebsiella oxytoca, Klebsiella pneumoniae, Proteus mirabilis, Pseudomonas aeruginosa, and Serratia marcescens.
1.4 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of ZERBAXA and other antibacterial drugs, ZERBAXA should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of ZERBAXA for injection is 1.5 gram (g) (ceftolozane 1 g and tazobactam 0.5 g) for cIAI and cUTI and 3 g (ceftolozane 2 g and tazobactam 1 g) for HABP/VABP administered every 8 hours by intravenous infusion over 1 hour in patients 18 years or older and with a creatinine clearance (CrCl) greater than 50 mL/min. The duration of therapy should be guided by the severity and site of infection and the patient's clinical and bacteriological progress as shown in Table 1.
Table 1: Dosage of ZERBAXA by Infection in Patients with CrCl Greater than 50 mL/min Infection Dose Frequency Infusion Time (hours) Duration of Treatment - * Used in conjunction with metronidazole 500 mg intravenously every 8 hours
Complicated Intra-abdominal Infections* 1.5 g Every 8 Hours 1 4-14 days Complicated Urinary Tract Infections, Including Pyelonephritis 1.5 g Every 8 Hours 1 7 days Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP) 3 g Every 8 Hours 1 8-14 days 2.2 Dosage Adjustments in Patients with Renal Impairment
Dose adjustment is required for patients with CrCl 50 mL/min or less (Table 2). All doses of ZERBAXA are administered over 1 hour. For patients with changing renal function, monitor CrCl at least daily and adjust the dosage of ZERBAXA accordingly [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
Table 2: Dosage of ZERBAXA in Adult Patients with CrCl 50 mL/min or less Estimated CrCl
(mL/min)*Complicated Intra-abdominal Infections and Complicated Urinary Tract Infections, Including Pyelonephritis Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP) - * CrCl estimated using Cockcroft-Gault formula
30 to 50 750 mg (500 mg and 250 mg) intravenously every 8 hours 1.5 g (1 g and 0.5 g) intravenously every 8 hours 15 to 29 375 mg (250 mg and 125 mg) intravenously every 8 hours 750 mg (500 mg and 250 mg) intravenously every 8 hours End-stage renal disease (ESRD) on hemodialysis (HD) A single loading dose of 750 mg (500 mg and 250 mg) followed by a 150 mg (100 mg and 50 mg) maintenance dose administered every 8 hours for the remainder of the treatment period (on hemodialysis days, administer the dose at the earliest possible time following completion of dialysis) A single loading dose of 2.25 g (1.5 g and 0.75 g) followed by a 450 mg (300 mg and 150 mg) maintenance dose administered every 8 hours for the remainder of the treatment period (on hemodialysis days, administer the dose at the earliest possible time following completion of dialysis) 2.3 Preparation of Solutions
ZERBAXA does not contain a bacteriostatic preservative. Aseptic technique must be followed in preparing the infusion solution.
Preparation of doses:
Constitute each vial of ZERBAXA with 10 mL of sterile water for injection or 0.9% Sodium Chloride for Injection, USP and gently shake to dissolve. The final volume is approximately 11.4 mL per vial. Caution: The constituted solution is not for direct injection.
To prepare the required dose, withdraw the appropriate volume determined from Table 3 from the reconstituted vial(s). Add the withdrawn volume to an infusion bag containing 100 mL of 0.9% Sodium Chloride for Injection, USP or 5% Dextrose Injection, USP. For doses above 1.5 g, reconstitute a second vial in the same manner as the first one, withdraw an appropriate volume (per Table 3), and add to the same infusion bag.
Table 3: Preparation of Doses ZERBAXA (ceftolozane and tazobactam) Dose Volume to Withdraw from Reconstituted Vial(s) 3 g (2 g and 1 g) Two vials of 11.4 mL each (entire contents from two vials) 2.25 g (1.5 g and 0.75 g) 11.4 mL from one vial (entire contents) and 5.7 mL from a second vial 1.5 g (1 g and 0.5 g) 11.4 mL (entire contents from one vial) 750 mg (500 mg and 250 mg) 5.7 mL 450 mg (300 mg and 150 mg) 3.5 mL 375 mg (250 mg and 125 mg) 2.9 mL 150 mg (100 mg and 50 mg) 1.2 mL Inspect drug products visually for particulate matter and discoloration prior to use. ZERBAXA infusions range from clear, colorless solutions to solutions that are clear and slightly yellow. Variations in color within this range do not affect the potency of the product.
2.4 Compatibility
Compatibility of ZERBAXA with other drugs has not been established. ZERBAXA should not be mixed with other drugs or physically added to solutions containing other drugs.
2.5 Storage of Constituted Solutions
Upon constitution with sterile water for injection or 0.9% sodium chloride injection, reconstituted ZERBAXA solution may be held for 1 hour prior to transfer and dilution in a suitable infusion bag.
Following dilution of the solution with 0.9% sodium chloride or 5% dextrose, ZERBAXA is stable for 24 hours when stored at room temperature or 7 days when stored under refrigeration at 2 to 8°C (36 to 46°F).
Constituted ZERBAXA solution or diluted ZERBAXA infusion should not be frozen.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Decreased Efficacy in Patients with Baseline Creatinine Clearance of 30 to 50 mL/min
In a subgroup analysis of a Phase 3 cIAI trial, clinical cure rates were lower in patients with baseline CrCl of 30 to 50 mL/min compared to those with CrCl greater than 50 mL/min (Table 4). The reduction in clinical cure rates was more marked in the ZERBAXA plus metronidazole arm compared to the meropenem arm. A similar trend was also seen in the cUTI trial. Monitor CrCl at least daily in patients with changing renal function and adjust the dosage of ZERBAXA accordingly [see Dosage and Administration (2.2)].
Table 4: Clinical Cure Rates in a Phase 3 Trial of cIAI by Baseline Renal Function (MITT Population) Baseline Renal Function ZERBAXA plus metronidazole
n/N (%)Meropenem
n/N (%)CrCl greater than 50 mL/min 312/366 (85.2) 355/404 (87.9) CrCl 30 to 50 mL/min 11/23 (47.8) 9/13 (69.2) 5.2 Hypersensitivity Reactions
Serious and occasionally fatal hypersensitivity (anaphylactic) reactions have been reported in patients receiving beta-lactam antibacterial drugs.
Before initiating therapy with ZERBAXA, make careful inquiry about previous hypersensitivity reactions to other cephalosporins, penicillins, or other beta-lactams. If this product is to be given to a patient with a cephalosporin, penicillin, or other beta-lactam allergy, exercise caution because cross sensitivity has been established. If an anaphylactic reaction to ZERBAXA occurs, discontinue the drug and institute appropriate therapy.
5.3 Clostridium difficile-associated Diarrhea
Clostridium difficile-associated diarrhea (CDAD) has been reported for nearly all systemic antibacterial agents, including ZERBAXA, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon and may permit overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. CDAD must be considered in all patients who present with diarrhea following antibacterial use. Careful medical history is necessary because CDAD has been reported to occur more than 2 months after the administration of antibacterial agents.
If CDAD is confirmed, discontinue antibacterials not directed against C. difficile, if possible. Manage fluid and electrolyte levels as appropriate, supplement protein intake, monitor antibacterial treatment of C. difficile, and institute surgical evaluation as clinically indicated.
-
6 ADVERSE REACTIONS
The following serious reactions are described in greater detail in the Warnings and Precautions section:
- Hypersensitivity reactions [see Warnings and Precautions (5.2)]
- Clostridium difficile-associated diarrhea [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and also may not reflect rates observed in practice.
Complicated Intra-abdominal Infections and Complicated Urinary Tract Infections, Including Pyelonephritis
ZERBAXA was evaluated in Phase 3 comparator-controlled clinical trials of cIAI and cUTI, which included a total of 1015 patients treated with ZERBAXA (1.5 g every 8 hours, adjusted based on renal function where appropriate) and 1032 patients treated with comparator (levofloxacin 750 mg daily in cUTI or meropenem 1 g every 8 hours in cIAI) for up to 14 days. The mean age of treated patients was 48 to 50 years (range 18 to 92 years), across treatment arms and indications. In both indications, about 25% of the subjects were 65 years of age or older. Most patients (75%) enrolled in the cUTI trial were female, and most patients (58%) enrolled in the cIAI trial were male. Most patients (>70%) in both trials were enrolled in Eastern Europe and were White.
The most common adverse reactions (5% or greater in either indication) occurring in patients receiving ZERBAXA were nausea, diarrhea, headache, and pyrexia. Table 5 lists adverse reactions occurring in 1% or greater of patients receiving ZERBAXA in Phase 3 cIAI and cUTI clinical trials.
Table 5: Adverse Reactions Occurring in 1% or Greater of Patients Receiving ZERBAXA in Phase 3 cIAI and cUTI Clinical Trials Preferred Term Complicated Intra-abdominal Infections Complicated Urinary Tract Infections, Including Pyelonephritis ZERBAXA*
(N=482)
n (%)Meropenem
(N=497)
n (%)ZERBAXA*
(N=533)
n (%)Levofloxacin
(N=535)
n (%)- * The ZERBAXA for injection dose was 1.5 g intravenously every 8 hours, adjusted to match renal function where appropriate. In the cIAI trials, ZERBAXA was given in conjunction with metronidazole.
Nausea 38 (7.9) 29 (5.8) 15 (2.8) 9 (1.7) Headache 12 (2.5) 9 (1.8) 31 (5.8) 26 (4.9) Diarrhea 30 (6.2) 25 (5) 10 (1.9) 23 (4.3) Pyrexia 27 (5.6) 20 (4) 9 (1.7) 5 (0.9) Constipation 9 (1.9) 6 (1.2) 21 (3.9) 17 (3.2) Insomnia 17 (3.5) 11 (2.2) 7 (1.3) 14 (2.6) Vomiting 16 (3.3) 20 (4) 6 (1.1) 6 (1.1) Hypokalemia 16 (3.3) 10 (2) 4 (0.8) 2 (0.4) ALT increased 7 (1.5) 5 (1) 9 (1.7) 5 (0.9) AST increased 5 (1) 3 (0.6) 9 (1.7) 5 (0.9) Anemia 7 (1.5) 5 (1) 2 (0.4) 5 (0.9) Thrombocytosis 9 (1.9) 5 (1) 2 (0.4) 2 (0.4) Abdominal pain 6 (1.2) 2 (0.4) 4 (0.8) 2 (0.4) Anxiety 9 (1.9) 7 (1.4) 1 (0.2) 4 (0.7) Dizziness 4 (0.8) 5 (1) 6 (1.1) 1 (0.2) Hypotension 8 (1.7) 4 (0.8) 2 (0.4) 1 (0.2) Atrial fibrillation 6 (1.2) 3 (0.6) 1 (0.2) 0 Rash 8 (1.7) 7 (1.4) 5 (0.9) 2 (0.4) Treatment discontinuation due to adverse events occurred in 2.0% (20/1015) of patients receiving ZERBAXA and 1.9% (20/1032) of patients receiving comparator drugs. Renal impairment (including the terms renal impairment, renal failure, and renal failure acute) led to discontinuation of treatment in 5/1015 (0.5%) subjects receiving ZERBAXA and none in the comparator arms.
Increased Mortality
In the cIAI trials (Phase 2 and 3), death occurred in 2.5% (14/564) of patients receiving ZERBAXA and in 1.5% (8/536) of patients receiving meropenem. The causes of death varied and included worsening and/or complications of infection, surgery and underlying conditions.
Less Common Adverse Reactions in Phase 3 cIAI and cUTI Clinical Trials
The following selected adverse reactions were reported in ZERBAXA-treated subjects at a rate of less than 1%:
- Cardiac disorders: tachycardia, angina pectoris
- Gastrointestinal disorders: gastritis, abdominal distension, dyspepsia, flatulence, ileus paralytic
- General disorders and administration site conditions: infusion site reactions
- Infections and infestations: candidiasis including oropharyngeal and vulvovaginal, fungal urinary tract infection
- Investigations: increased serum gamma-glutamyl transpeptidase (GGT), increased serum alkaline phosphatase, positive Coombs test
- Metabolism and nutrition disorders: hyperglycemia, hypomagnesemia, hypophosphatemia
- Nervous system disorders: ischemic stroke
- Renal and urinary system: renal impairment, renal failure
- Respiratory, thoracic and mediastinal disorders: dyspnea
- Skin and subcutaneous tissue disorders: urticaria
- Vascular disorders: venous thrombosis
Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP)
ZERBAXA was evaluated in a Phase 3 comparator-controlled clinical trial for HABP/VABP, which included a total of 361 patients treated with ZERBAXA (3 g every 8 hours, adjusted based on renal function where appropriate) and 359 patients treated with comparator (meropenem 1 g every 8 hours) for up to 14 days. The mean age of treated patients was 60 years (range 18 to 98 years), across treatment arms. About 44% of the subjects were 65 years of age or older. Most patients (71%) enrolled in the trial were male. All subjects were mechanically ventilated at randomization and 92% were in an intensive care unit (ICU) at randomization. The median APACHE II score was 17, and 33% of subjects had a baseline APACHE II score of ≥20, indicating a high severity of illness for many patients enrolled in this trial.
Table 6 lists adverse reactions occurring in 2% or greater of patients receiving ZERBAXA in a Phase 3 HABP/VABP clinical trial.
Table 6: Adverse Reactions Occurring in 2% or Greater of Patients Receiving ZERBAXA in a Phase 3 HABP/VABP Clinical Trial Adverse Reactions ZERBAXA*
N=361
n (%)Meropenem
N=359
n (%)- * The ZERBAXA for injection dose was 3 g intravenously every 8 hours, adjusted to match renal function where appropriate.
- † Includes alanine aminotransferase increased, aspartate aminotransferase increased, hepatic enzyme increased, hypertransaminasaemia, liver function test abnormal.
- ‡ Includes acute renal failure, anuria, azotemia, oliguria, prerenal failure, renal failure, renal impairment.
- § Includes cerebellar hemorrhage, cerebral hematoma, cerebral hemorrhage, hemorrhage intracranial, hemorrhagic stroke, hemorrhagic transformation stroke, intraventricular hemorrhage, subarachnoid hemorrhage, subdural hematoma.
- ¶ Includes Clostridium difficile colitis, Clostridium difficile infection, Clostridium test positive.
Hepatic transaminase increased† 43 (11.9) 26 (7.2) Renal impairment/renal failure‡ 32 (8.9) 22 (6.1) Diarrhea 23 (6.4) 25 (7.0) Intracranial hemorrhage§ 16 (4.4) 5 (1.4) Vomiting 12 (3.3) 10 (2.8) Clostridium difficile colitis¶ 10 (2.8) 2 (0.6) Treatment discontinuation due to adverse reactions occurred in 1.1% (4/361) of patients receiving ZERBAXA and 1.4% (5/359) of patients receiving meropenem.
Laboratory Values
The development of a positive direct Coombs test may occur during treatment with ZERBAXA. The incidence of seroconversion to a positive direct Coombs test was 0.2% in patients receiving ZERBAXA and 0% in patients receiving the comparator in the cUTI and cIAI clinical trials. The incidence of seroconversion to a positive direct Coombs test was 31.2% in patients receiving ZERBAXA and 3.6% in patients receiving meropenem in the HABP/VABP clinical trial. In clinical trials, there was no evidence of hemolysis in patients who developed a positive direct Coombs test in any treatment group.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no data available on ZERBAXA, ceftolozane or tazobactam use in pregnant women to allow assessment of a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. Available data from published prospective cohort studies, case series, and case reports over several decades have not identified an association of cephalosporin use during pregnancy with major birth defects, miscarriage, or other adverse maternal or fetal outcomes (see Data). Neither ceftolozane nor tazobactam produced embryo-fetal toxicity when administered to rodents during the period of organogenesis at ceftolozane doses approximately 3.5 times higher in mice and 2 times higher in rats than the maximum recommended human dose (MRHD) of 2 grams every 8 hours based on plasma AUC comparison or at tazobactam doses approximately 10 times higher in rats than the MRHD of 1 gram every 8 hours based on body surface area comparison. In pre-postnatal studies, where pregnant rats were administered intravenous ceftolozane or intraperitoneal tazobactam in gestation and through the lactation period, ceftolozane was associated with a decrease in auditory startle response in first generation offspring at a dose lower than the MRHD based on AUC comparison, and tazobactam was associated with reduced maternal body weight gain and increased still births at a dose equivalent to approximately 4 times the MRHD and reduced fetal body weights in first generation offspring at a dose approximately equivalent to the MRHD based on body surface area comparison (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Human Data
While available studies with multiple cephalosporins cannot definitively establish the absence of risk, published data from prospective cohort studies, case series, and case reports over several decades have not identified an association of cephalosporin use during pregnancy with major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Available studies have methodologic limitations, including small sample size, retrospective data collection, and inconsistent comparator groups.
Animal Data
Ceftolozane
Embryo-fetal development studies were performed in mice administered intravenous ceftolozane at doses of 300, 1000, and 2000 mg/kg/day during the period of organogenesis (Gestation Day 6 through 15) and in rats administered intravenous ceftolozane in doses of 100, 300, and 1000 mg/kg/day during the period of organogenesis (Gestation Day 6 through 17). In mice, ceftolozane was not associated with maternal or embryo-fetal toxicity with doses up to the highest dose of 2000 mg/kg/ day (approximately 3.5 times the MRHD of 2 grams every 8 hours based on plasma AUC comparison). In rats, no embryo-fetal toxicity was observed, but maternal body weight gain was reduced at a ceftolozane dose of 1000 mg/kg/day. No adverse maternal effects in rats were observed at a dose of 300 mg/kg/day and no adverse embryo-fetal effects were observed at a dose of 1000 mg/kg/day (respectively equivalent to approximately 0.7- and 2-times the MRHD based on plasma AUC comparison).
In a pre-postnatal study in rats, intravenous ceftolozane administered during pregnancy and lactation (Gestation Day 6 through Lactation Day 20) was associated with a decrease in auditory startle response in postnatal day 60 male pups at maternal doses greater than or equal to 300 mg/kg/day. No adverse effects were observed in rats at a dose of 100 mg/kg/day, a dose lower than the MRHD of 2 grams every 8 hours based on plasma AUC comparison.
Tazobactam
In an embryo-fetal study in rats, tazobactam was administered intravenously during the period of organogenesis (Gestation Day 7 through 17) at doses of 125, 500, and 3000 mg/kg/day. The high dose of 3000 mg/kg/day produced maternal toxicity (decreased food consumption and body weight gain) but was not associated with fetal toxicity. No adverse maternal effects were observed at a dose of 500 mg/kg/day and no adverse fetal effects were observed at a dose of 3000 mg/kg/day (respectively equivalent to approximately 2- and 10-times the MRHD of 1 gram every 8 hours based on body surface area comparison). In rats, tazobactam was shown to cross the placenta. Concentrations in the fetus were less than or equal to 10% of those found in maternal plasma.
In a pre-postnatal study in rats, tazobactam administered intraperitoneally in doses of 40, 320, and 1280 mg/kg/day at the end of gestation and during lactation (Gestation Day 17 through Lactation Day 21) was associated with decreased maternal food consumption and body weight gain at the end of gestation and significantly more stillbirths at the high dose of 1280 mg/kg/day. No effects on the physical development, neurological function, or fertility and reproductive ability of first generation (F1) pups were noted, but postnatal body weights for F1 pups delivered to dams receiving 320 and 1280 mg/kg/day tazobactam were significantly reduced 21 days after delivery. The second generation (F2) fetuses were normal for all doses of tazobactam. No adverse effects on maternal reproduction were observed at doses up to 320 mg/kg/day and F1 body weights were not reduced at a dose of 40 mg/kg/day (respectively equivalent to approximately 1.0 and 0.1 times the MRHD of 1 gram every 8 hours based on body surface area comparison).
8.2 Lactation
Risk Summary
There are no data on the presence of ceftolozane or tazobactam in human milk. There are no data on the effects of tazobactam or ceftolozane on the breastfed infant, or the effects on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ZERBAXA and any potential adverse effects on the breastfed child from ZERBAXA or from the underlying maternal conditions.
8.5 Geriatric Use
Of the 1015 patients treated with ZERBAXA in the Phase 3 cIAI and cUTI clinical trials, 250 (24.6%) were 65 years or older, including 113 (11.1%) 75 years or older. The incidence of adverse events in both treatment groups was higher in older subjects (65 years or older) in the trials for both indications. In the cIAI trial, cure rates in the elderly (aged 65 years and older) in the ZERBAXA plus metronidazole arm were 69/100 (69%) and in the comparator arm were 70/85 (82.4%). This finding in the elderly population was not observed in the cUTI trial.
Of the 361 patients treated with ZERBAXA in the Phase 3 HABP/VABP clinical trial, 160 (44.3%) were 65 years or older, including 83 (23%) 75 years or older. The incidence of adverse events in both treatment groups was higher in older subjects (65 years or older). In the trial, Day 28 all-cause mortality rates in the elderly (aged 65 years and older) were comparable between treatment arms:50/160 (31.3%) in the ZERBAXA arm and 54/160 (33.8%) in the comparator arm.
ZERBAXA is substantially excreted by the kidney and the risk of adverse reactions to ZERBAXA may be greater in patients with renal impairment. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection and it may be useful to monitor renal function. Adjust dosage for elderly patients based on renal function [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
8.6 Patients with Renal Impairment
Dosage adjustment is required in patients with CrCl 50 mL/min or less, including patients with ESRD on HD [see Dosage and Administration (2.2), Warnings and Precautions (5.1) and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
In the event of overdose, discontinue ZERBAXA and provide general supportive treatment. ZERBAXA can be removed by hemodialysis. Approximately 66% of ceftolozane, 56% of tazobactam, and 51% of the tazobactam metabolite M1 were removed by dialysis. No information is available on the use of hemodialysis to treat overdosage.
-
11 DESCRIPTION
ZERBAXA (ceftolozane and tazobactam) is an antibacterial combination product consisting of the cephalosporin antibacterial drug ceftolozane sulfate and the beta-lactamase inhibitor tazobactam sodium for intravenous administration.
Ceftolozane sulfate is a semi-synthetic antibacterial drug of the beta-lactam class for parenteral administration. The chemical name of ceftolozane sulfate is 1H-Pyrazolium, 5-amino-4-[[[(2-aminoethyl)amino]carbonyl]amino]-2-[[(6R,7R)-7-[[(2Z)-2-(5-amino-1,2,4-thiadiazol-3-yl)-2-[(1-carboxy-1-methylethoxy)imino]acetyl]amino]-2-carboxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl]methyl]-1-methyl-,sulfate (1:1). The molecular formula is C23H31N12O8S2+∙HSO4- and the molecular weight is 764.77.
Figure 1: Chemical structure of ceftolozane sulfate
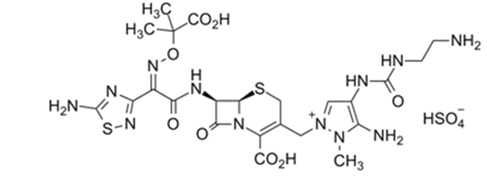
Tazobactam sodium, a derivative of the penicillin nucleus, is a penicillanic acid sulfone. Its chemical name is sodium (2S,3S,5R)-3-methyl-7-oxo-3-(1H-1,2,3-triazol-1-ylmethyl)-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylate-4,4-dioxide. The chemical formula is C10H11N4NaO5S and the molecular weight is 322.3.
Figure 2: Chemical structure of tazobactam sodium
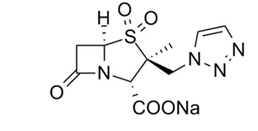
ZERBAXA 1.5 g (ceftolozane and tazobactam) for injection is a white to yellow sterile powder for reconstitution consisting of ceftolozane 1 g (equivalent to 1.147 g of ceftolozane sulfate) and tazobactam 0.5 g (equivalent to 0.537 g of tazobactam sodium) per vial, packaged in single-dose glass vials. The product contains sodium chloride (487 mg/vial) as a stabilizing agent, citric acid (21 mg/vial), and L-arginine (approximately 600 mg/vial) as excipients.
-
12 CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
As with other beta-lactam antibacterial agents, the percent time of dosing interval that the plasma concentration of ceftolozane exceeds the minimum inhibitory concentration (MIC) of the infecting organism has been shown to be the best predictor of efficacy in animal models of infection. The percent time of dosing interval that the plasma concentration of tazobactam exceeds a threshold concentration has been determined to be the parameter that best predicts the efficacy of tazobactam in in vitro and in vivo models. The exposure-response analyses in efficacy and safety clinical trials for cIAI, cUTI, and HABP/VABP support the recommended dose regimens of ZERBAXA.
Cardiac Electrophysiology
In a randomized, positive and placebo-controlled crossover thorough QTc study, 51 healthy subjects were administered a single therapeutic dose of ZERBAXA 1.5 gram (ceftolozane 1 g and tazobactam 0.5 g) and a supratherapeutic dose of ZERBAXA 4.5 gram (ceftolozane 3 g and tazobactam 1.5 g). No significant effects of ZERBAXA on heart rate, electrocardiogram morphology, PR, QRS, or QT interval were detected.
12.3 Pharmacokinetics
Ceftolozane and tazobactam pharmacokinetics are similar following single- and multiple-dose administrations. The Cmax and AUC of ceftolozane and tazobactam increase in proportion to dose.
The mean steady-state population pharmacokinetic parameters of ZERBAXA in patients with cIAI and cUTI receiving 1-hour intravenous infusions of ZERBAXA 1.5 g (ceftolozane 1 g and tazobactam 0.5 g) or patients with HABP/VABP receiving 1-hour intravenous infusions of ZERBAXA 3 g (ceftolozane 2 g and tazobactam 1 g) every 8 hours are summarized in Table 7.
Table 7: Mean (SD) Steady-State Plasma Population Pharmacokinetic Parameters of ZERBAXA (ceftolozane and tazobactam) after Multiple Intravenous 1-hour Infusions of ZERBAXA 1.5 g (ceftolozane 1 g and tazobactam 0.5 g) or 3 g (ceftolozane 2 g and tazobactam 1 g) Every 8 Hours in Patients with CrCl Greater than 50 mL/min PK parameters ZERBAXA 1.5 g (ceftolozane 1 g and tazobactam 0.5 g) in cIAI and cUTI Patients ZERBAXA 3 g (ceftolozane 2 g and tazobactam 1 g) in HABP/VABP Patients Ceftolozane
(n=317)Tazobactam
(n=244)Ceftolozane
(n=247)Tazobactam
(n=247)Cmax (mcg/mL) 65.7 (27) 17.8 (9) 105 (46) 26.4 (13) AUC0-8,ss (mcg∙h/mL) 186 (74) 35.8 (57) 392 (236) 73.3 (76) Distribution
The binding of ceftolozane and tazobactam to human plasma proteins is approximately 16% to 21% and 30%, respectively. The mean (CV%) steady-state volume of distribution of ZERBAXA in healthy adult males (n = 51) following a single intravenous dose of ZERBAXA 1.5 g (ceftolozane 1 g and tazobactam 0.5 g) was 13.5 L (21%) and 18.2 L (25%) for ceftolozane and tazobactam, respectively, similar to extracellular fluid volume.
Following 1-hour intravenous infusions of ZERBAXA 3 g (ceftolozane 2 g and tazobactam 1 g) or adjusted based on renal function every 8 hours in ventilated patients with confirmed or suspected pneumonia (N=22), mean pulmonary epithelial lining fluid-to-free plasma AUC ratios of ceftolozane and tazobactam were approximately 50% and 62%, respectively, and are similar to those in healthy subjects (approximately 61% and 63%, respectively) receiving ZERBAXA 1.5 g (ceftolozane 1 g and tazobactam 0.5 g). Minimum ceftolozane and tazobactam epithelial lung lining fluid concentrations in ventilated subjects at the end of the dosing interval were 8.2 mcg/mL and 1.0 mcg/mL, respectively.
Elimination
Ceftolozane is eliminated from the body by renal excretion with a mean half-life of approximately 3 to 4 hours. Tazobactam is eliminated by renal excretion and metabolism with a plasma mean half-life of approximately 2 to 3 hours. The elimination half-life (t1/2) of ceftolozane or tazobactam is independent of dose.
Metabolism
Ceftolozane does not appear to be metabolized to any appreciable extent and is not a substrate for CYP enzymes. The beta-lactam ring of tazobactam is hydrolyzed to form the pharmacologically inactive tazobactam metabolite M1.
Excretion
Ceftolozane, tazobactam and the tazobactam metabolite M1 are excreted by the kidneys. Following administration of a single ZERBAXA 1.5 g (ceftolozane 1 g and tazobactam 0.5 g) intravenous dose to healthy male adults, greater than 95% of ceftolozane was excreted in the urine as unchanged parent drug. More than 80% of tazobactam was excreted as the parent compound with the remainder excreted as the tazobactam M1 metabolite. After a single dose of ZERBAXA, renal clearance of ceftolozane (3.41 – 6.69 L/h) was similar to plasma CL (4.10 to 6.73 L/h) and similar to the glomerular filtration rate for the unbound fraction, suggesting that ceftolozane is eliminated by the kidney via glomerular filtration. Tazobactam is a substrate for OAT1 and OAT3 transporters and its elimination has been shown to be inhibited by probenecid, an inhibitor of OAT1/3.
Specific Populations
Dose adjustment is not warranted on the basis of age (18 years and older), gender, or race/ethnicity. No significant differences in the pharmacokinetics of ceftolozane and tazobactam were observed based on age (18 years and older), gender, weight, or race/ethnicity.
Patients with Renal Impairment
The ceftolozane dose normalized geometric mean AUC increased up to 1.26-fold, 2.5-fold, and 5-fold in subjects with CrCl 80-51 mL/min, 50-30 mL/min, and 29-15 mL/min, respectively, compared to healthy subjects with normal renal function. The respective tazobactam dose normalized geometric mean AUC increased approximately up to 1.3-fold, 2-fold, and 4-fold. To maintain similar systemic exposures to those with normal renal function, dosage adjustment is required [see Dosage and Administration (2.2)].
In subjects with ESRD on HD, approximately two-thirds of the administered ZERBAXA dose is removed by HD. A single loading dose of ZERBAXA followed by a maintenance dose administered every 8 hours for the remainder of the treatment period is recommended in patients with ESRD on HD. On HD days, administer the dose at the earliest possible time following completion of HD. [See Dosage and Administration (2.2).]
Patients with Augmented Renal Function
Following a single 1-hour intravenous infusion of ZERBAXA 3 g (ceftolozane 2 g and tazobactam 1 g) to critically-ill patients with CrCl greater than or equal to 180 mL/min (N=10), mean terminal half-life values of ceftolozane and tazobactam were 2.6 hours and 1.5 hours, respectively. No dose adjustment of ZERBAXA is recommended for HABP/VABP patients with augmented renal function [see Clinical Studies (14.3)].
Patients with Hepatic Impairment
As ZERBAXA does not undergo hepatic metabolism, the systemic clearance of ZERBAXA is not expected to be affected by hepatic impairment.
No dose adjustment is recommended for ZERBAXA in subjects with hepatic impairment.
Geriatric Patients
In a population pharmacokinetic analysis of ZERBAXA, no clinically relevant differences in exposure were observed with regard to age.
No dose adjustment of ZERBAXA based on age is recommended. Dosage adjustment for ZERBAXA in geriatric patients should be based on renal function [see Dosage and Administration (2.2)].
Drug Interactions
No drug-drug interaction was observed between ceftolozane and tazobactam in a clinical study in 16 healthy subjects. In vitro and in vivo data indicate that ZERBAXA is unlikely to cause clinically relevant drug-drug interactions related to CYPs and transporters at therapeutic concentrations.
Drug Metabolizing Enzymes
In vivo data indicated that ZERBAXA is not a substrate for CYPs. Thus, clinically relevant drug-drug interactions involving inhibition or induction of CYPs by other drugs are unlikely to occur.
In vitro studies demonstrated that ceftolozane, tazobactam and the M1 metabolite of tazobactam did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4 and did not induce CYP1A2, CYP2B6, or CYP3A4 at therapeutic plasma concentrations. In vitro induction studies in primary human hepatocytes demonstrated that ceftolozane, tazobactam, and the tazobactam metabolite M1 decreased CYP1A2 and CYP2B6 enzyme activity and mRNA levels in primary human hepatocytes as well as CYP3A4 mRNA levels at supratherapeutic plasma concentrations. Tazobactam metabolite M1 also decreased CYP3A4 activity at supratherapeutic plasma concentrations. A clinical drug-drug interaction study was conducted and results indicated drug interactions involving CYP1A2 and CYP3A4 inhibition by ZERBAXA are not anticipated.
Membrane Transporters
Ceftolozane and tazobactam were not substrates for P-gp or BCRP, and tazobactam was not a substrate for OCT2, in vitro at therapeutic concentrations.
Tazobactam is a known substrate for OAT1 and OAT3. Co-administration of tazobactam with the OAT1/OAT3 inhibitor probenecid has been shown to prolong the half-life of tazobactam by 71%. Co-administration of ZERBAXA with drugs that inhibit OAT1 and/or OAT3 may increase tazobactam plasma concentrations.
In vitro data indicate that ceftolozane did not inhibit P-gp, BCRP, OATP1B1, OATP1B3, OCT1, OCT2, MRP, BSEP, OAT1, OAT3, MATE1, or MATE2-K in vitro at therapeutic plasma concentrations.
In vitro data indicate that neither tazobactam nor the tazobactam metabolite M1 inhibit P-gp, BCRP, OATP1B1, OATP1B3, OCT1, OCT2, or BSEP transporters at therapeutic plasma concentrations. In vitro, tazobactam inhibited human OAT1 and OAT3 transporters with IC50 values of 118 and 147 mcg/mL, respectively. A clinical drug-drug interaction study was conducted and results indicated clinically relevant drug interactions involving OAT1/OAT3 inhibition by ZERBAXA are not anticipated.
12.4 Microbiology
Mechanism of Action
Ceftolozane belongs to the cephalosporin class of antibacterial drugs. The bactericidal action of ceftolozane results from inhibition of cell wall biosynthesis, and is mediated through binding to penicillin-binding proteins (PBPs). Ceftolozane is an inhibitor of PBPs of P. aeruginosa (e.g., PBP1b, PBP1c, and PBP3) and E. coli (e.g., PBP3).
Tazobactam sodium has little clinically relevant in vitro activity against bacteria due to its reduced affinity to penicillin-binding proteins. It is an irreversible inhibitor of some beta-lactamases (e.g., certain penicillinases and cephalosporinases), and can bind covalently to some chromosomal and plasmid-mediated bacterial beta-lactamases.
Resistance
Mechanisms of beta-lactam resistance may include the production of beta-lactamases, modification of PBPs by gene acquisition or target alteration, up-regulation of efflux pumps, and loss of outer membrane porin.
Clinical isolates may produce multiple beta-lactamases, express varying levels of beta-lactamases, or have amino acid sequence variations, and other resistance mechanisms that have not been identified.
Culture and susceptibility information and local epidemiology should be considered in selecting or modifying antibacterial therapy.
ZERBAXA demonstrated in vitro activity against Enterobacteriaceae in the presence of some extended-spectrum beta-lactamases (ESBLs) and other beta-lactamases of the following groups: TEM, SHV, CTX-M, and OXA. ZERBAXA is not active against bacteria that produce serine carbapenemases [K. pneumoniae carbapenemase (KPC)], and metallo-beta-lactamases.
In ZERBAXA clinical trials, some isolates of Enterobacteriaceae with minimum inhibitory concentration to ZERBAXA of ≤2 mcg/mL produced beta-lactamases. These isolates produced one or more beta-lactamases of the following enzyme groups: CTX-M, OXA, TEM, or SHV.
Some of these beta-lactamases were also produced by isolates of Enterobacteriaceae with minimum inhibitory concentration to ZERBAXA >2 mcg/mL.
ZERBAXA demonstrated in vitro activity against P. aeruginosa isolates tested that had chromosomal AmpC, loss of outer membrane porin (OprD), or up regulation of efflux pumps (MexXY, MexAB).
Isolates resistant to other cephalosporins may be susceptible to ZERBAXA, although cross-resistance may occur.
Interaction with Other Antimicrobials
In vitro synergy studies suggest no antagonism between ZERBAXA and other antibacterial drugs (e.g., meropenem, amikacin, aztreonam, levofloxacin, tigecycline, rifampin, linezolid, daptomycin, vancomycin, and metronidazole).
Antimicrobial Activity
ZERBAXA has been shown to be active against the following bacteria, both in vitro and in clinical infections [see Indications and Usage (1)].
Complicated Intra-abdominal Infections
Gram-negative bacteria:
-
Enterobacter cloacae
Escherichia coli
Klebsiella oxytoca
Klebsiella pneumoniae
Proteus mirabilis
Pseudomonas aeruginosa
Gram-positive bacteria:
-
Streptococcus anginosus
Streptococcus constellatus
Streptococcus salivarius
Anaerobic bacteria:
- Bacteroides fragilis
Complicated Urinary Tract Infections, Including Pyelonephritis
Gram-negative bacteria:
-
Escherichia coli
Klebsiella pneumoniae
Proteus mirabilis
Pseudomonas aeruginosa
Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP)
Gram-negative bacteria:
-
Enterobacter cloacae
Escherichia coli
Haemophilus influenzae
Klebsiella oxytoca
Klebsiella pneumoniae
Proteus mirabilis
Pseudomonas aeruginosa
Serratia marcescens
The following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for ceftolozane and tazobactam against isolates of similar genus or organism group. However, the efficacy of ZERBAXA in treating clinical infections due to these bacteria has not been established in adequate and well-controlled clinical trials.
Gram-negative bacteria:
-
Citrobacter koseri
Klebsiella aerogenes
Morganella morganii
Proteus vulgaris
Providencia rettgeri
Providencia stuartii
Serratia liquefaciens
Gram-positive bacteria:
-
Streptococcus agalactiae
Streptococcus intermedius
-
Enterobacter cloacae
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term carcinogenicity studies in animals have not been conducted with ZERBAXA, ceftolozane, or tazobactam.
ZERBAXA was negative for genotoxicity in an in vitro mouse lymphoma assay and an in vivo rat bone-marrow micronucleus assay. In an in vitro chromosomal aberration assay in Chinese hamster ovary cells, ZERBAXA was positive for structural aberrations.
Ceftolozane was negative for genotoxicity in an in vitro microbial mutagenicity (Ames) assay, an in vitro chromosomal aberration assay in Chinese hamster lung fibroblast cells, an in vitro mouse lymphoma assay, an in vitro HPRT assay in Chinese hamster ovary cells, an in vivo mouse micronucleus assay, and an in vivo unscheduled DNA synthesis (UDS) assay.
Tazobactam was negative for genotoxicity in an in vitro microbial mutagenicity (Ames) assay, an in vitro chromosomal aberration assay in Chinese hamster lung cells, an in vitro mammalian point-mutation (Chinese hamster ovary cell HPRT) assay, an in vivo mouse bone-marrow micronucleus assay, and an in vivo UDS assay.
Ceftolozane was administered in a fertility study at intravenous doses of 100, 300, and 1000 mg/kg/day to male rats for 28 days before mating and through the mating period and to female rats for 14 days before mating, through the mating period, and until the 7th day of gestation. Ceftolozane had no adverse effect on fertility in male or female rats at doses up to 1000 mg/kg/day (approximately 1.4 times the maximum recommended human dose (MRHD) of 2 grams every 8 hours based on AUC comparison).
In a rat fertility study, intraperitoneal tazobactam doses of 40, 160, and 640 mg/kg/day were administered twice-daily to male rats beginning 70 days before mating and through the mating period, and to female rats beginning 14 days before mating, during the mating period, and until Gestation Day 21. Male and female fertility parameters were not affected at doses less than or equal to 640 mg/kg/day (approximately 2 times the MRHD of 1 gram every 8 hours based on body surface comparison).
-
14 CLINICAL STUDIES
14.1 Complicated Intra-abdominal Infections
A total of 979 adults hospitalized with cIAI were randomized and received study medications in a multinational, double-blind study comparing ZERBAXA 1.5 g (ceftolozane 1 g and tazobactam 0.5 g) intravenously every 8 hours plus metronidazole (500 mg intravenously every 8 hours) to meropenem (1 g intravenously every 8 hours) for 4 to 14 days of therapy. Complicated intra-abdominal infections included appendicitis, cholecystitis, diverticulitis, gastric/duodenal perforation, perforation of the intestine, and other causes of intra-abdominal abscesses and peritonitis. The majority of patients (75%) were from Eastern Europe; 6.3% were from the United States.
The primary efficacy endpoint was clinical response, defined as complete resolution or significant improvement in signs and symptoms of the index infection at the test-of-cure (TOC) visit which occurred 24 to 32 days after the first dose of study drug. The primary efficacy analysis population was the microbiological intent-to-treat (MITT) population, which included all patients who had at least 1 baseline intra-abdominal pathogen regardless of the susceptibility to study drug. The key secondary efficacy endpoint was clinical response at the TOC visit in the microbiologically evaluable (ME) population, which included all protocol-adherent MITT patients.
The MITT population consisted of 806 patients; the median age was 52 years and 57.8% were male. The most common diagnosis was appendiceal perforation or peri-appendiceal abscess, occurring in 47% of patients. Diffuse peritonitis at baseline was present in 34.2% of patients.
ZERBAXA plus metronidazole was non-inferior to meropenem with regard to clinical cure rates at the TOC visit in the MITT population. Clinical cure rates at the TOC visit are displayed by patient population in Table 8. Clinical cure rates at the TOC visit by pathogen in the MITT population are presented in Table 9.
Table 8: Clinical Cure Rates in a Phase 3 Trial of Complicated Intra-Abdominal Infections Analysis Population ZERBAXA plus metronidazole*
n/N (%)Meropenem†
n/N (%)Treatment Difference
(95% CI)‡- * ZERBAXA 1.5 g intravenously every 8 hours + metronidazole 500 mg intravenously every 8 hours
- † 1 gram intravenously every 8 hours
- ‡ The 95% confidence interval (CI) was calculated as an unstratified Wilson Score CI.
MITT
323/389 (83) 364/417 (87.3) -4.3 (-9.2, 0.7) ME 259/275 (94.2) 304/321 (94.7) -0.5 (-4.5, 3.2) Table 9: Clinical Cure Rates by Pathogen in a Phase 3 Trial of Complicated Intra-abdominal Infections (MITT Population) Organism Group Pathogen ZERBAXA plus metronidazole
n/N (%)Meropenem
n/N (%)Aerobic Gram-negative Escherichia coli 216/255 (84.7) 238/270 (88.1) Klebsiella pneumoniae 31/41 (75.6) 27/35 (77.1) Pseudomonas aeruginosa 30/38 (79) 30/34 (88.2) Enterobacter cloacae 21/26 (80.8) 24/25 (96) Klebsiella oxytoca 14/16 (87.5) 24/25 (96) Proteus mirabilis 11/12 (91.7) 9/10 (90) Aerobic Gram-positive Streptococcus anginosus 26/36 (72.2) 24/27 (88.9) Streptococcus constellatus 18/24 (75) 20/25 (80) Streptococcus salivarius 9/11 (81.8) 9/11 (81.8) Anaerobic Gram-negative Bacteroides fragilis 42/47 (89.4) 59/64 (92.2) Bacteroides ovatus 38/45 (84.4) 44/46 (95.7) Bacteroides thetaiotaomicron 21/25 (84) 40/46 (87) Bacteroides vulgatus 12/15 (80) 24/26 (92.3) In a subset of the E. coli and K. pneumoniae isolates from both arms of the cIAI Phase 3 trial that met pre-specified criteria for beta-lactam susceptibility, genotypic testing identified certain ESBL groups (e.g., TEM, SHV, CTX-M, OXA) in 53/601 (9%). Cure rates in this subset were similar to the overall trial results. In vitro susceptibility testing showed that some of these isolates were susceptible to ZERBAXA (MIC ≤ 2 mcg/mL), while some others were not susceptible (MIC >2 mcg/mL). Isolates of a specific genotype were seen in patients who were deemed to be either successes or failures.
14.2 Complicated Urinary Tract Infections, Including Pyelonephritis
A total of 1068 adults hospitalized with cUTI (including pyelonephritis) were randomized and received study medications in a multinational, double-blind study comparing ZERBAXA 1.5 g (ceftolozane 1 g and tazobactam 0.5 g) intravenously every 8 hours to levofloxacin (750 mg intravenously once daily) for 7 days of therapy. The primary efficacy endpoint was defined as complete resolution or marked improvement of the clinical symptoms and microbiological eradication (all uropathogens found at baseline at ≥105 were reduced to <104 CFU/mL) at the test-of-cure (TOC) visit 7 (± 2) days after the last dose of study drug. The primary efficacy analysis population was the microbiologically modified intent-to-treat (mMITT) population, which included all patients who received study medication and had at least 1 baseline uropathogen. The key secondary efficacy endpoint was the composite microbiological and clinical cure response at the TOC visit in the microbiologically evaluable (ME) population, which included protocol-adherent mMITT patients with a urine culture at the TOC visit.
The mMITT population consisted of 800 patients with cUTI, including 656 (82%) with pyelonephritis. The median age was 50.5 years and 74% were female. Concomitant bacteremia was identified in 62 (7.8%) patients at baseline; 608 (76%) patients were enrolled in Eastern Europe and 14 (1.8%) patients were enrolled in the United States.
ZERBAXA demonstrated efficacy with regard to the composite endpoint of microbiological and clinical cure at the TOC visit in both the mMITT and ME populations (Table 10). Composite microbiological and clinical cure rates at the TOC visit by pathogen in the mMITT population are presented in Table 11.
In the mMITT population, the composite cure rate in ZERBAXA-treated patients with concurrent bacteremia at baseline was 23/29 (79.3%).
Although a statistically significant difference was observed in the ZERBAXA arm compared to the levofloxacin arm with respect to the primary endpoint, it was likely attributable to the 212/800 (26.5%) patients with baseline organisms non-susceptible to levofloxacin. Among patients infected with a levofloxacin-susceptible organism at baseline, the response rates were similar (Table 10).
Table 10: Composite Microbiological and Clinical Cure Rates in a Phase 3 Trial of Complicated Urinary Tract Infections Analysis Population ZERBAXA*
n/N (%)Levofloxacin†
n/N (%)Treatment Difference
(95% CI)‡- * ZERBAXA 1.5 g intravenously every 8 hours
- † 750 mg intravenously once daily
- ‡ The 95% confidence interval was based on the stratified Newcombe method.
mMITT 306/398 (76.9) 275/402 (68.4) 8.5 (2.3, 14.6) Levofloxacin resistant baseline pathogen(s) 60/100 (60) 44/112 (39.3) No levofloxacin resistant baseline pathogen(s) 246/298 (82.6) 231/290 (79.7) ME 284/341 (83.3) 266/353 (75.4) 8.0 (2.0, 14.0) Table 11: Composite Microbiological and Clinical Cure Rates in a Phase 3 Trial of Complicated Urinary Tract Infections, in Subgroups Defined by Baseline Pathogen (mMITT Population) Pathogen ZERBAXA
n/N (%)Levofloxacin
n/N (%)Escherichia coli 247/305 (81) 228/324 (70.4) Klebsiella pneumoniae 22/33 (66.7) 12/25 (48) Proteus mirabilis 11/12 (91.7) 6/12 (50) Pseudomonas aeruginosa 6/8 (75) 7/15 (46.7) In a subset of the E. coli and K. pneumoniae isolates from both arms of the cUTI Phase 3 trial that met pre-specified criteria for beta-lactam susceptibility, genotypic testing identified certain ESBL groups (e.g., TEM, SHV, CTX-M, OXA) in 104/687 (15%). Cure rates in this subset were similar to the overall trial results. In vitro susceptibility testing showed that some of these isolates were susceptible to ZERBAXA (MIC ≤2 mcg/mL), while some others were not susceptible (MIC >2 mcg/mL). Isolates of a specific genotype were seen in patients who were deemed to be either successes or failures.
14.3 Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP)
A total of 726 adult patients hospitalized with HABP/VABP were enrolled in a multinational, double-blind study (NCT02070757) comparing ZERBAXA 3 g (ceftolozane 2 g and tazobactam 1 g) intravenously every 8 hours to meropenem (1 g intravenously every 8 hours) for 8 to 14 days of therapy. All patients had to be intubated and on mechanical ventilation at randomization.
Efficacy was assessed based on all-cause mortality at Day 28 and clinical cure, defined as complete resolution or significant improvement in signs and symptoms of the index infection at the test-of-cure (TOC) visit which occurred 7 to 14 days after the end of treatment. The analysis population was the intent-to-treat (ITT) population, which included all randomized patients.
Following a diagnosis of HABP/VABP and prior to receipt of first dose of study drug, if required, patients could have received up to a maximum of 24 hours of active non-study antibacterial drug therapy in the 72 hours preceding the first dose of study drug. Patients who had failed prior antibacterial drug therapy for the current episode of HABP/VABP could be enrolled if the baseline lower respiratory tract (LRT) culture showed growth of a Gram-negative pathogen while the patient was on the antibacterial therapy and all other eligibility criteria were met. Empiric therapy at baseline with linezolid or other approved therapy for Gram-positive coverage was required in all patients pending baseline LRT culture results. Adjunctive Gram-negative therapy was optional and allowed for a maximum of 72 hours in centers with a prevalence of meropenem-resistant P. aeruginosa more than 15%.
Of the 726 patients in the ITT population, the median age was 62 years and 44% of the population was 65 years of age and older, with 22% of the population 75 years of age and older. The majority of patients were white (83%), male (71%) and were from Eastern Europe (64%). The median APACHE II score was 17 and 33% of subjects had a baseline APACHE II score of greater than or equal to 20. All subjects were on mechanical ventilation and 519 (71%) had VABP. At randomization, 92% of subjects were in the ICU, 77% had been hospitalized for 5 days or longer, and 49% were ventilated for 5 days or longer. A total of 258 of 726 (36%) patients had CrCl less than 80 mL/min at baseline; among these, 99 (14%) had CrCl less than 50 mL/min. Patients with end-stage renal disease (CrCl less than 15 mL/min) were excluded from the trial. Approximately 13% of subjects were failing their current antibacterial drug therapy for HABP/VABP, and bacteremia was present at baseline in 15% of patients. Key comorbidities included diabetes mellitus, congestive heart failure, and chronic obstructive pulmonary disease at rates of 22%, 16%, and 12%, respectively. In both treatment groups, most subjects (63.1%) received between 8 and 14 days of study therapy as specified in the protocol.
Table 12 presents the results for Day 28 all-cause mortality and clinical cure at the TOC visit overall and by ventilated HABP and VABP.
Table 12: Day 28 All-cause Mortality and Clinical Cure Rates at TOC from a Phase 3 Study of Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP) (ITT Population) Endpoint ZERBAXA
n/N (%)Meropenem
n/N (%)Treatment Difference
(95% CI)*- * The CI for overall treatment difference was based on the stratified Newcombe method with minimum risk weights. The CI for treatment difference of each primary diagnosis was based on the unstratified Newcombe method.
Day 28 All-cause Mortality 87/362 (24.0) 92/364 (25.3) 1.1 (-5.13, 7.39) VABP 63/263 (24.0) 52/256 (20.3) -3.6 (-10.74, 3.52) Ventilated HABP 24/99 (24.2) 40/108 (37.0) 12.8 (0.18, 24.75) Clinical Cure at TOC Visit 197/362 (54.4) 194/364 (53.3) 1.1 (-6.17, 8.29) VABP 147/263 (55.9) 146/256 (57.0) -1.1 (-9.59, 7.35) Ventilated HABP 50/99 (50.5) 48/108 (44.4) 6.1 (-7.44, 19.27) In the ITT population, Day 28 all-cause mortality and clinical cure rates in patients with CrCl greater than or equal to 150 mg/mL were similar between ZERBAXA and meropenem. In patients with bacteremia at baseline, Day 28 all-cause mortality rates were 23/64 (35.9%) for ZERBAXA-treated patients and 13/41 (31.7%) for meropenem-treated patients; clinical cure rates were 30/64 (46.9%) and 15/41 (36.6%), respectively.
Per pathogen Day 28 all-cause mortality and clinical cure at TOC were assessed in the microbiologic intention to treat population (mITT), which consisted of all randomized subjects who had a baseline lower respiratory tract (LRT) pathogen that was susceptible to both study treatments. In the mITT population, Klebsiella pneumoniae (113/425, 26.6%) and Pseudomonas aeruginosa (103/425, 24.2%) were the most prevalent pathogens isolated from baseline LRT cultures.
Day 28 all-cause mortality and clinical cure rates at TOC by pathogen in the mITT population are presented in Table 13. In the mITT population, clinical cure rates in patients with a Gram-negative pathogen at baseline were 139/215 (64.7%) for ZERBAXA and 115/204 (56.4%) for meropenem, respectively.
Table 13: Day 28 All-cause Mortality and Clinical Cure Rates at TOC by Baseline Pathogen from a Phase 3 Study of Hospital-acquired Bacterial Pneumonia and Ventilator-associated Bacterial Pneumonia (HABP/VABP) (mITT population) Baseline Pathogen Category Day 28 All-cause Mortality Clinical Cure at TOC Baseline Pathogen ZERBAXA
n/N (%)Meropenem
n/N (%)ZERBAXA
n/N (%)Meropenem
n/N (%)Pseudomonas aeruginosa 12/47 (25.5) 10/56 (17.9) 29/47 (61.7) 34/56 (60.7) Enterobacteriaceae 27/161 (16.8) 42/157 (26.8) 103/161 (64.0) 87/157 (55.4) Enterobacter cloacae 2/15 (13.3) 8/14 (57.1) 8/15 (53.3) 4/14 (28.6) Escherichia coli 10/50 (20.0) 11/42 (26.2) 32/50 (64.0) 26/42 (61.9) Klebsiella oxytoca 3/14 (21.4) 3/12 (25.0) 9/14 (64.3) 7/12 (58.3) Klebsiella pneumoniae 7/51 (13.7) 13/62 (21.0) 34/51 (66.7) 39/62 (62.9) Proteus mirabilis 5/22 (22.7) 5/18 (27.8) 13/22 (59.1) 11/18 (61.1) Serratia marcescens 3/14 (21.4) 1/12 (8.3) 8/14 (57.1) 7/12 (58.3) Haemophilus influenzae 0/20 (0) 2/15 (13.3) 17/20 (85.0) 8/15 (53.3) In a subset of Enterobacteriaceae isolates from both arms of the trial that met pre-specified criteria for beta-lactam susceptibility, genotypic testing identified certain ESBL groups (e.g., TEM, SHV, CTX-M, OXA) in 101/425 (23.8%). Day 28 all-cause mortality and clinical cure rates in this subset were similar to the overall trial results.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
ZERBAXA 1.5 g (ceftolozane and tazobactam) for injection is supplied in single-dose vials containing ceftolozane 1 g (equivalent to 1.147 g of ceftolozane sulfate) and tazobactam 0.5 g (equivalent to 0.537 g of tazobactam sodium) per vial. Vials are supplied in cartons containing 10 vials.
(NDC: 67919-030-01)
-
17 PATIENT COUNSELING INFORMATION
Serious Allergic Reactions
Advise patient that allergic reactions, including serious allergic reactions, could occur and that serious reactions require immediate treatment. Ask patient about any previous hypersensitivity reactions to ZERBAXA, other beta-lactams (including cephalosporins) or other allergens [see Warnings and Precautions (5.2)].
Potentially Serious Diarrhea
Advise patient that diarrhea is a common problem caused by antibacterial drugs. Sometimes, frequent watery or bloody diarrhea may occur and may be a sign of a more serious intestinal infection. If severe watery or bloody diarrhea develops, tell patient to contact his or her healthcare provider [see Warnings and Precautions (5.3)].
Antibacterial Resistance
Patients should be counseled that antibacterial drugs including ZERBAXA should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When ZERBAXA is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by ZERBAXA or other antibacterial drugs in the future [see Warnings and Precautions (5.4)].
-
SPL UNCLASSIFIED SECTION
Manufactured for: Merck Sharp & Dohme Corp., a subsidiary of
MERCK & CO., INC., Whitehouse Station, NJ 08889, USAManufactured by: Steri-Pharma, LLC
Syracuse, NY 13202, USAFor patent information: www.merck.com/product/patent/home.html
Copyright © 2015-2019 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
All rights reserved.uspi-mk7625a-iv-1906r004
-
PRINCIPAL DISPLAY PANEL - 1.5 g Vial Carton
NDC: 67919-030-01
10 Single-Dose vialsZerbaxa®
1.5 g per vial*
(ceftolozane and tazobactam) for injection*Ceftolozane 1 gram (equivalent to 1.147 g ceftolozane sulfate)
and Tazobactam 0.5 g (equivalent to 0.537 g tazobactam sodium)For Intravenous Infusion
Rx only
SterileGTIN 00367919030017

-
INGREDIENTS AND APPEARANCE
ZERBAXA
ceftolozane and tazobactam injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 67919-030 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ceftolozane sulfate (UNII: 7R247U84HY) (ceftolozane - UNII:37A4IES95Q) ceftolozane 1 g in 10 mL tazobactam sodium (UNII: UXA545ABTT) (tazobactam - UNII:SE10G96M8W) tazobactam 0.5 g in 10 mL Inactive Ingredients Ingredient Name Strength Arginine (UNII: 94ZLA3W45F) 600 mg in 10 mL sodium chloride (UNII: 451W47IQ8X) 487 mg in 10 mL anhydrous citric acid (UNII: XF417D3PSL) 21 mg in 10 mL Product Characteristics Color WHITE (white to yellow) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 67919-030-01 10 in 1 CARTON 12/19/2014 1 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA206829 12/19/2014 Labeler - Merck Sharp & Dohme Corp. (001317601) Registrant - Cubist Pharmaceuticals LLC (808394928)
Trademark Results [ZERBAXA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ZERBAXA 85904044 4744146 Live/Registered |
MERCK SHARP & DOHME CORP. 2013-04-15 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.