SKYRIZI- risankizumab-rzaa kit
Skyrizi by
Drug Labeling and Warnings
Skyrizi by is a Prescription medication manufactured, distributed, or labeled by AbbVie Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SKYRIZI safely and effectively. See full prescribing information for SKYRIZI.
SKYRIZI® (risankizumab-rzaa) injection, for subcutaneous use
Initial U.S. Approval: 2019
INDICATIONS AND USAGE
SKYRIZI is an interleukin-23 antagonist indicated for the treatment of moderate-to-severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy. (1)
DOSAGE AND ADMINISTRATION
- 150 mg (two 75 mg injections) administered by subcutaneous injection at Week 0, Week 4 and every 12 weeks thereafter. (2.1)
DOSAGE FORMS AND STRENGTHS
- Injection: 75 mg/0.83 mL in each single-dose prefilled syringe. (3)
CONTRAINDICATIONS
- None (4)
WARNINGS AND PRECAUTIONS
- Infections: SKYRIZI may increase the risk of infection. Instruct patients to seek medical advice if signs or symptoms of clinically important infection occur. If such an infection develops, do not administer SKYRIZI until the infection resolves. (5.1)
- Tuberculosis (TB): Evaluate for TB prior to initiating treatment with SKYRIZI. (5.2)
ADVERSE REACTIONS
Most common adverse reactions (≥ 1%) are upper respiratory infections, headache, fatigue, injection site reactions, and tinea infections. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AbbVie Inc. at 1-800-633-9110 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Avoid use of live vaccines in patients treated with SKYRIZI. (7.1)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
2.2 Tuberculosis Assessment Prior to Initiation of SKYRIZI
2.3 Important Administration Instructions
2.4 Preparation for Use of SKYRIZI Prefilled Syringes
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Infections
5.2 Pre-treatment Evaluation for Tuberculosis
5.3 Immunizations
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
7 DRUG INTERACTIONS
7.1 Live Vaccinations
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage
The recommended dose is 150 mg (two 75 mg injections) administered by subcutaneous injection at Week 0, Week 4, and every 12 weeks thereafter.
2.2 Tuberculosis Assessment Prior to Initiation of SKYRIZI
Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with SKYRIZI [see Warnings and Precautions (5.2)].
2.3 Important Administration Instructions
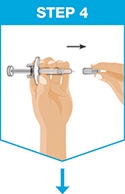
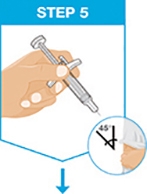
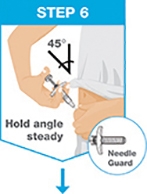
Administer SKYRIZI subcutaneously. Inject two separate 75 mg single-dose prefilled syringes for the full 150 mg dose. Discard prefilled syringes after use. Do not reuse.
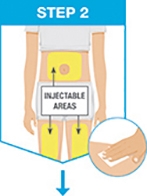
For each dose, administer the injections at different anatomic locations (such as thighs or abdomen), and not into areas where the skin is tender, bruised, erythematous, indurated or affected by psoriasis. Administration of SKYRIZI in the upper, outer arm may only be performed by a healthcare professional or caregiver.
If a dose is missed, administer the dose as soon as possible. Thereafter, resume dosing at the regular scheduled time.
SKYRIZI is intended for use under the guidance and supervision of a healthcare professional. Patients may self-inject SKYRIZI after training in subcutaneous injection technique. Provide proper training to patients and/or caregivers on the subcutaneous injection technique of SKYRIZI. Instruct the patient that two separate 75 mg single-dose injections are required to achieve the 150 mg dose.
The SKYRIZI “Instructions for Use” contains more detailed instructions on the preparation and administration of SKYRIZI [see Instructions for Use]. Instruct the patient to read the Instructions for Use before administration.
2.4 Preparation for Use of SKYRIZI Prefilled Syringes
Before injecting, patients may remove the carton from the refrigerator and allow to reach room temperature out of direct sunlight (15 to 30 minutes) without removing the prefilled syringes from the carton.
Visually inspect SKYRIZI for particulate matter and discoloration prior to administration. SKYRIZI is a colorless to slightly yellow and clear to slightly opalescent solution. It may contain a few translucent to white particles. Do not use if the solution contains large particles or is cloudy or discolored.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Infections
SKYRIZI may increase the risk of infections. In clinical studies, infections occurred in 22.1% of the SKYRIZI group compared to 14.7% of the placebo group through 16 weeks of treatment. Upper respiratory tract infections and tinea infections occurred more frequently in the SKYRIZI group than in the placebo group. Subjects with known chronic or acute infections were not enrolled in clinical studies [see Adverse Reactions (6.1)].
The rate of serious infections for the SKYRIZI group and the placebo group was ≤ 0.4%. Treatment with SKYRIZI should not be initiated in patients with any clinically important active infection until the infection resolves or is adequately treated.
In patients with a chronic infection or a history of recurrent infection, consider the risks and benefits prior to prescribing SKYRIZI. Instruct patients to seek medical advice if signs or symptoms of clinically important infection occur. If a patient develops such an infection or is not responding to standard therapy, monitor the patient closely and do not administer SKYRIZI until the infection resolves.
5.2 Pre-treatment Evaluation for Tuberculosis
Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with SKYRIZI. Across the Phase 3 psoriasis clinical studies, of the 72 subjects with latent TB who were concurrently treated with SKYRIZI and appropriate TB prophylaxis during the studies, none developed active TB during the mean follow-up of 61 weeks on SKYRIZI. Two subjects taking isoniazid for treatment of latent TB discontinued treatment due to liver injury. Of the 31 subjects from the IMMHANCE study with latent TB who did not receive prophylaxis during the study, none developed active TB during the mean follow-up of 55 weeks on SKYRIZI. Consider anti-TB therapy prior to initiating SKYRIZI in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Monitor patients for signs and symptoms of active TB during and after SKYRIZI treatment. Do not administer SKYRIZI to patients with active TB.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of labeling:
- Infections [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse drug reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 2234 subjects were treated with SKYRIZI in clinical development studies in plaque psoriasis. Of these, 1208 subjects with psoriasis were exposed to SKYRIZI for at least one year.
Data from placebo- and active-controlled studies were pooled to evaluate the safety of SKYRIZI for up to 16 weeks. In total, 1306 subjects were evaluated in the SKYRIZI 150 mg group.
Table 1 summarizes the adverse drug reactions that occurred at a rate of at least 1% and at a higher rate in the SKYRIZI group than the placebo group during the 16-week controlled period of pooled clinical studies.
Table 1. Adverse Drug Reactions Occurring in ≥ 1% of Subjects on SKYRIZI through Week 16 Adverse Drug Reactions SKYRIZI
N = 1306
n (%)Placebo
N = 300
n (%)Upper respiratory infectionsa 170 (13.0) 29 (9.7) Headacheb 46 (3.5) 6 (2.0) Fatiguec 33 (2.5) 3 (1.0) Injection site reactionsd 19 (1.5) 3 (1.0) Tinea infectionse 15 (1.1) 1 (0.3) a Includes: respiratory tract infection (viral, bacterial or unspecified), sinusitis (including acute), rhinitis, nasopharyngitis, pharyngitis (including viral), tonsillitis
b Includes: headache, tension headache, sinus headache, cervicogenic headache
c Includes: fatigue, asthenia
d Includes: injection site bruising, erythema, extravasation, hematoma, hemorrhage, infection, inflammation, irritation, pain, pruritus, reaction, swelling, warmth
e Includes: tinea pedis, tinea cruris, body tinea, tinea versicolor, tinea manuum, tinea infection, onychomycosisAdverse drug reactions that occurred in < 1% but > 0.1% of subjects in the SKYRIZI group and at a higher rate than in the placebo group through Week 16 were folliculitis and urticaria.
Specific Adverse Drug Reactions
In the first 16 weeks, infections occurred in 22.1% of the SKYRIZI group (90.8 events per 100 subject-years) compared to 14.7% of the placebo group (56.5 events per 100 subject-years) and did not lead to discontinuation of SKYRIZI. The rates of serious infections for the SKYRIZI group and the placebo group were ≤0.4%. Serious infections in the SKYRIZI group included cellulitis, osteomyelitis, sepsis and herpes zoster. In ULTIMMA-1 and ULTIMMA-2, through Week 52, the rate of infections (73.9 events per 100 subject-years) was similar to the rate observed during the first 16 weeks of treatment.
Through Week 52, no new adverse reactions were identified and the rates of the adverse reactions were similar to those observed during the first 16 weeks of treatment. During this period, serious infections that led to study discontinuation included pneumonia.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other products, including other risankizumab products, may be misleading.
By Week 52, approximately 24% (263/1079) of subjects treated with SKYRIZI at the recommended dose developed antibodies to risankizumab-rzaa. Of the subjects who developed antibodies to risankizumab-rzaa, approximately 57% (14% of all subjects treated with SKYRIZI) had antibodies that were classified as neutralizing. Higher antibody titers in approximately 1% of subjects treated with SKYRIZI were associated with lower risankizumab-rzaa concentrations and reduced clinical response.
-
7 DRUG INTERACTIONS
7.1 Live Vaccinations
Avoid use of live vaccines in patients treated with SKYRIZI [see Warnings and Precautions (5.2)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Limited available data with SKYRIZI use in pregnant women are insufficient to evaluate a drug associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcome. Human IgG is known to cross the placental barrier; therefore, SKYRIZI may be transmitted from the mother to the developing fetus.
In an enhanced pre- and post-natal developmental toxicity study, pregnant cynomolgus monkeys were administered subcutaneous doses of 5 and 50 mg/kg risankizumab-rzaa once weekly during the period of organogenesis up to parturition. At the 50 mg/kg dose [20 times the maximum recommended human dose (MRHD); 2.5 mg/kg based on administration of a 150 mg dose to a 60 kg individual], increased fetal/infant loss was noted in pregnant monkeys (see Data). No risankizumab-rzaa-related effects on functional or immunological development were observed in infant monkeys from birth through 6 months of age. The clinical significance of these findings for humans is unknown.
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
An enhanced pre- and post-natal developmental toxicity study was conducted in cynomolgus monkeys. Pregnant cynomolgus monkeys were administered weekly subcutaneous doses of risankizumab-rzaa of 5 or 50 mg/kg from gestation day 20 to parturition and the cynomolgus monkeys (mother and infants) were monitored for 6 months after delivery. No maternal toxicity was noted in this study. There were no treatment-related effects on growth and development, malformations, developmental immunotoxicology or neurobehavioral development. However, a dose-dependent increase in fetal/infant loss was noted in the risankizumab-rzaa-treated groups (32% and 43% in the 5 mg/kg and 50 mg/kg groups, respectively) compared to the vehicle control group (19%). The increased fetal/infant loss in the 50 mg/kg group was considered to be related to risankizumab-rzaa treatment. The no observed adverse effect level (NOAEL) for maternal toxicity was identified as 50 mg/kg (20 times the MRHD, based on mg/kg comparison) and the NOAEL for developmental toxicity was identified as 5 mg/kg (2 times the MRHD, based on mg/kg comparison). In the infants, mean serum concentrations increased in a dose-dependent manner and were approximately 17-86% of the respective maternal concentrations. Following delivery, most adult female cynomolgus monkeys and all infants from the risankizumab-rzaa‑treated groups had measurable serum concentrations of risankizumab-rzaa up to 91 days postpartum. Serum concentrations were below detectable levels at 180 days postpartum.
8.2 Lactation
There are no data on the presence of risankizumab-rzaa in human milk, the effects on the breastfed infant, or the effects on milk production. Maternal IgG is known to be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for SKYRIZI and any potential adverse effects on the breastfed infant from SKYRIZI or from the underlying maternal condition.
8.4 Pediatric Use
The safety and efficacy of SKYRIZI in pediatric patients less than 18 years of age have not yet been established.
8.5 Geriatric Use
Of the 2234 subjects with plaque psoriasis exposed to SKYRIZI, 243 subjects were 65 years or older and 24 subjects were 75 years or older. No overall differences in risankizumab-rzaa exposure, safety or effectiveness were observed between older and younger subjects who received SKYRIZI. However, the number of subjects aged 65 years and older was not sufficient to determine whether they respond differently from younger subjects.
- 10 OVERDOSAGE
-
11 DESCRIPTION
Risankizumab-rzaa, an interleukin-23 antagonist, is a humanized immunoglobulin G1 (IgG1) monoclonal antibody. Risankizumab-rzaa is produced by recombinant DNA technology in Chinese hamster ovary cells.
SKYRIZI (risankizumab-rzaa) injection is a sterile, preservative-free, colorless to slightly yellow, and clear to slightly opalescent solution for subcutaneous use.
Each prefilled syringe delivers 0.83 mL containing 75 mg risankizumab-rzaa, disodium succinate hexahydrate (0.88 mg), polysorbate 20 (0.17 mg), sorbitol (34 mg), succinic acid (0.049 mg), and Water for Injection, USP.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Risankizumab-rzaa is a humanized immunoglobulin G1 (IgG1) monoclonal antibody that selectively binds to the p19 subunit of human interleukin 23 (IL-23) cytokine and inhibits its interaction with the IL-23 receptor. IL-23 is a naturally occurring cytokine that is involved in inflammatory and immune responses.
Risankizumab-rzaa inhibits the release of pro-inflammatory cytokines and chemokines.
12.2 Pharmacodynamics
No formal pharmacodynamics studies have been conducted with risankizumab-rzaa.
12.3 Pharmacokinetics
Risankizumab-rzaa plasma concentrations increased dose-proportionally from 90 to 180 mg and from 18 to 300 mg (0.6 to 1.2 and 0.12 to 2.0 times the approved recommended dosage) following subcutaneous administration in subjects with plaque psoriasis and healthy volunteers, respectively. Steady-state concentrations were achieved by Week 16 following subcutaneous administration of risankizumab-rzaa at Weeks 0, 4, and every 12 weeks thereafter. At the 150 mg dose, the estimated steady-state peak concentration (Cmax) and trough concentration (Ctrough) were approximately 12 mcg/mL and 2 mcg/mL, respectively.
The absolute bioavailability of risankizumab-rzaa was estimated to be 89% following subcutaneous injection. Cmax was reached by 3-14 days.
The estimated steady-state volume of distribution (inter-subject CV%) was 11.2 L (34%) in subjects with plaque psoriasis.
The estimated systemic clearance (inter-subject CV%) was 0.31 L/day (24%) and terminal elimination half-life was approximately 28 days in subjects with plaque psoriasis.
The metabolic pathway of risankizumab-rzaa has not been characterized. As a humanized IgG1 monoclonal antibody, risankizumab-rzaa is expected to be degraded into small peptides and amino acids via catabolic pathways in a manner similar to endogenous IgG.
No clinically significant differences in the pharmacokinetics of risankizumab-rzaa were observed based on age (≥18 years). No specific studies have been conducted to determine the effect of renal or hepatic impairment on the pharmacokinetics of risankizumab-rzaa.
Risankizumab-rzaa clearance and volume of distribution increase and plasma concentrations decrease as body weight increases; however, no dose adjustment is recommended based on body weight.
No clinically significant changes in exposure of caffeine (CYP1A2 substrate), warfarin (CYP2C9 substrate), omeprazole (CYP2C19 substrate), metoprolol (CYP2D6 substrate), or midazolam (CYP3A substrate) were observed when used concomitantly with risankizumab-rzaa 150 mg administered subcutaneously at Weeks 0, 4, 8 and 12 (more frequent than the approved recommended dosing frequency) in subjects with plaque psoriasis.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity and mutagenicity studies have not been conducted with SKYRIZI.
No effects on male fertility parameters were observed in sexually mature male cynomolgus monkeys subcutaneously treated with 50 mg/kg risankizumab-rzaa (at 20 times the clinical exposure at the MRHD, based on mg/kg comparison) once weekly for 26 weeks.
-
14 CLINICAL STUDIES
Four multicenter, randomized, double-blind studies [ULTIMMA-1 (NCT02684370), ULTIMMA-2 (NCT02684357), IMMHANCE (NCT02672852), and IMMVENT (NCT02694523)] enrolled 2109 subjects 18 years of age and older with moderate to severe plaque psoriasis who had a body surface area (BSA) involvement of ≥10%, a static Physician’s Global Assessment (sPGA) score of ≥3 (“moderate”) in the overall assessment (plaque thickness/induration, erythema, and scaling) of psoriasis on a severity scale of 0 to 4, and a Psoriasis Area and Severity Index (PASI) score ≥12.
Overall, subjects had a median baseline PASI score of 17.8 and a median BSA of 20.0%. Baseline sPGA score was 4 (“severe”) in 19% of subjects. A total of 10% of study subjects had a history of diagnosed psoriatic arthritis.
Across all studies, 38% of subjects had received prior phototherapy, 48% had received prior non-biologic systemic therapy, and 42% had received prior biologic therapy for the treatment of psoriasis.
In ULTIMMA-1 and ULTIMMA-2, 997 subjects were enrolled (including 598 subjects randomized to the SKYRIZI 150 mg group, 200 subjects randomized to the placebo group, and 199 to the biologic active control group). Subjects received treatment at Weeks 0, 4, and every 12 weeks thereafter.
Both studies assessed the responses at Week 16 compared to placebo for the two co-primary endpoints:
- the proportion of subjects who achieved an sPGA score of 0 (“clear”) or 1 (“almost clear”)
- the proportion of subjects who achieved at least a 90% reduction from baseline PASI (PASI 90)
Secondary endpoints included the proportion of subjects who achieved PASI 100, sPGA 0, and PSS 0 at Week 16.
The results are presented in Table 2.
Table 2. Efficacy Results at Week 16 in Adults with Plaque Psoriasis in ULTIMMA-1 and ULTIMMA‑2 ULTIMMA-1 ULTIMMA-2 SKYRIZI
(N=304)
n (%)Placebo
(N=102)
n (%)SKYRIZI
(N=294)
n (%)Placebo
(N=98)
n (%)sPGA 0 or 1 (“clear or almost clear”)a 267 (88) 8 (8) 246 (84) 5 (5) PASI 90a 229 (75) 5 (5) 220 (75) 2 (2) sPGA 0 (“clear”) 112 (37) 2 (2) 150 (51) 3 (3) PASI 100 109 (36) 0 (0) 149 (51) 2 (2) a Co-primary endpoints Examination of age, gender, race, body weight, baseline PASI score and previous treatment with systemic or biologic agents did not identify differences in response to SKYRIZI among these subgroups at Week 16.
In ULTIMMA-1 and ULTIMMA-2 at Week 52, subjects receiving SKYRIZI achieved sPGA 0 (58% and 60%, respectively), PASI 90 (82% and 81%, respectively), and PASI 100 (56% and 60%, respectively).
Improvements in signs and symptoms related to pain, redness, itching and burning at Week 16 compared to placebo were observed in both studies as assessed by the Psoriasis Symptom Scale (PSS). In ULTIMMA-1 and ULTIMMA-2, about 30% of the subjects who received SKYRIZI achieved PSS 0 (“none”) at Week 16 compared to 1% of the subjects who received placebo.
IMMHANCE enrolled 507 subjects (407 randomized to SKYRIZI 150 mg and 100 to placebo). Subjects received treatment at Weeks 0, 4, and every 12 weeks thereafter.
At Week 16, SKYRIZI was superior to placebo on the co-primary endpoints of sPGA 0 or 1 (84% SKYRIZI and 7% placebo) and PASI 90 (73% SKYRIZI and 2% placebo). The respective response rates for SKYRIZI and placebo at Week 16 were: sPGA 0 (46% SKYRIZI and 1% placebo); PASI 100 (47% SKYRIZI and 1% placebo); and PASI 75 (89% SKYRIZI and 8% placebo).
Maintenance and Durability of Response
In ULTIMMA-1 and ULTIMMA-2, among the subjects who received SKYRIZI and had PASI 100 at Week 16, 80% (206/258) of the subjects who continued on SKYRIZI had PASI 100 at Week 52. For PASI 90 responders at Week 16, 88% (398/450) of the subjects had PASI 90 at Week 52.
In IMMHANCE, subjects who were originally on SKYRIZI and had sPGA 0 or 1 at Week 28 were re-randomized to continue SKYRIZI every 12 weeks or withdrawal of therapy. At Week 52, 87% (97/111) of the subjects re-randomized to continue treatment with SKYRIZI had sPGA 0 or 1 compared to 61% (138/225) who were re-randomized to withdrawal of SKYRIZI.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
SKYRIZI (risankizumab-rzaa) injection is a sterile, preservative-free, colorless to slightly yellow and clear to slightly opalescent solution. The product is supplied in a 1 mL glass syringe with a fixed 29 gauge ½ inch needle with needle guard.
- NDC: 0074-2042-02: one carton containing two prefilled syringes and two alcohol pads
-
17 PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Medication Guide and Instructions for Use) before starting SKYRIZI therapy and to reread the Medication Guide each time the prescription is renewed. Advise patients of the potential benefits and risks of SKYRIZI.
Inform patients that SKYRIZI may lower the ability of their immune system to fight infections. Instruct patients of the importance of communicating any history of infections to the healthcare provider and contacting their healthcare provider if they develop any symptoms of an infection [see Warnings and Precautions (5.1)].
Instruct patients or caregivers to perform the first self-injected dose under the supervision and guidance of a qualified healthcare professional for training in preparation and administration of SKYRIZI, including choosing anatomical sites for administration, and proper subcutaneous injection technique [see Instructions for Use].
Instruct patients or caregivers to administer two 75 mg single-dose syringes to achieve the 150 mg dose of SKYRIZI [see Instructions for Use].
Instruct patients or caregivers in the technique of needle and syringe disposal [see Instructions for Use].
Manufactured by:
AbbVie Inc.
North Chicago, IL 60064, USAUS License Number 1889
SKYRIZI® is a registered trademark of AbbVie Biotechnology Ltd.
© 2019-2020 AbbVie Inc.
20063596-R1 03/2020 -
MEDICATION GUIDE
-
INSTRUCTIONS FOR USE
Instructions for Use
SKYRIZI® (sky-RIZZ-ee)
(risankizumab-rzaa)
injection, for subcutaneous use
Refer to the Medication Guide for product information.
SKYRIZI Single-Dose Prefilled Syringe
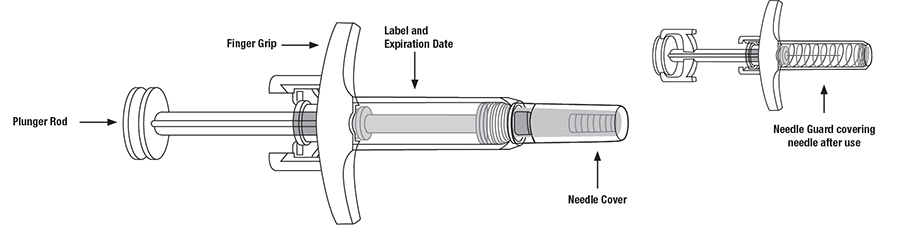
- Keep SKYRIZI in the original carton to protect from light until time to use.
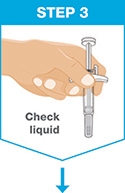
- The liquid should look clear to slightly yellow and may contain tiny white or clear particles.
- DO NOT use SKYRIZI if the liquid is cloudy or contains flakes or large particles.
- DO NOT use SKYRIZI if the expiration date (EXP:) shown on the carton and prefilled syringe has passed.
- DO NOT use SKYRIZI if the liquid has been frozen (even if thawed).
- DO NOT shake SKYRIZI.
- DO NOT use SKYRIZI if the syringe has been dropped or damaged.
- DO NOT use SKYRIZI if carton perforations are broken. Return product to the pharmacy.
- DO NOT remove the needle cover until right before giving the injections.
Keep SKYRIZI and all medicines out of the reach of children.
Please Read Complete Instructions For Use Before Using SKYRIZI Syringe
- Receive training on how to inject SKYRIZI before giving injections. Call your healthcare provider or (866) SKYRIZI or (866) 759-7494 if you need help.
- Mark your calendar ahead of time to remember when to take SKYRIZI.
-
Leave the carton at room temperature and out of direct sunlight for 15 to 30 minutes to warm.
- DO NOT remove the syringes from the carton while allowing SKYRIZI to reach room temperature.
- DO NOT warm SKYRIZI in any other way (for example, DO NOT warm it in a microwave or in hot water).
- The liquid should look clear to slightly yellow and may contain tiny white or clear particles.
- DO NOT use SKYRIZI if the liquid is cloudy or contains flakes or large particles.
- DO NOT use SKYRIZI if the expiration date (EXP:) shown on the carton and prefilled syringe has passed.
- DO NOT use SKYRIZI if the syringe has been dropped or damaged.
- DO NOT use SKYRIZI if carton perforations are broken. Return product to the pharmacy.
- Store SKYRIZI in your refrigerator between 36° F to 46°F (2° to 8°C).
- DO NOT shake SKYRIZI.
- Keep SKYRIZI in the original carton to protect from light until time to use.
- SKYRIZI is not made with natural rubber latex.
- DO NOT use if liquid has been frozen (even if thawed).
Keep SKYRIZI and all medicines out of the reach of children.
Call your healthcare provider or (866) SKYRIZI or (866) 759-7494 if you need help or do not know how to proceed.
Used SKYRIZI Prefilled Syringe Disposal
If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes.
For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA’s website at: www.fda.gov/safesharpsdisposal.
Manufactured by: AbbVie Inc., North Chicago, IL 60064, U.S.A.
US License Number 1889
SKYRIZI® is a registered trademark of AbbVie Biotechnology Ltd.
© 2019-2020 AbbVie Inc.
20061783This Instructions for Use has been approved by the U.S. Food and Drug Administration.
-
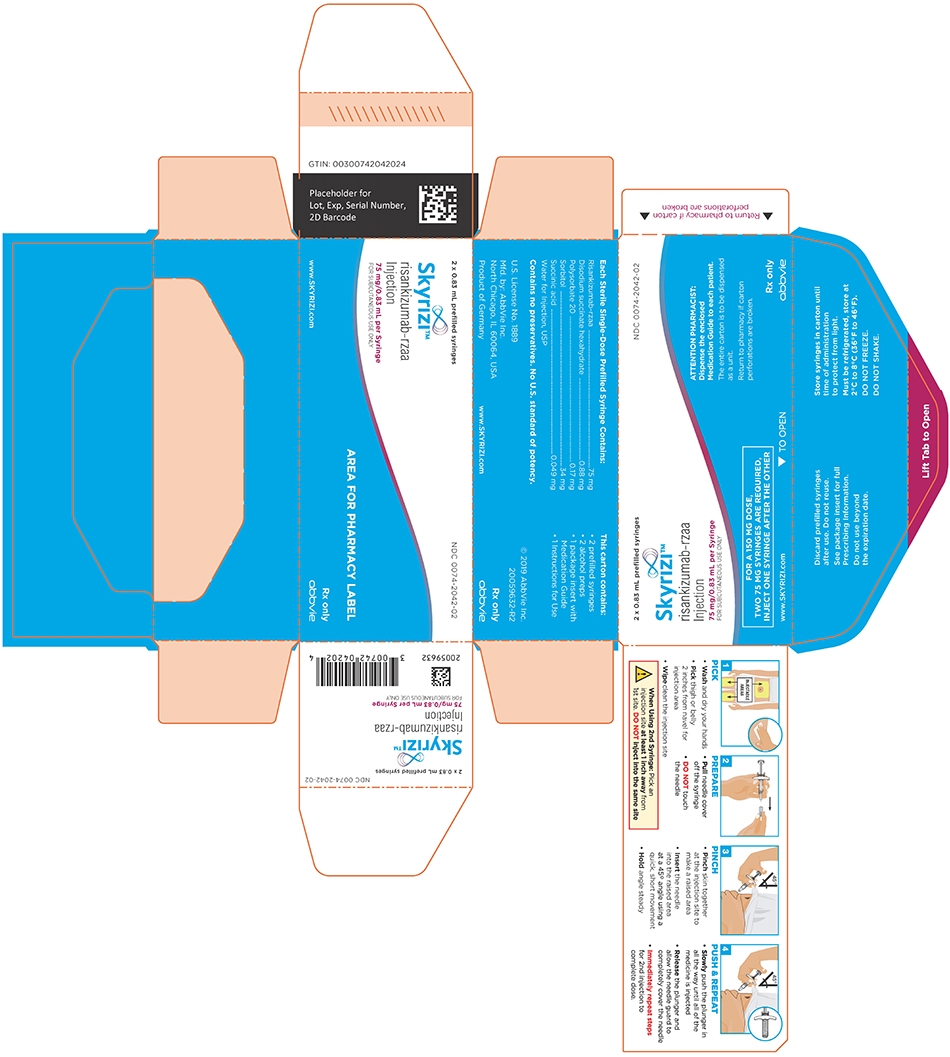
PRINCIPAL DISPLAY PANEL
NDC: 0074-2042-02
2 x 0.83 mL prefilled syringes
FOR A 150 MG DOSE,
TWO 75 MG SYRINGES ARE REQUIRED,
INJECT ONE SYRINGE AFTER THE OTHERATTENTION PHARMACIST:
Dispense the enclosed Medication Guide to each patient.The entire carton is to be dispensed as a unit.
Return to pharmacy if carton perforations are broken.
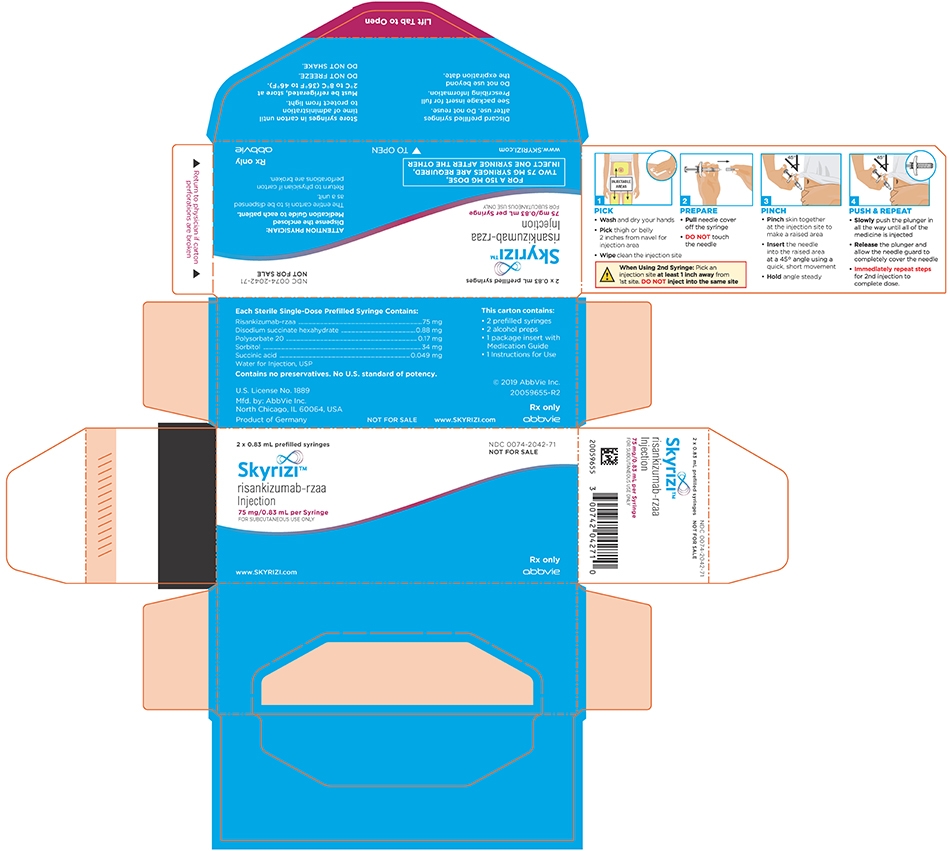
NDC: 0074-2042-71
2 x 0.83 mL prefilled syringes
FOR A 150 MG DOSE,
TWO 75 MG SYRINGES ARE REQUIRED,
INJECT ONE SYRINGE AFTER THE OTHERATTENTION PHYSICIAN:
Dispense the enclosed Medication Guide to each patient.The entire carton is to be dispensed as a unit.
Return to physician if carton perforations are broken.
-
INGREDIENTS AND APPEARANCE
SKYRIZI
risankizumab-rzaa kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0074-2042 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0074-2042-02 1 in 1 CARTON 04/23/2019 2 NDC: 0074-2042-71 1 in 1 CARTON 04/23/2019 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 2 SYRINGE 1.66 mL Part 2 2 PACKET 2 mL Part 1 of 2 SKYRIZI
risankizumab-rzaa injection, solutionProduct Information Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RISANKIZUMAB (UNII: 90ZX3Q3FR7) (RISANKIZUMAB - UNII:90ZX3Q3FR7) RISANKIZUMAB 75 mg in 0.83 mL Inactive Ingredients Ingredient Name Strength SUCCINIC ACID (UNII: AB6MNQ6J6L) 0.049 mg in 0.83 mL SORBITOL (UNII: 506T60A25R) 34 mg in 0.83 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.17 mg in 0.83 mL WATER (UNII: 059QF0KO0R) SODIUM SUCCINATE HEXAHYDRATE (UNII: U16QOD6C4E) 0.88 mg in 0.83 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 0.83 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761105 04/23/2019 Part 2 of 2 ALCOHOL SWAB
isopropyl alcohol swabProduct Information Route of Administration TOPICAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ISOPROPYL ALCOHOL (UNII: ND2M416302) (ISOPROPYL ALCOHOL - UNII:ND2M416302) ISOPROPYL ALCOHOL 0.70 mL in 1 mL Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 mL in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date OTC monograph not final part333A 04/23/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761105 04/23/2019 Labeler - AbbVie Inc. (078458370)




Trademark Results [Skyrizi]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 SKYRIZI 87320935 5857879 Live/Registered |
AbbVie Biotechnology Ltd. 2017-02-01 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.