IMCIVREE- setmelanotide solution
Imcivree by
Drug Labeling and Warnings
Imcivree by is a Prescription medication manufactured, distributed, or labeled by Rhythm Pharmaceuticals, Inc. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use IMCIVREE safely and effectively. See full prescribing information for IMCIVREE.
IMCIVREE® (setmelanotide) injection, for subcutaneous use
Initial U.S. Approval: 2020RECENT MAJOR CHANGES
INDICATIONS AND USAGE
IMCIVREE is a melanocortin 4 (MC4) receptor agonist indicated to reduce excess body weight and maintain reduction long term in adults and pediatric patients aged (1):
- 4 years and older with acquired hypothalamic obsesity (HO).
- 2 years and older with Bardet-Biedl syndrome (BBS).
- 2 years and older with pro-opiomelanocortin (POMC), proprotein convertase subtilisin/kexin type 1 (PCSK1), or leptin receptor (LEPR) deficiency confirmed by an genetic test demonstrating variants in POMC, PCSK1, or LEPRgenes that are interpreted as pathogenic, likely pathogenic, or of uncertain significance (VUS).
Limitations of Use:
IMCIVREE is not indicated for the treatment of patients with the following conditions as IMCIVREE would not be expected to be effective:
- Obesity due to suspected POMC, PCSK1, or LEPR-deficiency with POMC, PCSK1, or LEPRvariants classified as benign or likely benign. ( 1)
- Other types of obesity not related to acquired HO, BBS or POMC, PCSK1 or LEPR deficiency, including obesity associated with other genetic syndromes and general (polygenic) obesity. ( 1)
DOSAGE AND ADMINISTRATION
- Select patients for treatment who have a clinical diagnosis of acquired HO or BBS or who have genetically determined or suspected deficiency of POMC, PCSK1, or LEPR. ( 2.1)
- Recommended starting dosage injected subcutaneously for:
- Adults and pediatric patients aged 4 years and older with acquired HO is 0.5 mg (0.05 mL) once daily for 2 weeks. ( 2.2)
- Adults and pediatric patients aged 12 years and older with BBS or POMC, PCSK1, or LEPR deficiency is 2 mg (0.2 mL) once daily for 2 weeks. ( 2.3)
- Pediatric patients aged 6 to less than 12 years with BBS or POMC, PCSK1, or LEPR deficiency is 1 mg (0.1 mL) once daily for 2 weeks. ( 2.3)
- Pediatric patients aged 2 to less than 6 years with BBS or POMC, PCSK1, or LEPR deficiency is 0.5 mg (0.05 mL) once daily for 2 weeks. ( 2.4)
- Recommended maintenance dosage for adults and pediatric patients aged 6 years and older for all indications is 3 mg (0.3 mL) injected subcutaneously once daily. ( 2.2, 2.3)
- Recommended maintenance dose for pediatric patients with acquired HO aged 4 years to less than 6 years and for pediatric patients with BBS or POMC, PCSK1, or LEPR deficiency aged 2 to less than 6 years is determined by body weight. ( 2.2, 2.3)
- For recommended dosage in patients with renal impairment, see Full Prescribing Information. ( 2.4)
- For titration and administration recommendations, see Full Prescribing Information. ( 2.2, 2.3, 2.4, 2.5)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
Prior serious hypersensitivity to setmelanotide or any of the excipients in IMCIVREE ( 4)
WARNINGS AND PRECAUTIONS
- Disturbance in Sexual Arousal:Spontaneous penile erections in males and sexual adverse reactions in females have occurred. Inform patients that these events may occur and instruct patients who have an erection lasting longer than 4 hours to seek emergency medical attention. ( 5.1)
- Depression and Suicidal Ideation:Depression and suicidal ideation have occurred. Monitor patients for new onset or worsening depression or suicidal thoughts or behaviors. Consider discontinuing IMCIVREE if patients experience suicidal thoughts or behaviors, or clinically significant or persistent depression symptoms occur. ( 5.2)
- Hypersensitivity Reactions:Serious hypersensitivity reactions (e.g., anaphylaxis) have been reported. If suspected, advise patients to promptly seek medical attention and discontinue IMCIVREE. ( 5.3).
- Skin Hyperpigmentation, Darkening of Pre-Existing Nevi, and Development of New Melanocytic Nevi:Generalized increased skin pigmentation, darkening of pre-existing nevi, and development of new nevi have occurred. Perform a full body skin examination prior to initiation and periodically during treatment to monitor pre-existing and new pigmentary lesions. ( 5.4)
- Acute Adrenal Insufficiency in Patients with Acquired HO: Monitor patients for signs of acute adrenal insufficiency. ( 5.5)
-
Sodium Imbalance in Patients with Acquired HO and Central Diabetes Insipidus: Monitor patients for signs and symptoms of hyponatremia and
hypernatremia. (5.6)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥20% in at least 1 indication) included skin hyperpigmentation, injection site reactions, nausea, headache, diarrhea, abdominal pain, vomiting, depression, and spontaneous penile
erection. ( 6.1)To report SUSPECTED ADVERSE REACTIONS, contact Rhythm Pharmaceuticals at 1-833-789-6337 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
- Lactation:not recommended when breastfeeding ( 8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Limitations of Use:
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Dosage in Patients with Acquired HO
2.3 Recommended Dosage in Patients with BBS or POMC, PCSK1, or LEPR Deficiency
2.4 Recommended Dosage in Patients with Renal Impairment
2.5 Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Disturbance in Sexual Arousal
5.2 Depression and Suicidal Ideation
5.3 Hypersensitivity Reactions
5.4 Skin Hyperpigmentation, Darkening of Pre-Existing Nevi, and Development of New Melanocytic Nevi
5.5 Acute Adrenal Insufficiency in Patients with Acquired HO
5.6 Sodium Imbalance in Patients with Acquired HO and Central Diabetes Insipidus
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Acquired HO (Adults and Pediatric Patients Aged 4 Years and Older)
14.2 Bardet-Biedl Syndrome (Adults and Pediatric Patients Aged 6 Years and Older)
14.3 POMC, PCSK1, and LEPR Deficiency (Adults and Pediatric Patients Aged 6 Years and Older)
14.4 POMC, PCSK1, and LEPR Deficiency and BBS (Pediatric Patients Aged 2 to Less Than 6 Years)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
IMCIVREE is indicated to reduce excess body weight and maintain weight reduction long term in adults and pediatric patients aged [ see Dosage and Administration (2.1)]:
- 4 years and older with acquired hypothalamic obesity (HO)
- 2 years and older with syndromic or monogenic obesity due to:
o Bardet-Biedl syndrome (BBS)
o Pro-opiomelanocortin (POMC), proprotein convertase subtilisin/kexin type 1 (PCSK1), or leptin receptor (LEPR) deficiency confirmed by genetic testing demonstrating variants in POMC, PCSK1, or LEPR genes that are interpreted as pathogenic, likely pathogenic, or of uncertain significance (VUS).
1.1 Limitations of Use:
IMCIVREE is not indicated for the treatment of patients with the following conditions as IMCIVREE would not be expected to be effective:
Obesity due to suspected POMC, PCSK1, or LEPR deficiency with POMC, PCSK1, or LEPR variants classified as benign or likely benign.
Other types of obesity not related to acquired HO, BBS, or POMC, PCSK1, or LEPR deficiency, including obesity associated with other genetic syndromes and general (polygenic) obesity -
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Acquired HO
- Select patients for treatment with IMCIVREE who have acquired HO [see Clinical Studies (14.1)].
BBS
- Select patients for treatment with IMCIVREE who have a clinical diagnosis of BBS [see Clinical Studies (14.2, 14.4)].Consider genetic confirmation in pediatric patients aged <6 years.
POMC, PCSK1, or LEPR Deficiency
- Select patients for treatment with IMCIVREE who have genetically determined or suspected deficiency of POMC, PCSK1, or LEPR [see Clinical Studies (14.3, 14.4)].
- Treat patients with variants in POMC, PCSK1, or LEPR genes that are interpreted as pathogenic, likely pathogenic, or of uncertain significance (VUS) in the clinical context of the patient [see Clinical Studies (14.3, 14.4)].
- An FDA-approved test for the detection of variants in the POMC, PCSK1, or LEPR genes is not available.
2.2 Recommended Dosage in Patients with Acquired HO
- Monitor patients for gastrointestinal (GI) adverse reactions during dosage initiation and titration [see Adverse Reactions (6.1)].
- If the starting dosage is:
- Not tolerated, discontinue the product.
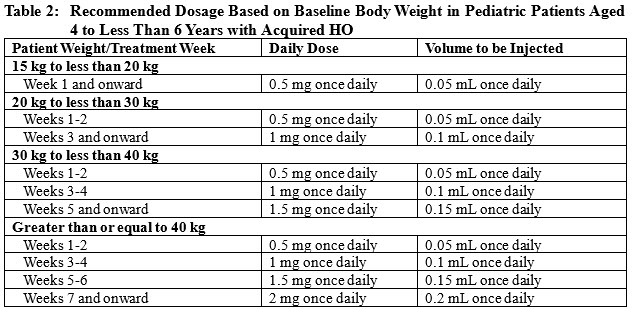
- Tolerated for 2 weeks, increase the dosage as presented in Table 1 or Table 2.
Adults and Pediatric Patients Aged 6 Years and Older
- The recommended starting dosage is 0.5 mg (0.05 mL) injected subcutaneously once daily for 2 weeks.
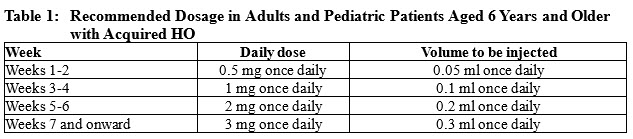
Pediatric Patients Aged 4 to Less Than 6 Years
- The recommended starting dosage is 0.5 mg (0.05 mL) injected subcutaneously once daily for 2 weeks.
2.3 Recommended Dosage in Patients with BBS or POMC, PCSK1, or LEPR Deficiency
Adults and Pediatric Patients Aged 12 Years and Older
- The recommended starting dosage is 2 mg (0.2 mL) injected subcutaneously once daily for 2 weeks in adults and pediatric patients aged 12 years and older.
- Monitor patients for GI adverse reactions during dosage initiation and titration [see Adverse Reactions (6.1)].
- If the starting dosage is:
- Not tolerated, reduce the dosage to 1 mg (0.1 mL) once daily. If the 1 mg once daily dosage is tolerated for at least 1 week, increase the dosage to 2 mg (0.2 mL) once daily.
- Tolerated for 2 weeks, increase the dosage to 3 mg (0.3 mL) once daily. If the 3 mg once daily dosage is not tolerated, decrease the dosage to 2 mg (0.2 mL) once daily.
- The recommended maintenance dosage is 3 mg (0.3 mL) injected subcutaneously once daily.
Pediatric Patients Aged 6 to Less Than 12 Years
- The recommended starting dosage is 1 mg (0.1 mL) injected subcutaneously once daily for 2 weeks in pediatric patients aged 6 to less than 12 years.
- Monitor patients for GI adverse reactions during dosage initiation and titration [see Adverse Reactions (6.1)].
- If the starting dosage is:
- Not tolerated, reduce the dosage to 0.5 mg (0.05 mL) once daily. If the 0.5 mg once daily dosage is tolerated for at least 1 week, increase the dosage to 1 mg (0.1 mL) once daily.
- Tolerated for 2 weeks, increase the dosage to 2 mg (0.2 mL) once daily. If the 2 mg daily dosage is:
- Not tolerated, reduce the dosage to 1 mg (0.1 mL) once daily.
- Tolerated, increase the dosage to 3 mg (0.3 mL) once daily.
- The recommended maintenance dosage is 3 mg (0.3 mL) injected subcutaneously once daily.
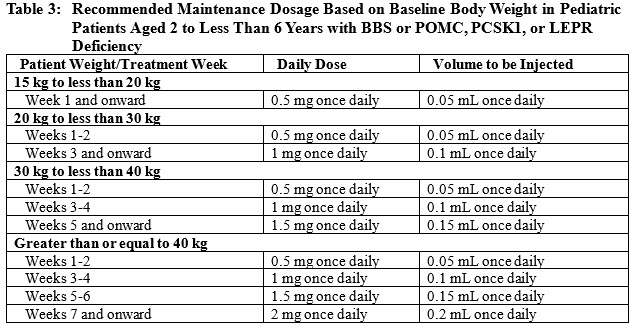
Pediatric Patients Aged 2 to Less Than 6 Years
- The recommended starting dosage is 0.5 mg (0.05 mL) injected subcutaneously once daily for 2 weeks in pediatric patients aged 2 to less than 6 years.
- Monitor patients for GI adverse reactions during dosage initiation and titration [see Adverse Reactions (6.1)].
- If the starting dosage is:
-
- Not tolerated, discontinue the product.
- Tolerated for 2 weeks, increase the dosage based on baseline body weight, as presented in Table 3.
2.4 Recommended Dosage in Patients with Renal Impairment
Recommended Dosage in Patients with End Stage Renal Disease [estimated glomerular filtration (eGFR) less than 15 mL/min/1.73 m 2]
IMCIVREE is not recommended for use in patients with end stage renal disease.
Recommended Dosage in Patients with Severe Renal Impairment (eGFR of 15 to 29 mL/min/1.73 m 2)
Adults and Pediatric Patients Aged 4 Years and Older with Acquired HO
IMCIVREE is not recommended for use in adults and pediatric patients aged 4 years and older with acquired HO and severe renal impairment.
Adults and Pediatric Patients Aged 12 Years and Older with BBS or POMC, PCSK1, or LEPR Deficiency
- The recommended starting dosage is 0.5 mg (0.05 mL) injected subcutaneously once daily for 2 weeks in adults and pediatric patients aged 12 years and older with severe renal impairment.
- Monitor patients for GI adverse reactions during dosage initiation and titration [see Adverse Reactions (6.1)].
- If the recommended starting dosage is [see Use in Specific Populations (8.6)]:
- Not tolerated, discontinue IMCIVREE.
- Tolerated for 2 weeks, increase the dosage to 1 mg (0.1 mL) once daily. If the 1 mg daily dosage is tolerated for at least 1 week, increase the dosage to 1.5 mg (0.15 mL) once daily. The recommended maintenance dosage is 1.5 mg (0.15 mL) injected subcutaneously once daily [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
Pediatric Patients Ages 6 Years to Less Than 12 Years with BBS or POMC, PCSK1, or LEPR Deficiency
- The recommended starting dosage is 0.5 mg (0.05 mL) injected subcutaneously once daily for 2 weeks in pediatric patients aged 6 to less than 12 years with severe renal impairment.
- Monitor patients for GI adverse reactions during dosage initiation and titration [see Adverse Reactions (6.1)].
- If the recommended starting dosage is [see Use in Specific Populations (8.6)]:
-
- Not tolerated, discontinue IMCIVREE.
- Tolerated for 2 weeks, increase the dosage to 1 mg (0.1 mL) injected subcutaneously once daily. The recommended maintenance dosage is 1 mg (0.1 mL) injected subcutaneously once daily [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
Pediatric Patients Aged 2 to Less Than 6 Years Weighing at Least 20 kg with BBS or POMC, PCSK1, or LEPR Deficiency
- The recommended starting dosage is 0.5 mg (0.05 mL) injected subcutaneously once daily for 2 weeks in pediatric patients aged 2 to less than 6 years with severe renal impairment and weight of at least 20 kg.
- The use of IMCIVREE in pediatric patients aged 2 to less than 6 years with weight less than 20 kg and severe renal impairment is not recommended [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
-
- Monitor patients for GI adverse reactions during dosage initiation and titration [see Adverse Reactions (6.1)].
- If the recommended starting dosage is [see Use in Specific Populations (8.6)]:
- Not tolerated, discontinue IMCIVREE.
- Tolerated for 2 weeks, increase the dosage based on baseline body weight, as presented in Table 4 [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
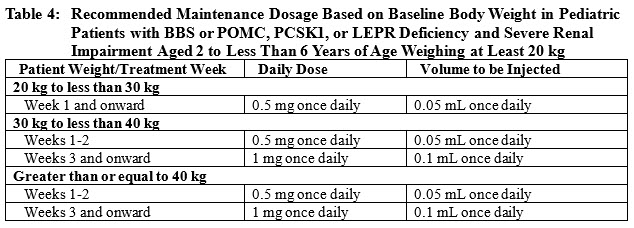
- The recommended maintenance dosage is 1 mg (0.1 mL) injected subcutaneously once daily [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)]. Monitor patients for adverse reactions [see Adverse Reactions (6.1)].
Recommended Dosage in Patients with Mild (eGFR of 60 to 89 mL/min/1.73 m 2) or Moderate (eGFR of 30 to 59 mL/min/1.73 m 2) Renal Impairment
The recommended dosage in patients with acquired HO, BBS, or POMC, PCSK1, or LEPR Deficiency and mild or moderate renal impairment is the same as in those with normal kidney function [see Dosage and Administration (2.2, 2.3)].
2.5 Administration Instructions
- Prior to initiation of IMCIVREE, train patients and their caregivers on proper injection technique. Instruct them to use a 1-mL syringe with a 28-gauge or 29-gauge needle appropriate for subcutaneous injection.
- Remove IMCIVREE from the refrigerator approximately 15 minutes prior to administration. Alternatively, warm IMCIVREE prior to administration by rolling the vial gently between the palms of the hands for 60 seconds.
- Inspect IMCIVREE visually before use. It should appear clear to slightly opalescent, colorless to slightly yellow. Do not use if particulate matter or discoloration is seen.
- Administer IMCIVREE once daily, at the beginning of the day, without regard to meals.
- Inject IMCIVREE subcutaneously in the abdomen, thigh, or arm, rotating to a different site each day. Do not administer IMCIVREE intravenously or intramuscularly.
- If a dose is missed, resume the once daily regimen as prescribed with the next scheduled dose.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Disturbance in Sexual Arousal
Sexual adverse reactions may occur in patients treated with IMCIVREE. Spontaneous penile erections and increased frequency of penile erections in males occurred in clinical trials with IMCIVREE [see Adverse Reactions (6.1)].
Inform patients that these events may occur and instruct patients who have an erection lasting longer than 4 hours to seek emergency medical attention.
5.2 Depression and Suicidal Ideation
Some drugs that target the central nervous system, such as IMCIVREE, may cause depression or suicidal ideation [see Adverse Reactions (6.1)]. Patients with a history of depression or suicidal ideation may be at increased risk for recurrent episodes while taking IMCIVREE.
Monitor patients for new onset or worsening of depression, suicidal thoughts or behavior, or any unusual changes in mood or behavior. Consider discontinuing IMCIVREE if patients experience suicidal thoughts or behaviors or if clinically significant or persistent depression symptoms occur.5.3 Hypersensitivity Reactions
Serious hypersensitivity reactions, including anaphylaxis, have been reported with IMCIVREE. These reactions generally occurred within minutes to hours after injecting IMCIVREE [see Adverse Reactions ( 6.2)]. If hypersensitivity reactions occur, advise patients to promptly seek medical attention and discontinue use of IMCIVREE. IMCIVREE is contraindicated in patients with a prior serious hypersensitivity reaction to setmelanotide or any of the excipients in IMCIVREE.
5.4 Skin Hyperpigmentation, Darkening of Pre-Existing Nevi, and Development of New Melanocytic Nevi
Generalized or focal increases in skin pigmentation occurred in the majority of IMCIVREE-treated patients in clinical trials [ see Adverse Reactions (6.1) and Clinical Pharmacology (12.1)]. This effect is reversible upon discontinuation of the drug.
IMCIVREE may also cause the development of new melanocytic nevi or darkening of pre-existing nevi due to its pharmacologic effect.
Perform a full body skin examination prior to initiation and periodically during treatment with IMCIVREE to monitor pre-existing and new skin pigmented lesions.5.5 Acute Adrenal Insufficiency in Patients with Acquired HO
In a clinical trial of adults and pediatric patients aged 4 years and older with acquired HO and secondary adrenal insufficiency, serious adverse reactions related to acute adrenal insufficiency were reported by 5% of IMCIVREE-treated patients and no placebo-treated patients. In patients with secondary adrenal insufficiency, monitor for clinical signs of acute adrenal insufficiency.
5.6 Sodium Imbalance in Patients with Acquired HO and Central Diabetes Insipidus
In a clinical trial of adults and pediatric patients aged 4 years and older with acquired HO and concomitant central diabetes insipidus (DI)/arginine vasopressin (AVP) deficiency, hyponatremia was reported in 6% of IMCIVREE-treated patients and 2% of placebo-treated patients, and hypernatremia was reported in 5% of IMCIVREE-treated patients and 4% of placebo-treated patients.
In patients with acquired HO and concomitant DI/AVP deficiency, monitor serum sodium levels with changes in fluid intake and hydration status. Adjust the doses of concomitant therapies for DI/AVP deficiency as needed.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Disturbance in Sexual Arousal [see Warnings and Precautions (5.1)]
- Depression and Suicidal Ideation [see Warnings and Precautions (5.2)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.3)]
- Skin Hyperpigmentation, Darkening of Pre-Existing Nevi, and Development of New Melanocytic Nevi [see Warnings and Precautions (5.4)]
- Acute Adrenal Insufficiency in Patients with Acquired HO [ see Warnings and Precautions (5.5)]
- Sodium Imbalance in Patients with Acquired HO and Central Diabetes Insipidus [ see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Acquired HO (Adults and Pediatric Patients Aged 4 Years and Older)
The safety of IMCIVREE was evaluated in a randomized, double-blind, placebo-controlled clinical trial which included a dose titration period of 4 to 8 weeks and a 52-week treatment period, in 142 patients aged 4 years and older with acquired HO (Trial 1) [see Clinical Studies (14.1)]. The trial duration was 56 to 60 weeks.
Table 5 summarizes the adverse reactions that occurred in 5% or more of the IMCIVREE-treated patients and more frequently than placebo-treated patients in Trial 1.
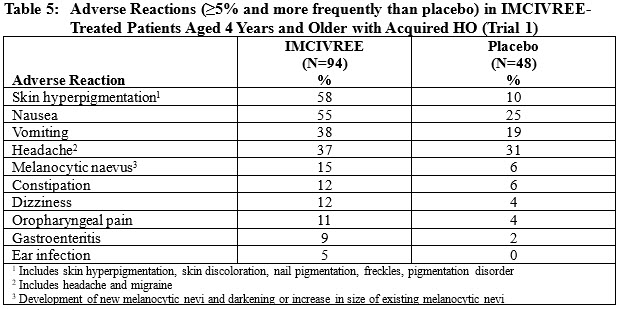
Other Adverse Reactions in Patients Aged 4 Years and Older with Acquired HODisturbance in Sexual Arousal
Spontaneous penile erections and increased frequency of penile erections were reported in 7% of IMCIVREE-treated patients and 4% of placebo-treated patients.
Acute Adrenal Insufficiency
Serious adverse reactions related to acute adrenal insufficiency were reported by 5% of IMCIVREE-treated patients and no placebo-treated patients.
Sodium Imbalance
Among patients with acquired HO and concomitant central diabetes insipidus (DI)/arginine vasopressin (AVP) deficiency, hyponatremia was reported in 6% of IMCIVREE-treated patients and 2% of placebo-treated patients, and hypernatremia was reported in 5% of IMCIVREE-treated patients and 4% of placebo-treated patients.
Bardet-Biedl Syndrome (Adults and Pediatric Patients Aged 6 Years and Older)
The safety of IMCIVREE was evaluated in a clinical trial, which included a 14‑week, randomized, double-blind, placebo-controlled period followed by a 52-week open-label treatment period, in 44 patients aged 6 years and older with obesity and a clinical diagnosis of BBS (Trial 2) [see Clinical Studies (14.2)]. The trial duration was 66 weeks.
During the 14-week placebo-controlled period in Trial 2, the most common reported adverse reactions in IMCIVREE-treated patients when compared to placebo-treated patients were hyperpigmentation disorders (67% vs 0%, respectively) and vomiting (11% vs 0%, respectively).
Adverse reactions were also evaluated during the 52-week active-treatment period, defined as the period from randomization to Week 52 in patients initially randomized to IMCIVREE, and from Week 14 to Week 66 in patients initially randomized to placebo. Table 6 summarizes the adverse reactions that occurred in 2 or more IMCIVREE-treated patients in Trial 2 during the 52-week active treatment period.
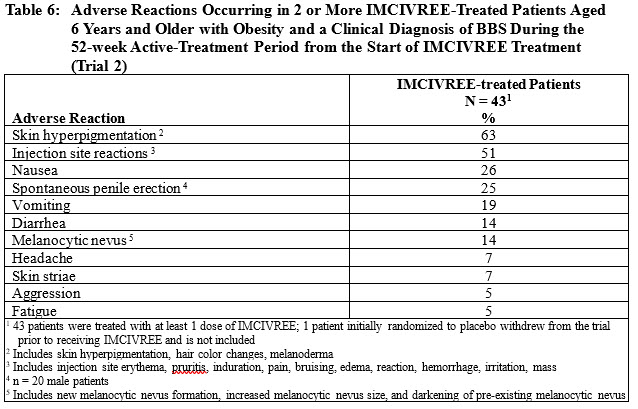
POMC, PCSK1, and LEPR Deficiency (Adults and Pediatric Patients Aged 6 Years and Older)The safety of IMCIVREE was evaluated in two 52-week, open-label clinical trials of 27 patients aged 6 years and older with obesity due to POMC, PCSK1, or LEPR deficiency with POMC, PCSK1, or LEPRgenes that are interpreted as pathogenic, likely pathogenic, or of uncertain significance (Trial 3 and Trial 4) [see Clinical Studies (14.3)].
Table 7 summarizes the adverse reactions that occurred in the open-label trials during the first 52 weeks of treatment in 3 or more patients treated with IMCIVREE.
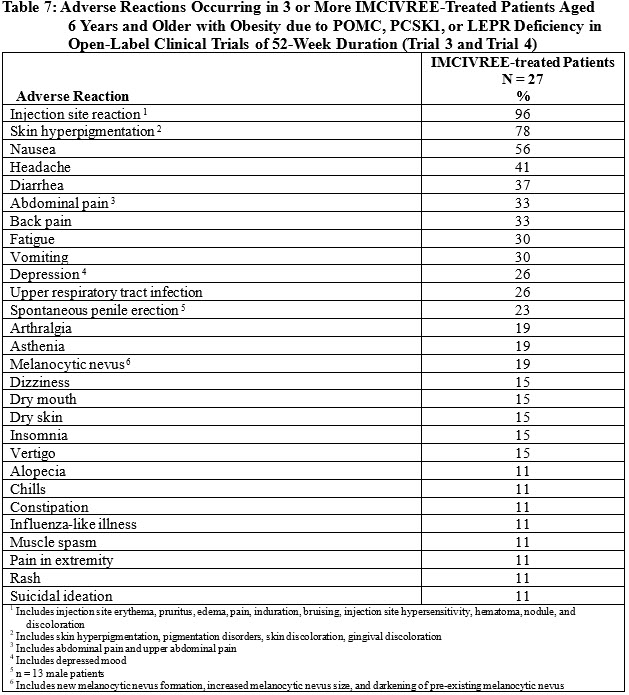
POMC, PCSK1, and LEPR Deficiency and BBS (Pediatric Patients Aged 2 to less than 6 Years)The safety of IMCIVREE was evaluated in one 52-week, open-label clinical trial of 12 patients aged 2 to less than 6 years with obesity due to POMC, PCSK1, or LEPR deficiency with POMC, PCSK1, or LEPRgenes that are interpreted as pathogenic, likely pathogenic, or of uncertain significance, or obesity due to BBS (Trial 5) [see Clinical Studies (14.4)]. No patients with PCSK1 were enrolled in the trial.
Table 8 summarizes the adverse reactions that occurred in the open-label trial during 52 weeks of treatment in 3 or more patients treated with IMCIVREE.
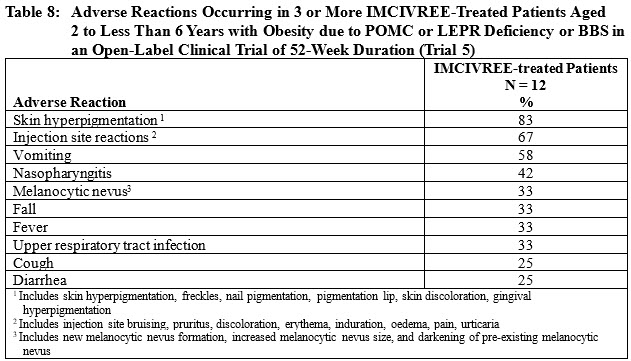
Other Adverse Reactions in Patients Aged 2 to less than 6 Years with Obesity due to POMC or LEPR Deficiency or BBSDisturbance in Sexual Arousal
Spontaneous penile erections were reported in 8% of IMCIVREE-treated patients.
Depression
Depressed mood was reported in 8% of IMCIVREE-treated patients
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of IMCIVREE. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Hypersensitivity, including anaphylaxis
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Discontinue IMCIVREE when pregnancy is recognized unless the benefits of therapy outweigh the potential risks to the fetus.
IMCIVREE contains the preservative benzyl alcohol. Because benzyl alcohol is rapidly metabolized by a pregnant woman, benzyl alcohol exposure in the fetus is unlikely.
There are no available data with IMCIVREE in pregnant women to inform a drug-associated risk for major birth defects and miscarriage, or adverse maternal or fetal outcomes. For the general US population, weight loss offers no potential benefit to a pregnant woman and may result in fetal harm (see Clinical Considerations).In animal reproduction studies, setmelanotide subcutaneously administered to pregnant rats from before mating to the end of organogenesis was not teratogenic at doses 11 times the maximum recommended human dose (MRHD) of 3 mg. Setmelanotide subcutaneously administered to pregnant rabbits during the period of organogenesis was not teratogenic at clinical doses. Setmelanotide administered subcutaneously to pregnant rats during organogenesis through lactation did not result in adverse developmental effects at doses 7 times the MRHD ( see Data).
The estimated background risk of birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Maternal obesity increases the risk for congenital malformations, including neural tube defects, cardiac malformations, oral clefts, and limb reduction defects. In addition, weight loss during pregnancy may result in fetal harm including increased risk of small for gestational age. Appropriate weight gain based on pre-pregnancy weight is currently recommended for all pregnant women, including those who are already overweight or have obesity, due to the obligatory weight gain that occurs in maternal tissues during pregnancy.
Data
Animal Data
Embryo-fetal development was evaluated in female rats administered setmelanotide subcutaneously during mating to end of major organogenesis (14 days prior to mating to gestation day 17) at doses of 0.5, 3, and 5 mg/kg/day, resulting in exposures up to 11 times the human exposure at MRHD of 3 mg, based on AUC. Dose-related decreases in maternal food intake and body weight gain were observed during the premating period but not during gestation. No evidence of embryo-fetal toxicity was observed.
Embryo-fetal development was evaluated in pregnant rabbits subcutaneously administered setmelanotide during organogenesis (gestation days 7 to 19) at doses of 0.05, 0.1, and 0.2 mg/kg/day, resulting in clinically relevant exposures at the MRHD, based on AUC. Decreases in maternal food consumption and body weight were observed at all doses. Increases in embryo-fetal resorptions and post-implantation losses were observed at ≥0.1 mg/kg/day in the presence of significant maternal toxicity, and fetal body weights were 7% lower than controls at 0.2 mg/kg/day.
Pre- and post-natal development was evaluated in rats subcutaneously administered setmelanotide during organogenesis and continuing until weaning (gestation day 6 to lactation day 21) at doses of 0.5, 3.0, and 5.0 mg/kg/day, which resulted in exposures up to 7 times the human exposure at the MRHD, based on AUC. Pup body weights at birth were 9% lower than controls at 3.0 and 5.0 mg/kg/day, which was consistent with reduced maternal body weight gain and food consumption during gestation. No adverse setmelanotide-related effects on pup survival, growth, maturation, visual function, neurobehavioral performance, or reproductive performance were observed up to the highest dose.
8.2 Lactation
Risk Summary
Treatment with IMCIVREE is not recommended for use while breastfeeding.
IMCIVREE from multiple-dose vials contains the preservative benzyl alcohol. Because benzyl alcohol is rapidly metabolized by a lactating woman, benzyl alcohol exposure in the breastfed infant is unlikely.
There is no information on the presence of setmelanotide or its metabolites in human milk, the effects on the breastfed infant, or the effects on milk production. However, setmelanotide is present in the milk of rats ( see Data). When a drug is present in rat milk, it is likely that the drug will be present in human milk.
Data
Dose-related setmelanotide concentrations were observed in milk 2 hours after subcutaneous injection in the preweaning phase of a pre- and post-natal development study in rats. No quantifiable setmelanotide concentrations were detected in plasma from nursing pups on postnatal Day 11.
8.4 Pediatric Use
Acquired HO
The safety and effectiveness of IMCIVREE have been established to reduce excess body weight and maintain weight reduction long term in pediatric patients aged 4 years and older with acquired HO. Use of IMCIVREE for this indication is supported by evidence from a 56- to 60‑week randomized, double-blind, placebo-controlled trial that included 76 pediatric patients with acquired HO aged 4 to 17 years [see Clinical Studies (14.1)].
Adverse reactions with IMCIVREE treatment in pediatric patients aged 4 to 17 years with acquired HO were generally similar to those reported in adults [ see Adverse Reactions(6.1)]. Perform a full body skin examination prior to initiation and periodically during treatment with IMCIVREE to monitor pre-existing and new skin pigmented lesions [ see Warnings and Precautions (5.4)].
The safety and effectiveness of IMCIVREE have not been established in pediatric patients younger than 4 years of age with acquired HO.
BBS or POMC, PCSK1, or LEPR Deficiency
The safety and effectiveness of IMCIVREE have been established to reduce excess body weight and maintain weight reduction long term in pediatric patients aged 2 years and older with obesity due to:
- BBS [see Clinical Studies (14.2, 14.4)]
- POMC, PCSK1, or LEPR deficiency with variants in POMC, PCSK1, or LEPRgenes that are interpreted as pathogenic, likely pathogenic, or of uncertain significance (VUS) [see Clinical Studies (14.3, 14.4)]
Use of IMCIVREE for these indications is supported by evidence from one 66-week trial, which included a 14-week, randomized, double-blind, placebo-controlled period followed by a 52-week open-label period, and included 22 pediatric patients with BBS aged 6 to 17 years (Trial 2); from two 1-year, open-label trials that included 9 pediatric patients with POMC, PCSK1, or LEPR deficiency aged 6 to 17 years (Trial 3 and Trial 4); and one 1-year, open-label trial that included 12 pediatric patients with POMC or LEPR deficiency or BBS aged 2 to less than 6 years (Trial 5) [see Clinical Studies (14.2, 14.3, 14.4)].
Adverse reactions with IMCIVREE treatment in pediatric patients aged 2 to less than 6 years with BBS, POMC, PCSK1, or LEPR deficiency were generally similar to those reported in adults and in pediatric patients aged 6 years and older. Pediatric patients with BBS, POMC, PCSK1, or LEPR deficiency treated with IMCIVREE had greater incidences of vomiting, skin hyperpigmentation, and new or darkening nevi compared to adults with BBS, POMC, PCSK1, or LEPR deficiency treated with IMCIVREE [see Adverse Reactions(6.1)]. Perform a full body skin examination prior to initiation and periodically during treatment with IMCIVREE to monitor pre-existing and new skin pigmented lesions [see Warnings and Precautions (5.4)].
The safety and effectiveness of IMCIVREE have not been established in pediatric patients younger than 2 years of age with BBS, POMC, PCSK1, or LEPR deficiency.
8.5 Geriatric Use
Clinical trials of IMCIVREE did not include patients aged 65 and over. It is not known whether geriatric patients would respond differently than younger adult patients.
8.6 Renal Impairment
Patients with severe renal impairment have a higher exposure of setmelanotide relative to patients with normal kidney function. IMCIVREE is not recommended for use in adults and pediatric patients aged 4 years and older with acquired HO and severe renal impairment.
For patients with BBS or POMC, PCSK1, or LEPR deficiency, reduce the recommended starting and maintenance dosage of IMCIVREE in adults and pediatric patients 2 years of age and older with severe renal impairment (eGFR 15 29 mL/min/1.73 m 2). The use of IMCIVREE in pediatric patients aged 2 to less than 6 years with weight less than 20 kg and severe renal impairment is not recommended [ see Dosage and Administration (2.4), Clinical Pharmacology (12.3)].
The recommended dosage in patients with mild (eGFR of 60 89 mL/min/1.73 m 2) or moderate renal impairment (eGFR of 30-59 mL/min/1.73 m 2) is the same as those with normal kidney function [ see Clinical Pharmacology (12.3)].
IMCIVREE is not recommended for use in patients with end stage renal disease (eGFR less than 15 mL/min/1.73 m 2). - 10 OVERDOSAGE
-
11 DESCRIPTION
IMCIVREE contains setmelanotide acetate, a melanocortin 4 (MC4) receptor agonist. Setmelanotide is an 8 amino acid cyclic peptide analog of endogenous melanocortin peptide α‑MSH (alpha-melanocyte stimulating hormone).
The chemical name for setmelanotide acetate is acetyl-L-arginyl-L-cysteinyl-D-alanyl-L-histidinyl-D-phenylalanyl-L-arginyl-L-tryptophanyl-L-cysteinamide cyclic (2→8)-disulfide acetate. Its molecular formula is C 49H 68N 18O 9S 2(anhydrous, free-base), and molecular mass is 1117.3 Daltons (anhydrous, free-base).
The chemical structure of setmelanotide acetate is:
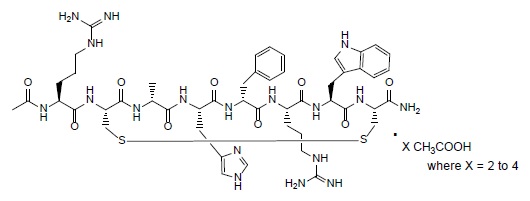
IMCIVREE (setmelanotide) injection is a sterile clear to slightly opalescent, colorless to slightly yellow solution for subcutaneous use. Each 1 mL vial of IMCIVREE contains 10 mg of setmelanotide provided as setmelanotide acetate, which is a salt with 2 to 4 molar equivalents of acetate, and the following inactive ingredients: 10 mg benzyl alcohol, 8 mg carboxymethylcellulose sodium (average MWt 90,500), 1 mg edetate disodium dihydrate, 100 mg N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl- glycero-3- phosphoethanolamine sodium salt, 11 mg mannitol, 5 mg phenol, hydrochloric acid, sodium hydroxide and Water for Injection. The pH of IMCIVREE is 5 to 6.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Setmelanotide is an MC4 receptor agonist with 20-fold less activity at the melanocortin 3 (MC3) and melanocortin 1 (MC1) receptors. MC4 receptors in the brain are involved in regulation of hunger, satiety, and energy expenditure. Based on nonclinical evidence, setmelanotide may re establish MC4 receptor pathway activity to reduce food intake and promote weight loss through decreased caloric intake and increased energy expenditure in patients with obesity due to acquired HO, BBS, or POMC, PCSK1, or LEPR deficiency associated with insufficient activation of the MC4 receptor. The MC1 receptor is expressed on melanocytes, and activation of this receptor leads to accumulation of melanin and increased skin pigmentation independently of ultraviolet light [see Warnings and Precautions (5.4) and Adverse Reactions (6.1)].
12.2 Pharmacodynamics
At a dose 2.3 times the maximum recommended dose, IMCIVREE does not prolong the QT interval to any clinically relevant extent.
Energy Expenditure
Short-term administration of IMCIVREE in 12 otherwise healthy patients with obesity increased resting energy expenditure and shifted substrate oxidation to fat. The safety and effectiveness of IMCIVREE have not been established in such patients and IMCIVREE is not approved to treat such patients [see Indications and Usage (1)].
12.3 Pharmacokinetics
The mean steady state setmelanotide Cmax,ss, AUCtau, and trough concentration for a 3-mg dose administered subcutaneously once daily was 31 ng/mL, 373 h*ng/mL, and 5 ng/mL, respectively simulated using individual PK model parameters from 109 adult patients with normal renal function. Steady-state plasma concentrations of setmelanotide were achieved within 2 days with daily dosing of 1-3 mg setmelanotide. The accumulation of setmelanotide in the systemic circulation during once-daily dosing over 12 weeks was approximately 30%. Setmelanotide AUC and Cmax increased proportionally following multiple-dose subcutaneous administration in the proposed dose range (1-3 mg).
Absorption
After subcutaneous injection of IMCIVREE, plasma concentrations of setmelanotide reached maximum concentrations at a median tmax of 8 h after dosing.
Distribution
The mean apparent volume of distribution of setmelanotide after subcutaneous administration of IMCIVREE 3 mg once daily was estimated to be 75.2 L. Protein binding of setmelanotide is 79.1%.
Elimination
The effective elimination half-life (t½) of setmelanotide was approximately 11 hours. The total apparent steady state clearance of setmelanotide following subcutaneous administration of IMCIVREE 3 mg once daily was estimated to be 7.15 L/h in a typical male patient weighing 120 kg (actual body weight) with normal renal function.
Metabolism
Setmelanotide is expected to be metabolized into small peptides by catabolic pathways.
Excretion
Approximately 39% of the administered setmelanotide dose was excreted unchanged in urine during the 24-hour dosing interval following subcutaneous administration of 3 mg once daily.
Specific Populations
No clinically significant differences in the pharmacokinetics of setmelanotide were observed based on sex or disease. The effect of age 65 years or older, pregnancy, or hepatic impairment on the pharmacokinetics of setmelanotide is unknown.
Pediatric Patients
IMCIVREE has been evaluated in pediatric patients aged 2 to less than 6 years, 6 to less than 12 years, and aged 12 to 17 years. Simulations were performed using population pharmacokinetic analysis for pediatric patients aged 2 to less than 6 years, following the maximum recommended doses in each of the body weight groups - 2 mg, 1.5 mg, 1 mg, and 0.5 mg in patients weighing ≥40 kg, 30 to <40 kg, 20 to <30 kg, and 15 to <20 kg, respectively. The analyses suggest that AUC and Cmax in pediatric patients aged 2 to less than 6 years are 52% and 75% higher in patients weighing ≥40 kg, 45% and 63% higher in patients weighing 30 to <40 kg, 24% and 38% higher in patients weighing 20 to <30 kg, and 17% and 14% lower in patients weighing 15 to <20 kg as compared to patients greater than or equal to 18 years (3 mg dose). For patients aged 6 to less than 12 years, the setmelanotide AUC and Cmax were 88% and 89% higher compared to patients greater than or equal to 18 years. For patients aged 12 to 17 years, the setmelanotide AUC and Cmax were both 26% higher as compared to patients aged greater than or equal to 18 years [ see Dosage and Administration (2.2, 2.3)].
Patients with Renal Impairment
Exposure parameters, AUC0-t and AUC0-inf, were approximately 13%-15%, 34%-35%, and 86% 96% higher for patients with mild, moderate, and severe renal impairment, respectively, as compared to patients with normal renal function [ see Dosage and Administration (2.4)].
Renal impairment did not appear to affect plasma protein binding. The average fraction unbound (fu) was approximately 0.2 and was independent of renal function.
Drug Interaction Studies
In vitro assessment of drug-drug interactions
Setmelanotide has low potential for pharmacokinetic drug-drug interactions related to cytochrome P450 (CYP), transporters and plasma protein binding.
In vivo assessment of drug-drug interactions
No clinical studies evaluating the drug-drug interaction potential of setmelanotide have been conducted.12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the trials described below with the incidence of ADA in other trials, including those of IMCIVREE or of other setmelanotide products.
Across 5 clinical trials with an exposure time of at least 52 weeks, 4 of 245 subjects (1.6%) were positive for antibodies against setmelanotide. Anti-setmelanotide antibodies prevalence by indication: BBS 3/50 (6.0%), AS 1/8 (12.5%), POMC deficiency (0/16), PCSK1 deficiency (0/2), LEPR deficiency (0/21), and acquired HO (0/142). Reported titers among ADA‑positive subjects were generally low.
In 5 clinical trials with an exposure time of at least 52 weeks anti-α-MSH antibodies were measured in 17 of 245 subjects (6.9%). Anti-α-MSH antibodies by genetic deficiency: BBS 5/50 (10.0%), AS 1/8 (12.5%), POMC 1/16 (6.3%), LEPR 6/21 (28.6%), PCSK1 0/2 (0%), and acquired HO 4/142 (2.8%). The reported titer of ADA against α-MSH was generally low.
In patients with acquired HO, BBS, or POMC, PCSK1, or LEPR deficiency, there is insufficient information to characterize the ADA response to setmelanotide or α-MSH and the effects of ADA on pharmacokinetics, pharmacodynamics, safety, or effectiveness of setmelanotide products. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Setmelanotide was not carcinogenic in Tg.rasH2 mice at doses up to 10 mg/kg/day when given subcutaneously for 26 weeks.
Setmelanotide was not mutagenic or clastogenic in a bacterial reverse mutation test, an in vitrochromosome aberration test in human lymphocyte cultures, or an in vivobone marrow micronucleus study in rats.
There were no effects on the fertility of male rats subcutaneously administered up to 3.0 mg/kg/day setmelanotide, which represents 9 times the MRHD of 3 mg, based on AUC. No effects on the fertility of female rats were observed with subcutaneous administration up to 5 mg/kg/day setmelanotide, which represents 11 times the MRHD of 3 mg, based on AUC.
-
14 CLINICAL STUDIES
14.1 Acquired HO (Adults and Pediatric Patients Aged 4 Years and Older)
Clinical Trial Overview
The efficacy of IMCIVREE for weight reduction in adults and pediatric patients aged 4 years and older with acquired HO was assessed in a randomized, double-blinded, placebo-controlled 56- to 60-week clinical trial [Trial 1 ( NCT05774756)]. The trial enrolled patients 4 years and older with acquired HO due to hypothalamic injury or dysfunction. Adult patients had a BMI of ≥30 kg/m 2andpediatric patients had a BMI ≥95 thpercentile for age and sex.
In Trial 1, eligible patients were randomized to either setmelanotide or placebo and entered an up to 8-week dose titration period followed by a 52-week treatment period. Efficacy analyses were conducted for 142 patients.
A total of 142 patients with acquired HO were randomized and analyzed; 47% were adults, 31% were aged 12 to less than 18 years, and 23% were 4 to less than 12 years; 40% were male; 75% were White, 11% were Asian, 5% were Black or African American, and 9% had an unknown or not reported race; 11% were Hispanic or Latino ethnicity and less than 1% had an unknown or not reported ethnicity; and the mean BMI was 36 kg/m 2(range: 21-70 kg/m 2).
Effect of IMCIVREE on BMI in Patients with Obesity and a Clinical Diagnosis of Acquired HO
The proportion of patients who discontinued trial drug in Trial 1 were 10.6% of the IMCIVREE-treated group and 12.5% of the placebo-treated group.
The primary efficacy parameter was mean percent change in BMI from baseline after 52 weeks on a therapeutic regimen of setmelanotide compared to placebo. After 52 weeks of treatment at the therapeutic dose, the mean percent change in BMI compared to placebo was -18.40% (Table 9), and greater proportions of patients treated with IMCIVREE achieved at least 5%, 10%, and 15% BMI reduction compared to placebo (Table 9 and Figure 1).

The cumulative frequency distributions of change in BMI are shown in Figure 1 for Trial 1. One way to interpret this figure is to select a change in BMI of interest on the horizontal axis and note the corresponding proportions of patients (vertical axis) in each treatment group who achieved at least that degree of BMI reduction. For example, a vertical line arising from ‑10% change in BMI in Trial 1 intersects the IMCIVREE and placebo curves at approximately 61%, and 5%, respectively, which correspond to the values shown in Table 1.
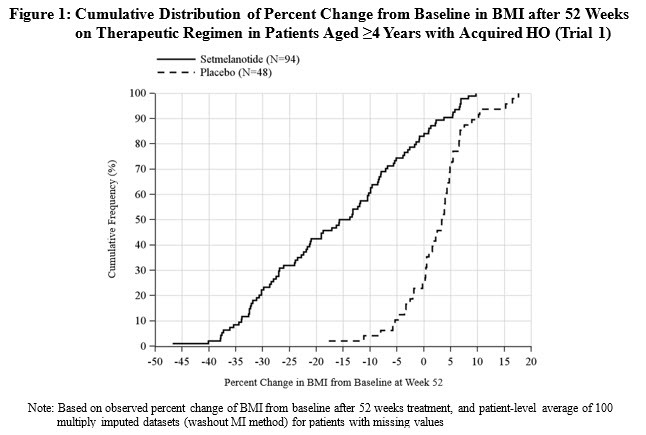
Note: Based on observed percent change of BMI from baseline after 52 weeks treatment, and patient-level average of 100 multiply imputed datasets (washout MI method) for patients with missing values
A reduction of BMI Z-score and BMI 95 thpercentile for pediatric patients less than 18 years of age was observed.
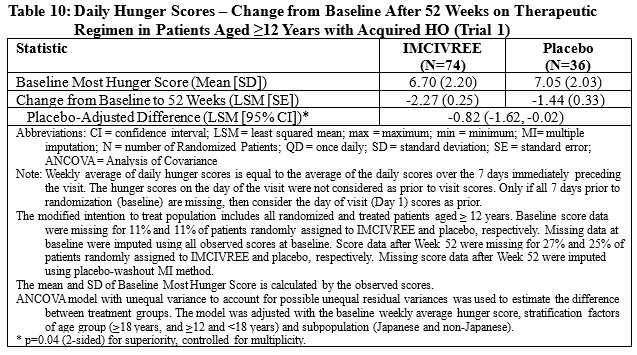
In Trial 1, patients 12 years and older who were able to self-report their hunger (n=110), recorded their daily maximal hunger in a diary, which was then assessed by the Daily Hunger Questionnaire Item 2. Hunger was scored on an 11-point scale from 0 (“not hungry at all”) to 10 (“hungriest possible”). After 52 weeks of treatment at the therapeutic dose, IMCIVREE resulted in a statistically significant reduction in hunger compared to placebo (Table 10).
IMCIVREE resulted in a reduction in waist circumference and general numeric improvements in blood pressure, lipids, and glycemic parameters compared with placebo.
14.2 Bardet-Biedl Syndrome (Adults and Pediatric Patients Aged 6 Years and Older)
Clinical Trial Overview
The efficacy of IMCIVREE for weight reduction in adults and pediatric patients aged 6 years and older with obesity and a clinical diagnosis of Bardet-Biedl syndrome (BBS) were assessed in a 66-week clinical trial, which included a 14 week randomized, double-blind, placebo-controlled period and a 52-week open-label period (Trial 2 [ NCT03746522]). The trial enrolled patients aged 6 years and above with obesity and a clinical diagnosis of BBS. Adult patients had a BMI of ≥30 kg/m2 and pediatric patients had weight ≥97th percentile using growth chart assessments.
In Trial 2, eligible patients entered a 14-week, randomized, double-blind, placebo-controlled treatment period (Period 1) in which patients received IMCIVREE or placebo, followed by a 52 week open-label treatment period (Period 2) in which all patients received IMCIVREE. To maintain the blind during Period 1, dose titration to a fixed dose of 3 mg given subcutaneously once daily was performed during the first 2 weeks of both Period 1 and Period 2.
Efficacy analyses were conducted in 44 patients at the end of Period 1 (Week 14, placebo-controlled data) and in 31 patients during the active-treatment period, defined as the period from randomization to Week 52 in patients initially randomized to IMCIVREE, and from Week 14 to Week 66 in patients initially randomized to placebo. Analyses of the active-treatment period include patients who had either completed 52 weeks from the start of IMCIVREE treatment or discontinued the trial early at the time of the prespecified data cutoff.
A total of 44 patients with obesity and a clinical diagnosis of BBS were enrolled; 50% were adults, 32% were aged 12 to <18 years, and 18% were aged 6 to <12 years; 46% were male; 77% were White, 5% were Black or African American, 2% were Asian, and 16% had an unknown or not reported race; 2% were Hispanic or Latino ethnicity and 14% had an unknown or not reported ethnicity; and the mean BMI was 41.5 kg/m2 (range: 24.4-66.1 kg/m2) at baseline.
Effect of IMCIVREE on BMI in Patients with Obesity and a Clinical Diagnosis of BBS
In patients aged ≥6 years with obesity and a clinical diagnosis of BBS in Trial 2, the mean percent change in BMI after 52 weeks of IMCIVREE treatment was -7.9% (Table 11), 61.3% of patients achieved a ≥5% BMI decrease from baseline, and 38.7% had a ≥10% decrease in BMI (Table 12).Table 11: Percent Change from Baseline in BMI after 52 Weeks from the Start of IMCIVREE Treatment in Patients Aged ≥6 Years with Obesity and a Clinical Diagnosis of BBS (Trial 2)*
Abbreviations: CI = confidence interval; SD = standard deviation
*BBS patients (N=31) who completed 52 weeks from the start of IMCIVREE treatment or discontinued the trial early. Five patients who discontinued trial early were defined as 0 percent change.
Statistic Result Baseline BMI (kg/m 2) Mean (SD) 41.8 (9.0) Median 41.5 Min, Max 24.4, 61.3 BMI after 52 Weeks (kg/m 2) Mean (SD) 38.6 (9.2) Median 39.1 Min, Max 20.4, 60.9 95% CI 35.2, 41.9 Percent Change from Baseline to 52 Weeks (%) Mean (SD) -7.9 (6.7) Median -8.8 Min, Max -25.4, 5.3 95% CI -10.4, -5.5 Table 12: Proportion of IMCIVREE-Treated Patients Aged ≥6 Years with Obesity and a Clinical Diagnosis of BBS Who Achieved at Least 5% and 10% BMI Decrease from Baseline After 52 Weeks from the Start of IMCIVREE Treatment (Trial 2) Abbreviations: CI = confidence interval; SD = standard deviation
*BBS patients (N=31) who completed 52 weeks from the start of IMCIVREE treatment or discontinued the trial early. Five patients who discontinued trial early were defined as not achieving 5% or 10% reduction.
Parameter Statistic Result Patients* Achieving at Least 5% BMI Loss at 52 Weeks % 61.3 95% CI 42.2, 78.2 Patients* Achieving at Least 10% BMI Loss at 52 Weeks % 38.7 95% CI 21.8, 57.8 During the 14-week double-blind, placebo-controlled portion of Trial 2 (Period 1), there was a statistically significant difference in BMI reduction between the IMCIVREE-treated group and the placebo-treated group ( Table 13).
Table 13. Percent Change from Baseline in BMI after 14 Weeks of Treatment in Patients Aged ≥6 Years with Obesity and a Clinical Diagnosis of BBS (Trial 2)*
Abbreviations: CI = confidence interval; SD = standard deviation
*BBS subjects who completed the 14-week double-blind, placebo-controlled period (N=44).
Parameter IMCIVREE
(N = 22)Placebo
(N = 22)Baseline BMI (SD) 41.4 (10.0) 41.6 (10.1) BMI at 14 Weeks (SD) 39.5 (9.9) 41.6 (9.9) Percent Change from Baseline to 14 Weeks (SD) -4.6 (4.1) -0.1 (2.3) Placebo-Adjusted Difference -4.5 95% CI -6.5, -2.5 Effect of IMCIVREE on Hunger in Patients with Obesity and a Clinical Diagnosis of BBS
In Trial 2, patients 12 years and older who were able to self-report their hunger (n=14), recorded their daily maximal hunger in a diary, which was then assessed by the Daily Hunger Questionnaire Item 2. Hunger was scored on an 11-point scale from 0 (“not hungry at all”) to 10 (“hungriest possible”). Weekly means of daily maximal hunger scores after 52 weeks from the start of IMCIVREE treatment are summarized in Table 14.
Hunger scores decreased in IMCIVREE-treated patients during the 14-week placebo-controlled period and during the open-label treatment period.
Table 14: Daily Hunger Scores – Change from Baseline in IMCIVREE-Treated Patients Aged ≥12 Years with Obesity and a Clinical Diagnosis of BBS After 52 Weeks From the Start of IMCIVREE Treatment (Trial 2)
Abbreviations: BBS = Bardet-Biedl syndrome; CI=confidence interval; Max=maximum; Min=minimum; NC=Not calculated; SD=Standard Deviation.
Note: Baseline is the last assessment prior to initiation of setmelanotide in both trials.
Note: The Daily Hunger Questionnaire is not administered to patients <12 years or to patients with cognitive impairment as assessed by the Investigator.
Timepoint Statistic Result Baseline N 14 Mean (SD) 6.99 (1.893) Median 7.29 Min, Max 4.0, 10.0 Week 52 N 14 Mean (SD) 4.87 (2.499) Median 4.43 Min, Max 2.0, 10.0 Change at Week 52 N 14 Mean (SD) -2.12 (2.051) Median -1.69 Min, Max -6.7, 0.0 Supportive of IMCIVREE’s effect on weight loss, there were general numeric improvements in blood pressure, lipids, and waist circumference. However, because of the limited number of patients studied and the lack of a control group, the treatment effects on these parameters could not be accurately quantified.
14.3 POMC, PCSK1, and LEPR Deficiency (Adults and Pediatric Patients Aged 6 Years and Older)
Overview of Clinical Trials
The efficacy of IMCIVREE for weight reduction in adults and pediatric patients 6 years of age and older with obesity due to POMC, PCSK1, or LEPR deficiency were assessed in 2 identically designed, 1-year, open-label trials, each with an 8‑week, double-blind withdrawal period.
- Trial 3 ( NCT02896192) enrolled patients aged 6 years and above with obesity and genetically confirmed or suspected POMC or PCSK1 deficiency.
- Trial 4 ( NCT03287960) enrolled patients aged 6 years and above with obesity and genetically confirmed or suspected LEPR deficiency.
The trials enrolled patients with homozygous or presumed compound heterozygous pathogenic, likely pathogenic variants, or VUS for either the POMCor PCSK1genes (Trial 3) or the LEPRgene (Trial 4). In both trials, the local genetic testing results were centrally confirmed using Sanger sequencing. Patients with double heterozygous variants in 2 different genes were not eligible for treatment with IMCIVREE. In both trials, adult patients had a body mass index (BMI) of ≥30 kg/m 2. Weight in pediatric patients was ≥95 th percentile using growth chart assessments.
IMCIVREE dose titration occurred over a 2- to 12-week period, followed by a 10-week, open-label treatment period with IMCIVREE. Patients who achieved at least a 5-kilogram weight loss (or at least 5% weight loss if baseline body weight was <100 kg) at the end of the open-label treatment period continued into a double-blind withdrawal period lasting 8 weeks, including 4 weeks of IMCIVREE followed by 4 weeks of placebo (investigators and patients were blinded to this sequence). Following the withdrawal sequence, patients re-initiated treatment with IMCIVREE at their therapeutic dose for up to 32 weeks.
Efficacy analyses were conducted in 21 patients (10 in Trial 3 and 11 in Trial 4) who had completed at least 1 year of treatment at the time of a prespecified data cutoff. Six additional patients enrolled in the trials (4 in Trial 3 and 2 in Trial 4) who had not yet completed 1 year of treatment at the time of the cutoff were not included in the efficacy analyses.
Of the 21 patients included in the efficacy analysis in Trials 3 and 4, 62% were adults and 38% were pediatric patients aged 16 years or younger.
- In Trial 3, 50% of patients were female, 70% were White, and the median BMI was 40 kg/m 2(range: 26.6‑53.3) at baseline.
- In Trial 4, 73% of patients were female, 91% were White, and the median BMI was 46.6 kg/m 2(range: 35.8‑64.6) at baseline.
Effect of IMCIVREE on Body Weightin Patients with Obesity due to POMC, PCSK1, or LEPR Deficiency
In Trial 3, 80% of patients with obesity due to POMC or PCSK1 deficiency met the primary endpoint, achieving a ≥10% weight loss after 1 year of treatment with IMCIVREE.
In Trial 4, 46% of patients with obesity due to LEPR deficiency achieved a ≥10% weight loss after 1 year of treatment with IMCIVREE (Table 15).
Table 15: Body Weight (kg) – Proportion of IMCIVREE-Treated Patients with Obesity due to POMC, PCSK1, or LEPR Deficiency Who Achieved at Least 10% Weight Loss from Baseline at 1 Year in Trials 3 and 4 Abbreviations: CI = confidence interval
Note: The analysis set includes patients who received at least 1 dose of trial drug and had at least 1 baseline assessment.
1From the Clopper-Pearson (exact) method
2Testing the null hypothesis: Proportion =5%
Parameter Statistic Trial 3
(POMC or PCSK1)
(N=10)Trial 4
(LEPR)
(N=11)Patients Achieving at Least 10% Weight Loss at Year 1 n (%) 8 (80%) 5 (46%) 95% CI 1 (44.4%, 97.5%) (16.8%, 76.6%) P-value 2 <0.0001 0.0002 When treatment with IMCIVREE was withdrawn in the 16 patients who had lost at least 5 kg (or 5% of body weight if baseline body weight was <100 kg) during the 10 week open-label period in Trials 3 and 4, these patients gained an average of 5.5 kg in Trial 3 and 5.0 kg in Trial 4 over 4 weeks. Re initiation of treatment with IMCIVREE resulted in subsequent weight loss (see Figure 2).
Table 16: Percent Change from Baseline in Weight in IMCIVREE-Treated Patients Aged ≥6 Years with Obesity due to POMC, PCSK1, or LEPR Deficiency at 1 Year in Trials 3 and 4 (Full Analysis Set) Abbreviations: CI = confidence interval; SD = standard deviation
Note: This analysis includes patients who received at least 1 dose of trial drug, had at least 1 baseline assessment.
1ANCOVA model containing baseline body weight as a covariate
2Testing the null hypothesis: mean percent change=0
Parameter Statistic Trial 3
(POMC or PCSK1)
(N=10)Trial 4
(LEPR)
(N=11)Baseline Body Weight (kg) Mean (SD) 118.7 (37.5) 133.3 (26.0) Median 115.0 132.3 Min, Max 55.9, 186.7 89.4, 170.4 1-Year Body Weight (kg) Mean (SD) 89.8 (29.4) 119.2 (27.0) Median 84.1 120.3 Min, Max 54.5, 150.5 81.7, 149.9 Percent Change from Baseline to 1 Year (%) Mean (SD) -23.1 (12.1) -9.7 (8.8) Median -26.7 -9.8 Min, Max -35.6, -1.2 -23.3, 0.1 LS Mean 1 -23.12 -9.65 95% CI 1 (-31.9, -14.4) (-16.0, -3.3) P-value 2 0.0003 0.0074 Figure 2: Mean Percent Change in Body Weight from Baseline in Patients Aged ≥6 Years with Obesity due to POMC, PCSK1, or LEPR Deficiency by Visit (Trial 3 [N=9] and Trial 4 [N=7])
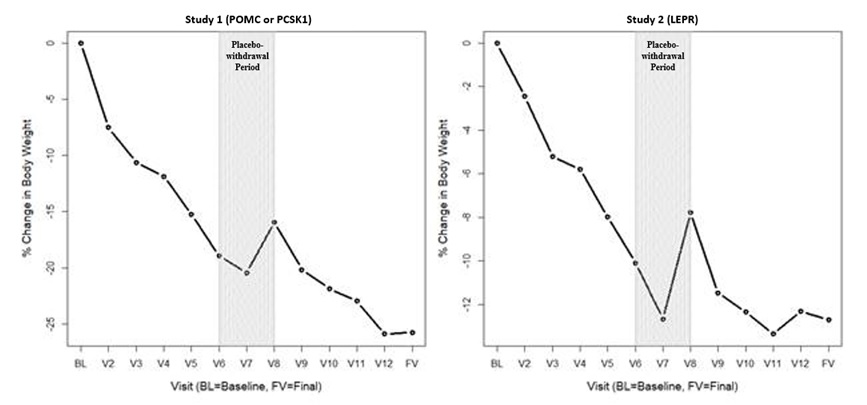
BL=Baseline (day of first dose)
V2 to V3 = variable dose titration period (2 to 12 weeks)
V3 to V6 = 10-week open-label treatment period
V6 to V8 = 8-week placebo withdrawal period (4 weeks active, 4 weeks placebo)
V8 to V12 = 32-week open-label treatment period
FV = Final visit; time point for primary efficacy analysis
Note: This figure includes patients who had lost at least 5 kg (or 5% of body weight if baseline body weight was <100 kg) during the 10-week open-label period.
Effect of IMCIVREE on Hunger in Patients with Obesity due to POMC, PCSK1, or LEPR Deficiency
In Trials 3 and 4, patients 12 years and older self-reported their daily maximal hunger in a diary, assessed by the Daily Hunger Questionnaire Item 2. Hunger was scored on an 11-point numeric rating scale from 0 (“not hungry at all”) to 10 (“hungriest possible”). Weekly means of daily hunger scores at Baseline and Week 52 are summarized in Table 17.
Table 17: Daily Hunger Scores – Change from Baseline at 1 Year in IMCIVREE-Treated Patients Aged ≥12 Years with Obesity due to POMC, PCSK1, or LEPR Deficiency in Trials 3 and 4 with Available Hunger Data
Note: This analysis includes patients aged 12 years and older who received at least 1 dose of study drug and had available data. Three patients in Study 2 had missing hunger data at Week 52.
Hunger score was captured in a daily diary and was averaged to calculate a weekly score for analysis. Hunger ranged from 0 to 10 on an 11-point scale where 0 = “not hungry at all” and 10 = “hungriest possible.”
Parameter Statistic Hunger in 24 Hours Trial 3
(POMC or PCSK1(N=8)Trial 4
(LEPR)
(N=8)Baseline Hunger Score Median 7.9 7.0 Median 7.0, 9.1 5.0, 8.4 1-Year Hunger Score Min, Max 5.5 4.4 Min, Max 2.5, 8.0 2.1, 8.0 Change from Baseline to 1 Year Median -2.0 -3.4 Min, Max -6.5, -0.1 -4.7, 1.0 Hunger scores generally worsened during the double-blind, placebo withdrawal period among those patients who had experienced an improvement from baseline, and scores improved when IMCIVREE was reinitiated. Supportive of IMCIVREE’s effect on weight loss, there were general numeric improvements in blood pressure, lipids, glycemic parameters, and waist circumference. However, because of the limited number of patients studied and the lack of a control group, the treatment effects on these parameters could not be accurately quantified.
14.4 POMC, PCSK1, and LEPR Deficiency and BBS (Pediatric Patients Aged 2 to Less Than 6 Years)
Clinical Trial Overview
The efficacy of IMCIVREE for weight reduction in pediatric patients aged 2 to less than 6 years with obesity due to POMC, PCSK1, or LEPR deficiency or BBS were assessed in a 52-week clinical trial [Trial 5 (NCT04966741)]. Patients with PCSK1 deficiency were eligible but noneenrolled. POMC and LEPR deficiency were confirmed by genetic testing demonstrating biallelic variants interpreted as pathogenic, likely pathogenic, or of undetermined significance; BBS was diagnosed clinically with genetic confirmation. Obesity was defined as baseline BMI ≥97thpercentile for age and sex and body weight ≥20 kg.
In Trial 5, IMCIVREE dose titration occurred over an 8-week period, followed by a 44-week open-label treatment period with IMCIVREE.
Twelve (12) patients were enrolled (3 patients with POMC deficiency, 4 patients with LEPR deficiency, and 5 patients with BBS); 58% were male; 58% were White, 8% were Asian, and 33% had an unknown or not reported race; 8% were Hispanic or Latino ethnicity and 17% had an unknown or not reported ethnicity; and the mean BMI was 29.9 kg/m2 (range: 19-43 kg/m2) at baseline.
Efficacy analyses were conducted in all 12 patients at the end of treatment.
Effect of IMCIVREE on BMI in Patients Aged 2 to Less Than 6 Years with POMC or LEPR Deficiency or BBS
In Trial 5, 8% of patients discontinued trial drug.
The mean percent change in BMI after 52 weeks of IMCIVREE treatment was -33.8%, -13.1%, and -9.7% in patients with POMC deficiency, LEPR deficiency, and BBS, respectively (Table 18).
Table 18: Percent Change from Baseline in BMI after 52 Weeks of IMCIVREE Treatment in Patients Aged 2 to Less Than 6 Years with Obesity due to POMC Deficiency, LEPR Deficiency, or BBS (Trial 5)
Statistic
POMC
(N=3)LEPR
(N=4)BBS
(N=5)Baseline BMI (kg/m 2)
N
3
4
5
Mean (SD)
27.8 (1.6)
39.3 (4.8)
23.7 (3.5)
Median
28.4
41.2
23.0
Min, Max
26.0, 28.9
32.2, 42.5
19.3, 29.0
BMI at Week 52 (kg/m 2)
N
3
4
5
Mean (SD)
18.3 (1.2)
34.0 (5.0) 2
21.4 (3.3)
Median
18.0
32.7
22.2
Min, Max
17.3, 19.7
29.5, 41.1
17.9, 25.2
Percent Change from Baseline to 52 Weeks (%)
Mean (SE)
-33.8 (4.7)
-13.1 (5.4) 3
-9.7 (4.0)
Median
-37.6
-15.1
-9.0
Min, Max
-39.3, -24.3
-22.1, 0
-21.6, 2.5
95% CI 1
-54.1, -13.4
-30.4, 4.2
-20.7, 1.3
Abbreviations: CI = confidence interval; SD = standard deviation
1Two-sided 95% CI is calculated with Student’s t-distribution.
2Using last observation carried forward (LOCF), the mean BMI (SD), median, min, and max at Week 52 are 34.9 (6.8), 32.7 29.5, and 44.9, respectively. Using observed data only, the mean BMI (SD), median, min, and max at Week 52 are 31.6 (1.9), 32.2, 29.5, and 33.1, respectively.
3Week 52 results are based off baseline observation carried forward (BOCF) for 1 patient lost to follow up after Week 8. Results using last observation carried forward are -10.8 (-34.4, 12.8) and using observed data excluding 1 patient lost to follow up are –17.4 (-37.2, 2.4).
Supportive of IMCIVREE’s effect on weight loss, general numeric improvements in waist circumference were observed.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
IMCIVREE injection is supplied as:
- 10 mg/mL, clear to slightly opalescent, colorless to slightly yellow solution in a 1-mL multiple-dose vial
- Package of 1 multiple-dose vial: NDC: 72829-010-01
Store unopened IMCIVREE vials in the refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton. After removal from the refrigerator, vials may be kept at temperatures ranging from refrigerated to room temperature (2°C to 25°C [36°F to 77°F]) for up to 30 days with brief excursions up to 30°C (86°F). After the vial is punctured (opened), discard after 30 days. See Table 19 for a summary of storage conditions for IMCIVREE. Store vials in the original carton.
Table 19 Recommended Storage for IMCIVREE Vials
1If necessary, IMCIVREE may be stored at room temperature (≤30°C [≤86°F]) and then returned to refrigerated conditions
Storage Condition Unopened Vial Opened Vial 2°C to 8°C (36°F to 46°F) Until the expiration date Up to 30 days, OR
Until the expiration date
(whichever is earlier)2°C to 25°C (36°F to 77°F) with excursions permitted up to 30°C (86°F) 1 Up to 30 days, OR
Until the expiration date
(whichever is earlier)Up to 30 days, OR
Until the expiration date
(whichever is earlier)>30°C (>86°F) Discard and do not use Discard and do not use -
17 PATIENT COUNSELING INFORMATION
Advise the patient and caregiver to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Disturbance in Sexual Arousal
Inform patients and caregivers that sexual adverse reactions, including spontaneous erection, may occur in patients treated with IMCIVREE. Advise patients to seek emergency medical treatment if an erection lasts longer than 4 hours [see Warnings and Precautions (5.1)].
Depression and Suicidal Ideation
Inform patients and caregivers that IMCIVREE may cause depression or suicidal ideation. Advise patients and caregivers to report any new or worsening symptoms of depression, suicidal thoughts or behaviors, or unusual changes in mood or behavior [see Warnings and Precautions (5.2)].
Hypersensitivity Reactions
Inform patients and caregivers that serious hypersensitivity reactions have been reported with use of IMCIVREE. Advise patients and caregivers on the symptoms of hypersensitivity reactions and instruct them to stop taking IMCIVREE and seek medical advice promptly if such symptoms occur [see Warnings and Precautions (5.3)].
Skin Hyperpigmentation, Darkening of Pre-Existing Nevi, and Development of New Melanocytic Nevi
Inform patients and caregivers that skin darkening occurs in the majority of patients treated with IMCIVREE because of its mechanism of action. This change is reversible upon discontinuation of IMCIVREE. Inform patients and caregivers that the development of new melanocytic nevi may occur. Inform patients and caregivers that they should have a full body skin examination before starting and during treatment with IMCIVREE to monitor these changes [see Warnings and Precautions (5.4)].
Acute Adrenal Insufficiency in Patients with Acquired HO
Inform patients with adrenal insufficiency and their caregivers to contact their healthcare provider for any significant changes in fatigue or lethargy, mental status, dizziness, fever, or signs of infection which may require an increase in steroid dosing occur during treatment with IMCIVREE [see Warnings and Precautions (5.5)].
Sodium Imbalance in Patients with Acquired HO and Central Diabetes Insipidus
Inform patients with diabetes insipidus and their caregivers to contact their healthcare provider if changes in fluid intake or urine output or other signs of dehydration, mental status changes (e.g., confusion, lethargy), or significant nausea and vomiting occur which may require adjustments in concomitant therapies during treatment with IMCIVREE [see Warnings and Precautions (5.6)].
Pregnancy
Advise patients who may become pregnant to inform their healthcare provider of a known or suspected pregnancy [see Use in Specific Populations (8.1)].
Lactation
Advise patients thattreatment with IMCIVREE is not recommended while breastfeeding [see Use in Specific Populations (8.2)].
Administration
Instruct patients and caregivers how to prepare and administer the correct dose of IMCIVREE and assess their ability to inject subcutaneously to ensure the proper administration of IMCIVREE. Instruct patients to use a 1 mL syringe with a 28- or 29-gauge needle appropriate for subcutaneous injection [see Dosage and Administration (2.5)].
Manufactured for:
Rhythm Pharmaceuticals, Inc.
222 Berkeley Street, Suite 1200
Boston, MA 02116© 2026, Rhythm Pharmaceuticals, Inc. All rights reserved.
IMCIVREE is a registered trademark of Rhythm Pharmaceuticals, Inc.
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
IMCIVREE™ [im-SIV-ree]
(setmelanotide)
injection, for subcutaneous useWhat is IMCIVREE?
- IMCIVREE is a prescription medicine used in adults and children:
- 4 years of age and older with acquired hypothalamic obesity (HO) to help them lose weight and keep the weight off.
- 2 years of age and older with obesity due to Bardet-Biedl syndrome (BBS) to help them lose weight and keep the weight off.
- 2 years of age and older with obesity due to the genetic conditions proopiomelanocortin (POMC), proprotein convertase subtilisin/kexin type 1 (PCSK1), or leptin receptor (LEPR) deficiency, to help them lose weight and keep the weight off.
- Your healthcare provider should order a genetic test to confirm POMC, PCSK1, or LEPR deficiency before you start using IMCIVREE.
- IMCIVREE is not for use in people with the following conditions because it may not work:
- Obesity due to suspected POMC, PCSK1, or LEPR deficiency not confirmed by genetic testing or with benign or likely benign genetic testing results.
- Other types of obesity not related to acquired HO, BBS, or POMC, PCSK1, or LEPR deficiency, including obesity associated with other genetic conditions and general obesity.
It is not known if IMCIVREE is safe and effective in children under 2 years of age.
Do not use IMCIVREE if you have had a serious allergic reaction to setmelanotide or any of the ingredients in IMCIVREE. Serious allergic reactions, including a severe allergic reaction called anaphylaxis, can happen when you use IMCIVREE. See the end of this Patient Information leaflet for a complete list of ingredients in IMCIVREE.
Before using IMCIVREE, tell your healthcare provider about all your medical conditions, including if you:
- have or have had areas of darkened skin, including skin discoloration (hyperpigmentation).
- have or have had depression, or suicidal thoughts or behavior.
- have had a prior allergic reaction to setmelanotide or any of the ingredients in IMCIVREE.
- have kidney problems.
- have adrenal insufficiency.
- have diabetes insipidus.
- are pregnant or planning to become pregnant. Losing weight while pregnant may harm your unborn baby. Your healthcare provider may stop your treatment with IMCIVREE if you become pregnant. Tell your healthcare provider if you become pregnant or think you might be pregnant during treatment with IMCIVREE.
- are breastfeeding or plan to breastfeed. It is not known if IMCIVREE passes into your breastmilk. You should not breastfeed during treatment with IMCIVREE.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How should I use IMCIVREE?
- See the detailed Instructions for Use to learn how to prepare and inject IMCIVREE.
- IMCIVREE is given as an injection under your skin (subcutaneous) by you or a caregiver.
- A healthcare provider should show you or your caregiver how to prepare and inject your dose of IMCIVREE before injecting for the first time. Do not try to inject IMCIVREE unless you have been trained by a healthcare provider.
- Use IMCIVREE exactly as prescribed by your healthcare provider.
- IMCIVREE should be injected 1 time each day when you first wake up. IMCIVREE can be given with or without food.
- If you miss a dose of IMCIVREE, inject your next dose at the regularly scheduled time the next day.
What are the possible side effects of IMCIVREE?
IMCIVREE may cause serious side effects, including:
- Male and female sexual function problems. IMCIVREE can cause an erection that happens without any sexual activity in males (spontaneous penile erection) and unwanted sexual reactions (changes in sexual arousal that happen without any sexual activity) in females. If you have an erection lasting longer than 4 hours, get emergency medical help right away.
- Depression and suicidal thoughts or actions. You or a caregiver should call your healthcare provider right away if you have any new or worsening symptoms of depression, suicidal thoughts or behaviors, or any unusual changes in mood or behavior.
-
Serious allergic reactions. Stop taking IMCIVREE and get medical help right away if you have any symptoms of a serious allergic reaction including:
- swelling of your face, lips, tongue or throat
- problems breathing or swallowing
- severe rash or itching
- fainting or feeling dizzy
- rapid heartbeat
- Increased skin pigmentation, darkening of skin lesions (moles or nevi) you already have, and development of new skin lesions. These changes happen because of how IMCIVREE works in the body and will go away when you stop using IMCIVREE. You should have a full body skin exam before starting and during treatment with IMCIVREE to check for skin changes.
- Adrenal insufficiency. If you have acquired HO and adrenal insufficiency, your healthcare provider should evaluate your adrenal function before starting IMCIVREE. You or your caregiver should call your healthcare provider if you have changes in feeling tired or exhausted (fatigue), lack of energy (lethargy), mental status, or dizziness, or fever, or signs of infection during treatment with IMCIVREE. Your healthcare provider should monitor and adjust any medicines that may be affected during treatment.
- Low sodium levels in the blood. If you have acquired HO and diabetes insipidus, you or your caregiver should watch for signs of dehydration. You or your caregiver should contact your healthcare provider if you have changes in fluid intake or urine output, confusion, lethargy, or nausea and vomiting during treatment with IMCIVREE. Your healthcare provider should closely monitor sodium levels in your blood and adjust your other medicines if needed.
The most common side effects of IMCIVREE include:
- darkening of the skin
- injection site reactions
- nausea
- headache
- diarrhea
- stomach pain
- vomiting
- depression
- erection that happens without any sexual activity in males
These are not all the possible side effects of IMCIVREE.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
You may also report side effects to Rhythm Pharmaceuticals at 1-833-789-6337.General information about the safe and effective use of IMCIVREE.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use IMCIVREE for a condition for which it was not prescribed. Do not give IMCIVREE to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about IMCIVREE that is written for health professionals.
What are the ingredients in IMCIVREE?
Active ingredient: setmelanotide
Inactive ingredients: benzyl alcohol, carboxymethylcellulose sodium, edetate disodium dihydrate, N-(carbonyl-methoxypolyethylene glycol 2000)-1, 2-distearoyl- glycero-3-phosphoethanolamine sodium salt, mannitol, phenol, hydrochloric acid, sodium hydroxide and water for injection.
- IMCIVREE is a prescription medicine used in adults and children:
-
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE
IMCIVREE™ [im-SIV-ree]
(setmelanotide)
injection, for subcutaneous useThis Instructions for Use contains information on how to inject IMCIVREE. Read and follow these instructions before injecting IMCIVREE.
Important Information You Need to Know Before Injecting IMCIVREE
- IMCIVREE is for injection under the skin only (subcutaneous injection). Do not inject IMCIVREE into a vein or muscle.
- The IMCIVREE vial may be used to give more than 1 dose of medicine (multiple-dose vial) but is for single patient use only and should not be shared with other patients.
- Inject IMCIVREE 1 time each day when you first wake up.
- Take IMCIVREE with or without food.
- IMCIVREE is given by you or a caregiver. A healthcare provider will show you or your caregiver how to inject your dose of IMCIVREE before you inject for the first time. Ask your healthcare provider or call IMCIVREE Guidance, Partnership, Support (GPS) at 1-844-YOUR-GPS (1-844-968-7477) if you have questions.
Important note:
- Only use the syringes and needles provided to you for use with IMCIVREE.
- Always use a new syringe and needle for each injection to prevent contamination.
- Throw away used syringes and needles in a puncture-resistant, disposable sharps container as soon as you finish giving the injection. See “Disposing of IMCIVREE”at the end of these instructions.
- Do not reuse or share your needles with other people.
- Do not recap the needle. Recapping the needle can lead to a needle stick injury.
Understanding Your IMCIVREE Dose
Calculate the number of doses of IMCIVREE in each vial:
- Each unopened IMCIVREE vial contains 10 milligrams (mg) of medicine in 1 milliliter (mL) of solution.
- The vial will contain both medicine and air. Most of the vial will be filled with air.
- Your healthcare provider will determine your dose of medicine in milligrams (mg).
- The IMCIVREE vial may be used to give more than 1 dose of medicine (multiple-dose vial).
- Use Figure Ato see how many times you may use each vial based on your prescribed dose.
- Do not use more doses from a single vial than listed in Figure A
Preparing to Inject IMCIVREE
Step 1 Gather your supplies.
- Gather the supplies you will need for your injection ( Figure B).
- Place your supplies on a clean, flat work surface.
Step 2 Check your IMCIVREE vial.
- Check the expiration (Exp) date on the vial label ( Figure C).
- Check the liquid. The liquid should look clear to almost clear and colorless to slightly yellow. The liquid should be free of particles.
-
Do not use if:
- The expiration (Exp) date has passed.
- The liquid is cloudy.
- There are particles floating in the vial.
- The plastic cap on a new vial is broken or missing.
Step 3 Prepare your IMCIVREE vial.
- Allow the vial to reach room temperature.
- Remove the vial from the refrigerator 15 minutes before injection.
- You can also warm the vial by rolling it gently between the palms of your hands for 60 seconds ( Figure D).
- Do not try to warm the vial by using a heat source such as hot water or a microwave.
- Do not shake the vial.
- Wash your hands with soap and warm water.
- If using a new vial, remove the plastic cap ( Figure E) and throw away this plastic cap in the trash. Do not put the plastic cap back on the vial.
- Clean the top of the vial rubber stopper with 1 alcohol wipe ( Figure F). Throw away the alcohol wipe in the trash.
- Do not remove the vial rubber stopper.
Step 4 Prepare the syringe.
When measuring your dose, be sure to read the markings starting from the end closest to the black rubber stopper ( Figure G).
- Fill the syringe with air.
- Keep the protective needle cap on the syringe.
- Pull back on the plunger until the end of the black rubber stopper stops at your dose. Fill the syringe with air equal to the amount of the medicine to be given ( Figure H).
- Remove the protective needle cap from the syringe.
- Remove the protective needle cap by pulling it straight off and away from your body.
- Insert the needle.
- Place the vial on the clean, flat work surface.
- With the vial in the upright position, place the syringe directly over the vial and insert the needle straight down into the center of the vial rubber stopper ( Figure I).
- Push the air from the syringe into the vial.
- Fill the syringe with IMCIVREE
- Keep the needle in the vial and slowly turn the vial upside down.
- Make sure to keep the tip of the needle in the medicine ( Figure J).
- Slowly pull back on the plunger to fill the syringe with the amount of IMCIVREE needed for your prescribed dose.
- Be careful not to pull the plunger out of the syringe.
- Do not use more than 1 vial of IMCIVREE to give a single dose. Use a new vial that has enough medicine for your prescribed dose.
- Check for large air bubbles (
Figure K).
- Keep the needle in the vial and check the syringe for large air bubbles.
What to do if you see large air bubbles:
Large air bubbles can reduce the dose of medicine you receive. To remove large air bubbles:
- Gently tap the side of the syringe with your finger to move the air bubbles to the top of the syringe.
- Move the tip of the needle above the medicine and slowly push the plunger up to push the large air bubbles back into the vial.
- After the large air bubbles are removed, pull back on the plunger again (more slowly this time) to fill the syringe with your prescribed dose of medicine.
- Withdraw the needle
- Return the vial to an upright position and place it on the clean, flat work surface.
- While holding the vial with 1 hand and the barrel of the syringe between the fingertips of your other hand, pull the needle straight out of the vial ( Figure L).
- Set the syringe down on the clean, flat work surface.
- Make sure the needle does not touch the surface.
- Do not recap the needle
Injecting IMCIVREE
Step 5 Prepare your injection site.
- Choose the area where you will give the injection. Choose from the following recommended injection sites:
- Be sure to choose an area on the belly (abdomen) at least 2 inches from the belly button.
- Do not inject into the belly button, ribs, and hip bones, as well as scars or moles.
- Do not inject in an area that is red, swollen, or irritated
- Clean the injection site with the second alcohol wipe using a circular motion.
- Do not touch, fan, or blow on the cleaned area.
- Allow the skin to dry for about 10 seconds.
Rotate your injection site each day.
You should use a different injection site each time you give an injection, at least 1 inch away from the area you used for your previous injection. You may want to use a calendar or diary to record your injection sites.
Step 6 Place your hands for the injection.
- With 1 hand, pinch about 2 inches of skin at the injection site between your thumb and index (pointer) finger ( Figure P). Pinching the skin is important to help make sure that you inject the medicine under the skin (into fatty tissue) but not any deeper (into the muscle).
Step 7 Inject and release.
- With your other hand, place the syringe between the thumb and index (pointer) finger.
- Hold the middle of the syringe (where the markings are printed) at a 90-degree angle to your body and push the needle straight into the injection site ( Figure Q). Make sure that you push the needle all the way into the skin.
- Do not hold or push on the plunger while inserting the needle.
- Slowly push the plunger down to inject the medicine ( Figure R)
- Keep the needle in your skin and count to 5 to make sure that all the medicine is given.
- Let go of the pinched skin and remove the needle.
- Use the gauze pad to gently apply pressure to the injection site.
- Do not recap the needle.
Tips for giving injections to children
When giving a child an injection, it can help to have the child do other things. Have the child:
- squeeze something soft like a ball or stuffed animal.
- slowly breathe in and out.
- sing a song, count, or name favorite colors or animals.
Storing IMCIVREE
- Store unopened and opened vials in the original carton to protect them from light.
- Store opened vials of IMCIVREE in the refrigerator between 36°F to 46°F (2°C to 8°C). If needed, opened vials may be removed from the refrigerator and stored at temperatures ranging from refrigerated to room temperature (36°F to 77°F (2°C to 25°C)). Vials may be returned to the refrigerator. Throw away all opened vials 30 days after first opening, even if some medicine is still left in the vial.
- Unopened vials of IMCIVREE may be stored in the refrigerator between 36°F to 46°F (2°C to 8°C) until the expiration date. If needed, unopened vials may also be removed from the refrigerator and stored at temperatures ranging from refrigerated to room temperature (36°F to 77°F (2°C to 25°C)) for up to 30 days. Vials may be returned to the refrigerator. Throw away IMCIVREE if it has been more than 30 days since the vial was first removed from the refrigerator.
- If necessary, vials may be stored at room temperature below 86°F (30°C) and may be returned to refrigerated conditions. Throw away IMCIVREE that has been stored above 86°F (30°C).
- Write the date on the carton when you first open the vial.
- Keep IMCIVREE, needles, syringes, and all medicines out of the reach of children.
- Alcohol wipes, used gauze pads, and vials can be thrown away in the trash. Throw away (dispose of) used syringes and needles in a puncture-resistant container, such as an FDA-cleared sharps disposal container immediately after use (
Figure S).
Do not throw away (dispose of) syringes and needles in your household trash.
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used syringes and needles. For more information about safe sharps disposal and for specific information about sharps disposal in the state that you live in, go to the FDA’s website at: http://www.fda.gov/safesharpsdisposal. - Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this.
- Do not recycle your used sharps disposal container.
Note: Keep your sharps disposal container out of the reach of children and pets.
For more information about IMCIVREE, including how to inject IMCIVREE, go to www.IMCIVREE.com or call 1-844-YOUR-GPS (1-844-968-7477).
IMCIVREE is Manufactured for:
Rhythm Pharmaceuticals, Inc
222 Berkeley Street, Suite 1200
Boston, MA 02116Made in France
IMCIVREE is a trademark of Rhythm Pharmaceuticals, Inc.
©2020 Rhythm Pharmaceuticals, Inc.This Patient Information and Instructions for Use have been approved by the U.S. Food and Drug Administration.
Revised [June 2025] - PRINCIPAL DISPLAY PANEL - NDC: 72829-010-01 - Carton Label
- PRINCIPAL DISPLAY PANEL - NDC: 72829-010-01 - Vial Label
-
INGREDIENTS AND APPEARANCE
IMCIVREE
setmelanotide solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72829-010 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SETMELANOTIDE (UNII: N7T15V1FUY) (SETMELANOTIDE - UNII:N7T15V1FUY) SETMELANOTIDE 10 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM N-(CARBONYL-METHOXYPOLYETHYLENE GLYCOL 2000)-1,2-DISTEAROYL-SN-GLYCERO-3-PHOSPHOETHANOLAMINE (UNII: 3L6NN8ZZKU) 100 mg in 1 mL MANNITOL (UNII: 3OWL53L36A) 11 mg in 1 mL CARBOXYMETHYLCELLULOSE (UNII: 05JZI7B19X) 8 mg in 1 mL PHENOL (UNII: 339NCG44TV) 5 mg in 1 mL BENZYL ALCOHOL (UNII: LKG8494WBH) 10 mg in 1 mL EDETATE DISODIUM (UNII: 7FLD91C86K) 1 mg in 1 mL HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72829-010-01 1 in 1 CARTON 12/14/2020 1 1 mL in 1 VIAL, MULTI-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA213793 12/14/2020 Labeler - Rhythm Pharmaceuticals, Inc (061479535)
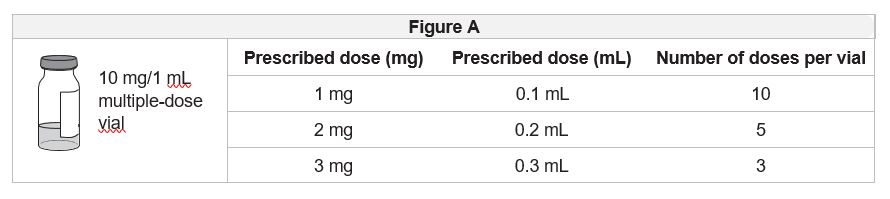
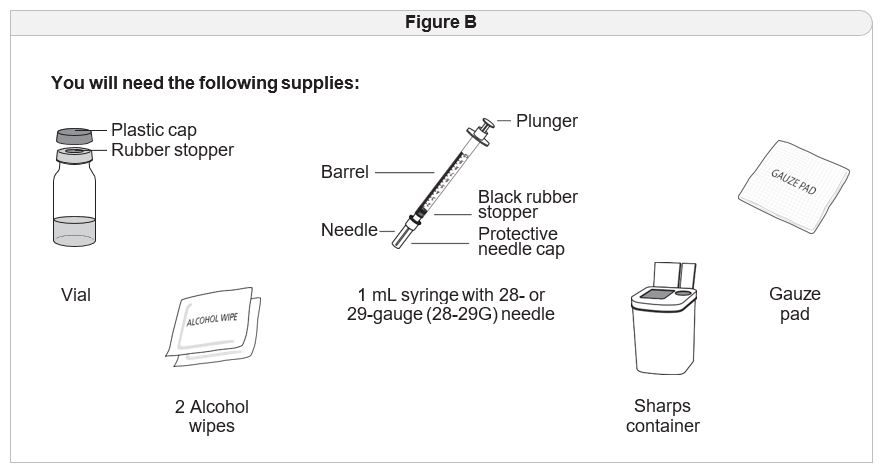
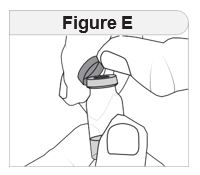
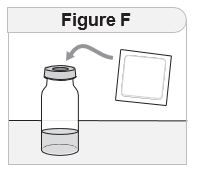
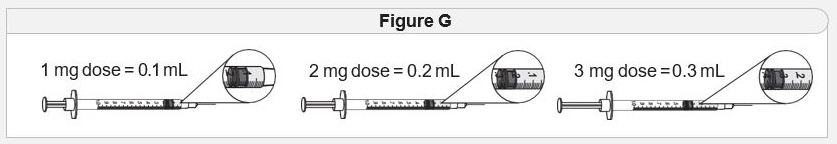
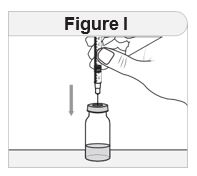
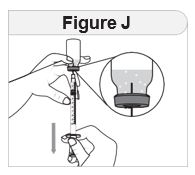
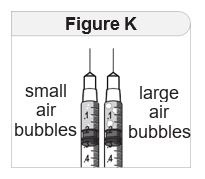
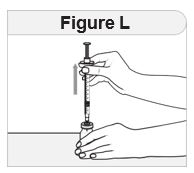
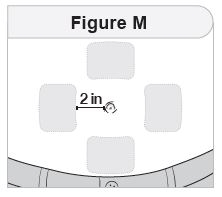
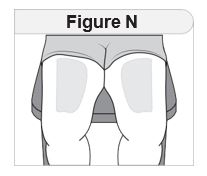
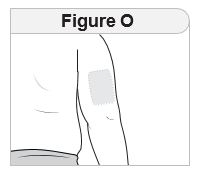
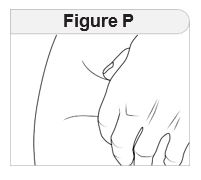
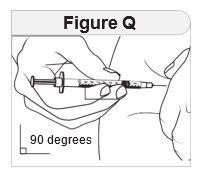
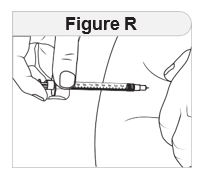


Trademark Results [Imcivree]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 IMCIVREE 90178986 not registered Live/Pending |
Rhythm Pharmaceuticals, Inc. 2020-09-14 |
 IMCIVREE 88226182 not registered Live/Pending |
Rhythm Pharmaceuticals, Inc. 2018-12-12 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.