VYVGART HYTRULO (efgartigimod alfa and hyaluronidase- human recombinant injection, solution
VYVGART Hytrulo by
Drug Labeling and Warnings
VYVGART Hytrulo by is a Prescription medication manufactured, distributed, or labeled by argenx US, Patheon Italia SpA, Lonza Biologics Tuas Pte.Ltd., Avid Bioservices Inc., Catalent Indiana, LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VYVGART HYTRULO safely and effectively. See full prescribing information for VYVGART HYTRULO.
VYVGART HYTRULO® (efgartigimod alfa and hyaluronidase-qvfc) injection, for subcutaneous use
Initial U.S. Approval: 2023RECENT MAJOR CHANGES
INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
- See Full Prescribing Information for instructions on dosage, preparation, and administration. (2.1, 2.2, 2.3, 2.4, 2.5)
- Evaluate the need to administer age-appropriate vaccines according to immunization guidelines before initiation of a new treatment cycle with VYVGART HYTRULO. (2.1)
- Important Administration Information
- gMG: recommended dose and dose schedule
- Administer in cycles of once weekly injections for 4 weeks. (2.3)
- Prefilled syringe: 1,000 mg efgartigimod alfa and 10,000 units hyaluronidase administered over 20 to 30 seconds. (2.3)
- Vial: 1,008 mg efgartigimod alfa and 11,200 units hyaluronidase over 30 to 90 seconds. (2.3)
- Administer subsequent treatment cycles based on clinical evaluation. (2.3)
- CIDP: recommended dose and dose schedule
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
VYVGART HYTRULO is contraindicated in patients with serious hypersensitivity to efgartigimod alfa products, to hyaluronidase, or to any of the excipients of VYVGART HYTRULO. (4)
WARNINGS AND PRECAUTIONS
- Infections: Delay administration of VYVGART HYTRULO to patients with an active infection. Monitor for signs and symptoms of infection in patients treated with VYVGART HYTRULO. If serious infection occurs, administer appropriate treatment and consider withholding VYVGART HYTRULO until the infection has resolved. (5.1)
- Hypersensitivity Reactions: Anaphylaxis, hypotension leading to syncope, angioedema, dyspnea, rash, and urticaria have occurred in patients treated with VYVGART HYTRULO or intravenous efgartigimod alfa-fcab product. If a hypersensitivity reaction occurs, the healthcare professional should institute appropriate measures if needed or the patient should seek medical attention. (4, 5.2)
- Infusion/injection-Related Reactions: If a severe infusion/injection-related reaction occurs, initiate appropriate therapy; consider the risks and benefits of readministering. If a mild to moderate infusion/injection-related reaction occurs, consider rechallenging with close clinical observation, slower infusion/injection rates, and pre-medications. (5.3)
ADVERSE REACTIONS
The most common adverse reactions (≥ 10%) in patients with gMG treated with efgartigimod alfa-fcab were respiratory tract infections, headache, and urinary tract infection.
Injection site reactions were common (≥ 15%) in patients with gMG and CIDP who were treated with VYVGART HYTRULO (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact argenx at 1-833-argx411 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Closely monitor for reduced effectiveness of medications that bind to the human neonatal Fc receptor. When concomitant long-term use of such medications is essential for patient care, consider discontinuing VYVGART HYTRULO and using alternative therapies. (7)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Vaccination
2.2 Important Dosage and Administration Instructions
2.3 Recommended Dosage for gMG
2.4 Recommended Dosage for CIDP
2.5 Preparation and Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Infections
5.2 Hypersensitivity Reactions
5.3 Infusion/Injection-Related Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of VYVGART HYTRULO on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Generalized Myasthenia Gravis
14.2 Chronic Inflammatory Demyelinating Polyneuropathy
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Vaccination
Evaluate the need to administer age-appropriate vaccines according to immunization guidelines before initiation of a new treatment cycle with VYVGART HYTRULO. Because VYVGART HYTRULO causes transient reduction in IgG levels, vaccination with live vaccines is not recommended during treatment with VYVGART HYTRULO [see Dosage and Administration (2.3) and Warnings and Precautions (5.1)].
2.2 Important Dosage and Administration Instructions
VYVGART HYTRULO is for subcutaneous use only. Do not administer intravenously. Do not dilute VYVGART HYTRULO.
Single-Dose Prefilled Syringe
VYVGART HYTRULO prefilled syringe may be administered by patients and/or caregivers after proper instruction in subcutaneous injection technique [see Instructions for Use].
Single-Dose Vial
VYVGART HYTRULO vial is to be administered with a winged infusion set by a healthcare professional only [see Dosage and Administration (2.4)].
2.3 Recommended Dosage for gMG
Single-Dose Prefilled Syringe
The recommended dosage of VYVGART HYTRULO prefilled syringe is 1,000 mg / 10,000 units (1,000 mg efgartigimod alfa and 10,000 units hyaluronidase) administered subcutaneously over approximately 20 seconds to 30 seconds in cycles of once weekly injections for 4 weeks.
Single-Dose Vial
The recommended dosage of VYVGART HYTRULO vial is 1,008 mg / 11,200 units (1,008 mg efgartigimod alfa and 11,200 units hyaluronidase) administered subcutaneously over approximately 30 seconds to 90 seconds in cycles of once weekly injections for 4 weeks.
General Dosage Considerations
Administer subsequent treatment cycles according to clinical evaluation.
If a scheduled dose is missed, VYVGART HYTRULO may be administered up to 3 days after the scheduled time point. Thereafter, resume the original dosing schedule until the treatment cycle is completed.
2.4 Recommended Dosage for CIDP
Single-Dose Prefilled Syringe
The recommended dosage of VYVGART HYTRULO prefilled syringe is 1,000 mg / 10,000 units (1,000 mg efgartigimod alfa and 10,000 units hyaluronidase) administered subcutaneously over approximately 20 to 30 seconds as once weekly injections.
Single-Dose Vial
The recommended dosage of VYVGART HYTRULO vial is 1,008 mg / 11,200 units (1,008 mg efgartigimod alfa and 11,200 units hyaluronidase) administered subcutaneously over approximately 30 seconds to 90 seconds as once weekly injections.
General Dosage Considerations
If a scheduled injection is missed, VYVGART HYTRULO may be administered up to 3 days after the scheduled time point. Thereafter, resume the original dosing schedule.
Not all patients respond to VYVGART HYTRULO. Consider the appropriateness of continuing treatment in patients who experience worsening of neurological symptoms after starting VYVGART HYTRULO [see Adverse Reactions (6.2) and Clinical Studies (14.2)].
2.5 Preparation and Administration Instructions
Single-Dose Prefilled Syringe
- Take the VYVGART HYTRULO prefilled syringe out of the refrigerator at least 30 minutes before injecting to allow it to reach room temperature [see How Supplied/Storage and Handling (16)]. Do not use external heat sources to bring the syringe to room temperature.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Visually inspect that the prefilled syringe solution is yellowish, clear to opalescent and devoid of particulate matter. Do not use if visible particles are present.
- Each prefilled syringe is for one-time use only.
- To administer VYVGART HYTRULO prefilled syringe, use a safety needle that is 25G, 5/8 inches in length, and thin wall type. The safety needle is not included in the carton.
- Choose an injection site on the abdomen (at least 2 inches away from the navel).
- Do not inject into areas where the skin is irritated, red, bruised, infected, tender, hard, or into areas where there are moles or scars.
- Rotate injection sites for subsequent administrations.
- Inject VYVGART HYTRULO prefilled syringe subcutaneously into a pinched skin area at an angle of 45 to 90 degrees for approximately 20 seconds to 30 seconds.
- Discard any unused portions of medicine remaining in the syringe.
- Localized injection site reactions may occur after VYVGART HYTRULO is administered [see Adverse Reactions (6.1)].
- Monitor for clinical signs and symptoms of hypersensitivity reactions for at least 30 minutes after administration. If a hypersensitivity reaction occurs, the patient should seek medical attention and the healthcare professional should institute appropriate measures, if needed [see Warnings and Precautions (5.2)].
For detailed instructions on the preparation and administration of VYVGART HYTRULO prefilled syringe see INSTRUCTIONS FOR USE.
Single-Dose Vial
Use aseptic technique when preparing and administering VYVGART HYTRULO vial. Do not shake the vial. Each vial is for one-time use only. Avoid exposure to direct sunlight.
Preparation
- Take the VYVGART HYTRULO vial out of the refrigerator at least 15 minutes before injecting to allow it to reach room temperature [see How Supplied/Storage and Handling (16)]. Do not use external heat sources.
- Check that the VYVGART HYTRULO solution is yellowish, clear to opalescent.
- Parenteral medicine products should be inspected visually for particulate matter prior to administration, whenever solution and container permit. Do not use if opaque particles or other foreign particles are present.
- Withdraw the entire content of VYVGART HYTRULO from the vial using a polypropylene syringe and an 18G stainless steel transfer needle.
- Remove large air bubbles, if present.
- Each vial contains overfill to compensate for liquid loss during preparation and to compensate for the priming volume of the winged infusion set.
- VYVGART HYTRULO vial does not contain preservatives. Administer immediately after preparation.
Administration
- To administer VYVGART HYTRULO vial use a winged infusion set made of polyvinyl chloride (PVC), 25G, 12 inches tubing, maximum priming volume of 0.4 mL.
- Remove the transfer needle from the syringe and connect the syringe to the winged infusion set.
- Prior to administration, fill the tubing of the winged infusion set by gently pressing the syringe plunger until the plunger is at 5.6 mL. There should be solution at the end of the winged infusion set needle.
- Choose an injection site on the abdomen (at least 2 inches to 3 inches away from the navel).
- Do not inject in areas where the skin is red, bruised, tender, hard, or into areas where there are moles or scars.
- Rotate injection sites for subsequent administrations.
- Inject VYVGART HYTRULO vial subcutaneously into a pinched skin area at an angle of about 45 degrees over 30 seconds to 90 seconds.
- Localized injection site reactions may occur after VYVGART HYTRULO is administered. [see Adverse Reactions (6.1)].
- Discard any unused portions of medicine remaining in the vial, the syringe and the winged infusion set.
- Healthcare professionals should monitor for clinical signs and symptoms of hypersensitivity reactions for at least 30 minutes after administration. If a hypersensitivity reaction occurs, the healthcare professional should institute appropriate measures if needed or the patient should seek medical attention [see Warnings and Precautions (5.2)].
-
3 DOSAGE FORMS AND STRENGTHS
Injection: 1,000 mg efgartigimod alfa and 10,000 units hyaluronidase per 5 mL (200 mg/2,000 units per mL) as yellowish, clear to opalescent solution, in a single-dose prefilled syringe.
Injection: 1,008 mg efgartigimod alfa and 11,200 units hyaluronidase per 5.6 mL (180 mg/2,000 units per mL) as yellowish, clear to opalescent solution, in a single-dose vial.
-
4 CONTRAINDICATIONS
VYVGART HYTRULO is contraindicated in patients with serious hypersensitivity to efgartigimod alfa products, to hyaluronidase, or to any of the excipients of VYVGART HYTRULO. Reactions have included anaphylaxis and hypotension leading to syncope [see Warnings and Precautions (5.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Infections
VYVGART HYTRULO may increase the risk of infection. The most common infections observed in Study 1 were urinary tract infection (10% of efgartigimod alfa-fcab-treated patients compared to 5% of placebo-treated patients) and respiratory tract infections (33% of efgartigimod alfa-fcab- treated patients compared to 29% of placebo-treated patients) [see Adverse Reactions (6.1) and Clinical Studies (14)]. A higher frequency of patients who received efgartigimod alfa-fcab compared to placebo were observed to have below normal levels for white blood cell counts (12% versus 5%, respectively), lymphocyte counts (28% versus 19%, respectively), and neutrophil counts (13% versus 6%, respectively). The majority of infections and hematologic abnormalities were mild to moderate in severity. Delay VYVGART HYTRULO administration in patients with an active infection until the infection is resolved. During treatment with VYVGART HYTRULO, monitor for clinical signs and symptoms of infections. If serious infection occurs, administer appropriate treatment and consider withholding VYVGART HYTRULO until the infection has resolved.
Immunization
Evaluate the need to administer age-appropriate vaccines according to immunization guidelines before initiation of a new treatment cycle with VYVGART HYTRULO. The safety of immunization with live vaccines and the immune response to vaccination during treatment with VYVGART HYTRULO are unknown. Because VYVGART HYTRULO causes a reduction in IgG levels, vaccination with live vaccines is not recommended during treatment with VYVGART HYTRULO.
5.2 Hypersensitivity Reactions
In clinical trials, hypersensitivity reactions, including rash, angioedema, and dyspnea were observed in patients treated with VYVGART HYTRULO or intravenous efgartigimod alfa-fcab. Urticaria was also observed in patients treated with VYVGART HYTRULO. Hypersensitivity reactions were mild or moderate, occurred within one hour to three weeks of administration.
Anaphylaxis and hypotension leading to syncope have been reported in postmarketing experience with intravenous efgartigimod alfa-fcab. Anaphylaxis and hypotension occurred during or within an hour of administration and led to infusion discontinuation and in some cases to permanent treatment discontinuation.
Monitor for clinical signs and symptoms of hypersensitivity reactions for at least 30 minutes after administration [see Dosage and Administration (2.4)]. If a hypersensitivity reaction occurs, the healthcare professional should institute appropriate measures if needed or the patient should seek medical attention. VYVGART HYTRULO is contraindicated in patients with a history of serious hypersensitivity to efgartigimod alfa products, to hyaluronidase, or to any of the excipients of VYVGART HYTRULO [see Contraindications (4)].
5.3 Infusion/Injection-Related Reactions
Infusion-related reactions have been reported with intravenous efgartigimod alfa-fcab in postmarketing experience. The most frequent symptoms and signs were hypertension, chills, shivering, and thoracic, abdominal, and back pain. Infusion-related reactions occurred during or within an hour of administration and led to infusion discontinuation. If a severe infusion/injection-related reaction occurs, initiate appropriate therapy. Consider the risks and benefits of readministering VYVGART HYTRULO following a severe infusion/injection-related reaction. If a mild to moderate infusion/injection-related reaction occurs, patients may be rechallenged with close clinical observation, slower infusion/injection rates, and pre-medications.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Infections [see Warnings and Precautions (5.1)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
- Infusion/Injection-Related Reactions [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Experience in Patients with gMG
The safety of efgartigimod alfa in adult patients with gMG was established in a double blinded placebo- controlled study with efgartigimod alfa-fcab administered intravenously (Study 1) and its open-label extension, and in an active-controlled study of VYVGART HYTRULO administered subcutaneously (Study 3) and its open-label extension [see Clinical Studies (14.1)].
Adverse Reactions with Efgartigimod Alfa-fcab Intravenous in Adult Patients with gMG
In clinical studies, the safety of efgartigimod alfa-fcab administered intravenously in adult patients with gMG has been evaluated in 364 patients who received at least one dose of efgartigimod alfa-fcab.
In Study 1, 84 patients received efgartigimod alfa-fcab 10 mg/kg [see Clinical Studies (14)]. Of these 84 patients, approximately 75% were female, 82% were White, 11% were Asian, and 8% were of Hispanic or Latino ethnicity. The mean age at study entry was 46 years (range 19 to 78).
The minimum time to initiate a subsequent cycle, specified by study protocol, was 28 days from the last administration of the previous treatment cycle. On average, efgartigimod alfa-fcab-treated patients received 2 cycles in Study 1. The mean and median times to the second treatment cycle were 54 days and 50 days from the last administration of the first treatment cycle, respectively, for efgartigimod alfa-fcab-treated patients.
Adverse reactions reported in at least 5% of patients treated with efgartigimod alfa-fcab and more frequently than placebo are summarized in Table 1. The most common adverse reactions (reported in at least 10% of efgartigimod alfa-fcab-treated patients) were respiratory tract infection, headache, and urinary tract infection.
Table 1: Adverse Reactions in at least 5% of Patients with gMG Treated with Efgartigimod Alfa-fcab Intravenously (EFG IV) and More Frequently than in Placebo-Treated Patients in Study 1 (Safety Population) Adverse reaction EFG IV
(N=84)
%Placebo
(N=83)
%- * Headache includes migraine and procedural headache.
- † Paresthesia includes oral hypoesthesia, hypoesthesia, and hyperesthesia.
Respiratory tract infection 33 29 Headache* 32 29 Urinary tract infection 10 5 Paraesthesia† 7 5 Myalgia 6 1 In a placebo-controlled study in patients with gMG who are anti-acetylcholine receptor (AChR) antibody negative (Study 2), 119 patients received one cycle of once-weekly administrations for 4 weeks of either efgartigimod alfa-fcab 10 mg/kg (n=58) or placebo (n=61). The overall safety profile in Study 2 was consistent with the known safety profile of efgartigimod alfa-fcab in patients with gMG except for nausea, which occurred in 7% of anti-AChR antibody negative patients who received efgartigimod alfa-fcab compared to 5% of patients who received placebo.
The safety of initiating subsequent cycles between 7 and 28 days from the last administration of the previous treatment cycle was assessed in another study in 60 patients (78% anti-AChR antibody positive and 22% anti-AChR antibody negative). Of these patients, 63% were exposed to treatment for over a year when cycles were initiated less than 4 weeks after the last administration. Safety in these patients was similar to that seen in Study 1.
Adverse Reactions with VYVGART HYTRULO in Patients with gMG
In an active-controlled study in patients with gMG (Study 3), 110 patients were randomized and received one cycle of once weekly administrations for 4 weeks (4 administrations total), of either VYVGART HYTRULO subcutaneously (n=55) or efgartigimod alfa-fcab intravenously (n=55) at recommended doses [see Dosage and Administration (2.2)]. The open-label extension of Study 3 included 128 patients who switched from efgartigimod alfa-fcab IV to VYVGART HYTRULO.
The most common adverse reactions (reported in at least 10% of VYVGART HYTRULO-treated patients) were injection site reactions and headache.
In Study 3, injection site reactions occurred in 38% of patients receiving VYVGART HYTRULO. These were injection site rash, erythema, pruritus, bruising, pain, and urticaria.
In Study 3 and its open-label extension (n = 168), all injection site reactions were mild to moderate in severity and did not lead to treatment discontinuation. The majority occurred within 24 hours after administration and resolved spontaneously. Most injection site reactions occurred during the first treatment cycle, and the incidence decreased with each subsequent cycle.
Clinical Experience in Patients with CIDP
Adverse Reactions with VYVGART HYTRULO in Patients with CIDP
In a placebo-controlled study in patients with CIDP (Study 4, stage B), 221 patients were randomized to receive once-weekly administration of either VYVGART HYTRULO 1,008 mg /11, 200 units subcutaneously (n=111) or placebo (n=110) [see Clinical Studies (14.2)]. The mean duration of treatment with VYVGART HYTRULO in stage B was 25 weeks. The overall safety profile observed in patients with CIDP treated with VYVGART HYTRULO was consistent with the known safety profile of VYVGART HYTRULO and of efgartigimod alfa-fcab administered intravenously.
In Study 4, injection site reactions occurred in 15% of patients treated with VYVGART HYTRULO compared to 6% of patients who received placebo. The most common of these injection site reactions were injection site bruising and injection site erythema. All injection site reactions were mild to moderate in severity. Most injection site reactions occurred during the first 3 months of treatment.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of efgartigimod alfa products. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorders: Hypersensitivity reactions including anaphylaxis and hypotension, and infusion/injection-related reactions [see Warnings and Precautions (5.2, 5.3)].
Neurological: There have been reports of worsening of symptoms and signs of CIDP when transitioning from intravenous immunoglobulin treatments to VYVGART HYTRULO [see Drug Interactions (7.1)].
-
7 DRUG INTERACTIONS
7.1 Effect of VYVGART HYTRULO on Other Drugs
Concomitant use of VYVGART HYTRULO with medications that bind to the human neonatal Fc receptor (FcRn) (e.g., immunoglobulin products, monoclonal antibodies, or antibody derivates containing the human Fc domain of the IgG subclass) may lower systemic exposures and reduce effectiveness of such medications. Closely monitor for reduced effectiveness of medications that bind to the human neonatal Fc receptor. When concomitant long-term use of such medications is essential for patient care, consider discontinuing VYVGART HYTRULO and using alternative therapies.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to VYVGART HYTRULO during pregnancy. Healthcare providers and patients may call 1-855-272-6524 or go to https://www.Vyvgartpregnancy.com to enroll in or to obtain information about the registry.
Risk Summary
There are no available data on the use of VYVGART HYTRULO or efgartigimod alfa containing products during pregnancy. There was no evidence of adverse developmental outcomes following the intravenous administration of efgartigimod alfa at up to 100 mg/kg/day in rats and rabbits (see Data).
The background rate of major birth defects and miscarriage in the indicated population is unknown. In the U.S. general population, the estimated background rate of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Monoclonal antibodies are increasingly transported across the placenta as pregnancy progresses, with the largest amount transferred during the third trimester. Therefore, efgartigimod alfa may be transmitted from the mother to the developing fetus.
As VYVGART HYTRULO is expected to reduce maternal IgG antibody levels, reduction in passive protection to the newborn is anticipated. Risk and benefits should be considered prior to administering live vaccines to infants exposed to VYVGART HYTRULO in utero [see Warnings and Precautions (5.1)].
Data
Animal Data
VYVGART HYTRULO for subcutaneous injection contains efgartigimod alfa and hyaluronidase [see Description (11)].
Efgartigimod alfa:
- - Intravenous administration of efgartigimod alfa (0, 30, or 100 mg/kg/day) to pregnant rats and rabbits throughout organogenesis resulted in no adverse effects on embryofetal development in either species. Maternal efgartigimod alfa exposures at the highest no-effect doses were approximately 8 and 62 times, respectively, that in humans at the recommended human dose (RHD) of 1008 mg.
- - Intravenous administration of efgartigimod alfa (0, 30, or 100 mg/kg/day) to rats throughout gestation and lactation resulted in no adverse effects on pre- or postnatal development. Maternal exposures at the highest no-effect dose were approximately 13 times that in humans at the RHD.
Hyaluronidase:
- - In a study in which hyaluronidase (human recombinant) was administered by subcutaneous injection to pregnant mice throughout organogenesis, increased embryofetal mortality and decreased fetal body weights were observed at the highest doses tested. The no-effect dose for adverse effects on embryofetal development in the mouse was approximately 1800 times the dose of hyaluronidase at the recommended human dose (RHD) of VYVGART HYTRULO (1,008 mg efgartigimod alfa and 11,200 U hyaluronidase), on a U/kg basis.
- - There were no adverse effects on pre- and postnatal development following subcutaneous administration of hyaluronidase (human recombinant) to mice throughout gestation and lactation at doses up to 5,000 times the dose of hyaluronidase at the RHD of VYVGART HYTRULO, on a U/kg basis.
8.2 Lactation
Risk Summary
There is no information regarding the presence of efgartigimod alfa or hyaluronidase, from administration of VYVGART HYTRULO, in human milk, the effects on the breastfed infant, or the effects on milk production. Maternal IgG is known to be present in human milk.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for VYVGART HYTRULO and any potential adverse effects on the breastfed infant from VYVGART HYTRULO or from the underlying maternal condition.
8.5 Geriatric Use
Clinical studies of VYVGART HYTRULO did not include sufficient numbers of patients aged 65 and older to determine whether they respond differently from younger adult patients.
8.6 Renal Impairment
No dose adjustment of VYVGART HYTRULO is needed for patients with mild renal impairment. There are insufficient data to evaluate the impact of moderate renal impairment (eGFR 30-59 mL/min/1.73 m2) and severe renal impairment (eGFR <30 mL/min/1.73 m2) on pharmacokinetic parameters of VYVGART HYTRULO [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
VYVGART HYTRULO is a fixed-combination drug product containing efgartigimod alfa and hyaluronidase (human recombinant).
Efgartigimod alfa, a neonatal Fc receptor blocker, is a human immunoglobulin G1 (IgG1) -derived Fc fragment (fragment, crystallized) of the za allotype, produced in Chinese hamster ovary (CHO) cells. The efgartigimod alfa Fc fragment is a homodimer consisting of two identical peptide chains each consisting of 227 amino acids linked together by two interchain disulfide bonds with affinity for FcRn. The molecular weight of efgartigimod alfa is approximately 54 kDa.
Hyaluronidase (human recombinant) is an endoglycosidase used to increase the dispersion and absorption of co-administered drugs when administered subcutaneously. Hyaluronidase (human recombinant) is a glycosylated single-chain protein produced by Chinese hamster ovary cells containing a DNA plasmid encoding for a soluble fragment of human hyaluronidase (PH20). Hyaluronidase (human recombinant) has a molecular weight of approximately 61 kDa.
VYVGART HYTRULO (efgartigimod alfa and hyaluronidase-qvfc) injection is a sterile, preservative free, yellowish, clear to opalescent solution supplied in a single-dose prefilled syringe or vial for subcutaneous injection.
Each 5 mL single-dose prefilled syringe contains 1,000 mg of efgartigimod alfa and 10,000 units of hyaluronidase (human recombinant). Each mL contains 200 mg of efgartigimod alfa, 2,000 units of hyaluronidase (human recombinant) and arginine hydrochloride (10.5 mg), histidine (1.4 mg), L-histidine hydrochloride monohydrate (2.2 mg), methionine (1.5 mg), polysorbate 80 (0.4 mg), sodium chloride (4.1 mg), sucrose (20.5 mg), and Water for Injection, USP, at a pH of 6.0.
Each 5.6 mL single-dose vial contains 1,008 mg of efgartigimod alfa and 11,200 units of hyaluronidase (human recombinant). Each mL contains 180 mg of efgartigimod alfa, 2,000 units of hyaluronidase (human recombinant) and arginine hydrochloride (10.5 mg), histidine (1.4 mg), L- histidine hydrochloride monohydrate (2.2 mg), methionine (1.5 mg), polysorbate 80 (0.4 mg), sodium chloride (4.1 mg), sucrose (20.5 mg), and Water for Injection, USP, at a pH of 6.0.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
VYVGART HYTRULO is a coformulation of efgartigimod alfa and hyaluronidase.
Efgartigimod alfa is a human IgG1 antibody fragment that binds to the neonatal Fc receptor (FcRn), resulting in the reduction of circulating IgG.
Hyaluronidase increases permeability of the subcutaneous tissue by depolymerizing hyaluronan. This effect is transient, and permeability of the subcutaneous tissue is restored within 24 to 48 hours.
12.2 Pharmacodynamics
In Study 1 [see Clinical Studies (14)], the pharmacological effect of efgartigimod alfa-fcab was assessed by measuring the decrease in serum IgG levels and anti-AChR autoantibody levels. In patients testing positive for anti-AChR antibodies and who were treated with efgartigimod alfa-fcab intravenous, there was a reduction in total IgG levels relative to baseline. Decrease in anti-AChR autoantibody levels followed a similar pattern. A decrease in anti-AChR autoantibodies was associated with a clinical response in anti-AChR antibody positive patients, as measured by the change from baseline in MG- ADL total score.
In Study 2 [see Clinical Studies (14)], the pharmacological effect of efgartigimod alfa-fcab was assessed by measuring the decrease in serum IgG levels and anti-muscle-specific tyrosine kinase (MuSK) autoantibody levels. In patients testing negative for anti-AChR antibodies who were treated with efgartigimod alfa-fcab IV, there was a reduction in total IgG levels relative to baseline. In patients testing positive for anti-MuSK antibodies, decreases in anti-MuSK autoantibodies levels followed a similar pattern as total IgG levels.
In Study 3, the pharmacological effect of VYVGART HYTRULO administered subcutaneously (SC) at 1,008 mg / 11,200 Units was compared to efgartigimod alfa-fcab administered intravenously at 10 mg/kg (EFG IV) in gMG patients. The maximum mean reduction in anti-AChR autoantibody level was observed at week 4, with a mean reduction of 62.2% and 59.7% in the VYVGART HYTRULO SC and efgartigimod alfa-fcab IV arm, respectively. The decrease in total IgG levels followed a similar pattern. The 90% confidence intervals for the geometric mean ratios of anti-AChR autoantibody reduction at day 29 and AUEC0-4w (area under the effect-time curve from time 0 to 4 weeks post dose) were within the range of 80% to 125%, indicating no clinically significant difference between the two formulations.
12.3 Pharmacokinetics
Efgartigimod alfa exposures were approximately dose-proportional up to the highest subcutaneously tested dose of VYVGART HYTRULO (1750 mg, 1.75 times the recommended dosage).
Absorption
The rate and extent of absorption from a single-dose prefilled syringe and single-dose vial are similar.
Metabolism and Elimination
Efgartigimod alfa and hyaluronidase are expected to be degraded by proteolytic enzymes into small peptides and amino acids.
The terminal half-life is 80 to 120 hours (3 to 5 days).
After a single intravenous dose of 10 mg/kg efgartigimod alfa-fcab in healthy subjects, less than 0.1% of the administered dose was recovered in urine.
Specific Populations
Age, Sex and Race
A population pharmacokinetics analysis assessing the effects of age, body weight, sex, and race did not suggest any clinically significant impact of these covariates on efgartigimod alfa exposures.
Body Weight
A population pharmacokinetics analysis suggests that the influence of body weight on efgartigimod alfa exposure after administration of VYVGART HYTRULO SC 1008 mg was limited and not clinically relevant.
Patients with Renal Impairment
No dedicated pharmacokinetic study has been performed in patients with renal impairment.
Population PK analyses of data from the VYVGART HYTRULO clinical studies indicated that patients with mild renal impairment (eGFR 60-89 mL/min/1.73 m2) had 11 to 20% increase in exposure relative to the exposure in patients with normal renal function [see Use in Specific Populations (8.6)].
Drug Interaction Studies
Clinical drug interactions studies have not been performed with efgartigimod alfa.
P450 Enzymes
Efgartigimod alfa is not metabolized by cytochrome P450 enzymes; therefore, interactions with concomitant medications that are substrates, inducers, or inhibitors of cytochrome P450 enzymes are unlikely.
Drug Interactions with Other Drugs or Biological Products
Efgartigimod alfa may decrease concentrations of compounds that bind to the human FcRn [see Drug Interactions (7.1)].
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of VYVGART HYTRULO or of other efgartigimod products.
In Study 3, in up to 10 weeks following the initiation of a treatment period with 4 weekly administrations, the incidence of anti-efgartigimod alfa antibodies was 35% (19/55) following treatment with VYVGART HYTRULO and 20% (11/55) in patients receiving intravenous efgartigimod alfa-fcab. For both IV and SC arms, neutralizing anti-efgartigimod alfa antibodies were detected in 4% (2/55) of patients.
In Study 4, in up to 12 weeks of treatment in stage A and 48 weeks in stage B, the incidence of anti-efgartigimod alfa antibodies was 6% (20/317) in stage A and 2% (2/111) in stage B, following treatment with VYVGART HYTRULO. Neutralizing anti-efgartigimod alfa antibodies were detected in 0.3% (1/317) of patients in stage A and in no patient in stage B.
Some neutralizing antibodies may not be detected by the assay. The available data are too limited to make definitive conclusions regarding immunogenicity and the effect on pharmacokinetics, safety, or efficacy of VYVGART HYTRULO.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
VYVGART HYTRULO for subcutaneous injection contains efgartigimod alfa and hyaluronidase [see Description (11)].
Carcinogenesis and Mutagenesis
No studies have been conducted to assess the carcinogenic potential of efgartigimod alfa.
No studies have been conducted to assess the genotoxic potential of efgartigimod alfa.
No carcinogenicity or genotoxicity studies were conducted for human recombinant hyaluronidase.
Impairment of Fertility
Intravenous administration of efgartigimod alfa (0, 30, or 100 mg/kg/day) to male and female rats prior to and during mating and continuing in females through gestation day 7 resulted in no adverse effects on fertility. Efgartigimod alfa exposures at the highest no-effect dose were approximately 12 times that in humans at the recommended human dose of 1008 mg. There were no effects on reproductive tissues in monkeys following subcutaneous administration of hyaluronidase (human recombinant) doses up to approximately 1,200 times the dose of hyaluronidase at the recommended human dose (RHD) of VYVGART HYTRULO (1,008 mg efgartigimod alfa and 11,200 U hyaluronidase) on a U/kg basis for 39 weeks. No systemic exposure to hyaluronidase was observed at doses up to approximately 120 times the dose of hyaluronidase at the RHD of VYVGART HYTRULO, on a U/kg basis.
-
14 CLINICAL STUDIES
14.1 Generalized Myasthenia Gravis
The efficacy of efgartigimod alfa-fcab for the treatment of adult patients with generalized myasthenia gravis (gMG) was established in two multicenter, randomized, double-blind, placebo-controlled trials, one in adults who are anti-AChR antibody positive (Study 1) and one in adults who are anti-AChR antibody negative (Study 2). In Study 3, VYVGART HYTRULO demonstrated a comparable pharmacodynamic effect as compared to the efgartigimod alfa-fcab intravenous formulation, which established the efficacy of VYVGART HYTRULO [see Clinical Pharmacology (12.2)].
Study 1 (Efgartigimod Alfa-fcab Intravenous)
The efficacy of efgartigimod alfa-fcab intravenous (EFG IV) for the treatment of gMG in adults who are anti-AChR antibody positive was established in a 26-week, multicenter, randomized, double-blind, placebo-controlled trial (Study 1; NCT03669588).
Study 1 enrolled patients who met the following criteria at screening:
- - Myasthenia Gravis Foundation of America (MGFA) Clinical Classification Class II to IV
- - MG-Activities of Daily Living (MG-ADL) total score of ≥ 5
- - On stable dose of MG therapy prior to screening, that included acetylcholinesterase (AChE) inhibitors, steroids, or non-steroidal immunosuppressive therapies (NSISTs), either in combination or alone
- - IgG levels of at least 6 g/L
A total of 167 patients were enrolled in Study 1 and were randomized to receive either EFG IV 10 mg/kg (1,200 mg for those weighing 120 kg or more) (n=84) or placebo (n=83). Baseline characteristics were similar between treatment groups. Patients had a median age of 46 years at screening (range: 19 to 81 years) and a median time since diagnosis of 7 years. Seventy-one percent were female, and 84% were White. Median MG-ADL total score was 9, and median Quantitative Myasthenia Gravis (QMG) total score was 16. The majority of patients (n=65 for EFG IV; n=64 for placebo) were positive for anti-AChR antibodies.
At baseline, over 80% of patients in each group received AChE inhibitors, over 70% in each treatment group received steroids, and approximately 60% in each treatment group received NSISTs, at stable doses.
Patients were treated with 10 mg/kg EFG IV administered as an intravenous infusion over one hour once weekly for 4 weeks. In patients weighing 120 kg or more, EFG IV was administered as 1200 mg per infusion. Subsequent treatment cycles were administered based on clinical evaluation, but no sooner than 28 days from the last administration of the previous treatment cycle.
The efficacy of EFG IV was measured using the Myasthenia Gravis-Specific Activities of Daily Living scale (MG-ADL) which assesses the impact of gMG on daily functions of 8 signs or symptoms that are typically affected in gMG. Each item is assessed on a 4-point scale where a score of 0 represents normal function and a score of 3 represents loss of ability to perform that function. A total score ranges from 0 to 24, with the higher scores indicating more impairment. In this study, an MG-ADL responder was defined as a patient with a 2-point or greater reduction in the total MG-ADL score compared to the treatment cycle baseline for at least 4 consecutive weeks, with the first reduction occurring no later than 1 week after the last infusion of the cycle.
The primary efficacy endpoint was the comparison of the percentage of MG-ADL responders during the first treatment cycle between treatment groups in the anti-AChR antibody positive population. A statistically significant difference favoring EFG IV was observed in the MG-ADL responder rate during the first treatment cycle (67.7% in the EFG IV-treated group vs 29.7% in the placebo-treated group [p <0.0001]).
The efficacy of EFG IV was also measured using the Quantitative Myasthenia Gravis (QMG) total score which is a 13-item categorical grading system that assesses muscle weakness. Each item is assessed on a 4-point scale where a score of 0 represents no weakness and a score of 3 represents severe weakness. A total possible score ranges from 0 to 39, where higher scores indicate more severe impairment. In this study, a QMG responder was defined as a patient who had a 3-point or greater reduction in the total QMG score compared to the treatment cycle baseline for at least 4 consecutive weeks, with the first reduction occurring no later than 1 week after last infusion of the cycle.
The secondary endpoint was the comparison of the percentage of QMG responders during the first treatment cycle between both treatment groups in the anti-AChR antibody positive patients. A statistically significant difference favoring EFG IV was observed in the QMG responder rate during the first treatment cycle (63.1% in the EFG IV-treated group vs 14.1% in the placebo-treated group [p <0.0001]).
The results are presented in Table 2.
Table 2: MG-ADL and QMG Responders During Cycle 1 in anti-AChR antibody Positive Patients (mITT Analysis Set) EFG IV
n=65
%Placebo
n=64
%P-value Odds Ratio (95% CI) EFG IV= Efgartigimod alfa-fcab intravenous; MG-ADL=Myasthenia Gravis Activities of Daily Living; QMG =Quantitative Myasthenia Gravis; mITT=modified intent-to-treat; n=number of patients for whom the observation was reported; CI = confidence interval;
Logistic regression stratified for anti-AChR antibody status (if applicable), Japanese/Non-Japanese and standard of care, with baseline MG-ADL as covariate / QMG as covariates
Two-sided exact p-valueMG-ADL Responders 67.7 29.7 < 0.0001 4.951 (2.213, 11.528) QMG Responders 63.1 14.1 < 0.0001 10.842 (4.179, 31.200) Figure 1 shows the mean change from baseline on the MG-ADL during cycle 1.
Figure 1: Mean Change in Total MG-ADL From Cycle 1 Baseline Over Time in anti-AChR-antibody Positive Patients (mITT Analysis Set) 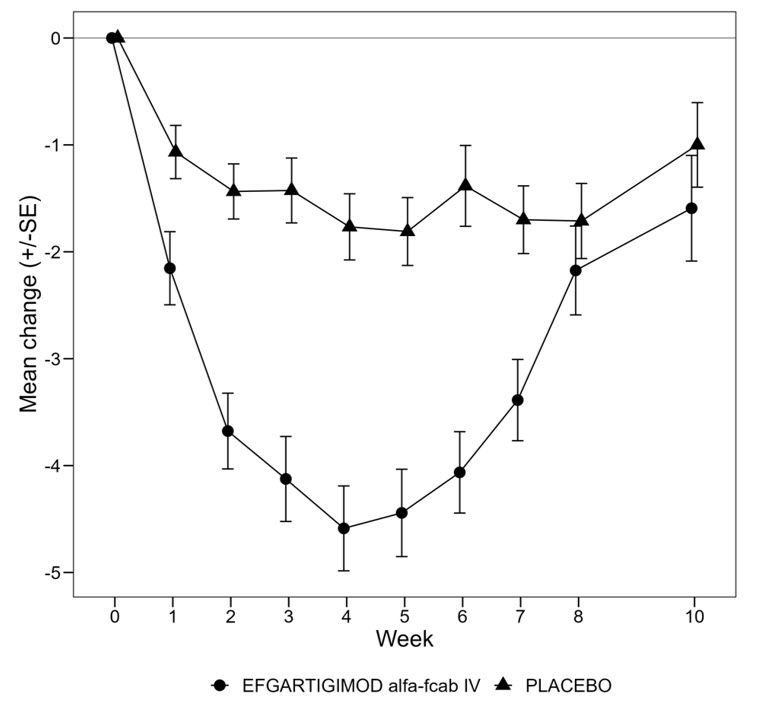
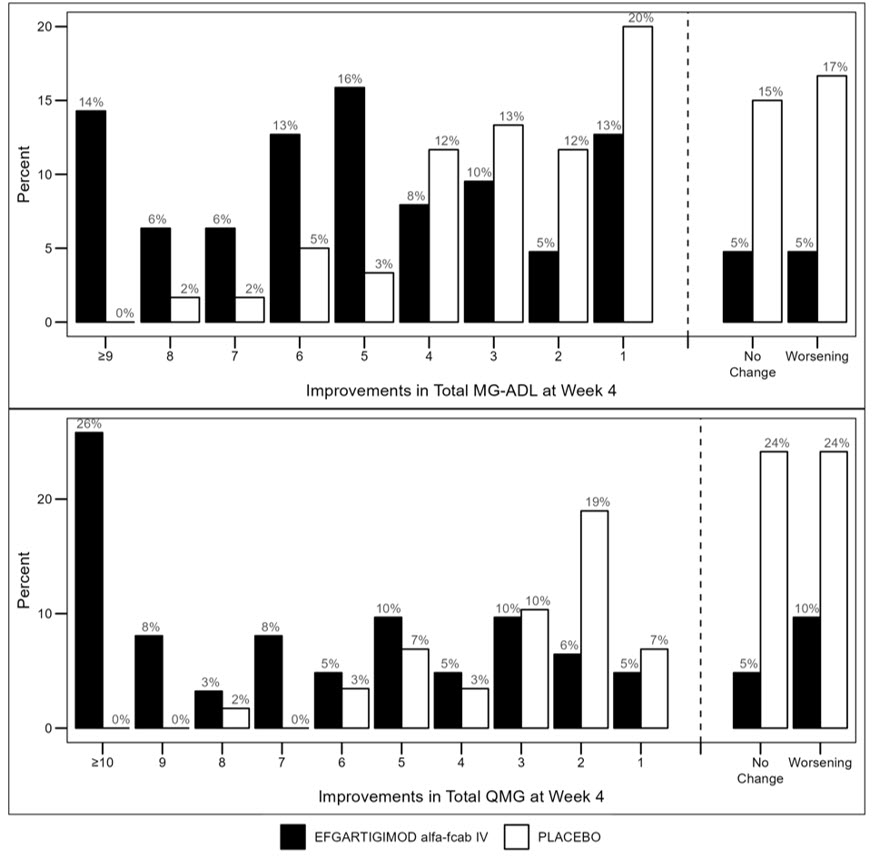
Figure 2 shows the distribution of response on the MG-ADL and QMG during cycle 1, four weeks after the first infusion with EFG IV.
Figure 2: Percentage of Patients with MG-ADL and QMG Total Score Change 4 Weeks Post Initial Infusion of the First Cycle in anti-AChR antibody Positive Patients
Study 2 (Efgartigimod Alfa-fcab Intravenous)
The efficacy of efgartigimod alfa-fcab intravenous (EFG IV) for the treatment of adult patients with gMG who are anti-AChR antibody negative was established in an 8-week, randomized double-blind, placebo-controlled study (Study 2 Part A; NCT06298552). Patients were treated with EFG IV with the recommended dosage regimen for intravenous administration. Subsequent treatment cycles were administered in the open-label Part B of the study per the recommended dosage regimen for two cycles followed by variable inter-treatment periods no shorter than 7 days for additional cycles for up to two years.
Study 2 enrolled patients with gMG who met the following criteria at screening:
- MGFA Clinical Classification Class II to IV
- MG-ADL total score of at least 5
- On stable doses of MG therapy prior to screening, that included AChE inhibitors, steroids or NSISTs, either in combination or alone
- IgG levels of at least 4 g/L
In Study 2 Part A, total of 119 patients were randomized 1:1 to receive once-weekly infusions for 4 weeks of either EFG IV 10 mg/kg (1,200 mg for those weighing 120 kg or more) (n=58) or placebo (n=61).
Baseline characteristics were similar between treatment groups. The median age was 50 years at screening (range: 21 to 80 years; median 49 years for patients treated with EFG IV and 52 years for placebo) and the median time since diagnosis was 4 years. Seventy-six percent of patients were female, and 80% were White, 14% were Asian, and 3% were Black or African American. At baseline, the median MG-ADL total score was 9 and median QMG total score was 14.
At baseline, over 75% of patients in each treatment group received AChE inhibitors and over 60% in each treatment group received steroids at stable doses. There were more patients receiving NSISTs in the EFG IV arm (53%) than those in the placebo arm (36%).
Sixty-one percent (n = 73) of patients were triple seronegative (i.e., anti-AChR, anti-MuSK, and anti- low-density lipoprotein receptor-related protein 4 [LRP4] antibodies negative). Thirty-four percent (n = 40) of patients were positive for anti-MuSK antibodies, and 5% (n = 6) of patients were positive for anti-LRP4 antibodies.
The primary efficacy endpoint was the comparison of the mean change from baseline at Week 4 between treatment groups in the MG-ADL total score. A statistically significant difference favoring EFG IV was observed (LS mean difference [95% CI] = - 1.46 [- 2.50 to -0.41]; p-value = 0.0068; see Figure 3).
Figure 3 shows the mean change from baseline to Week 8 in MG-ADL total score in Study 2 Part A.
Figure 3: Mean Change From Baseline in MG-ADL Total Score Over 8 Weeks in Study 2
Part ASE = standard error, MG-ADL = Myasthenia Gravis Activities of Daily Living, EFG IV = Efgartigimod IV NOTE: Arrows indicate the timepoints at which the treatment was given. The dotted line at week 4 displays the start of the observation period, i.e., no shorter than 7 days from the last administration 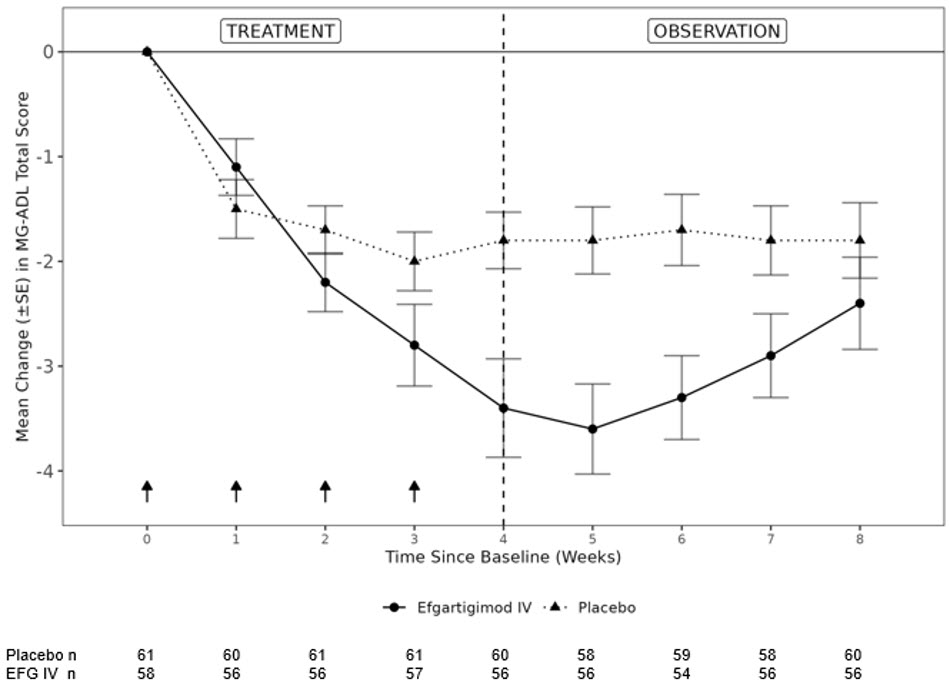
14.2 Chronic Inflammatory Demyelinating Polyneuropathy
The efficacy of VYVGART HYTRULO for the treatment of adults with chronic inflammatory demyelinating polyneuropathy (CIDP) was established in a two stage, multicenter study (Study 4; NCT04281472). Study 4 included a run-in period of up to 12 weeks after withdrawal of existing treatment for CIDP in order to identify patients with active disease, followed by an open-label period to identify VYVGART HYTRULO responders (stage A) who then entered a randomized, double-blind, placebo-controlled, withdrawal period (stage B).
Study 4 enrolled male and female patients age 18 years and older, who at the time of screening, had a documented diagnosis of definite or probable CIDP using the European Federation of Neurological Societies/Peripheral Nerve Society (EFNS/PNS; 2010) criteria for progressing or relapsing forms.
The Inflammatory Neuropathy Cause and Treatment disability score (INCAT) is a scale used to assess the impact of CIDP on daily upper and lower limb function, and is composed of the arm score and leg score (0 to 5 points for each). A total score on the INCAT ranges from 0 to 10 points with a higher number representing more disability. The adjusted INCAT (aINCAT) disability score, identical to the INCAT disability score but with changes in the upper limb function from 0 (normal) to 1 (minor symptoms) excluded, was used to assess efficacy for VYVGART HYTRULO for the treatment of CIDP.
Stage A
In stage A, a total of 322 patients received up to 12 once weekly subcutaneous injections of VYVGART HYTRULO 1008 mg / 11,200 units until evidence of improvement occurred at two consecutive study visits. Improvement was defined as aINCAT improvement ≥1 point, I-RODS improvement ≥4 points, or mean grip strength improvement ≥ 8 kPa. Stage A included 228 patients currently receiving standard-of-care therapy and 94 patients who had either not received prior treatment for CIDP or were not treated with standard-of-care therapy for at least 6 months before study entry. Sixty-nine percent of patients (n=221) who had documented improvement at two consecutive visits during Stage A then entered Stage B. A total of 36 patients (11%) who demonstrated lack of efficacy and 19 patients (6%) that had CIDP worsening as determined by reasonable clinical judgement of investigator, during Stage A did not enter Stage B.
Stage B
In stage B, a total of 221 patients were randomized to receive once weekly subcutaneous injections of VYVGART HYTRULO 1008 mg / 11,200 units (n=111) or placebo (n=110).
Baseline characteristics of patients in stage B were similar between treatment groups. Patients had a median age of 55 years (range: 20 to 82 years), a median time since CIDP diagnosis of 2.2 years, and median INCAT score of 3.0. Sixty-four percent were male and 65% were White, 30% Asian, and 1% African American.
Stage B included 146 patients currently receiving standard-of-care therapy and 75 patients who had either not received prior treatment for CIDP or were not treated with standard-of-care therapy for at least 6 months before study entry.
The primary endpoint was the time to clinical deterioration defined as a 1-point increase in aINCAT at two consecutive visits or a >1-point increase in aINCAT at one visit. Patients who had clinical deterioration or completed week 48 in Stage B without clinical deterioration were withdrawn from the placebo-controlled portion of the study. The study stopped when 88 events of clinical deterioration occurred for the primary endpoint analysis.
Patients who received VYVGART HYTRULO experienced a longer time to clinical deterioration (i.e., increase of ≥1 point in aINCAT score) compared to patients who received placebo, which was statistically significant, as demonstrated by a hazard ratio of 0.394 (95% CI [0.253; 0.614] p<0.0001). The results are presented in Table 3 and Figure 4.
Table 3: Time to First Increase of ≥1 Point in aINCAT Score In Patients With CIDP In Study 4 Stage B Stage B VYVGART HYTRULO Placebo (N=111) (N=110) N=number of patients in the analysis set; %: percentage; aINCAT: adjusted Inflammatory Neuropathy Cause and Treatment Time to 1st aINCAT increase (clinical deterioration) in days Hazard ratio (95% CI) 0.394 (0.253; 0.614)
p-value <0.0001Figure 4: Time to the First aINCAT Increase (Kaplan-Meier Curve) In Patients With CIDP In Study 4 Stage B

Note: The time to clinical deterioration is defined as the time in days from the first VYVGART HYTRULO or placebo administration in Stage B to the first occurrence of either: an increase in aINCAT score of 1 point compared with Stage B baseline if confirmed at the next visit or an increase in aINCAT score of >1 point compared with Stage B baseline.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
VYVGART HYTRULO (efgartigimod alfa and hyaluronidase-qvfc) injection is a preservative free, sterile, yellowish, clear to opalescent solution supplied as a single-dose prefilled syringe or vial.
Single-Dose Prefilled Syringe
Each single-dose prefilled syringe contains 1,000 mg efgartigimod alfa and 10,000 units hyaluronidase per 5 mL (200 mg/2,000 units per mL). It is available in cartons of one or four single-dose prefilled syringes (NDC: 73475-1221-1 or NDC: 73475-1221-4). The safety needle that is 25G, 5/8 inches length, and thin wall type is not included in the carton.
Store VYVGART HYTRULO prefilled syringe refrigerated at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light until time of use. Do not freeze. Do not shake.
If needed, prefilled syringes may be stored at room temperature at up to 30°C (86°F) in the original carton for a single period of up to 30 days after removing from refrigerator or until expiration date on the carton, whichever occurs first. Record the date removed from the refrigerator on the carton.
Single-Dose Vial
Each single-dose vial contains 1,008 mg efgartigimod alfa and 11,200 units hyaluronidase per 5.6 mL (180 mg/2,000 units per mL). It is available in carton of one single-dose vial (NDC: 73475-3102-3).
Store VYVGART HYTRULO vials refrigerated at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light until time of use. Do not freeze. Do not shake.
If needed, unopened vials may be stored in the original carton for up to 3 days at room temperature at 20°C to 25°C (68°F to 77°F) for a single period before administration or returned to refrigeration. Do not store the vial at room temperature more than one time. Record the date removed from and the date returned to the refrigerator on the carton.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read FDA-approved patient labeling (Patient Information and Instructions for Use).
Infections
Instruct patients to communicate any history of infections to the healthcare provider and to contact their healthcare provider if they develop any symptoms of an infection. Advise patients to complete age-appropriate vaccines according to immunization guidelines prior to initiation of a new treatment cycle with VYVGART HYTRULO. Administration of live vaccines is not recommended during treatment with VYVGART HYTRULO [see Warnings and Precautions (5.1)].
Hypersensitivity Reactions
Inform patients that hypersensitivity reactions, including angioedema and anaphylaxis, have occurred in patients who were treated with efgartigimod alfa products. Inform patients about the signs and symptoms of these reactions, and advise patients to contact their healthcare provider immediately if these occur [see Warnings and Precautions (5.2)].
Infusion/injection-Related Reactions
Advise patients of the potential risk of infusion/injection-related reactions, which can include hypertension, chills, shivering, and chest, abdominal, and back pain [see Warnings and Precautions (5.3)].
Neurological Symptoms
Instruct patients to communicate any worsening of neurological symptoms to their prescriber. Symptom progression may indicate a lack of efficacy in these patients.
Instruction on Injection Technique
If a patient or caregiver is to administer subcutaneous VYVGART HYTRULO prefilled syringe, instruct them in proper injection technique (e.g., site selection and duration of injection) [see Instructions for Use].
Pregnancy Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to VYVGART HYTRULO during pregnancy. Encourage participation and advise patients about how they may enroll in the registry [see Use in Specific Populations (8.1)].
-
SPL UNCLASSIFIED SECTION
Manufactured by:
argenx BV
Industriepark 7
9052 Zwijnaarde, Belgium
U.S. License No. 2217Halozyme, Inc.
12390 El Camino Real
San Diego, CA 92130
U.S. License No. 2187Distributed by:
argenx US, Inc.
33 Arch Street
Boston, MA 02110VYVGART HYTRULO is a registered trademark of argenx BV.
© 2026 argenx BVFor patent information: www.argenx.com/patents
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
VYVGART HYTRULO® [viv' gart hye troo' loe]
(efgartigimod alfa and hyaluronidase-qvfc)
injection, for subcutaneous useThis Patient Information has been approved by the U.S. Food and Drug Administration Revised: 5/2026 What is VYVGART HYTRULO?
VYVGART HYTRULO is a prescription medicine used to treat adults with:- generalized myasthenia gravis (gMG).
- chronic inflammatory demyelinating polyneuropathy (CIDP).
Do not take VYVGART HYTRULO if you: - are allergic to efgartigimod alfa, hyaluronidase, or any of the ingredients in VYVGART HYTRULO. See the end of this leaflet for a complete list of ingredients in VYVGART HYTRULO. VYVGART HYTRULO can cause serious allergic reactions and a decrease in blood pressure leading to fainting.
Before taking VYVGART HYTRULO, tell your healthcare provider about all of your medical conditions, including if you: - have an infection or fever.
- have recently received or are scheduled to receive any vaccinations.
- have any history of allergic reactions.
- have kidney (renal) problems.
- are pregnant or plan to become pregnant. It is not known whether VYVGART HYTRULO will harm your unborn baby.
- Pregnancy Exposure Registry. There is a pregnancy exposure registry for women who use VYVGART HYTRULO during pregnancy. The purpose of this registry is to collect information about your health and your baby. Your healthcare provider can enroll you in this registry. You may also enroll yourself or get more information about the registry by calling 1-855-272-6524 or going to https://www.Vyvgartpregnancy.com
- are breastfeeding or plan to breastfeed. It is not known if VYVGART HYTRULO passes into your breast milk.
How should I take VYVGART HYTRULO? - Take VYVGART HYTRULO exactly as your healthcare provider tells you to take it.
- VYVGART HYTRULO is injected under your skin (subcutaneously) only. Do not inject into a vein (intravenously).
- For gMG, VYVGART HYTRULO is injected 1 time weekly for 4 weeks (treatment cycle).Your healthcare provider will tell you when to administer your future treatment cycles.
- For CIDP, VYVGART HYTRULO is injected 1 time weekly.
- VYVGART HYTRULO comes in a single-dose prefilled syringe and single-dose vial.
- Only a healthcare provider should inject VYVGART HYTRULO using the single-dose vial.
- If you are prescribed the VYVGART HYTRULO prefilled syringe, you or your caregiver should receive training on the right way to prepare and inject VYVGART HYTRULO prefilled syringe.
- Do not try to inject VYVGART HYTRULO prefilled syringe until you have been shown the right way by a healthcare provider. See the detailed Instructions for Use that comes with VYVGART HYTRULO prefilled syringe.
- Monitor for signs and symptoms of an allergic reaction for at least 30 minutes after administration. See "What are the possible side effects of VYVGART HYTRULO?"
- If you miss a dose of VYVGART HYTRULO, you may give the injection for up to 3 days after the missed dose. After that, continue with the normal time between doses (dosing schedule).
What are the possible side effects of VYVGART HYTRULO?
VYVGART HYTRULO can cause side effects which can be serious, including:- Infection. VYVGART HYTRULO may increase the risk of infection. If you have an active infection, your healthcare provider should delay your treatment with VYVGART HYTRULO until your infection is gone. Tell your healthcare provider right away if you get any of the following signs and symptoms of an infection:
- fever
- chills
- frequent and painful urination
- cough
- pain and blockage of nasal passages
- wheezing
- shortness of breath
- sore throat
- excess phlegm
- nasal discharge
- Allergic reactions (hypersensitivity reactions). VYVGART HYTRULO can cause allergic reactions that can be severe. These reactions can happen during, shortly after, or weeks after your VYVGART HYTRULO injection. Tell your healthcare provider or get emergency help right away if you have any of the following symptoms of an allergic reaction:
- rash
- swelling of the face, lips, throat, or tongue
- shortness of breath
- hives
- trouble breathing
- low blood pressure
- fainting
- Infusion or injection-related reactions. VYVGART HYTRULO can cause infusion or injection-related reactions. These reactions can happen during or shortly after your VYVGART HYTRULO injection. Tell your healthcare provider if you have any of the following symptoms of an infusion or injection-related reaction:
- high blood pressure
- chills
- shivering
- chest, stomach, or back pain
The most common side effects of VYVGART HYTRULO include: - respiratory tract infection
- injection site reactions
- headache
- urinary tract infection
These are not all the possible side effects of VYVGART HYTRULO. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store VYVGART HYTRULO?
Refrigerated- Store VYVGART HYTRULO in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Warm VYVGART HYTRULO by letting it sit on a flat surface for at least 30 minutes for the prefilled syringe and 15 minutes for the vial. Do not warm VYVGART HYTRULO any other way (e.g., with an external heat source).
- Store VYVGART HYTRULO in the original carton to protect from light until you are ready to use it.
- Do not freeze VYVGART HYTRULO.
- Do not shake VYVGART HYTRULO.
- Do not use VYVGART HYTRULO past the expiration date.
- VYVGART HYTRULO is for one-time use only (single-dose).
- Remove from the refrigerator only the dose you are preparing to inject. Future doses should remain refrigerated until ready to use.
- If needed, VYVGART HYTRULO may be stored at room temperature. Record the date you removed VYVGART HYTRULO from the refrigerator.
- Single-dose prefilled syringe may be stored at room temperature (up to 86°F (30°C)) for up to 30 days. When removed from the refrigerator and brought to room temperature VYVGART HYTRULO must be used within 30 days or thrown away.
- Single-dose vial may be stored at room temperature (between 68°F to 77°F (20°C to 25°C)) for up to 3 days. When removed from the refrigerator and brought to room temperature, VYVGART HYTRULO must be used within 3 days or thrown away.
- Do not put VYVGART HYTRULO back into the refrigerator after it has warmed to room temperature.
General information about the safe and effective use of VYVGART HYTRULO.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use VYVGART HYTRULO for a condition for which it was not prescribed. Do not give VYVGART HYTRULO to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about VYVGART HYTRULO that is written for health professionals.What are the ingredients in VYVGART HYTRULO?
Active ingredients: efgartigimod alfa and hyaluronidase (human recombinant)
Inactive ingredients:- single-dose prefilled syringe: arginine hydrochloride, histidine, L-histidine hydrochloride monohydrate, methionine, polysorbate 80, sodium chloride, sucrose, and water for injection, USP
- single-dose vial: arginine hydrochloride, histidine, L-histidine hydrochloride monohydrate, methionine, polysorbate 80, sodium chloride, sucrose and water for injection, USP
- argenx BV Industriepark 7, 9052 Zwijnaarde, Belgium, U.S. License No. 2217
- Halozyme, Inc. 12390 El Camino Real, San Diego, CA 92130, U.S. License No. 2187
VYVGART HYTRULO is a registered trademark of argenx BV.
© 2026 argenx BV -
INSTRUCTIONS FOR USE
VYVGART HYTRULO [viv' gart hye troo' loe]
(efgartigimod alfa and hyaluronidase-qvfc)
injection, for subcutaneous use
1,000 mg and 10,000 units/5 mLThis Instructions for Use contains information on how to inject VYVGART HYTRULO.
Important Information You Need to Know Before Injecting VYVGART HYTRULO
- VYVGART HYTRULO is for under the skin (subcutaneous) injection only.
- Be sure to read and understand this Instructions for Use before injecting VYVGART HYTRULO. Your healthcare provider should show you or your caregiver how to prepare and inject VYVGART HYTRULO the correct way before using it for the first time. Ask your healthcare provider if you have any questions.
- The prefilled syringe is for one-time-use only and cannot be reused.
- Do not use VYVGART HYTRULO if it has been at room temperature for longer than 30 days.
- Do not use the prefilled syringe if it is expired.
- Do not use the prefilled syringe if it is cracked, broken, or damaged. Return damaged prefilled syringes to the specialty pharmacy.
- Do not use a prefilled syringe if the liquid medicine is discolored or contains particles. The liquid medicine should look clear to yellowish in color. A little cloudiness is normal.
- Do not shake the prefilled syringe.
- You will need supplies that are not provided with VYVGART HYTRULO (see Figure J).
Storing VYVGART HYTRULO Prefilled Syringe
- Store VYVGART HYTRULO in the refrigerator between 36°F to 46°F (2°C to 8°C) (see Figure A).
- Keep the prefilled syringes in the original carton to protect them from light until ready to use.
- Keep VYVGART HYTRULO and all medicines out of the reach of children.

Do not freeze the prefilled syringes or store them in direct sunlight.
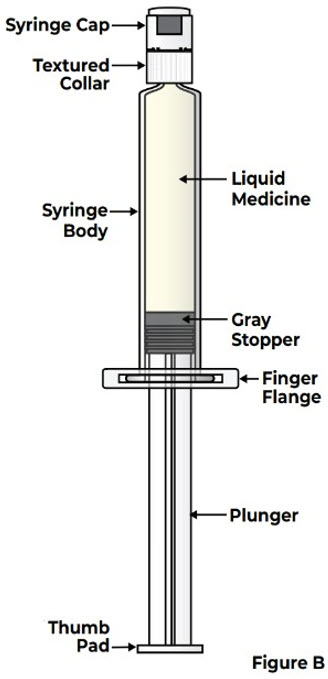
Do not use a prefilled syringe that has been frozen or left in direct sunlight.Prefilled Syringe Parts
Safety Needle (not provided with VYVGART HYTRULO prefilled syringe) that is 25G, 5/8 inch length, thin wall 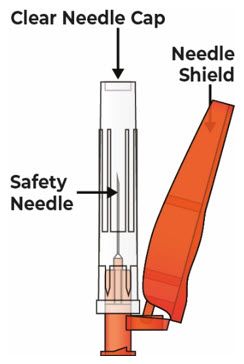
Gather and Check the Prefilled Syringe
1 Remove the carton from the refrigerator 1.1 Remove the carton containing the prefilled syringe from the refrigerator (see Figure C). 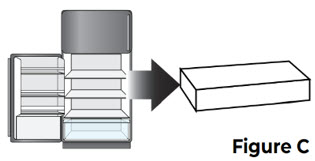
1.2 Remove 1 prefilled syringe from the carton (see Figure D) and place any remaining prefilled syringes back into the refrigerator for later use. 
1.3 Remove the prefilled syringe from the tray (see Figure E). 
2 Check the prefilled syringe before use 2.1 Check the expiration date on the prefilled syringe label (see Figure F). 
Do not use the prefilled syringe if the expiration date has passed. 2.2 Check the condition of the prefilled syringe and the prefilled syringe cap (see Figure G). 
Do not use the prefilled syringe if it is cracked, broken, damaged, or if the prefilled syringe cap is missing. 2.3 Check the appearance of the liquid medicine in the prefilled syringe (see Figure H).
The liquid medicine should look clear to yellowish in color. A little cloudiness is normal.
Do not use the prefilled syringe if the liquid medicine is discolored or contains particles. Prepare for the Injection
3 Allow the prefilled syringe to warm to room temperature 3.1 Place the prefilled syringe on a clean flat surface and let it sit for at least 30 minutes, to allow it to warm to room temperature (see Figure I). 
Do not attempt to warm the prefilled syringe in any other way.
Do not use the VYVGART HYTRULO prefilled syringe if it has been at room temperature for longer than 30 days.4 Gather supplies and wash your hands 4.1 Gather the following supplies that are not provided with the prefilled syringe (see Figure J). 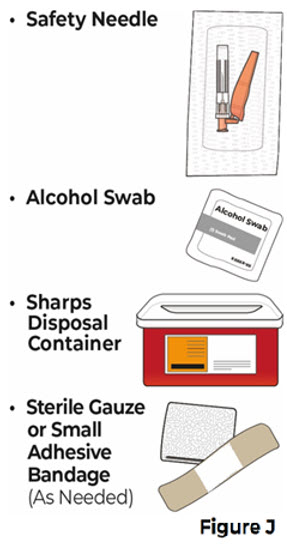
4.2 Wash your hands with soap and water (see Figure K). 
5 Snap off the prefilled syringe cap and attach the safety needle 5.1 Carefully open the safety needle package and remove the needle (see Figure L).
Throw away the packaging into household trash.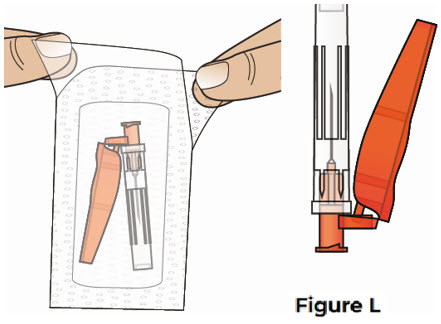
5.2 Bend the prefilled syringe cap to one side to snap it off and remove it from the prefilled syringe (see Figure M).
Throw away the prefilled syringe cap into the household trash.
Do not touch the tip of the prefilled syringe after the prefilled syringe cap has been removed. 5.3 Hold the prefilled syringe by the syringe body in one hand, and attach the safety needle to the prefilled syringe by twisting it to the right (clockwise) until you feel resistance (see Figure N). 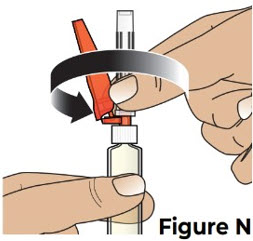
The safety needle is now attached to the prefilled syringe (see Figure O). 
6 Choose and clean the injection site on the stomach (abdomen) 6.1 Choose an injection site on the stomach (abdomen) at least 2 inches away from the belly button (navel) (see Figure P).
Change (rotate) the injection site the next time you inject VYVGART HYTRULO.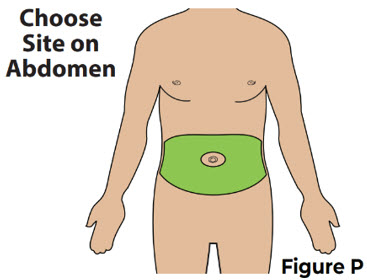
Do not inject into skin that is irritated, red, bruised, infected, or tender. Do not inject into skin that is hard, scarred, or has moles.
Do not inject into a vein. VYVGART HYTRULO is for under the skin (subcutaneous) injection only.6.2 Clean the chosen injection site with an alcohol swab and let it air dry (see Figure Q). 
Do not blow on or touch the injection site after it has been cleaned. Inject VYVGART HYTRULO
7 Pull back the needle shield and remove the clear needle cap 7.1 Pull the needle shield back (see Figure R).
Note: The needle shield will be used after the injection to cover the needle and protect from needle-stick injuries.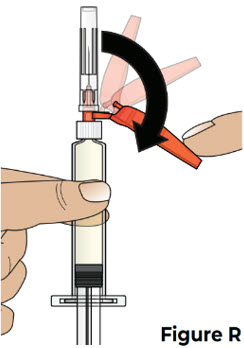
7.2 Hold the prefilled syringe body and remove the clear needle cap by pulling it straight off of the needle (see Figure S).
Throw away the clear needle cap into the household trash.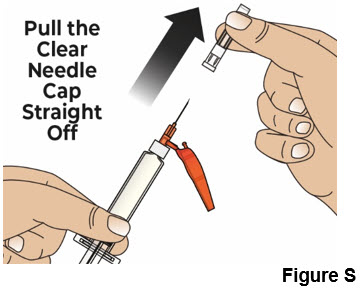
Do not recap the needle. 8 Give the injection 8.1 Pinch the cleaned injection site (Figure T). 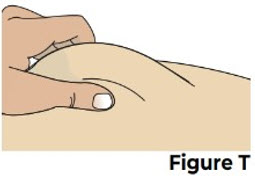
While pinching the skin, insert the needle at a 45 to 90 degree angle all the way into the pinched skin (see Figure U).
Then release the pinched skin.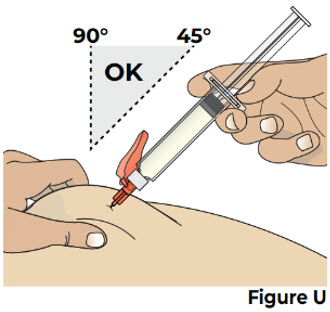
Do not pinch the skin too tightly as this can cause bruising. 8.2 Slowly press the thumb pad down all the way until it stops to inject the liquid medicine (see Figure V).
It will take about 20 to 30 seconds to inject all of the liquid medicine.
You will feel resistance as you press down. Inject more slowly in case there is discomfort.
It is ok if you need to pause or change your grip during the injection.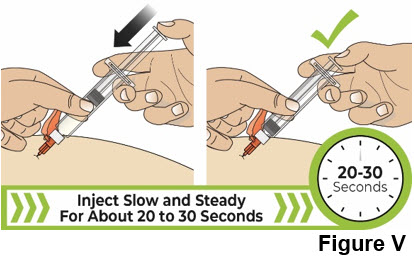
Do not try to force the thumb pad down quickly as this will make the thumb pad harder to press. 8.3 After all the liquid medicine is injected, remove the needle from the skin by pulling it straight out without changing the angle that it was inserted (see Figure W). 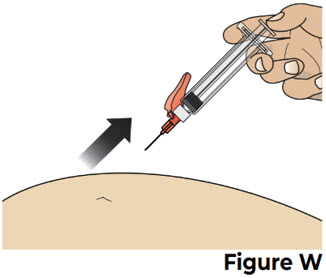
Throw away (dispose of) the Used Prefilled Syringe
9 Cover the needle and throw away the used prefilled syringe 9.1 Carefully push the needle shield over the needle until it snaps into place and covers the needle (see Figure X).
This helps to prevent needle-stick injuries.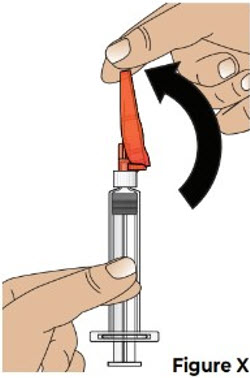
Do not recap the needle using the clear needle cap. Only use the needle shield to cover the needle. 9.2 Throw away the used prefilled syringe, with the needle still attached, into an FDA-cleared sharps disposal container right away after use (see Figure Y). 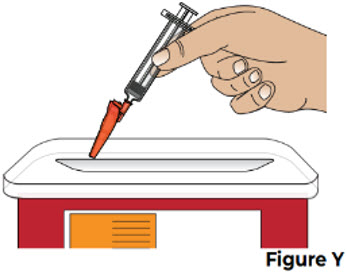
Do not throw away (dispose of) loose needles and syringes in your household trash. 10 Treat the injection site 10.1 If there is a small amount of blood or liquid at the injection site, press a sterile gauze over the injection site until the bleeding stops (see Figure Z).
If needed, you may apply a small adhesive bandage.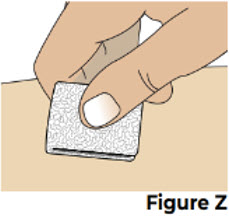
Additional Disposal Information
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is: - made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
Always keep the sharps disposal container out of the reach of children.Additional Information
This Instructions for Use has been approved by the U.S. Food and Drug Administration Issued: 4/2025 For more information, go to www.vyvgart.com or call 1-833-898-4278.
Manufactured by:
argenx BV
Industriepark 7
9052 Zwijnaarde, Belgium
U.S. License No. 2217
Halozyme, Inc.
12390 El Camino Real
San Diego, CA 92130
U.S. License No. 2187
Distributed by:
argenx US, Inc.
33 Arch Street
Boston, MA 02110 -
PRINCIPAL DISPLAY PANEL - 1,008 mg and 11,2000 units/5.6 mL Vial Carton
NDC: 73475-3102-3
VYVGART® Hytrulo
(efgartigimod alfa and
hyaluronidase-qvfc)INJECTION
1,008 mg and 11,200 units/5.6 mL
(180 mg and 2,000 units/mL)For subcutaneous injection
over 30 to 90 secondsOne single-dose vial
Discard unused portion
Must be administered by
a healthcare providerRx only

-
PRINCIPAL DISPLAY PANEL - 5 mL Syringe Carton
NDC: 73475-1221-1
VYVGART Hytrulo®
(efgartigimod alfa and hyaluronidase-qvfc)INJECTION
1,000 mg and 10,000 units/5 mL
(200 mg and 2,000 units/mL)For subcutaneous injection over 20 to 30 seconds
One single-dose prefilled syringe
Rx only

-
INGREDIENTS AND APPEARANCE
VYVGART HYTRULO
efgartigimod alfa and hyaluronidase (human recombinant) injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 73475-3102 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EFGARTIGIMOD ALFA (UNII: 961YV2O515) (EFGARTIGIMOD ALFA - UNII:961YV2O515) EFGARTIGIMOD ALFA 180 mg in 1 mL HYALURONIDASE (HUMAN RECOMBINANT) (UNII: 743QUY4VD8) (HYALURONIDASE (HUMAN RECOMBINANT) - UNII:743QUY4VD8) HYALURONIDASE (HUMAN RECOMBINANT) 2000 U in 1 mL Inactive Ingredients Ingredient Name Strength ARGININE HYDROCHLORIDE (UNII: F7LTH1E20Y) 10.5 mg in 1 mL histidine (UNII: 4QD397987E) 1.4 mg in 1 mL HISTIDINE MONOHYDROCHLORIDE (UNII: 1D5Q932XM6) 2.2 mg in 1 mL methionine (UNII: AE28F7PNPL) 1.5 mg in 1 mL polysorbate 80 (UNII: 6OZP39ZG8H) 0.4 mg in 1 mL sodium chloride (UNII: 451W47IQ8X) 4.1 mg in 1 mL sucrose (UNII: C151H8M554) 20.5 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 73475-3102-3 1 in 1 CARTON 06/20/2023 1 5.6 mL in 1 VIAL, SINGLE-DOSE; Type 6: Drug/Biologic Combination Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761304 06/20/2023 VYVGART HYTRULO
efgartigimod alfa and hyaluronidase (human recombinant) injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 73475-1221 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EFGARTIGIMOD ALFA (UNII: 961YV2O515) (EFGARTIGIMOD ALFA - UNII:961YV2O515) EFGARTIGIMOD ALFA 200 mg in 1 mL HYALURONIDASE (HUMAN RECOMBINANT) (UNII: 743QUY4VD8) (HYALURONIDASE (HUMAN RECOMBINANT) - UNII:743QUY4VD8) HYALURONIDASE (HUMAN RECOMBINANT) 2000 U in 1 mL Inactive Ingredients Ingredient Name Strength ARGININE HYDROCHLORIDE (UNII: F7LTH1E20Y) 10.5 mg in 1 mL histidine (UNII: 4QD397987E) 1.4 mg in 1 mL HISTIDINE MONOHYDROCHLORIDE (UNII: 1D5Q932XM6) 2.2 mg in 1 mL methionine (UNII: AE28F7PNPL) 1.5 mg in 1 mL polysorbate 80 (UNII: 6OZP39ZG8H) 0.4 mg in 1 mL sodium chloride (UNII: 451W47IQ8X) 4.1 mg in 1 mL sucrose (UNII: C151H8M554) 20.5 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 73475-1221-1 1 in 1 CARTON 04/10/2025 1 5 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 2 NDC: 73475-1221-4 4 in 1 CARTON 04/10/2025 2 5 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761304 04/10/2025 Labeler - argenx US (116702819) Establishment Name Address ID/FEI Business Operations Patheon Italia SpA 434078638 MANUFACTURE(73475-3102) Establishment Name Address ID/FEI Business Operations Avid Bioservices Inc. 042535740 API MANUFACTURE(73475-3102, 73475-1221) Establishment Name Address ID/FEI Business Operations Vetter RVS 316126754 MANUFACTURE(73475-1221) Establishment Name Address ID/FEI Business Operations Lonza Biologics Portsmouth 093149750 API MANUFACTURE(73475-1221) Establishment Name Address ID/FEI Business Operations Catalent Indiana, LLC 172209277 API MANUFACTURE(73475-3102, 73475-1221)
Trademark Results [VYVGART Hytrulo]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 VYVGART HYTRULO 97792721 not registered Live/Pending |
argenx B.V. 2023-02-13 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.