BYDUREON- exenatide injection, suspension, extended release BYDUREON- exenatide kit
BYDUREON by
Drug Labeling and Warnings
BYDUREON by is a Prescription medication manufactured, distributed, or labeled by AstraZeneca Pharmaceuticals LP, AstraZeneca PLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BYDUREON safely and effectively. See full prescribing information for BYDUREON.
BYDUREON® (exenatide extended-release) for injectable suspension, for subcutaneous use
Initial U.S. Approval: 2005WARNING: RISK OF THYROID C-CELL TUMORS
See full prescribing information for complete boxed warning.
- Exenatide extended-release causes thyroid C-cell tumors at clinically relevant exposures in rats. It is unknown whether BYDUREON causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC) in humans, as the human relevance of exenatide extended-release-induced rodent thyroid C-cell tumors has not been determined (5.1, 13.1).
- BYDUREON is contraindicated in patients with a personal or family history of MTC or in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2). Counsel patients regarding the potential risk of MTC and the symptoms of thyroid tumors (4, 5.1).
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
BYDUREON is a glucagon-like peptide-1 (GLP-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. (1, 14)
Limitations of Use:
- Not recommended as first-line therapy for patients inadequately controlled on diet and exercise. (1)
- Should not be used to treat type 1 diabetes or diabetic ketoacidosis. (1)
- Use with prandial insulin has not been studied. (1)
- BYDUREON is an extended-release formulation of exenatide. Do not coadminister with other exenatide containing products. (1)
- Has not been studied in patients with a history of pancreatitis. Consider other antidiabetic therapies in patients with a history of pancreatitis. (1, 5.3)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Extended-release for injectable suspension available as: (3)
- Single-dose tray containing 2 mg of exenatide in single-dose vial
- Single-dose pen containing 2 mg of exenatide
CONTRAINDICATIONS
- Personal or family history of medullary thyroid carcinoma or in patients with Multiple Endocrine Neoplasia syndrome type 2. (4)
- Prior serious hypersensitivity reaction to exenatide or any of the product components. (4)
- History of drug-induced immune-mediated thrombocytopenia from exenatide products. (4)
WARNINGS AND PRECAUTIONS
- Never share a BYDUREON pen or needle with anyone else. (5.2)
- Acute Pancreatitis: Including fatal and non-fatal hemorrhagic or necrotizing pancreatitis has been reported. Discontinue promptly if pancreatitis is suspected. Do not restart if pancreatitis is confirmed. Consider other antidiabetic therapies if patient has history of pancreatitis. (5.3)
- Hypoglycemia: When used in combination with an insulin secretagogue (e.g., a sulfonylurea) or insulin, consider lowering dose of the secretagogue or insulin to reduce risk of hypoglycemia. (5.4)
- Acute Kidney Injury: May induce nausea and vomiting with transient hypovolemia and may worsen renal function. Postmarketing increased serum creatinine, renal impairment, worsened chronic renal failure and acute renal failure, sometimes requiring hemodialysis or kidney transplantation has been reported. Not recommended for use in patients with eGFR below 45 mL/min/1.73 m2. (5.5, 8.6, 12.3)
- Gastrointestinal Disease: Not recommended in patients with severe gastrointestinal disease (e.g., gastroparesis). (5.6)
- Immunogenicity: Patients may develop antibodies to exenatide. If there is worsening glycemic control or failure to achieve target glycemic control, consider alternative antidiabetic therapy. (5.7)
- Hypersensitivity: Serious hypersensitivity reactions (e.g., anaphylaxis and angioedema) have been reported. In such cases, patients are to discontinue BYDUREON and promptly seek medical advice. (5.8)
- Drug-induced Immune-mediated Thrombocytopenia: Serious bleeding which may be fatal has been reported. Discontinue BYDUREON promptly and avoid re-exposure to exenatide. (5.9)
- Injection-site Reactions: Serious injection-site reactions with or without subcutaneous nodules have been reported. (5.10)
- Acute Gallbladder Disease: If cholelithiasis or cholecystitis are suspected, gallbladder studies are indicated. (5.11)
ADVERSE REACTIONS
Most common (≥5%) and occurring more frequently than comparator in clinical trials: nausea, diarrhea, headache, vomiting, constipation, injection-site pruritus, injection-site nodule, and dyspepsia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca at 1-800-236-9933 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
- Pregnancy: Use during pregnancy only if the potential benefit justifies the potential risk to the fetus. (8.1)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 2/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: RISK OF THYROID C-CELL TUMORS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
2.2 Missed Dose
2.3 Administration Instructions
2.4 Initiating BYDUREON Therapy
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Thyroid C-cell Tumors
5.2 Never Share a BYDUREON Pen Between Patients
5.3 Acute Pancreatitis
5.4 Hypoglycemia with Concomitant Use of Insulin Secretagogues or Insulin
5.5 Acute Kidney Injury
5.6 Gastrointestinal Disease
5.7 Immunogenicity
5.8 Hypersensitivity
5.9 Drug-Induced Thrombocytopenia
5.10 Injection-Site Reactions
5.11 Acute Gallbladder Disease
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Glycemic Control Trials in Adults with Type 2 Diabetes Mellitus
14.2 EXSCEL Cardiovascular Outcomes Trial in Patients with Type 2 Diabetes
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: RISK OF THYROID C-CELL TUMORS
- Exenatide extended-release causes an increased incidence in thyroid C-cell tumors at clinically relevant exposures in rats compared to controls. It is unknown whether BYDUREON causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans, as the human relevance of exenatide extended-release-induced rodent thyroid C-cell tumors has not been determined [see Warnings and Precautions (5.1) and Nonclinical Toxicology (13.1)].
- BYDUREON is contraindicated in patients with a personal or family history of MTC and in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2). Counsel patients regarding the potential risk for MTC with the use of BYDUREON and inform them of symptoms of thyroid tumors (e.g., mass in the neck, dysphagia, dyspnea, persistent hoarseness). Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for detection of MTC in patients treated with BYDUREON [see Contraindications (4) and Warnings and Precautions (5.1)].
-
1 INDICATIONS AND USAGE
BYDUREON is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus [see Clinical Studies (14)].
Limitations of Use:
- BYDUREON is not recommended as first-line therapy for patients who have inadequate glycemic control on diet and exercise because of the uncertain relevance of the rat thyroid C-cell tumor findings to humans [see Warnings and Precautions (5.1)].
- BYDUREON is not a substitute for insulin. BYDUREON is not indicated for use in patients with type 1 diabetes mellitus or for the treatment of diabetic ketoacidosis, as it would not be effective in these settings.
- The concurrent use of BYDUREON with prandial insulin has not been studied.
- BYDUREON is an extended-release formulation of exenatide. BYDUREON should not be used with other products containing the active ingredient exenatide.
- BYDUREON has not been studied in patients with a history of pancreatitis. Consider other antidiabetic therapies in patients with a history of pancreatitis [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
The recommended dose of BYDUREON is 2 mg subcutaneously once every 7 days (weekly). The dose can be administered at any time of day, with or without meals.
The day of weekly administration can be changed if necessary as long as the last dose was administered 3 or more days before the new day of administration.
2.2 Missed Dose
If a dose is missed, administer the dose as soon as noticed, provided the next regularly scheduled dose is due at least 3 days later. Thereafter, patients can resume their usual dosing schedule of once every 7 days (weekly).
If a dose is missed and the next regularly scheduled dose is due 1 or 2 days later, do not administer the missed dose and instead resume BYDUREON with the next regularly scheduled dose.
2.3 Administration Instructions
- There are two presentations of BYDUREON (i.e., a single dose tray and a single dose pen) [see How Supplied/Storage and Handling (16)]. The BYDUREON “Instructions for Use” for each presentation contains detailed instructions on the preparation and administration of BYDUREON [see Instructions for Use].
- Each presentation of BYDUREON requires constitution prior to use to obtain a final concentration of 2 mg of exenatide per 0.65 mL of suspension.
- BYDUREON is intended for patient self-administration. Prior to initiation, train patients on proper mixing and injection technique to ensure the product is adequately mixed and a full dose is delivered.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. The suspension should appear white to off-white and cloudy. (BYDUREON contains microspheres which appear as white to off-white particles). Do not use if foreign particulate matter is present or if discoloration is observed. Refer patients to the accompanying Instructions for Use for disposal information [see Instructions for Use].
- Administer BYDUREON immediately after the dose is prepared as a subcutaneous injection in the abdomen, thigh, or upper arm region. Advise patients to use a different injection site each week when injecting in the same region.
- When using BYDUREON with insulin, always administer BYDUREON and insulin as separate injections. Do not mix these medications together into a single injection. It is acceptable to inject BYDUREON and insulin in the same body region but the injections should not be adjacent to each other.
- Do not administer BYDUREON intravenously or intramuscularly.
- Refer patients to the accompanying Instructions for Use for complete administration instructions with illustrations [see Instructions for Use].
2.4 Initiating BYDUREON Therapy
Prior treatment with an immediate- or extended-release exenatide product is not required when initiating BYDUREON therapy. Discontinue an immediate- or extended-release exenatide product prior to initiation of BYDUREON.
Patients changing from immediate-release exenatide to BYDUREON may experience transient (approximately 2 to 4 weeks) elevations in blood glucose concentrations.
-
3 DOSAGE FORMS AND STRENGTHS
Extended-release for injectable suspension available as:
- Single-dose tray which contains one single dose vial of 2 mg exenatide white to off-white powder, one vial connector, one prefilled diluent syringe, and two needles (one provided as a spare).
- Single-dose pen which contains 2 mg of exenatide white to off-white powder, diluent, and includes one needle. Each carton contains one spare needle.
-
4 CONTRAINDICATIONS
BYDUREON is contraindicated in patients with:
- A personal or family history of medullary thyroid carcinoma (MTC) or in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2).
- A prior serious hypersensitivity reaction to exenatide or to any of the product components. Serious hypersensitivity reactions including anaphylaxis and angioedema have been reported with BYDUREON [see Warnings and Precautions (5.8)].
- A history of drug-induced immune-mediated thrombocytopenia from exenatide products. Serious bleeding, which may be fatal, from drug-induced immune-mediated thrombocytopenia has been reported with exenatide use [see Warnings and Precautions (5.9)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Thyroid C-cell Tumors
In both genders of rats, exenatide extended-release caused a dose-related and treatment-duration–dependent increase in the incidence of thyroid C-cell tumors (adenomas and/or carcinomas) at clinically relevant exposures compared to controls [see Nonclinical Toxicology (13.1)]. A statistically significant increase in malignant thyroid C-cell carcinomas was observed in female rats receiving exenatide extended-release at 25-times clinical exposure compared to controls and higher incidences were noted in males above controls in all treated groups at ≥2-times clinical exposure. The potential of exenatide extended-release to induce C-cell tumors in mice has not been evaluated. Other GLP-1 receptor agonists have also induced thyroid C-cell adenomas and carcinomas in male and female mice and rats at clinically relevant exposures. It is unknown whether BYDUREON will cause thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans as the human relevance of exenatide extended-release-induced rodent thyroid C-cell tumors has not been determined.
Cases of MTC in patients treated with liraglutide, another GLP-1 receptor agonist, have been reported in the postmarketing period; the data in these reports are insufficient to establish or exclude a causal relationship between MTC and GLP-1 receptor agonist use in humans.
BYDUREON is contraindicated in patients with a personal or family history of MTC or in patients with MEN 2. Counsel patients regarding the potential risk of MTC with the use of BYDUREON and inform them of symptoms of thyroid tumors (e.g., a mass in the neck, dysphagia, dyspnea, persistent hoarseness).
Routine monitoring of serum calcitonin or using thyroid ultrasound is of uncertain value for early detection of MTC in patients treated with BYDUREON. Such monitoring may increase the risk of unnecessary procedures, due to the low specificity of serum calcitonin testing for MTC and a high background incidence of thyroid disease. Significantly elevated serum calcitonin may indicate MTC and patients with MTC usually have values >50 ng/L. If serum calcitonin is measured and found to be elevated, the patient should be further evaluated. Patients with thyroid nodules noted on physical examination or neck imaging should also be further evaluated.
5.2 Never Share a BYDUREON Pen Between Patients
BYDUREON pens must never be shared between patients. Pen-sharing poses a risk for transmission of blood-borne pathogens.
5.3 Acute Pancreatitis
Based on postmarketing data, exenatide has been associated with acute pancreatitis, including fatal and non-fatal hemorrhagic or necrotizing pancreatitis. After initiation of BYDUREON, observe patients carefully for signs and symptoms of pancreatitis (including persistent severe abdominal pain, sometimes radiating to the back, which may or may not be accompanied by vomiting). If pancreatitis is suspected, BYDUREON should promptly be discontinued and appropriate management should be initiated. If pancreatitis is confirmed, BYDUREON should not be restarted. Consider antidiabetic therapies other than BYDUREON in patients with a history of pancreatitis.
5.4 Hypoglycemia with Concomitant Use of Insulin Secretagogues or Insulin
The risk of hypoglycemia is increased when BYDUREON is used in combination with insulin secretagogues (e.g., sulfonylureas) or insulin. Patients may require a lower dose of the secretagogue or insulin to reduce the risk of hypoglycemia in this setting [see Adverse Reactions (6.1)].
5.5 Acute Kidney Injury
BYDUREON may induce nausea and vomiting with transient hypovolemia and may worsen renal function. There have been postmarketing reports of altered renal function with exenatide, including increased serum creatinine, renal impairment, worsened chronic renal failure and acute renal failure, sometimes requiring hemodialysis or kidney transplantation. Some of these events occurred in patients receiving one or more pharmacologic agents known to affect renal function or hydration status such as angiotensin converting enzyme inhibitors, nonsteroidal anti-inflammatory drugs, or diuretics. Some events occurred in patients who had been experiencing nausea, vomiting or diarrhea, with or without dehydration. Reversibility of altered renal function has been observed in many cases with supportive treatment and discontinuation of potentially causative agents, including BYDUREON. BYDUREON is not recommended for use in patients with an eGFR below 45 mL/min/1.73 m2 [see Use in Specific Populations (8.6)].
5.6 Gastrointestinal Disease
Exenatide has not been studied in patients with severe gastrointestinal disease, including gastroparesis. Because exenatide is commonly associated with gastrointestinal adverse reactions, including nausea, vomiting, and diarrhea, the use of BYDUREON is not recommended in patients with severe gastrointestinal disease.
5.7 Immunogenicity
Patients may develop antibodies to exenatide following treatment with BYDUREON. Anti-exenatide antibodies were measured in BYDUREON-treated patients in five of the six comparator-controlled 24- to 30-week studies of BYDUREON. In 6% of BYDUREON-treated patients, antibody formation was associated with an attenuated glycemic response. If there is worsening glycemic control or failure to achieve targeted glycemic control, consider alternative antidiabetic therapy [see Adverse Reactions (6.1) and (6.2)].
5.8 Hypersensitivity
There have been postmarketing reports of serious hypersensitivity reactions (e.g., anaphylaxis and angioedema) in patients treated with exenatide. If a hypersensitivity reaction occurs, the patient should discontinue BYDUREON and promptly seek medical advice [see Contraindications (4) and Adverse Reactions (6.3)]. Inform and closely monitor patients with a history of anaphylaxis or angioedema with another GLP-1 receptor agonist for allergic reactions, because it is unknown whether such patients will be predisposed to anaphylaxis with BYDUREON.
5.9 Drug-Induced Thrombocytopenia
Serious bleeding, which may be fatal, from drug-induced immune-mediated thrombocytopenia has been reported in the postmarketing setting with exenatide use. Drug-induced thrombocytopenia is an immune-mediated reaction, with exenatide-dependent anti-platelet antibodies. In the presence of exenatide, these antibodies cause platelet destruction. If drug-induced thrombocytopenia is suspected, discontinue BYDUREON immediately and do not re-expose the patient to exenatide. Upon discontinuation, thrombocytopenia can persist due to the prolonged exenatide exposure from BYDUREON (about 10 weeks) [see Adverse Reactions (6.3)].
5.10 Injection-Site Reactions
There have been postmarketing reports of serious injection-site reactions (e.g., abscess, cellulitis, and necrosis), with or without subcutaneous nodules, with the use of BYDUREON. Isolated cases required surgical intervention [see Adverse Reactions (6.1)].
5.11 Acute Gallbladder Disease
Acute events of gallbladder disease have been reported in GLP-1 receptor agonist trials. In the EXSCEL trial [see Clinical Studies (14.2)], 1.9% of BYDUREON-treated patients and 1.4% of placebo-treated patients reported an acute event of gallbladder disease, such as cholelithiasis or cholecystitis. If cholelithiasis is suspected, gallbladder studies and appropriate clinical follow-up are indicated.
-
6 ADVERSE REACTIONS
- The following serious adverse reactions are described below or elsewhere in the prescribing information:
- Risk of Thyroid C-cell Tumors [see Warnings and Precautions (5.1)]
- Never Share a BYDUREON Pen Between Patients [see Warnings and Precautions (5.2)]
- Acute Pancreatitis [see Warnings and Precautions (5.3)]
- Hypoglycemia [see Warnings and Precautions (5.4)]
- Acute Kidney Injury [see Warnings and Precautions (5.5)]
- Gastrointestinal Disease [see Warnings and Precautions (5.6)]
- Immunogenicity [see Warnings and Precautions (5.7)]
- Hypersensitivity [see Warnings and Precautions (5.8)]
- Drug-Induced Thrombocytopenia [see Warnings and Precautions (5.9)]
- Injection-Site Reactions [see Warnings and Precautions (5.10)]
- Acute Gallbladder Disease [see Warnings and Precautions (5.11)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data presented below are derived from six comparator-controlled trials of BYDUREON in patients who entered the studies not achieving adequate glycemic control on their current therapy [see Clinical Studies (14)]. In a double-blind 26-week trial, patients on diet and exercise were treated with BYDUREON 2 mg once every 7 days (weekly), sitagliptin 100 mg daily, pioglitazone 45 mg daily, or metformin 2000 mg daily. In a double-blind 26-week trial, patients on metformin were treated with BYDUREON 2 mg once every 7 days (weekly), sitagliptin 100 mg daily, or pioglitazone 45 mg daily. In an open-label 26-week trial, patients on metformin or metformin plus sulfonylurea were treated with BYDUREON 2 mg once every 7 days (weekly) or optimized insulin glargine. In two open-label 24- to 30-week studies, patients on diet and exercise or metformin, a sulfonylurea, a thiazolidinedione, or combination of oral agents were treated with BYDUREON 2 mg once every 7 days (weekly) or BYETTA 10 mcg twice daily. In an open-label 26-week trial, patients on metformin, a sulfonylurea, metformin plus a sulfonylurea, or metformin plus pioglitazone were treated with BYDUREON 2 mg every 7 days (weekly) or liraglutide 1.8 mg once daily.
Common Adverse Reactions
Tables 1 and 2 summarize adverse reactions with an incidence ≥5% reported in the six comparator-controlled 24- to 30-week trials of BYDUREON used as monotherapy or as add-on to metformin, a sulfonylurea, a thiazolidinedione, or combination of these oral antidiabetic agents.
Table 1: Adverse Reactions Reported in ≥5% of BYDUREON-Treated Patients with Type 2 Diabetes Mellitus in Monotherapy Trial 26-Week Monotherapy Trial
BYDUREON
2 mg
N = 248
%
Sitagliptin
100 mg
N = 163
%
Pioglitazone
30-45 (mean dose
40) mg
N = 163
%
Metformin
1000-2500 (mean dose 2077) mg
N = 246
%
Nausea
11.3
3.7
4.3
6.9
Diarrhea
10.9
5.5
3.7
12.6
Injection-site nodule
10.5
6.7
3.7
10.2
Constipation
8.5
2.5
1.8
3.3
Headache
8.1
9.2
8.0
12.2
Dyspepsia
7.3
1.8
4.9
3.3
N = number of intent-to-treat patients.
Note: Percentages are based on the number of intent-to-treat patients in each treatment group.
Table 2: Adverse Reactions Reported in ≥5% of BYDUREON-Treated Patients with Type 2 Diabetes Mellitus in 24- to 30-Week Add-On Combination Therapy Trials - * Patients in the sitagliptin, pioglitazone, and metformin treatment groups received weekly placebo injections.
26-Week Add-On to Metformin Trial
BYDUREON
2 mg
N = 160
%Sitagliptin
100 mg
N = 166
%Pioglitazone
45 mg
N = 165
%Nausea
24.4
9.6
4.8
Diarrhea
20.0
9.6
7.3
Vomiting
11.3
2.4
3.0
Headache
9.4
9.0
5.5
Constipation
6.3
3.6
1.2
Fatigue
5.6
0.6
3.0
Dyspepsia
5.0
3.6
2.4
Decreased appetite
5.0
1.2
0.0
Injection-site pruritus*
5.0
4.8
1.2
26-Week Add-On to Metformin or Metformin + Sulfonylurea Trial
BYDUREON
2 mg
N = 233
%Insulin Glargine Titrated
N = 223
%Nausea
12.9
1.3
Headache
9.9
7.6
Diarrhea
9.4
4.0
Injection-site nodule
6.0
0.0
30-Week Monotherapy or as Add-On to Metformin, a Sulfonylurea, a Thiazolidinedione, or Combination of Oral Agents Trial
BYDUREON
2 mg
N = 148
%BYETTA
10 mcg
N = 145
%Nausea
27.0
33.8
Diarrhea
16.2
12.4
Vomiting
10.8
18.6
Injection-site pruritus
18.2
1.4
Constipation
10.1
6.2
Gastroenteritis viral
8.8
5.5
Gastroesophageal reflux disease
7.4
4.1
Dyspepsia
7.4
2.1
Injection-site erythema
7.4
0.0
Fatigue
6.1
3.4
Headache
6.1
4.8
Injection-site hematoma
5.4
11.0
24-Week Monotherapy or as Add-On to Metformin, a Sulfonylurea, a Thiazolidinedione, or Combination of Oral Agents Trial
BYDUREON
2 mg
N = 129
%BYETTA
10 mcg
N = 123
%Nausea
14.0
35.0
Diarrhea
9.3
4.1
Injection-site erythema
5.4
2.4
26-Week Add-On to Metformin, a Sulfonylurea, Metformin + Sulfonylurea, or Metformin + Pioglitazone Trial
BYDUREON
2 mg
N = 461
%Injection-site nodule
10.4
Nausea
9.3
Diarrhea
6.1
N = number of intent-to-treat patients.
Note: Percentages are based on the number of intent-to-treat patients in each treatment group.Nausea was a common adverse reaction associated with initiation of treatment with BYDUREON and usually decreased over time.
Adverse Reactions Leading to Study Withdrawal
The incidence of withdrawal due to adverse reactions was 4.1% (N=57) for BYDUREON-treated patients, 4.9% (N=13) for BYETTA-treated patients, and 2.9% (N=46) for other comparator-treated patients in the six comparator-controlled 24- to 30-week trials. The most common classes of adverse reactions (0.5%) leading to withdrawal for BYDUREON-treated patients were, Gastrointestinal Disorders 1.6% (N=22) versus 4.1% (N=11) for BYETTA and 1.9% (N=30) for other comparators, and Administration Site Conditions 0.8% (N=11) versus 0.0% for BYETTA and 0.2% (N=3) for other comparators. The most frequent adverse reactions within each of these respective classes were, nausea 0.4% (N=6) for BYDUREON versus 1.5% (N=4) for BYETTA and 0.8% (N=12) for other comparators, and injection-site nodule, 0.4% (N=6) for BYDUREON versus 0.0% for BYETTA and 0.0% for other comparators.
Hypoglycemia
Table 3 summarizes the incidence of minor hypoglycemia in the six comparator-controlled 24- to 30-week trials of BYDUREON used as monotherapy or as add-on to metformin, a sulfonylurea, a thiazolidinedione, or combination of these oral antidiabetic agents. In these trials, an event was classified as minor hypoglycemia if there were symptoms of hypoglycemia with a concomitant glucose <54 mg/dL and the patient was able to self-treat.
Table 3: Incidence (% of Subjects) of Minor* Hypoglycemia in Clinical Trials in Patients with Type 2 Diabetes Mellitus - * Reported event that has symptoms consistent with hypoglycemia with a concomitant glucose <54 mg/dL and the patient was able to self-treat.
- † Insulin glargine was dosed to a target fasting glucose concentration of 72 to 100 mg/dL. The mean dose of insulin glargine was 10 units/day at baseline and 31 units/day at endpoint.
26-Week Monotherapy Trial
BYDUREON 2 mg (N = 248)
2.0%
Sitagliptin 100 mg (N = 163)
0.0%
Pioglitazone 30-45 (mean dose 40) mg (N = 163)
0.0%
Metformin 1000-2500 (mean dose 2077) mg (N = 246)
0.0%
26-Week Add-On to Metformin Trial
BYDUREON 2 mg (N = 160)
1.3%
Sitagliptin 100 mg (N = 166)
3.0%
Pioglitazone 45 mg (N = 165)
1.2%
26-Week Add-On to Metformin or Metformin + Sulfonylurea Trial
With Concomitant Sulfonylurea Use (N = 136)
BYDUREON 2 mg (N = 70)
20.0%
Titrated Insulin Glargine (N = 66)
43.9%
Without Concomitant Sulfonylurea Use (N = 320)
BYDUREON 2 mg (N = 163)
3.7%
Titrated Insulin Glargine† (N = 157)
19.1%
24-Week Monotherapy or Add-On to Metformin, a Sulfonylurea, a Thiazolidinedione, or Combination of Oral Agents Trial
With Concomitant Sulfonylurea Use (N = 74)
BYDUREON 2 mg (N = 40)
12.5%
BYETTA 10 mcg (N = 34)
11.8%
Without Concomitant Sulfonylurea Use (N = 178)
BYDUREON 2 mg (N = 89)
0.0%
BYETTA 10 mcg (N = 89)
0.0%
30-Week Monotherapy or Add-On to Metformin, a Sulfonylurea, a Thiazolidinedione, or Combination of Oral Agents Trial
With Concomitant Sulfonylurea Use (N = 107)
BYDUREON 2 mg (N = 55)
14.5%
BYETTA 10 mcg (N = 52)
15.4%
Without Concomitant Sulfonylurea Use (N = 186)
BYDUREON 2 mg (N = 93)
0.0%
BYETTA 10 mcg (N = 93)
1.1%
26-Week as Add-On to Metformin, a Sulfonylurea, Metformin + Sulfonylurea, or Metformin +
Pioglitazone TrialWith Concomitant Sulfonylurea Use (N = 590)
BYDUREON 2 mg (N = 294)
15.3%
Without Concomitant Sulfonylurea Use (N = 321)
BYDUREON 2 mg (N = 167)
3.6%
N = number of intent-to-treat patients.
Note: Percentages are based on the number of intent-to-treat patients in each treatment group.Injection-Site Adverse Reactions
In five comparator-controlled 24- to 30-week trials, injection-site reactions were observed more frequently in patients treated with BYDUREON (17.1%) than in patients treated with BYETTA (12.7%), titrated insulin glargine (1.8%), or those patients who received placebo injections (sitagliptin (10.6%), pioglitazone (6.4%), and metformin (13.0%) treatment groups). These reactions for patients treated with BYDUREON were more commonly observed in antibody-positive patients (14.2%) compared with antibody-negative patients (3.1%), with a greater incidence in those with higher titer antibodies [see Warnings and Precautions (5.7)]. Incidence of injection-site reactions for patients treated with BYETTA was similar for antibody-positive patients (5.8%) and antibody-negative patients (7.0%). One percent of patients treated with BYDUREON withdrew due to injection-site adverse reactions (injection-site mass, injection-site nodule, injection-site pruritus, and injection-site reaction).
Subcutaneous injection-site nodules may occur with the use of BYDUREON. In a separate 15-week study in which information on nodules were collected and analyzed, 24 out of 31 subjects (77%) experienced at least 1 injection-site nodule during treatment; 2 subjects (6.5%) reported accompanying localized symptoms. The mean duration of events was 27 days. The formation of subcutaneous nodules is consistent with the known properties of the microspheres used in BYDUREON.
Increase in Heart Rate
Increases in heart rate from baseline ranging from 1.5 to 4.5 beats per minute have been observed in comparator-controlled clinical trials.
Other Adverse Reactions
The following adverse reactions were also reported in three 30-week controlled trials of BYETTA (N=963) add-on to metformin and/or sulfonylurea, with an incidence of ≥1% and reported more frequently than with placebo: feeling jittery (9% BYETTA, 4% placebo), dizziness (9% BYETTA, 6% placebo), asthenia (4% BYETTA, 2% placebo), and hyperhidrosis (3% BYETTA, 1% placebo).
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, the incidence of antibodies to exenatide cannot be directly compared with the incidence of antibodies with other products.
Anti-exenatide antibodies were measured at prespecified intervals (4-14 weeks) in all BYDUREON-treated patients (N=918) in five of the comparator-controlled studies of BYDUREON. In these five trials, 452 BYDUREON-treated patients (49%) had low titer antibodies (≤125) to exenatide at any time during the trials and 405 BYDUREON-treated patients (45%) had low titer antibodies to exenatide at study endpoint (24-30 weeks). The level of glycemic control in these patients was generally comparable to that observed in the 379 BYDUREON-treated patients (43%) without antibody titers. An additional 107 BYDUREON-treated patients (12%) had higher titer antibodies at endpoint. Of these patients, 50 (6% overall) had an attenuated glycemic response to BYDUREON (<0.7% reduction in HbA1c); the remaining 57 (6% overall) had a glycemic response comparable to that of patients without antibodies [see Warnings and Precautions (5.7)]. In the 30-week trial in which anti-exenatide antibody assessments were performed at baseline and at 4-week intervals from Week 6 to Week 30, the mean anti-exenatide antibody titer in the BYDUREON-treated patients peaked at Week 6 then declined by 56% from this peak by Week 30.
A total of 246 patients with antibodies to exenatide in the BYETTA and BYDUREON clinical trials were tested for the presence of cross-reactive antibodies to GLP-1 and/or glucagon. No treatment-emergent cross-reactive antibodies were observed across the range of titers.
6.3 Postmarketing Experience
The following additional adverse reactions have been reported during post-approval use of BYDUREON or other formulations of exenatide. Because these events are reported voluntarily from a population of uncertain size, it is generally not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Allergy/Hypersensitivity: injection-site reactions, generalized pruritus and/or urticaria, macular or papular rash, angioedema; anaphylactic reaction.
Blood and Lymphatic Systems: drug-induced thrombocytopenia [see Warnings and Precautions (5.9)].
Drug Interactions: increased international normalized ratio (INR), sometimes associated with bleeding, with concomitant warfarin use [see Drug Interactions (7)].
Gastrointestinal: nausea, vomiting, and/or diarrhea resulting in dehydration; abdominal distension, abdominal pain, eructation, constipation, flatulence, acute pancreatitis, hemorrhagic and necrotizing pancreatitis sometimes resulting in death [see Indications and Usage (1)].
Neurologic: dysgeusia; somnolence
Renal and Urinary Disorders: altered renal function, including increased serum creatinine, renal impairment, worsened chronic renal failure or acute renal failure (sometimes requiring hemodialysis), kidney transplant and kidney transplant dysfunction.
Skin and Subcutaneous Tissue Disorders: alopecia
-
7 DRUG INTERACTIONS
Table 4: Clinically Relevant Interactions Affecting Drugs Co-Administered with BYDUREON and Other Exenatide-Containing Products Orally Administered Drugs (e.g., acetaminophen)
Clinical Impact
Exenatide slows gastric emptying. Therefore, BYDUREON has the potential to reduce the rate of absorption of orally administered drugs
[see Clinical Pharmacology (12.3)].
Intervention
Use caution when administering oral medications with BYDUREON where a slower rate of oral absorption may be clinically meaningful.
Warfarin
Clinical Impact
BYDUREON has not been studied with warfarin. However, in a drug interaction study, BYETTA did not have a significant effect on INR [see Clinical Pharmacology (12.3)]. There have been postmarketing reports for exenatide of increased INR with concomitant use of warfarin, sometimes associated with bleeding [see Adverse Reactions (6.3)].
Intervention
In patients taking warfarin, the INR should be monitored more frequently after initiating BYDUREON. Once a stable INR has been documented, the INR can be monitored at the intervals usually recommended for patients on warfarin.
Concomitant Use of Insulin Secretagogues or Insulin
Clinical Impact
Exenatide promotes insulin release from pancreatic beta-cells in the presence of elevated glucose concentrations. The risk of hypoglycemia is increased when exenatide is used in combination with insulin secretagogues (e.g., sulfonylureas) or insulin [see Adverse Reactions (6.1)].
Intervention
Patients may require a lower dose of the secretagogue or insulin to reduce the risk of hypoglycemia in this setting.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Limited data with exenatide, the active ingredient in BYDUREON, in pregnant women are not sufficient to determine a drug-associated risk for major birth defects or miscarriage. There are risks to the mother and fetus associated with poorly controlled diabetes in pregnancy (see Clinical Considerations). Based on animal reproduction studies, there may be risks to the fetus from exposure to BYDUREON during pregnancy. BYDUREON should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Animal reproduction studies identified increased adverse fetal and neonatal outcomes from exposure to exenatide extended-release during pregnancy, or from exposure to exenatide, during pregnancy and lactation, in association with maternal effects. In rats, exenatide extended-release administered during the period of organogenesis reduced fetal growth and produced skeletal ossification deficits at doses that approximate clinical exposures at the maximum recommended human dose (MRHD) of 2 mg/week. In mice, exenatide administered during gestation and lactation caused increased neonatal deaths at doses that approximate clinical exposures at the MRHD (see Data). Based on animal data, advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects is 6-10% in women with pre-gestational diabetes with an HbA1c >7 and has been reported to be as high as 20-25% in women with HbA1c >10. The estimated background risk of miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryofetal risk
Poorly controlled diabetes in pregnancy increases the maternal risk for diabetic ketoacidosis, preeclampsia, spontaneous abortions, preterm delivery and delivery complications. Poorly controlled diabetes increases the fetal risk for major birth defects, stillbirth, and macrosomia related morbidity.
Data
Animal Data
Pregnant rats given subcutaneous doses of 0.3, 1, or 3 mg/kg exenatide extended-release every 3 days during organogenesis had systemic exposures 3-, 7-, and 17-times human exposure, respectively, at the maximum recommended human dose (MRHD) of 2 mg/week BYDUREON based on plasma exenatide exposure (AUC) comparison. Reduced fetal growth at all doses and skeletal ossification deficits at 1 and 3 mg/kg occurred at doses that decreased maternal food intake and body weight gain.
In studies evaluating reproduction and development in pregnant mice and rabbits, maternal animals were administered exenatide, the active ingredient in BYDUREON, by subcutaneous injection twice a day. Differences in embryo-fetal developmental toxicity from subcutaneously injected exenatide extended-release and exenatide were not evaluated in mice, rats, or rabbits.
In pregnant mice given 6, 68, 460, or 760 mcg/kg/day exenatide during fetal organogenesis, skeletal variations associated with slowed fetal growth, including changes in number of rib pairs or vertebral ossifications sites, and wavy ribs were observed at 760 mcg/kg/day, a dose that produced maternal toxicity and yielded systemic exposure 200 times the human exposure resulting from the MRHD of BYDUREON based on AUC comparison.
In pregnant rabbits given 0.2, 2, 22, 156, or 260 mcg/kg/day exenatide during fetal organogenesis, irregular fetal skeletal ossifications were observed at 2 mcg/kg/day, a dose yielding systemic exposure up to 4 times the human exposure from the MRHD of BYDUREON based on AUC comparison.
In maternal mice given 6, 68, or 760 mcg/kg/day exenatide from gestation day 6 through lactation day 20 (weaning), an increased number of neonatal deaths were observed on postpartum days 2 to 4 in dams given 6 mcg/kg/day, a dose yielding a systemic exposure equivalent to the human exposure from the MRHD of BYDUREON based on AUC comparison.
8.2 Lactation
Risk Summary
There is no information regarding the presence of exenatide in human milk, the effects of exenatide on the breastfed infant, or the effects of exenatide on milk production. Exenatide, the active ingredient in BYDUREON, was present in the milk of lactating mice. However, due to species-specific differences in lactation physiology, the clinical relevance of these data is not clear (see Data).
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for exenatide and any potential adverse effects on the breastfed child from exenatide or from the underlying maternal condition.
Data
In lactating mice subcutaneously injected twice a day with exenatide, the active ingredient in BYDUREON, the concentration of exenatide in milk was up to 2.5% of the concentration in maternal plasma.
8.4 Pediatric Use
Safety and effectiveness of BYDUREON have not been established in pediatric patients. BYDUREON is not recommended for use in pediatric patients.
8.5 Geriatric Use
In five comparator-controlled 24- to 30-week trials, BYDUREON was studied in 132 patients (16.6%) who were at least 65 years old and 20 patients who were at least 75 years old. No differences in safety (N=152) and efficacy (N=52) were observed between these patients and the overall population, but the small sample size for patients ≥75 years old limits conclusions. In a large cardiovascular outcomes trial, BYDUREON was studied in 2959 patients (40.3%) who were at least 65 years old and of those, 605 patients (8.2%) were at least 75 years old. Use caution when initiating BYDUREON in elderly patients because they are more likely to have decreased renal function.
8.6 Renal Impairment
Pharmacokinetic studies of renally impaired patients receiving BYDUREON indicate that there is an increase in exposure in moderate and mild renally impaired patients as compared to patients with normal renal function. BYDUREON may induce nausea and vomiting with transient hypovolemia and may worsen renal function.
Monitor patients with mild renal impairment for adverse reactions that may lead to hypovolemia. BYDUREON is not recommended for use in patients with eGFR below 45 mL/min/1.73 m2 or end stage renal disease. If used in patients with renal transplantation, closely monitor for adverse reactions that may lead to hypovolemia [see Warnings and Precautions (5.5) and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
Effects of overdoses with BYETTA, another formulation of exenatide, included severe nausea, severe vomiting, and rapidly declining blood glucose concentrations, including severe hypoglycemia requiring parenteral glucose administration. In the event of overdose, appropriate supportive treatment should be initiated according to the patient's clinical signs and symptoms.
-
11 DESCRIPTION
BYDUREON (exenatide extended-release) for injectable suspension is a GLP-1 receptor agonist supplied as a sterile powder to be suspended in diluent and administered by subcutaneous injection. Exenatide is a 39-amino acid synthetic peptide amide with an empirical formula of C184H282N50O60S and a molecular weight of 4186.6 Daltons. The amino acid sequence for exenatide is shown below.
H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu-Ser-Lys-Gln-Met-Glu-Glu-Glu-Ala-Val-Arg-Leu-Phe-Ile-Glu-Trp-Leu-Lys-Asn-Gly-Gly-Pro-Ser-Ser-Gly-Ala-Pro-Pro-Pro-Ser-NH2
BYDUREON is a white to off-white powder that is available in a dosage strength of 2 mg exenatide per vial or per pen. Exenatide is incorporated in an extended-release microsphere formulation containing the 50:50 poly(D,L-lactide-co-glycolide) polymer (37.2 mg per dose) along with sucrose (0.8 mg per dose). The powder must be suspended in the diluent prior to injection.
The diluent for the BYDUREON vial is supplied in a prefilled syringe within each single-dose tray. The diluent for the BYDUREON Pen is contained within each single-dose pen. Each configuration contains sufficient diluent to deliver 0.65 mL. The diluent is a clear, colorless to pale-yellow solution composed of carboxymethylcellulose sodium (19 mg), polysorbate 20 (0.63 mg), sodium phosphate monobasic monohydrate (0.61 mg), sodium phosphate dibasic heptahydrate (0.51 mg), sodium chloride (4.1 mg), and water for injection. Sodium hydroxide may be added during manufacture of BYDUREON Pen for pH adjustment.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Incretins, such as glucagon-like peptide-1 (GLP-1), enhance glucose-dependent insulin secretion and exhibit other antihyperglycemic actions following their release into the circulation from the gut. BYDUREON is a GLP-1 receptor agonist that enhances glucose-dependent insulin secretion by the pancreatic beta-cell, suppresses inappropriately elevated glucagon secretion, and slows gastric emptying.
The amino acid sequence of exenatide partially overlaps that of human GLP-1. Exenatide is a GLP-1 receptor agonist that has been shown to bind and activate the human GLP-1 receptor in vitro. This leads to an increase in both glucose-dependent synthesis of insulin and in vivo secretion of insulin from pancreatic beta cells, by mechanisms involving cyclic AMP and/or other intracellular signaling pathways. Exenatide promotes insulin release from pancreatic beta-cells in the presence of elevated glucose concentrations.
12.2 Pharmacodynamics
Exenatide improves glycemic control through the actions described below.
Glucose-Dependent Insulin Secretion
The effect of exenatide infusion on glucose-dependent insulin secretion rates (ISR) was investigated in 11 healthy subjects. In these healthy subjects, on average, the ISR response was glucose-dependent (Figure 1). Exenatide did not impair the normal glucagon response to hypoglycemia.
Figure 1: Mean (SE) Insulin Secretion Rates During Infusion of Exenatide or Placebo by Treatment, Time, and Glycemic Condition in Healthy Subjects
SE = standard error.
Notes: 5 mmol = 90 mg/dL, 4 mmol/L = 72 mg/dL, 3.2 mmol/L = 58 mg/dL; Study medication infusion was started at time = 0 minutes.
Statistical assessments were for the last 30 minutes of each glycemic step, during which the target glucose concentrations were maintained.
*p <0.05, exenatide treatment relative to placebo.Glucagon Secretion
In patients with type 2 diabetes, exenatide moderates glucagon secretion and lowers serum glucagon concentrations during periods of hyperglycemia.
Gastric Emptying
Exenatide slows gastric emptying, thereby reducing the rate at which postprandial glucose appears in the circulation.
Fasting and Postprandial Glucose
In a clinical study in adults with type 2 diabetes mellitus, treatment with once weekly BYDUREON resulted in mean reductions in fasting plasma glucose of -45 mg/dL and 2-hour PPG concentrations of -95 mg/dL.
Cardiac Electrophysiology
The effect of exenatide at therapeutic (253 pg/mL) and supratherapeutic (627 pg/mL) concentrations, following an intravenous infusion on QTc interval was evaluated in a randomized, placebo- and active-controlled (moxifloxacin 400 mg) three-period crossover thorough QT study in 74 healthy subjects. The upper bound of the one-sided 95% confidence interval for the largest placebo adjusted, baseline-corrected QTc based on population correction method (QTcP) was below 10 ms. Therefore, exenatide was not associated with prolongation of the QTc interval at therapeutic and supratherapeutic concentrations.
12.3 Pharmacokinetics
Absorption
Following a single dose of BYDUREON, exenatide is released from the microspheres over approximately 10 weeks. There is an initial period of release of surface-bound exenatide followed by a gradual release of exenatide from the microspheres, which results in two subsequent peaks of exenatide in plasma at around Week 2 and Week 6 to 7, respectively, representing the hydration and erosion of the microspheres.
Following initiation of once every 7 days (weekly) administration of 2 mg BYDUREON, a gradual increase in the plasma exenatide concentration is observed over 6 to 7 weeks. After 6 to 7 weeks, mean exenatide concentrations of approximately 300 pg/mL were maintained over once every 7 days (weekly) dosing intervals indicating that steady state was achieved.
Distribution
The mean apparent volume of distribution of exenatide following subcutaneous administration of a single dose of BYETTA is 28.3 L and is expected to remain unchanged for BYDUREON.
Metabolism
Elimination
Nonclinical studies have shown that exenatide is predominantly eliminated by glomerular filtration with subsequent proteolytic degradation. The mean apparent clearance of exenatide in humans is 9.1 L/hour and is independent of the dose. Approximately 10 weeks after discontinuation of BYDUREON therapy, plasma exenatide concentrations generally fall below the minimal detectable concentration of 10 pg/mL.
Drug Interaction Studies
Acetaminophen
When 1000 mg acetaminophen tablets were administered, either with or without a meal, following 14 weeks of BYDUREON therapy (2 mg weekly), no significant changes in acetaminophen AUC were observed compared to the control period. Acetaminophen Cmax decreased by 16% (fasting) and 5% (fed) and Tmax was increased from approximately 1 hour in the control period to 1.4 hours (fasting) and 1.3 hours (fed).
The following drug interactions have been studied using BYETTA. The potential for drug-drug interaction with BYDUREON is expected to be similar to that of BYETTA.
Digoxin
Administration of repeated doses of BYETTA 30 minutes before oral digoxin (0.25 mg once daily) decreased the Cmax of digoxin by 17% and delayed the Tmax of digoxin by approximately 2.5 hours; however, the overall steady-state pharmacokinetic exposure (e.g., AUC) of digoxin was not changed.
Lovastatin
Administration of BYETTA (10 mcg twice daily) 30 minutes before a single oral dose of lovastatin (40 mg) decreased the AUC and Cmax of lovastatin by approximately 40% and 28%, respectively, and delayed the Tmax by about 4 hours compared with lovastatin administered alone. In the 30-week controlled clinical trials of BYETTA, the use of BYETTA in patients already receiving HMG CoA reductase inhibitors was not associated with consistent changes in lipid profiles compared to baseline.
Lisinopril
In patients with mild to moderate hypertension stabilized on lisinopril (5-20 mg/day), BYETTA (10 mcg twice daily) did not alter steady-state Cmax or AUC of lisinopril. Lisinopril steady-state Tmax was delayed by 2 hours. There were no changes in 24-hour mean systolic and diastolic blood pressure.
Oral Contraceptives
The effect of BYETTA (10 mcg twice daily) on single and on multiple doses of a combination oral contraceptive (30 mcg ethinyl estradiol plus 150 mcg levonorgestrel) was studied in healthy female subjects. Repeated daily doses of the oral contraceptive (OC) given 30 minutes after BYETTA administration decreased the Cmax of ethinyl estradiol and levonorgestrel by 45% and 27%, respectively, and delayed the Tmax of ethinyl estradiol and levonorgestrel by 3.0 hours and 3.5 hours, respectively, as compared to the oral contraceptive administered alone. Administration of repeated daily doses of the OC one hour prior to BYETTA administration decreased the mean Cmax of ethinyl estradiol by 15%, but the mean Cmax of levonorgestrel was not significantly changed as compared to when the OC was given alone. BYETTA did not alter the mean trough concentrations of levonorgestrel after repeated daily dosing of the oral contraceptive for both regimens. However, the mean trough concentration of ethinyl estradiol was increased by 20% when the OC was administered 30 minutes after BYETTA administration injection as compared to when the OC was given alone. The effect of BYETTA on OC pharmacokinetics is confounded by the possible food effect on OC in this study [see Drug Interactions (7)].
Warfarin
Administration of warfarin (25 mg) 35 minutes after repeated doses of BYETTA (5 mcg twice daily on days 1-2 and 10 mcg twice daily on days 3-9) in healthy volunteers delayed warfarin Tmax by approximately 2 hours. No clinically relevant effects on Cmax or AUC of S- and R-enantiomers of warfarin were observed. BYETTA did not significantly alter the pharmacodynamic properties (e.g., international normalized ratio) of warfarin [see Drug Interactions (7)].
Specific Populations
Patients with Renal Impairment
BYDUREON has not been studied in patients with severe renal impairment (creatinine clearance <30 mL/min) or end-stage renal disease receiving dialysis. Population pharmacokinetic analysis of renally impaired patients receiving 2 mg BYDUREON indicate that there is a 62% and 33% increase in exposure in moderate (N=10) and mild (N=56) renally impaired patients, respectively, as compared to patients with normal renal function (N=84).
In a study of BYETTA in subjects with end-stage renal disease receiving dialysis, mean exenatide exposure increased by 3.4-fold compared to that of subjects with normal renal function [see Use in Specific Populations (8.6)].
Patients with Hepatic Impairment
BYDUREON has not been studied in patients with acute or chronic hepatic impairment.
Age, Male and Female Patients, Race, and Body Weight
Age, gender, race and body weight did not alter the pharmacokinetics of BYDUREON in population pharmacokinetic analyses.
Pediatric Patients
BYDUREON has not been studied in pediatric patients [see Use in Specific Populations (8.4)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Thyroid C-cell tumors have been observed in rats and mice with GLP-1 receptor agonists.
A 2‑year carcinogenicity study was conducted with exenatide extended-release, the active component of BYDUREON, in male and female rats at doses of 0.3, 1.0, and 3.0 mg/kg (2-, 9-, and 26-times human systemic exposure at the maximum recommended human dose (MRHD) of 2 mg/week BYDUREON based on plasma exenatide AUC, respectively) administered by subcutaneous injection every other week. In this study there was an increased incidence of C-cell adenomas and C-cell carcinomas at all doses. An increase in benign fibromas was seen in the skin subcutis at injection sites of males given 3 mg/kg. No treatment-related injection-site fibrosarcomas were observed at any dose. The human relevance of these findings is currently unknown.
Carcinogenicity of exenatide extended-release has not been evaluated in mice.
Exenatide, the active ingredient in BYDUREON, was not mutagenic or clastogenic, with or without metabolic activation, in the Ames bacterial mutagenicity assay or chromosomal aberration assay in Chinese hamster ovary cells. Exenatide was negative in the in vivo mouse micronucleus assay.
In mouse fertility studies with exenatide, the active ingredient in BYDUREON, at twice-daily subcutaneous doses of 6, 68, or 760 mcg/kg/day, males were treated for 4 weeks prior to and throughout mating, and females were treated 2 weeks prior to mating and throughout mating until gestation day 7. No adverse effect on fertility was observed at 760 mcg/kg/day, a systemic exposure 148 times the human exposure resulting from the recommended dose of 2 mg/week, based on AUC.
-
14 CLINICAL STUDIES
BYDUREON has been studied as monotherapy and in combination with metformin, a sulfonylurea, a thiazolidinedione, a combination of metformin and a sulfonylurea, a combination of metformin and a thiazolidinedione, in combination with a SGLT2 inhibitor on a background of metformin, and in combination with basal insulin.
14.1 Glycemic Control Trials in Adults with Type 2 Diabetes Mellitus
Monotherapy
BYDUREON Monotherapy versus Metformin, Sitagliptin, and Pioglitazone
A 26-week, randomized, comparator-controlled trial was conducted to compare the safety and efficacy of BYDUREON to metformin, sitagliptin, and pioglitazone in patients with type 2 diabetes whose glycemic control was inadequate with diet and exercise (NCT00676338).
A total of 820 patients were studied: 552 (67%) were Caucasian, 102 (12%) were East Asian, 71 (9%) were West Asian, 65 (8%) were Hispanic, 25 (3.0%) were Black, 4 (0.5%) were Native American, and 1 was classified otherwise. The mean baseline HbA1c was 8.5%. Patients were randomly assigned to receive BYDUREON 2 mg once every seven days (weekly), titrated metformin from 1000 to 2500 mg/day, sitagliptin 100 mg/day or titrated pioglitazone from 30 to 45 mg/day, all dosed according to approved labeling.
The primary endpoint was change in HbA1c from baseline to Week 26 (or the last value at time of early discontinuation). Treatment with BYDUREON 2 mg once weekly (QW) resulted in mean HbA1c reduction that was statistically significantly greater compared to sitagliptin 100 mg/day. The mean reduction in HbA1c was non-inferior compared with metformin 1000-2500 mg/day (mean dose 2077 mg/day at study endpoint). Non-inferiority of BYDUREON 2 mg QW to pioglitazone 30-45 mg/day (mean dose 40 mg/day at study endpoint) in reducing HbA1c after 26 weeks of treatment was not demonstrated (the mean change from baseline in HbA1c after 26 weeks was -1.6% with BYDUREON and -1.7% with pioglitazone). The non-inferiority margin was set at +0.3% in this study. The results for the primary endpoint at 26 weeks are summarized in Table 5.
Table 5: Results of 26-Week Trial of BYDUREON Monotherapy versus Metformin, Sitagliptin, and Pioglitazone in Patients with Type 2 Diabetes Mellitus - * Least squares means were obtained using a mixed model repeated measure analysis with treatment, pooled country, visit, baseline HbA1c value, and treatment by visit interaction as fixed effects, and subject as a random effect.
- † p<0.001, treatment vs comparator.
BYDUREON
2 mg QW
Metformin
1000-2500
(mean dose
2077) mg/day
Sitagliptin
100 mg/day
Pioglitazone
30-45 (mean
dose 40)
mg/day
Intent-to-Treat Population (N)
248
246
163
163
HbA1c (%)
Mean Baseline
8.4
8.6
8.4
8.5
Mean Change at Week 26*
-1.6
-1.5
-1.2
-1.7
Difference from metformin*
[Bonferroni-adjusted 98.3% CI]
-0.05
[-0.26, 0.17]
Difference from sitagliptin*
[Bonferroni-adjusted 98.3% CI]
-0.39†
[-0.63, -0.16]
Difference from pioglitazone*
[Bonferroni-adjusted 98.3% CI]
0.16
[-0.08, 0.41]
N = number of patients in each treatment group.
Note: mean change is least squares mean change.
Note: The primary efficacy analysis was adjusted for multiple comparisons and a two-sided 98.3% confidence interval was utilized to assess difference between treatments.
Note: HbA1c change data at 26 weeks were available from 86%, 87%, 85%, and 82% of the randomized subjects in the BYDUREON, metformin, sitagliptin, and pioglitazone groups, respectively.
QW = once weekly.
The proportion of patients with a Week 26 value achieving HbA1c of less than 7% at Week 26 were 56%, 52%, 40%, and 55% for BYDUREON, metformin, sitagliptin, and pioglitazone, respectively. Patients who did achieve and HbA1c goal <7% and discontinued before Week 26 were not included as responders. The mean changes from baseline to Week 26 for fasting serum glucose were -41 mg/dL, -36 mg/dL, -20 mg/dL, and -46 mg/dL, and for body weight were -2.0 kg, -2.0 kg, -0.8 kg, and +1.5 kg for BYDUREON, metformin, sitagliptin, and pioglitazone, respectively.
Combination Therapy
BYDUREON versus Sitagliptin and Pioglitazone, All as Add-on to Metformin Therapy
A 26-week double-blind comparator-controlled trial was conducted to compare the safety and efficacy of BYDUREON to sitagliptin and pioglitazone in patients with type 2 diabetes whose glycemic control was inadequate with metformin therapy (NCT00637273).
A total of 491 patients were studied 168 (34.2%) were Caucasian, 143 (29.1%) were Hispanic, 119 (24.2%) were Asian, 52 (10.6%) were Black, 3 (0.6%) were Native American, and 6 (1.2%) were classified otherwise. The mean baseline HbA1c was 8.5%. Patients were randomly assigned to receive BYDUREON 2 mg once every 7 days (weekly), sitagliptin 100 mg/day or pioglitazone 45 mg/day, in addition to their existing metformin therapy.
The primary endpoint was change in HbA1c from baseline to Week 26 (or the last value at time of early discontinuation). In this study, treatment with BYDUREON 2 mg QW resulted in a statistically significant mean HbA1c reduction compared to sitagliptin 100 mg/day. There was a numerically greater reduction in HbA1c with BYDUREON compared to pioglitazone, but there was not sufficient evidence to conclude superiority of BYDUREON 2 mg QW to pioglitazone 45 mg/day in reducing HbA1c after 26 weeks of treatment. Results for the primary endpoint at 26 weeks are summarized in Table 6.
Table 6: Results of 26-Week Trial of BYDUREON versus Sitagliptin and Pioglitazone, All as Add-On to Metformin Therapy in Patients with Type 2 Diabetes Mellitus BYDUREON
2 mg QWSitagliptin
100 mg/dayPioglitazone
45 mg/day- * Least squares means were obtained using an ANCOVA model with treatment, baseline HbA1c stratum, and country as fixed effects. Missing Week 26 data (28%, 18%, and 24% for the BYDUREON, sitagliptin, and pioglitazone groups, respectively) were imputed by the LOCF technique.
Intent-to-Treat Population (N)
160
166
165
HbA1c (%)
Mean Baseline
8.6
8.5
8.5
Mean Change at Week 26*
−1.5
−0.9
−1.2
Difference from sitagliptin*
[95% CI]−0.63
[−0.89, −0.37]Difference from pioglitazone*
[95% CI]−0.32
[−0.57, −0.06]N = number of patients in each treatment group.
Note: mean change is least squares mean change.
QW = once weekly.
The proportion of patients with a Week 26 value achieving HbA1c of less than 7% at Week 26 were 46%, 30%, and 39% for BYDUREON, sitagliptin, and pioglitazone, respectively. Patients who did achieve an HbA1c goal <7% and discontinued before Week 26 were not included as responders. The mean changes from baseline to Week 26 for fasting serum glucose were ‑32 mg/dL, ‑16 mg/dL and ‑27 mg/dL, and for body weight were ‑2.3 kg, ‑0.8 kg and +2.8 kg for BYDUREON, sitagliptin, and pioglitazone, respectively.
BYDUREON versus Insulin Glargine, Both as Add-on to Metformin or Metformin + Sulfonylurea Therapy
A 26-week open-label comparator-controlled trial was conducted to compare the safety and efficacy of BYDUREON to titrated insulin glargine in patients with type 2 diabetes whose glycemic control was inadequate with metformin or metformin plus sulfonylurea therapy (NCT00641056).
A total of 456 patients were studied: 379 (83.1%) were Caucasian, 47 (10.3%) were Hispanic, 25 (5.5%) were East Asian, 3 (0.7%) were Black, and 2 (0.4%) were West Asian. Background therapy was either metformin (70%) or metformin plus sulfonylurea (30%). The mean baseline HbA1c was 8.3%. Patients were randomly assigned to receive BYDUREON 2 mg once every 7 days (weekly) or insulin glargine once daily in addition to their existing oral antidiabetic therapy. Insulin glargine was dosed to a target fasting glucose concentration of 72 to 100 mg/dL. The mean dose of insulin glargine was 10 units/day at baseline and 31 units/day at endpoint. At Week 26, 21% of insulin glargine treated patients were at fasting glucose goal.
The primary endpoint was change in HbA1c from baseline to Week 26 (or the last value at time of early discontinuation). Treatment with BYDUREON once weekly resulted in a mean reduction in HbA1c from baseline at 26 weeks of -1.5%. The mean reduction in HbA1c seen in insulin glargine arm at 26 weeks was ‑1.3%. The difference in observed effect size between BYDUREON and glargine in this trial excluded the pre-specified non-inferiority margin of +0.3%.
The proportion of patients with a Week 26 value achieving HbA1c of less than 7% at Week 26 were 57% and 48% for BYDUREON and insulin glargine, respectively. Patients who did achieve an HbA1c goal <7% and discontinued before Week 26 were not included as responders. The mean changes from baseline to Week 26 for fasting serum glucose in this study were ‑38 mg/dL and ‑50 mg/dL, and for body weight were ‑2.6 kg and +1.4 kg for BYDUREON and insulin glargine, respectively.
BYDUREON versus BYETTA, Both as Monotherapy or as Add-on to Metformin, a Sulfonylurea, a Thiazolidinedione, or Combination of Oral Agents
A 24-week, randomized, open-label trial was conducted to compare the safety and efficacy of BYDUREON to BYETTA in patients with type 2 diabetes and inadequate glycemic control with diet and exercise alone or with oral antidiabetic therapy, including metformin, a sulfonylurea, a thiazolidinedione, or combination of two of those therapies (NCT00877890).
A total of 252 patients were studied: 149 (59%) were Caucasian, 78 (31%) Hispanic, 15 (6%) Black, and 10 (4%) Asian. Patients were treated with diet and exercise alone (19%), a single oral antidiabetic agent (47%), or combination therapy of oral antidiabetic agents (35%). The mean baseline HbA1c was 8.4%. Patients were randomly assigned to receive BYDUREON 2 mg once every 7 days (weekly) or BYETTA (10 mcg twice daily), in addition to existing oral antidiabetic agents. Patients assigned to BYETTA initiated treatment with 5 mcg twice daily then increased the dose to 10 mcg twice daily after 4 weeks.
The primary endpoint was change in HbA1c from baseline to Week 24 (or the last value at time of early discontinuation). Treatment with BYDUREON 2 mg QW resulted in a statistically significantly greater mean HbA1c reduction compared to BYETTA 10 mcg twice daily. Change in body weight was a secondary endpoint. Twenty-four-week study results are summarized in Table 7.
Table 7: Results of 24-Week Trial of BYDUREON versus BYETTA, Both as Monotherapy or as Add-On to Metformin, a Sulfonylurea, a Thiazolidinedione, or Combination of Oral Agents in Patients with Type 2 Diabetes Mellitus BYDUREON
2 mg QWBYETTA
10 mcg twice daily*- * BYETTA 5 mcg twice daily before the morning and evening meals for 4 weeks followed by 10 mcg twice daily for 20 weeks.
- † Least squares (LS) means are adjusted for baseline HbA1c strata, background antihyperglycemic therapy, and baseline value of the dependent variable (if applicable).
- ‡ p<0.001, treatment vs comparator.
Intent-to-Treat Population (N)
129
123
HbA1c (%)
Mean Baseline
8.5
8.4
Mean Change at Week 24†
−1.6
−0.9
Difference from BYETTA† [95% CI]
−0.7 [−0.9, −0.4]‡
Percentage Achieving HbA1c <7% at Week 24 (%)
58‡
30
Fasting Plasma Glucose (mg/dL)
Mean Baseline
173
168
Mean Change at Week 24
−25
−5
Difference from BYETTA† [95% CI]
−20 [−31, −10]‡
N = number of patients in each treatment group.
Note: mean change is least squares mean change.
QW = once weekly.
Reductions from mean baseline (97/94 kg) in body weight were observed in both BYDUREON (−2.3 kg) and BYETTA (−1.4 kg) treatment groups.
BYDUREON versus Liraglutide, Both as Add-on to Metformin, a Sulfonylurea, Metformin + Sulfonylurea, or Metformin + Pioglitazone Therapy
A 26-week open-label comparator-controlled trial was conducted to compare the safety and efficacy of BYDUREON to liraglutide in patients with type 2 diabetes whose glycemic control was inadequate with metformin, a sulfonylurea, metformin plus sulfonylurea, or metformin plus pioglitazone therapy (NCT01029886).
A total of 911 patients were studied: 753 (82.7%) were Caucasian, 111 (12.2%) were Asian, 32 (3.5%) were American Indian or Alaska Native, 8 (0.9%) were Black, 6 (0.7%) were multiple races, and 1 (0.1%) was Pacific Islander. Background therapy was either a single oral antidiabetic agent (35%) or a combination of oral antidiabetic agents (65%). The mean baseline HbA1c was 8.4%. Patients were randomly assigned to receive BYDUREON 2 mg once every 7 days (weekly) or liraglutide uptitrated from 0.6 mg/day to 1.2 mg/day, then 1.8 mg/day in addition to their existing oral antidiabetic therapy. Each titration was to be completed after at least one week, but could be delayed if the patient had severe nausea or vomiting as established by the investigator. Patients not tolerating the 1.8 mg/day dose of liraglutide by Week 4 were discontinued from the study.
The primary endpoint was change in HbA1c from baseline to Week 26 (or the last value at time of early discontinuation). Treatment with BYDUREON once weekly resulted in a mean reduction in HbA1c from baseline at 26 weeks of -1.3%. The mean reduction in HbA1c seen in the liraglutide arm at 26 weeks was ‑1.5%. The HbA1c reduction with BYDUREON did not meet predefined non-inferiority criteria compared to liraglutide 1.8 mg/day. The non-inferiority margin was set at +0.25% in this study. Results for the primary endpoint at 26 weeks are summarized in Table 8.
Table 8: Results of 26-Week Trial of BYDUREON versus Liraglutide, Both as Add-On to Metformin, a Sulfonylurea, Metformin + Sulfonylurea, or Metformin + Pioglitazone Therapy in Patients with Type 2 Diabetes Mellitus BYDUREON
2 mg QWLiraglutide
1.8 mg/day- * Least squares means were obtained using a mixed model repeated measure analysis with treatment, country, OAD stratum, baseline HbA1c stratum, visit, baseline HbA1c, and treatment by visit interaction as fixed effects, and subject as a random effect.
Intent-to-Treat Population (N)
461
450
HbA1c (%)
Mean Baseline
8.5
8.4
Mean Change at Week 26*
−1.3
−1.5
Difference from liraglutide* [95% CI]
0.2 [0.08, 0.33]
N = number of patients in each treatment group.
Note: mean change is least squares mean change.
Note: HbA1c change data at 26 weeks were available from 85% and 86% of the randomized subjects in the BYDUREON and liraglutide groups, respectively.
QW = once weekly.
The proportion of patients with a Week 26 value achieving HbA1c of less than 7% at Week 26 were 48% and 56% for BYDUREON and liraglutide, respectively. Patients who did achieve an HbA1c goal <7% and discontinued before Week 26 were not included as responders. The mean changes from baseline to Week 26 for fasting serum glucose were ‑32 mg/dL and ‑38 mg/dL, and for body weight were ‑2.7 kg and ‑3.6 kg for BYDUREON and liraglutide, respectively.
BYDUREON in Combination with Dapagliflozin versus BYDUREON Alone and Dapagliflozin Alone, All as Add-On to Metformin
A 28‑week double‑blind comparator‑controlled trial was conducted to compare the efficacy of BYDUREON and dapagliflozin (an SGLT2 inhibitor) to BYDUREON alone and dapagliflozin alone in patients with type 2 diabetes with inadequate glycemic control with metformin therapy (NCT02229396).
A total of 694 patients were studied; 580 (83.6%) were Caucasian, 96 (13.8%) were Black, 5 (0.7%) were Asian, 2 (0.3%) were American Indian or Alaska Native and 11 (1.6%) were classified otherwise. The mean baseline HbA1c was 9.3%. All patients entered a 1‑week placebo lead–in period. Patients with HbA1c ≥8.0% and ≤12% and on metformin at a dose of at least 1,500 mg per day were randomly assigned to receive either BYDUREON 2 mg once every 7 days (weekly) plus dapagliflozin 10 mg once daily, BYDUREON 2 mg once weekly, or dapagliflozin 10 mg once daily.
The primary endpoint was change in HbA1c from baseline to Week 28. At Week 28, BYDUREON in combination with dapagliflozin provided statistically significantly greater reductions in HbA1c (‑1.77%) compared to BYDUREON alone (‑1.42%, p=0.012) and dapagliflozin alone (‑1.32%, p=0.001). BYDUREON in combination with dapagliflozin provided statistically significantly greater reductions in FPG (‑57.35 mg/dL) compared to BYDUREON alone (‑40.53, p <0.001) and dapagliflozin alone (‑44.72 mg/dL, p=0.006).
BYDUREON versus Placebo, Both as Add-On to Basal Insulin or Basal Insulin + Metformin Therapy
A 28‑week, double‑blind, placebo‑controlled trial was conducted to compare the safety and efficacy of BYDUREON to placebo when added to basal insulin glargine, with or without metformin, in patients with type 2 diabetes with inadequate glycemic control (NCT02229383).
A total of 460 patients were studied: 400 (87.0%) were White, 47 (10.2%) were Black or African American, 6 (1.3%) were Asian, 1 (0.2%) was American Indian or Alaska Native, 1 (0.2%) was Pacific Islander and 5 (1.1%) were classified otherwise. Patients on sulfonylurea therapy discontinued sulfonylurea. Patients on metformin continued on the same dose of metformin. All patients initially entered an 8‑week insulin dose‑titration phase. Insulin glargine was to be titrated every 3 days with an aim of achieving a target fasting plasma glucose concentration of 72 to 99 mg/dL. Following the titration period, patients with HbA1c ≥7.0% and ≤10.5% were then randomly assigned to receive either BYDUREON 2 mg once every 7 days (weekly) or placebo once every 7 days (weekly).
The primary endpoint was the change in HbA1c from baseline to Week 28. Compared to placebo, treatment with BYDUREON resulted in a statistically significant reduction in mean HbA1c from baseline to Week 28 (Table 9).
Table 9: Results of 28-Week Trial of BYDUREON versus Placebo, Both as Add-On to Insulin Glargine or Insulin Glargine + Metformin BYDUREON
2 mg QWPlacebo
QW- * Adjusted LS means and treatment group difference(s) in the change from baseline values at Week 28 using a multiple imputation method that models a “wash-out” for patients having missing data who discontinued treatment. ANCOVA was used with treatment, region, baseline HbA1c stratum (<9.0% or ≥9.0%), and baseline SU-use stratum (yes vs. no) as fixed factors, and baseline value as a covariate.
- † p-value <0.001 (adjusted for multiplicity).
- ‡ Categories are derived from continuous measurements. All patients with missing endpoint data are imputed as non-responders. Treatment comparison is based on Cochran-Mantel-Haenszel (CMH) test stratified by baseline HbA1c (<9.0% or ≥9.0%), and baseline SU-use stratum (yes vs. no). P-values are from the general association statistics.
Intent-to-Treat Population (N)
231
229
Mean HbA1c (%)
Mean Baseline
8.53
8.53
Mean Change at Week 28*
-0.88 (0.070)
-0.24 (0.069)
Difference from Placebo [95% CI]
-0.64†
[-0.83, -0.45]
Percentage Achieving HbA1c <7.0% at Week 28 (%)‡
32.5†
7.0
N = number of patients in each treatment group, CI=confidence interval, QW=once weekly.
Note: mean change is least squares mean change.
Analyses include measurements post rescue therapy and post premature discontinuation of study medication.
The mean change in fasting plasma glucose from baseline to Week 28 was -12.50 mg/dL for BYDUREON and -2.26 mg/dL for placebo. The mean change from baseline to Week 28 in body weight was -0.92 kg for BYDUREON and +0.38 kg for placebo.
14.2 EXSCEL Cardiovascular Outcomes Trial in Patients with Type 2 Diabetes
EXSCEL was a multinational, placebo-controlled, double-blind, randomized, parallel group pragmatic study that evaluated cardiovascular (CV) outcomes during treatment with BYDUREON in patients with type 2 diabetes and any level of CV risk when added to the current usual care (NCT01144338).
A total of 14,752 patients were randomized 1:1 to either BYDUREON 2 mg once weekly or placebo and followed as in routine clinical practice for a median of 38.7 months with a median treatment duration of 27.8 months. Ninety six percent of the patients in both treatment groups completed the study in accordance with the protocol, and the vital status was known at the end of the study for 98.9% and 98.8% of the patients in the BYDUREON and placebo group, respectively. The mean age at study entry was 62 years (21 to 92 years with 8.5% of the patients ≥75 years). Approximately 62.0% of the patients were male, 75.8% were Caucasian, 9.8% were Asian, 6.0% were Black, and 20.5% were Hispanic or Latino. The mean BMI was 32.7 kg/m2 and the mean duration of diabetes was 13.1 years. Approximately 49.3% had mild renal impairment (estimated glomerular filtration rate [eGFR] ≥60 to ≤89 mL/min/1.73 m2) and 21.6% had moderate renal impairment (eGFR ≥30 to ≤59 mL/min/1.73 m2).
The mean HbA1c was 8.1%. At baseline, 1.5% of patients were not treated with either oral antidiabetic medications or insulin, 42.3% were treated with one oral antidiabetic medication and 42.4% were treated with two or more oral antidiabetic medications. Usage of oral antidiabetic medications included metformin (76.6%), sulfonylurea (36.6%), DPP-4 inhibitors (14.9%), thiazolidinediones (3.9%), and SGLT-2 inhibitors (0.9%). Overall insulin usage was 46.3% (13.8% with insulin alone and 32.6% with insulin and one or more oral antidiabetic medications).
Overall, at baseline, 26.9% of patients did not have established cardiovascular (CV) disease, while 73.1% had established CV disease. The concomitant use of CV medications (e.g., ACE inhibitors, angiotensin receptor blockers, diuretics, beta blockers, calcium channel blockers, antithrombotic and anticoagulants, and lipid-lowering agents) was similar in the BYDUREON and placebo groups. At baseline, the mean systolic blood pressure was 135.5 mmHg, the mean diastolic blood pressure was 78.1 mmHg, the mean LDL was 95.0 mg/dL, and the mean HDL was 44.0 mg/dL.
The primary endpoint in EXSCEL was the time to first confirmed Major Adverse Cardiac Event (MACE) from randomization. MACE was defined as occurrence of either a cardiovascular (CV)-related death, or a nonfatal myocardial infarction (MI) or a nonfatal stroke. All-cause mortality, CV-related death, and fatal or nonfatal MI or stroke, hospitalization for acute coronary syndrome, and hospitalization for heart failure were also assessed as secondary endpoints.
A Cox proportional hazards model was used to test for non-inferiority against the pre-specified risk margin of 1.3 for the hazard ratio of MACE and superiority on MACE if non-inferiority was demonstrated. Type-1 error was controlled across multiples tests using a hierarchical testing strategy.
BYDUREON did not increase the risk of MACE in patients with type 2 diabetes mellitus (HR: 0.91; 95% CI: 0.832, 1.004; P<0.001 for non-inferiority; P=0.06 for superiority). See results in Table 10 and Figure 2. The incidence of MACE in patients with and without established CV disease was 13.4% in the BYDUREON group versus 14.6% in the placebo group and 6.0% (BYDUREON) versus 5.9% (placebo), respectively. Five hundred and seven (507) patients (6.9%) died in the BYDUREON group versus 584 (7.9%) in the placebo group.
Table 10: Analysis of Primary Composite Endpoint MACE and Its Components in Patients with Type 2 Diabetes BYDUREON
N=7356Placebo
N=7396HR*
(95% CI)- * HR (active/placebo) and CI are based on Cox proportional hazards regression model, stratified by established CV disease, with treatment group only as explanatory variable.
MACE Composite of CV death, nonfatal MI or nonfatal stroke (time to first confirmed event)
839 (11.4%)
905 (12.2%)
0.91
(0.832, 1.004)
Cardiovascular Death
340 (4.6%)
383 (5.2%)
0.88
(0.76, 1.02)
Nonfatal Myocardial Infarction
466 (6.3%)
480 (6.5%)
0.96
(0.85, 1.09)
Nonfatal Stroke
169 (2.3%)
193 (2.6%)
0.86
(0.70, 1.06)
N=number of patients in each treatment group, HR=hazard ratio, CI=confidence interval, CV=cardiovascular, MI=myocardial infarction.
Figure 2: Time to First Adjudicated MACE in Patients with Type 2 Diabetes
HR=hazard ratio, CI=confidence interval.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How supplied
BYDUREON (exenatide extended-release for injectable suspension) for once every 7 days (weekly) subcutaneous administration is supplied as:
BYDUREON single-dose tray, supplied in cartons that contain four single-dose trays (NDC: 0310-6520-04). Each single-dose tray contains:
- One single-dose vial containing 2 mg exenatide (as a white to off-white powder)
- One prefilled syringe delivering 0.65 mL diluent
- One vial connector
- Two custom needles (23G, 5/16") specific to this delivery system (one is a spare needle)
BYDUREON Pen, supplied in cartons that contain four single-dose pens and one spare needle (NDC: 0310-6530-04). Each single-dose pen contains:
- One single-dose pen containing 2 mg of exenatide (as a white to off-white powder) and delivering 0.65 mL diluent.
- One custom needle (23G, 9/32") specific to this delivery system.
Do not substitute needles or any other components provided with BYDUREON.
Storage and Handling
- BYDUREON should be stored in the refrigerator at 36°F to 46°F (2°C to 8°C), up to the expiration date or until preparing for use. BYDUREON should not be used past the expiration date. The expiration date can be found on the carton, on the cover of the single-dose tray, or on the pen label.
- Do not freeze BYDUREON. Do not use BYDUREON if it has been frozen. Protect from light.
- BYDUREON can be kept at room temperature not to exceed 77°F (25°C) [see USP Controlled Room Temperature] for no more than a total of 4 weeks, if needed.
- Use the diluent only if it is clear and free of particulate matter.
- After suspension, the mixture should be white to off-white and cloudy.
- BYDUREON must be administered immediately after the exenatide powder is suspended in the diluent.
- Use a puncture-resistant container to discard BYDUREON with the needle still attached. Do not reuse or share needles or syringes.
- Keep out of the reach of children.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Risk of Thyroid C-cell Tumors
Inform patients that exenatide extended-release causes benign and malignant thyroid C-cell tumors in rats and that the human relevance of this finding has not been determined. Counsel patients to report symptoms of thyroid tumors (e.g., a lump in the neck, hoarseness, dysphagia, or dyspnea) to their physician [see Boxed Warning and Warnings and Precautions (5.1)].
Never Share a BYDUREON Pen Between Patients
Advise patients that they must never share a BYDUREON pen with another person, because doing so carries a risk for transmission of blood-borne pathogens [see Warnings and Precautions (5.2)].
Risk of Pancreatitis
Inform patients treated with BYDUREON of the potential risk for pancreatitis. Explain that persistent severe abdominal pain that may radiate to the back, and which may or may not be accompanied by vomiting, is the hallmark symptom of acute pancreatitis. Instruct patients to discontinue BYDUREON promptly and contact their healthcare provider if persistent severe abdominal pain occurs [see Warnings and Precautions (5.3)].
Risk of Hypoglycemia
Inform patients that the risk of hypoglycemia is increased when BYDUREON is used in combination with an agent that induces hypoglycemia, such as a sulfonylurea or insulin [see Warnings and Precautions (5.4)]. Explain the symptoms, treatment, and conditions that predispose to the development of hypoglycemia. Review and reinforce instructions for hypoglycemia management when initiating BYDUREON therapy, particularly when concomitantly administered with a sulfonylurea or insulin [see Warnings and Precautions (5.4)].
Risk of Acute Kidney Injury
Inform patients treated with BYDUREON of the potential risk for worsening kidney function and explain the associated signs and symptoms of renal impairment, as well as the possibility of dialysis as a medical intervention if renal failure occurs [see Warnings and Precautions (5.5)].
Risk of Hypersensitivity Reactions
Inform patients that serious hypersensitivity reactions have been reported during postmarketing use of exenatide. Inform patients that if symptoms of hypersensitivity reactions occur, stop taking BYDUREON and seek medical advice promptly [see Warnings and Precautions (5.8)].
Risk of Drug-Induced Thrombocytopenia
Inform patients that drug-induced immune-mediated thrombocytopenia has been reported during use of exenatide. Inform patients that if symptoms of thrombocytopenia occur, e.g. bleeding, stop taking BYDUREON and seek medical advice promptly [see Warnings and Precautions (5.9)].
Risk of Injection-Site Reactions
Inform patients that there have been postmarketing reports of serious injection-site reactions with or without subcutaneous nodules, with the use of BYDUREON. Isolated cases of injection-site reactions required surgical intervention. Advise patients to seek medical advice if symptomatic nodules occur, or for any signs or symptoms of abscess, cellulitis, or necrosis [see Warnings and Precautions (5.10)].
Acute Gallbladder Disease
Inform patients of the potential risk for cholelithiasis or cholecystitis. Instruct patients to contact their physician if cholelithiasis or cholecystitis is suspected for appropriate clinical follow-up [see Warnings and Precautions (5.11)].
Instructions
Train patients on how to use BYDUREON properly prior to self-administration. Instruct patients on proper mixing and injection technique to ensure the product is adequately mixed and a full dose is delivered. Refer patients to the accompanying Instructions for Use for complete administration instructions with illustrations [see Dosage and Administration (2)].
Counsel patients that they should never share BYDUREON with another person, even if the needle is changed. Sharing of BYDUREON or needles between patients may pose a risk of transmission of infection.
If a patient is currently taking BYETTA, it should be discontinued upon starting BYDUREON. Inform patients formerly on BYETTA who start BYDUREON that they may experience transient elevations in blood glucose concentrations, which generally improve within the first 2 weeks after initiation of therapy [see Dosage and Administration (2.4) and Clinical Studies (14.1)].
Treatment with BYDUREON may also result in nausea, particularly upon initiation of therapy [see Adverse Reactions (6)].
Inform patients about the importance of proper storage of BYDUREON [see How Supplied/Storage and Handling (16)].
Instruct the patient to review the BYDUREON Medication Guide and the Instructions for Use each time the prescription is refilled.
Manufactured for:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
By:
Amylin Ohio LLC
West Chester, OH 45071
BYDUREON is a registered trademark of the AstraZeneca group of companies.
-
MEDICATION GUIDE
MEDICATION GUIDE
BYDUREON® (by-DUR-ee-on)
(exenatide extended-release)
for injectable suspension, for subcutaneous use
What is the most important information I should know about BYDUREON?
BYDUREON may cause serious side effects, including:
- Possible thyroid tumors, including cancer. Tell your healthcare provider if you get a lump or swelling in your neck, hoarseness, trouble swallowing, or shortness of breath. These may be symptoms of thyroid cancer. In studies with rats or mice, BYDUREON and medicines that work like BYDUREON caused thyroid tumors, including thyroid cancer. It is not known if BYDUREON will cause thyroid tumors or a type of thyroid cancer called medullary thyroid carcinoma (MTC) in people.
- Do not use BYDUREON if you or any of your family have ever had a type of thyroid cancer called medullary thyroid carcinoma (MTC), or if you have an endocrine system condition called Multiple Endocrine Neoplasia syndrome type 2 (MEN 2).
- Do not share your BYDUREON pen, prefilled syringe, or needles with another person. You may give another person an infection or get an infection from them.
What is BYDUREON?
- BYDUREON is an injectable prescription medicine that may improve blood sugar (glucose) in adults with type 2 diabetes mellitus and should be used along with diet and exercise.
- BYDUREON is not recommended as the first choice of medicine for treating diabetes.
- BYDUREON is not a substitute for insulin and is not for use in people with type 1 diabetes or people with diabetic ketoacidosis.
- It is not known if BYDUREON can be used with mealtime insulin.
- BYDUREON and BYDUREON BCise are long-acting forms of the medicine in BYETTA (exenatide). BYDUREON should not be used at the same time as BYETTA or BYDUREON BCise.
- It is not known if BYDUREON can be used in people who have had pancreatitis.
- It is not known if BYDUREON is safe and effective for use in children.
Who should not use BYDUREON?
Do not use BYDUREON if:
- you or any of your family have ever had a type of thyroid cancer called medullary thyroid carcinoma (MTC) or if you have an endocrine system condition called Multiple Endocrine Neoplasia syndrome type 2 (MEN 2).
- you have a history of low blood platelet count from using exenatide medicines (drug-induced thrombocytopenia).
- you are allergic to exenatide or any of the ingredients in BYDUREON. See the end of this Medication Guide for a complete list of ingredients in BYDUREON.
What should I tell my healthcare provider before using BYDUREON?
Before using BYDUREON, tell your healthcare provider about all of your medical conditions, including if you:
- have or have had problems with your pancreas or kidneys.
- have severe problems with your stomach, such as slowed emptying of your stomach (gastroparesis) or problems with digesting food.
- are pregnant or plan to become pregnant. BYDUREON may harm your unborn baby. Tell your healthcare provider if you become pregnant while using BYDUREON. Talk to your healthcare provider about the best way to control your blood sugar if you plan to become pregnant or while you are pregnant.
- are breastfeeding or plan to breastfeed. It is not known if BYDUREON passes into your breast milk. You should talk with your healthcare provider about the best way to feed your baby while using BYDUREON.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. BYDUREON may affect the way some medicines work and some medicines may affect the way BYDUREON works.
Before using BYDUREON, talk to your healthcare provider about low blood sugar and how to manage it. Tell your healthcare provider if you are taking other medicines to treat diabetes including insulin or sulfonylureas.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I use BYDUREON?
- Read the Instructions for Use that comes with BYDUREON.
- Use BYDUREON exactly as your healthcare provider tells you to.
- BYDUREON should be injected right away after you prepare your dose.
- Your healthcare provider should show you how to use BYDUREON before you use it for the first time.
- BYDUREON is injected under the skin (subcutaneously) of your stomach (abdomen), thigh, or upper arm. Do not inject BYDUREON into a muscle (intramuscularly) or vein (intravenously).
- Use BYDUREON 1 time each week on the same day each week at any time of the day.
- BYDUREON may be taken with or without food.
- If you miss a dose of BYDUREON, take the missed dose as soon as possible if there are at least 3 days (72 hours) until your next scheduled dose. If there are less than 3 days remaining, skip the missed dose and take your next dose on the regularly scheduled day. Do not take 2 doses of BYDUREON within 3 days of each other.
- You may change the day of the week as long as your last dose was given 3 or more days before.
- Do not mix insulin and BYDUREON together in the same injection.
- You may give an injection of BYDUREON and insulin in the same body area (such as, your stomach area), but not right next to each other.
- Change (rotate) your injection site with each weekly injection. Do not use the same site for each injection.
- Your dose of BYDUREON and other diabetes medicines may need to change because of: change in level of physical activity or exercise, weight gain or loss, increased stress, illness, change in diet, or because of other medicines you take.
What are the possible side effects of BYDUREON?
BYDUREON may cause serious side effects, including:
- See "What is the most important information I should know about BYDUREON?"
- inflammation of your pancreas (pancreatitis). Stop using BYDUREON and call your healthcare provider right away if you have severe pain in your stomach area (abdomen) that will not go away, with or without vomiting. You may feel the pain from your abdomen to your back.
- low blood sugar (hypoglycemia). Your risk for getting low blood sugar may be higher if you use BYDUREON with another medicine that can cause low blood sugar, such as a sulfonylurea or insulin. Signs and symptoms of low blood sugar may include:
- dizziness or light-headedness
- sweating
- confusion or drowsiness
- headache
- blurred vision
- slurred speech
- shakiness
- fast heartbeat
- anxiety, irritability, or mood changes
- hunger
- weakness
- feeling jittery
- kidney problems. In people who have kidney problems, diarrhea, nausea, and vomiting may cause a loss of fluids (dehydration) which may cause kidney problems to get worse or kidney failure.
- stomach problems. Other medicines like BYDUREON may cause severe stomach problems. It is not known if BYDUREON causes or worsens stomach problems.
- low blood platelet count (drug-induced thrombocytopenia). BYDUREON may cause the number of platelets in your blood to be reduced. When your platelet count is too low, your body cannot form blood clots. You could have serious bleeding that could lead to death. Stop using BYDUREON and call your healthcare provider right away if you have unusual bleeding or bruising. Your blood platelet count may continue to be low for about 10 weeks after stopping BYDUREON.
- serious allergic reactions. Stop using BYDUREON and get medical help right away if you have any symptoms of a serious allergic reaction, including itching, rash, or difficulty breathing.
- injection-site reactions. Serious injection-site reactions, with or without bumps (nodules), have happened in some people who use BYDUREON. Some of these injection-site reactions have required surgery. Call your healthcare provider if you have any symptoms of an injection-site reaction, including severe pain, swelling, blisters, an open wound, a dark scab.
- gallbladder problems. Gallbladder problems have happened in some people who take BYDUREON or other medicines like BYDUREON. Tell your healthcare provider right away if you get symptoms of gallbladder problems which may include: pain in the right or middle upper stomach area, nausea and vomiting, fever, or your skin or the white part of your eyes turns yellow.
The most common side effects of BYDUREON may include nausea, diarrhea, headache, vomiting, constipation, itching at the injection site, a small bump (nodule) at the injection site, indigestion.
Nausea is most common when you first start using BYDUREON but decreases over time in most people as their body gets used to the medicine.
Talk to your healthcare provider about any side effect that bothers you or does not go away.
These are not all the possible side effects of BYDUREON. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
Keep BYDUREON and all medicines out of the reach of children.
General information about the safe and effective use of BYDUREON.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use BYDUREON for a condition for which it was not prescribed. Do not give your BYDUREON to other people, even if they have the same symptoms you have. It may harm them.
You can ask your pharmacist or healthcare provider for information about BYDUREON that is written for health professionals.
What are the ingredients in BYDUREON?
Contents of the powder:
Active Ingredient: exenatide
Inactive Ingredients: polylactide-co-glycolide and sucrose
Contents of liquid (diluent):
Inactive Ingredients: carboxymethylcellulose sodium, polysorbate 20, sodium phosphate monobasic monohydrate, sodium phosphate dibasic heptahydrate, sodium chloride, water for injection. Sodium hydroxide may be added during manufacture of BYDUREON Pen for pH adjustment.
BYDUREON is a registered trademark and BYETTA is a registered trademark of the AstraZeneca group of companies. All other marks are the marks of their respective owners.
Manufactured for:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
By:
Amylin Ohio LLC
West Chester, OH 45071
For more information about BYDUREON, go to www.BYDUREON.com or call 1-877-700-7365.
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: February 2020
-
Instructions for Use
BYDUREON® (by-DUR-ee-on) Single-Dose Tray
(exenatide extended-release) for injectable suspensionBefore using Bydureon, your healthcare provider should show you how to use it the right way.
Read these Instructions for Use before you start using BYDUREON Single-Dose Tray and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
Getting ready
Never share your BYDUREON vials or needles with anyone else. You may give an infection to them or get an infection from them.
BYDUREON Single-Dose Tray is not for self-injection by people who are blind or cannot see well.
Supplies needed to give your BYDUREON Single-Dose Tray injection (not all supplies are included):
-
1 BYDUREON Single-Dose Tray that contains:
- 1 BYDUREON vial
- 1 Syringe
- 2 Needles
- 1 Vial connector
- alcohol swab
- a clean flat surface
- sharps container for throwing away used needles, vials, and syringes. See Step 4h "Disposing of used Needles and Syringes."
Your guide to your BYDUREON Single-Dose Tray
- Single-dose tray
Keep this flap open so you can refer to it as you go through the steps.
Your guide to the parts
- Single-dose tray
What is Inside
To take the correct dose, read each page so that you do every step in order.
This step-by-step guide is divided into 4 sections:
- Getting Started
- Connecting the Parts
- Mixing the Medicine and Filling the Syringe
- Injecting the Medicine
For Common Questions and Answers, see page X.
How to store your Single-Dose Trays of BYDUREON
- Store your BYDUREON trays in the refrigerator at 36°F to 46°F (2°C to 8°C).
- If needed, you can keep your BYDUREON tray out of the refrigerator at 68°F to 77°F (20°C to 25°C) for up to 4 weeks.
- Protect BYDUREON from light until you are ready to prepare and use your dose.
- Do not freeze BYDUREON trays.
- Do not use BYDUREON past the expiration date. The expiration date is labeled EXP and can be found on the paper cover of each tray.
- Keep BYDUREON, and all medicines, out of the reach of children.
1. Getting Started
1a) Take a Single-Dose Tray from the refrigerator.
1b) Wash your hands. Prepare to clean your injection site with soap and water or an alcohol swab prior to injecting your medicine.
2. Connecting the Parts
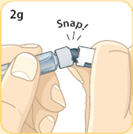
Break off the cap.
Be careful not to push in the plunger.
Just like you might break a stick, you are breaking off the cap.
3. Mixing the Medicine and Filling the Syringe
Important:
During these next steps, you will be mixing the medicine and filling the syringe. After you mix the medicine, you must inject it. You cannot save the mixed medicine to inject at a later time.
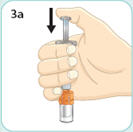
With your thumb, push down the plunger until it stops.
The plunger may feel like it is springing back a little.
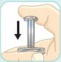
For steps 3a to 3f, keep pushing down on the plunger with your thumb.
4. Injecting the Medicine
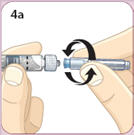
Pick up the needle. Twist the needle onto the syringe until snug. Do not remove the needle cover yet.
Important:
Read the next steps carefully and look closely at the pictures. This helps you get the correct dose of medicine.
Important:
It is normal to see a few bubbles in the mixture. The bubbles will not harm you or affect your dose.
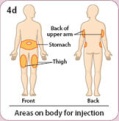
You can inject the medicine in your stomach area (abdomen), your thigh, or the back of your upper arm.
Each week you can use the same area of your body but choose a different injection site in that area.
Gently clean the site you choose with soap and water or an alcohol swab.
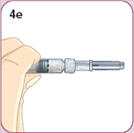
Now, pick up the syringe and hold it near the black dashed Dose Line.
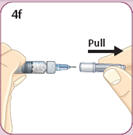
Pull the needle cover straight off. Do not twist.
Be careful not to push in the plunger.
When you remove the cover, you may see 1 or 2 drops of liquid. This is normal.
Insert the needle into your skin (subcutaneously). To inject your full dose, push down on the plunger with your thumb until it stops.
Withdraw the needle.
Be sure to use the injection technique recommended by your healthcare provider.

Disposing of used Needles and Syringes:
- Put your used needles and syringes in a FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles and syringes in your household trash.
-
If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and pens. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA’s website at: http://www.fda.gov/safesharpsdisposal.
Please keep these Instructions for Use for your next dose.
Common Questions and Answers
If your question is about:
See question number:
How soon to inject after mixing
1
Mixing the medicine
2
Air bubbles in syringe
3
Attaching the needle
4
Removing the needle cover
5
Plunger not lining up with black dashed Dose Line
6
Being unable to push the plunger down when injecting
7
- 1.
After I mix the medicine, how long can I wait before taking the injection?
You must take your injection of BYDUREON right after mixing it. If you do not inject BYDUREON right away, the medicine will start to form small clumps in the syringe. These clumps can clog the needle when you take the injection (see question 7).
- 2.
How do I know that the medicine is mixed well?
When the medicine is mixed well, it should look cloudy. There should not be any dry powder on the sides or bottom of the vial. If you do see any dry powder, shake hard while continuing to push down on the plunger with your thumb. (This question relates to the steps shown on pages X through X.)
- 3.
I'm ready to take the injection. What should I do if I see air bubbles in the syringe?
It is normal for air bubbles to be in the syringe. The air bubbles will not harm you or affect your dose. BYDUREON is injected into your skin (subcutaneously). Air bubbles are not a problem with this type of injection. (This question relates to step 3f shown on page X and step 4c shown on page X.)
- 4.
What should I do if I have trouble attaching the needle?
First, be sure you have removed the blue cap. Then, twist the needle onto the syringe until snug. To prevent losing medicine, do not push in the plunger while attaching the needle. (This question relates to step 4a on page X.)
- 5.
What should I do if I have trouble removing the needle cover?
With one hand, hold the syringe near the black dashed Dose Line. With your other hand, hold the needle cover. Pull the needle cover straight off. Do not twist it. (This question relates to step 4f on page X.)
- 6.
I am at step 4c. What should I do if the top of the plunger has been pushed past the black dashed Dose Line?
The black dashed Dose Line shows the correct dose. If the top of the plunger has been pushed past the line, you should continue from step 4d and take the injection. Before your next injection in 1 week, carefully review the instructions on pages X through X.
- 7.
When I inject, what should I do if I cannot push the plunger all the way down?
This means the needle has become clogged. Remove the needle from your skin and replace it with the spare needle from your tray. Then choose a different injection site and finish taking the injection.
To review how to:- Remove the blue cap of the needle, see page X
- Attach the needle, see page X
- Remove the needle cover and give the injection, see pages X and X
-
If you still cannot push the plunger all the way down, remove the needle from your skin. Use a puncture-resistant container to throw away the syringe with the needle still attached. It is important that you then call 1-877-700-7365.
To help prevent a clogged needle, always mix the medicine very well and inject right after mixing.
Where to learn more about BYDUREON
- Talk with your healthcare provider
- Read the Medication Guide that came with your BYDUREON. The Medication Guide can help answer your questions about BYDUREON, such as what it is used for, possible side effects, and when to take BYDUREON.
- Enroll in BYDUREON Support for FREE ongoing help managing your diabetes. Visit www.bydureon.com or call 1-877-700-7365.
These Instructions for Use have been approved by the U.S. Food and Drug Administration.
BYDUREON® is a registered trademark of the AstraZeneca group of companies.
Manufactured for:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850By:
Amylin Ohio LLC
West Chester, OH 45071Approved: October 2017
-
1 BYDUREON Single-Dose Tray that contains:
-
Instructions for Use
BYDUREON® (by-DUR-ee-on) Pen
(exenatide extended-release) for injectable suspensionFigure A
Before using Bydureon Pen, your healthcare provider should show you how to use it the right way.
Read the Instructions for Use before you start using Bydureon Pen and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
Getting ready
Never share your Bydureon Pen or needles with anyone else. You may give an infection to them or get an infection from them.
Bydureon Pen is not for self-injection by people who are blind or cannot see well.
Supplies needed to give your Bydureon Pen injection (not all supplies are included):
-
1 Bydureon single-use "Pen" tray that contains:
- 1 Bydureon Pen
- 1 custom needle
- a clean flat surface
- alcohol swab
- sharps container for throwing away used needles and Pens. See "Properly dispose of your Pen" at the end of these instructions.
How should I store Bydureon?
- Store your Bydureon Pens in the refrigerator at 36°F to 46°F (2°C to 8°C).
- Protect pens from light until you are ready to prepare and use your dose.
- Do not use pens past the expiration date.
- Do not freeze Bydureon. Do not use Bydureon if it has been frozen.
- Keep Bydureon in its sealed tray until ready for use.
- If needed, you can keep your Bydureon Pen out of the refrigerator at 68°F to 77°F (20°C to 25°C) for up to 4 weeks.
Keep Bydureon Pen, and all medicines, out of the reach of children.
Step 2: Mix your dose
Combine the medicine.- While holding the pen straight up, slowly turn the knob. Stop when you hear the click and the green label disappears.
Firmly tap your Pen to mix.
Check the Bydureon mix.
- Hold your Pen up to the light and look through both sides of the mixing window. The solution should have no clumps and be uniformly cloudy (see Figure B).
- To get your full dose, Bydureon must be mixed well.
- If Bydureon is not mixed well, keep tapping your Pen longer and more firmly until it is mixed well.
- Do not give your Bydureon injection unless your Bydureon is mixed well.
Stop. Do Not proceed unless your medicine is mixed well.
To get your full dose the medicine must be mixed well. If it’s not mixed well, tap longer and more firmly.
Check the Bydureon mix again.
- Compare both sides of the mixing window to the photos below by holding your Pen against the page. Pay attention to the bottom surface. If you do not see clumps you are ready to inject (see Figure C).
If you have any questions or are not sure if your Bydureon is mixed well, call 1-877-700-7365 for help.
Step 3: Inject your dose
Important: After the medicine is mixed well, you must inject your dose right away. You cannot save it for later use.
Remove the needle cover.
-
Pull the needle cover straight off. Do not twist the needle cover.
- You may see a few drops of liquid on the needle or in the cover.

Inject your Bydureon.
- Insert the needle into your skin.
- Press the injection button with your thumb until you hear a “click”. Keep holding the button down and slowly count to 10 to get your full dose.

Common Questions and Answers:
- 1.
How do I know that the Bydureon is mixed well?
The Bydureon is mixed well when the liquid looks cloudy from both sides of the window. You should not see any clumps in the liquid. It may help to hold your Pen up to the light to see in the window. If you see clumps of any size keep tapping your Pen firmly against the palm of your hand until mixed.
- 2.
I am having trouble mixing my dose. What should I do?
Remember, before preparing your dose, leave your Pen out of the refrigerator for at least 15 minutes. This will let your Pen warm up to room temperature. It will be easier to mix Bydureon if your Pen is at room temperature.
Be sure you are holding your Pen at the end with the knob and the orange label. This will help you grip your Pen better and tap it more firmly against your palm.
It may also help to tap the mixing window on both sides against your palm. If you see any clumps, keep tapping.
- 1.
After I mix Bydureon, how long can I wait before taking the injection?
You must inject your dose of Bydureon right after mixing it. If you do not inject your Bydureon right away, small clumps of medicine may form in your Pen and you may not get your full dose.
- 2.
I am ready to inject my dose. What should I do if I see air bubbles in the Pen?
It is normal for air bubbles to be in your Pen. Bydureon is injected into your skin (subcutaneously). Air bubbles will not harm you or affect your dose with this type of injection.
- 3.
What should I do if I cannot push the injection button all the way in when trying to inject my dose?
Check that you have fully screwed on the pen needle. Also be sure you twisted the knob until it stopped, the orange label disappeared, and the injection button appears.
If you still cannot push the button in, this may mean that the needle is clogged. Remove the needle from your skin and replace it with the spare needle from the carton. Review how to attach the needle. Then choose a different injection site and finish taking the injection.
If you still cannot push the button all the way in, remove the needle from your skin. Use a puncture-resistant container to throw away the pen with the needle still attached.
If you have problems giving your Bydureon Pen injection or have any questions call 1-877-700-7365 for more instructions.
- 4.
How do I know if I injected my full dose?
To be sure you get your full dose, press the injection button with your thumb until you hear a "click". After the "click", continue to hold the needle in your skin for 10 seconds. This will allow enough time for you to get your full dose.
- 5.
What if I do not have an FDA-cleared sharps disposal container?
Do not throw away (dispose of) loose needles and Pens in your household trash.-
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- o made of a heavy-duty plastic,
- o can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- o upright and stable during use,
- o leak-resistant, and
- o properly labeled to warn of hazardous waste inside the container.
-
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and Pens. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA’s website at: http://www.fda.gov/safesharpsdisposal
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
BYDUREON® is a registered trademark of the AstraZeneca group of companies.
Manufactured for:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850By:
Amylin Ohio LLC
West Chester, OH 45071Approved: October 2017
-
1 Bydureon single-use "Pen" tray that contains:
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL - 2 mg/vial
NDC: 0310-6520-04
Rx OnlyOnce-weekly
Bydureon®
exenatide extended-release
for injectable suspension2 mg/vial
Subcutaneous use only.
Dispense the enclosed
Medication Guide to each patient.
Total quality: 4 single-dose trays.
Each tray includes supplies to deliver a 2 mg dose.
Use 1 tray every week.
Store refrigerated: 36°F to 46°F (2°C to 8°C). Do not freeze.
Package Not Child-Resistant. Keep out of reach of children.
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL - 2 mg/pen
NDC: 0310-6530-04
Rx OnlyOnce-Weekly
Bydureon® Pen
exenatide extended-release
for injectable suspension
2 mg/pen
Subcutaneous use only.
Dispense the enclosed Medication Guide to each patient.
- Total quality: 4 single-dose pens
- Each pen contains a needle. There is one extra needle in the carton.
- Each pen includes supplies to deliver a 2 mg dose.
- Use 1 pen per week.
Follow the enclosed Instructions for Use to prepare and inject your dose.
For more information about BYDUREON, call 1-877-700-7365
or visit www.BYDUREON.com.
Store refrigerated: 36°F to 46°F (2°C to 8°C). Do not freeze.
Package Not Child-Resistant. Keep out of reach of children.
-
INGREDIENTS AND APPEARANCE
BYDUREON
exenatide injection, suspension, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0310-6530 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EXENATIDE (UNII: 9P1872D4OL) (EXENATIDE - UNII:9P1872D4OL) EXENATIDE 2 mg in 0.65 mL Inactive Ingredients Ingredient Name Strength POLY(DL-LACTIC-CO-GLYCOLIC ACID), (50:50; 63000 MW) (UNII: QD923V64H3) SUCROSE (UNII: C151H8M554) CARBOXYMETHYLCELLULOSE SODIUM, UNSPECIFIED FORM (UNII: K679OBS311) SODIUM CHLORIDE (UNII: 451W47IQ8X) POLYSORBATE 20 (UNII: 7T1F30V5YH) SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) SODIUM PHOSPHATE, DIBASIC, HEPTAHYDRATE (UNII: 70WT22SF4B) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0310-6530-04 1 in 1 CARTON 09/08/2014 1 4 in 1 TRAY 1 0.65 mL in 1 CARTRIDGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 2 NDC: 0310-6530-01 1 in 1 TRAY 09/08/2014 2 0.65 mL in 1 CARTRIDGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 3 NDC: 0310-6530-85 1 in 1 CARTON 09/08/2014 3 1 in 1 TRAY 3 0.65 mL in 1 CARTRIDGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022200 09/08/2014 BYDUREON
exenatide kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0310-6520 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0310-6520-04 4 in 1 CARTON 02/01/2015 11/30/2020 1 1 in 1 TRAY; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL 0.65 mL Part 2 1 SYRINGE 0.65 mL Part 1 of 2 BYDUREON
exenatide injection, powder, for suspensionProduct Information Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EXENATIDE (UNII: 9P1872D4OL) (EXENATIDE - UNII:9P1872D4OL) EXENATIDE 2 mg in 0.65 mL Inactive Ingredients Ingredient Name Strength POLY(DL-LACTIC-CO-GLYCOLIC ACID), (50:50; 63000 MW) (UNII: QD923V64H3) SUCROSE (UNII: C151H8M554) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 0.65 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022200 02/01/2015 Part 2 of 2 DILUENT
diluent injection, solutionProduct Information Route of Administration SUBCUTANEOUS Inactive Ingredients Ingredient Name Strength CARBOXYMETHYLCELLULOSE SODIUM, UNSPECIFIED FORM (UNII: K679OBS311) SODIUM CHLORIDE (UNII: 451W47IQ8X) POLYSORBATE 20 (UNII: 7T1F30V5YH) SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) SODIUM PHOSPHATE, DIBASIC, HEPTAHYDRATE (UNII: 70WT22SF4B) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 0.65 mL in 1 SYRINGE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022200 02/01/2015 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022200 02/01/2015 Labeler - AstraZeneca Pharmaceuticals LP (054743190) Registrant - AstraZeneca PLC (230790719)
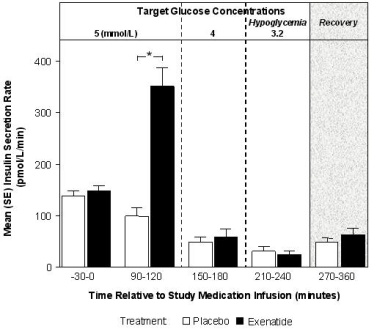
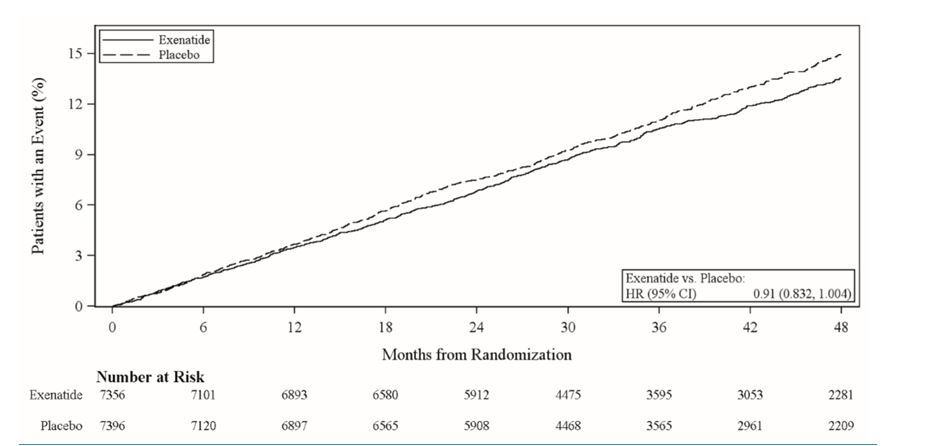
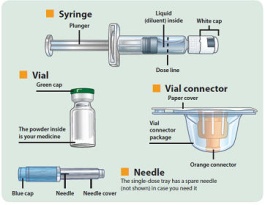
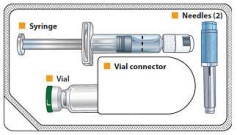
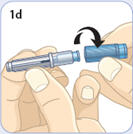
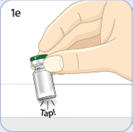
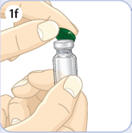
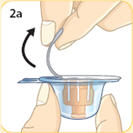
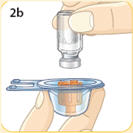
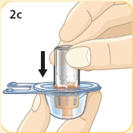
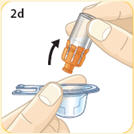

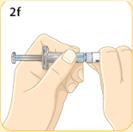
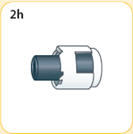

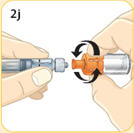
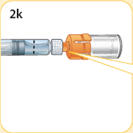

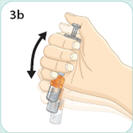

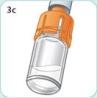
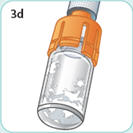
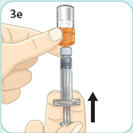
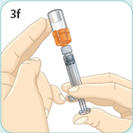
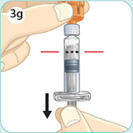
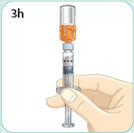
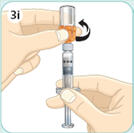

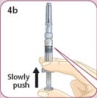


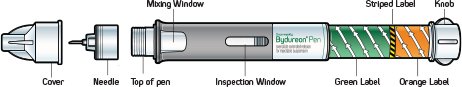





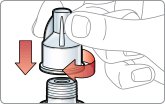
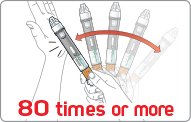


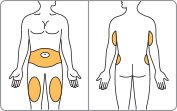
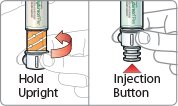




Trademark Results [BYDUREON]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 BYDUREON 85329947 4169150 Live/Registered |
AMYLIN PHARMACEUTICALS, LLC 2011-05-25 |
 BYDUREON 77331009 not registered Dead/Abandoned |
Amylin Pharmaceuticals, Inc 2007-11-15 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.