BUPRENORPHINE AND NALOXONE tablet
Buprenorphine and Naloxone by
Drug Labeling and Warnings
Buprenorphine and Naloxone by is a Prescription medication manufactured, distributed, or labeled by Rhodes Pharmaceuticals L.P., Purdue Pharmaceuticals L.P., Halo Pharmaceutical Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BUPRENORPHINE AND NALOXONE SUBLINGUAL TABLETS safely and effectively. See full prescribing information for BUPRENORPHINE AND NALOXONE SUBLINGUAL TABLETS.
BUPRENORPHINE and NALOXONE sublingual tablets for sublingual administration CIII
Initial U.S. Approval: 2002RECENT MAJOR CHANGES
Warnings and Precautions (5.2) 10/2019 INDICATIONS AND USAGE
Buprenorphine and naloxone sublingual tablet contains buprenorphine, a partial opioid agonist, and naloxone, an opioid antagonist, and is indicated for the maintenance treatment of opioid dependence. (1)
Buprenorphine and naloxone sublingual tablets should be used as part of a complete treatment plan that includes counseling and psychosocial support. (1)
DOSAGE AND ADMINISTRATION
- Prescription use of this product is limited under the Drug Addiction Treatment Act. (2.1)
- Administer buprenorphine and naloxone sublingual tablet sublingually as a single daily dose. (2.2)
- To avoid precipitating withdrawal, induction with buprenorphine sublingual tablets should be undertaken when objective and clear signs of withdrawal are evident. After induction, doses of buprenorphine and naloxone sublingual tablets should be progressively adjusted to a level that holds the patient in treatment and suppresses opioid withdrawal signs and symptoms (2.3)
- The recommended target dosage of buprenorphine and naloxone sublingual tablet for maintenance is 16/4 mg. (2.3)
- Administer buprenorphine and naloxone sublingual tablets as directed in the Full Prescribing Information. (2.3, 2.4)
- When discontinuing treatment, gradually taper to avoid signs and symptoms of withdrawal. (2.7)
DOSAGE FORMS AND STRENGTHS
Sublingual tablet:
- buprenorphine 2 mg/ naloxone 0.5 mg and
- buprenorphine 8 mg/ naloxone 2 mg. (3)
CONTRAINDICATIONS
- Hypersensitivity to buprenorphine or naloxone. (4)
WARNINGS AND PRECAUTIONS
- Addiction, Abuse, and Misuse: Buprenorphine can be abused in a similar manner to other opioids. Clinical monitoring appropriate to the patient’s level of stability is essential. Monitor patients for conditions indicative of diversion or progression of opioid dependence and addictive behaviors. Multiple refills should not be prescribed early in treatment or without appropriate patient follow-up visits. (5.1)
- Respiratory Depression: Life-threatening respiratory depression and death have occurred in association with buprenorphine use. Warn patients of the potential danger of self-administration of benzodiazepine or other CNS depressants while under treatment with buprenorphine and naloxone sublingual tablets. (5.2, 5.3).
- Unintentional Pediatric Exposure: Store buprenorphine and naloxone sublingual tablets safely out of the sight and reach of children. Buprenorphine can cause severe, possibly fatal, respiratory depression in children. (5.4)
- Neonatal Opioid Withdrawal Syndrome: Neonatal opioid withdrawal syndrome (NOWS) is an expected and treatable outcome of prolonged use of opioids during pregnancy. (5.5)
- Adrenal Insufficiency: If diagnosed, treat with physiologic replacement of corticosteroids, and wean patient off of the opioid. (5.6)
- Risk of Opioid Withdrawal with Abrupt Discontinuation: If treatment is temporarily interrupted or discontinued, monitor patients for withdrawal and treat appropriately. (5.7)
- Risk of Hepatitis, Hepatic Events: Monitor liver function tests prior to initiation and during treatment and evaluate suspected hepatic events. (5.8)
- Precipitation of Opioid Withdrawal Signs and Symptoms: An opioid withdrawal syndrome is likely to occur with parenteral misuse of buprenorphine and naloxone sublingual tablets by individuals physically dependent on full opioid agonists, or by sublingual administration before the agonist effects of other opioids have subsided. (5.10)
- Risk of Overdose in Opioid-Naïve Patients: Buprenorphine and naloxone sublingual tablet is not appropriate as an analgesic. There have been reported deaths of opioid naïve individuals who received a 2 mg sublingual dose. (5.11)
ADVERSE REACTIONS
Adverse events commonly observed with the administration of buprenorphine/naloxone are oral hypoesthesia, glossodynia, oral mucosal erythema, headache, nausea, vomiting, hyperhidrosis, constipation, signs and symptoms of withdrawal, insomnia, pain, and peripheral edema. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Rhodes Pharmaceuticals L.P. at 1-888-827-0616, FDA at 1-800-FDA-1088, or www.fda.gov/medwatch
DRUG INTERACTIONS
- Benzodiazepines: Use caution in prescribing buprenorphine and naloxone sublingual tablets for patients receiving benzodiazepines or other CNS depressants and warn patients against concomitant self-administration/misuse. (7)
- CYP3A4 Inhibitors and Inducers: Monitor patients starting or ending CYP3A4 inhibitors or inducers for potential over- or under- dosing. (7)
- Antiretrovirals: Patients who are on chronic buprenorphine treatment should have their dose monitored if NNRTIs are added to their treatment regimen. Monitor patients taking buprenorphine and atazanavir with and without ritonavir, and reduce dose of buprenorphine if warranted. (7)
- Serotonergic Drugs: Concomitant use may result in serotonin syndrome. Discontinue buprenorphine and naloxone sublingual tablets if serotonin syndrome is suspected. (7)
USE IN SPECIFIC POPULATIONS
- Lactation: Buprenorphine passes into mother’s milk. (8.2)
- Geriatric Patients: Monitor for sedation and respiratory depression. (8.5)
- Moderate and Severe Hepatic Impairment: Buprenorphine/naloxone products are not recommended in patients with severe hepatic impairment and may not be appropriate for patients with moderate hepatic impairment. (8.6)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 1/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Drug Addiction Treatment Act
2.2 Important Dosage and Administration Information
2.3 Maintenance
2.4 Method of Administration
2.5 Clinical Supervision
2.6 Unstable Patients
2.7 Discontinuing Treatment
2.8 Switching between Buprenorphine and Naloxone Sublingual Film and Buprenorphine and Naloxone Sublingual Tablets
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Addiction, Abuse, and Misuse
5.2 Risk of Life-Threatening Respiratory and Central Nervous System (CNS) Depression
5.3 Managing Risks from Concomitant Use of Benzodiazepine or Other CNS Depressants
5.4 Unintentional Pediatric Exposure
5.5 Neonatal Opioid Withdrawal Syndrome
5.6 Adrenal Insufficiency
5.7 Risk of Opioid Withdrawal with Abrupt Discontinuation
5.8 Risk of Hepatitis, Hepatic Events
5.9 Hypersensitivity Reactions
5.10 Precipitation of Opioid Withdrawal Signs and Symptoms
5.11 Risk of Overdose in Opioid Naïve Patients
5.12 Use in Patients with Impaired Hepatic Function
5.13 Impairment of Ability to Drive or Operate Machinery
5.14 Orthostatic Hypotension
5.15 Elevation of Cerebrospinal Fluid Pressure
5.16 Elevation of Intracholedochal Pressure
5.17 Effects in Acute Abdominal Conditions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-marketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
9.2 Abuse
9.3 Dependence
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Drug Addiction Treatment Act
Under the Drug Addiction Treatment Act (DATA) codified at 21 U.S.C. 823(g), prescription use of this product in the treatment of opioid dependence is limited to healthcare providers who meet certain qualifying requirements, and who have notified the Secretary of Health and Human Services (HHS) of their intent to prescribe this product for the treatment of opioid dependence and have been assigned a unique identification number that must be included on every prescription.
2.2 Important Dosage and Administration Information
Buprenorphine and naloxone sublingual tablet is administered sublingually as a single daily dose. Buprenorphine and naloxone sublingual tablets should be used in patients who have been initially inducted using buprenorphine sublingual tablets.
Medication should be prescribed in consideration of the frequency of visits. Provision of multiple refills is not advised early in treatment or without appropriate patient follow-up visits.
2.3 Maintenance
- The dosage of buprenorphine and naloxone sublingual tablets should be progressively adjusted in increments/decrements of 2/0.5 mg or 4/1 mg buprenorphine/naloxone to a level that holds the patient in treatment and suppresses opioid withdrawal signs and symptoms.
- The maintenance dose of buprenorphine and naloxone sublingual tablets is generally in the range of 4/1 mg buprenorphine/naloxone to 24/6 mg buprenorphine/naloxone per day depending on the individual patient. The recommended target dosage of buprenorphine and naloxone sublingual tablets is 16/4 mg buprenorphine/naloxone/day as a single daily dose. Dosages higher than 24/6 mg have not been demonstrated to provide any clinical advantage.
- When determining the prescription quantity for unsupervised administration, consider the patient’s level of stability, the security of his or her home situation, and other factors likely to affect the ability to manage supplies of take-home medication.
- There is no maximum recommended duration of maintenance treatment. Patients may require treatment indefinitely and should continue for as long as patients are benefiting and the use of buprenorphine and naloxone sublingual tablets contributes to the intended treatment goals.
2.4 Method of Administration
Buprenorphine and naloxone sublingual tablet must be administered whole. Do not cut, chew, or swallow buprenorphine and naloxone sublingual tablets. Advise patients not to eat or drink until the tablet is completely dissolved.
Buprenorphine and naloxone sublingual tablet should be placed under the tongue until it is dissolved. For doses requiring the use of more than two tablets, patients are advised to either place all the tablets at once or alternatively (if they cannot fit in more than two tablets comfortably), place two tablets at a time under the tongue. Either way, the patients should continue to hold the tablets under the tongue until they dissolve; swallowing the tablets reduces the bioavailability of the drug. To ensure consistency in bioavailability, patients should follow the same manner of dosing with continued use of the product.
Proper administration technique should be demonstrated to the patient.
2.5 Clinical Supervision
Treatment should be initiated with supervised administration, progressing to unsupervised administration as the patient’s clinical stability permits. Buprenorphine and naloxone sublingual tablets are subject to diversion and abuse. When determining the prescription quantity for unsupervised administration, consider the patient’s level of stability, the security of his or her home situation, and other factors likely to affect the ability to manage supplies of take-home medication.
Ideally patients should be seen at reasonable intervals (e.g., at least weekly during the first month of treatment) based upon the individual circumstances of the patient. Medication should be prescribed in consideration of the frequency of visits. Provision of multiple refills is not advised early in treatment or without appropriate patient follow-up visits. Periodic assessment is necessary to determine compliance with the dosing regimen, effectiveness of the treatment plan, and overall patient progress.
Once a stable dosage has been achieved and patient assessment (e.g., urine drug screening) does not indicate illicit drug use, less frequent follow-up visits may be appropriate. A once-monthly visit schedule may be reasonable for patients on a stable dosage of medication who are making progress toward their treatment objectives. Continuation or modification of pharmacotherapy should be based on the healthcare provider’s evaluation of treatment outcomes and objectives such as:
- Absence of medication toxicity
- Absence of medical or behavioral adverse effects
- Responsible handling of medications by the patient
- Patient’s compliance with all elements of the treatment plan (including recovery-oriented activities, psychotherapy, and/or other psychosocial modalities)
- Abstinence from illicit drug use (including problematic alcohol and/or benzodiazepine use)
If treatment goals are not being achieved, the healthcare provider should re-evaluate the appropriateness of continuing the current treatment.
2.6 Unstable Patients
Healthcare providers will need to decide when they cannot appropriately provide further management for particular patients. For example, some patients may be abusing or dependent on various drugs, or unresponsive to psychosocial intervention such that the healthcare provider does not feel that he/she has the expertise to manage the patient. In such cases, the healthcare provider may want to assess whether to refer the patient to a specialist or more intensive behavioral treatment environment. Decisions should be based on a treatment plan established and agreed upon with the patient at the beginning of treatment.
Patients who continue to misuse, abuse, or divert buprenorphine products or other opioids should be provided with, or referred to, more intensive and structured treatment.
2.7 Discontinuing Treatment
The decision to discontinue therapy with buprenorphine and naloxone sublingual tablets after a period of maintenance should be made as part of a comprehensive treatment plan. Advise patients of the potential to relapse to illicit drug use following discontinuation of opioid agonist/partial agonist medication-assisted treatment. Taper patients to reduce the occurrence of withdrawal signs and symptoms [see Warnings and Precautions (5.7)].
2.8 Switching between Buprenorphine and Naloxone Sublingual Film and Buprenorphine and Naloxone Sublingual Tablets
Patients being switched between buprenorphine and naloxone sublingual tablets and buprenorphine and naloxone sublingual film should be started on the same dosage as the previously administered product. However, dosage adjustments may be necessary when switching between products. Because of the potentially greater relative bioavailability of buprenorphine and naloxone sublingual film compared to buprenorphine and naloxone sublingual tablets, patients switching from buprenorphine and naloxone sublingual tablets to buprenorphine and naloxone sublingual film should be monitored for over-medication. Those switching from buprenorphine and naloxone sublingual film to buprenorphine and naloxone sublingual tablets should be monitored for withdrawal or other indications of under dosing. In clinical studies, pharmacokinetics of buprenorphine and naloxone sublingual film was similar to the respective dosage strengths of buprenorphine and naloxone sublingual tablets; although not all doses and dose combinations met bioequivalence criteria.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Buprenorphine and naloxone sublingual tablets is contraindicated in patients with a history of hypersensitivity to buprenorphine or naloxone as serious adverse reactions, including anaphylactic shock, have been reported [see Warnings and Precautions (5.9)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Addiction, Abuse, and Misuse
Buprenorphine and naloxone sublingual tablets contain buprenorphine, a schedule III controlled substance that can be abused in a manner similar to other opioids, legal or illicit. Prescribe and dispense buprenorphine with appropriate precautions to minimize risk of misuse, abuse, or diversion, and ensure appropriate protection from theft, including in the home. Clinical monitoring appropriate to the patient’s level of stability is essential. Multiple refills should not be prescribed early in treatment or without appropriate patient follow-up visits [see Drug Abuse and Dependence (9.2)].
5.2 Risk of Life-Threatening Respiratory and Central Nervous System (CNS) Depression
Buprenorphine has been associated with life-threatening respiratory depression and death. Many, but not all, post-marketing reports regarding coma and death involved misuse by self-injection or were associated with the concomitant use of buprenorphine and benzodiazepines or other CNS depressant, including alcohol. Warn patients of the potential danger of self-administration of benzodiazepines or other CNS depressants while under treatment with buprenorphine and naloxone sublingual tablets [see Warnings and Precautions (5.3), Drug Interactions (7)].
Use buprenorphine and naloxone sublingual tablets with caution in patients with compromised respiratory function (e.g., chronic obstructive pulmonary disease, cor pulmonale, decreased respiratory reserve, hypoxia, hypercapnia, or pre-existing respiratory depression).
Opioids can cause sleep-related breathing disorders including central sleep apnea (CSA) and sleep-related hypoxemia. Opioid use increases the risk of CSA in a dose-dependent fashion. In patients who present with CSA, consider decreasing the opioid dosage using best practices for opioid taper [see Dosage and Administration (2.7)].
5.3 Managing Risks from Concomitant Use of Benzodiazepine or Other CNS Depressants
Concomitant use of buprenorphine and benzodiazepines or other CNS depressants increases the risk of adverse reactions including overdose and death. Medication-assisted treatment of opioid use disorder, however, should not be categorically denied to patients taking these drugs. Prohibiting or creating barriers to treatment can pose an even greater risk of morbidity and mortality due to the opioid use disorder alone.
As a routine part of orientation to buprenorphine treatment, educate patients about the risks of concomitant use of benzodiazepines, sedatives, opioid analgesics, and alcohol.
Develop strategies to manage use of prescribed or illicit benzodiazepines or other CNS depressants at initiation of buprenorphine treatment, or if it emerges as a concern during treatment. Adjustments to induction procedures and additional monitoring may be required. There is no evidence to support dose limitations or arbitrary caps of buprenorphine as a strategy to address benzodiazepine use in buprenorphine-treated patients. However, if a patient is sedated at the time of buprenorphine dosing, delay or omit the buprenorphine dose if appropriate.
Cessation of benzodiazepines or other CNS depressants is preferred in most cases of concomitant use. In some cases, monitoring in a higher level of care for taper may be appropriate. In others, gradually tapering a patient off of a prescribed benzodiazepine or other CNS depressant or decreasing to the lowest effective dose may be appropriate.
For patients in buprenorphine treatment, benzodiazepines are not the treatment of choice for anxiety or insomnia. Before co-prescribing benzodiazepines, ensure that patients are appropriately diagnosed and consider alternative medications and non-pharmacologic treatments to address anxiety or insomnia. Ensure that other healthcare providers prescribing benzodiazepines or other CNS depressants are aware of the patient’s buprenorphine treatment and coordinate care to minimize the risks associated with concomitant use.
In addition, take measures to confirm that patients are taking their medications as prescribed and are not diverting or supplementing with illicit drugs. Toxicology screening should test for prescribed and illicit benzodiazepines [see Drug Interactions (7)].
5.4 Unintentional Pediatric Exposure
Buprenorphine can cause severe, possibly fatal, respiratory depression in children who are accidentally exposed to it. Store buprenorphine-containing medications safely out of the sight and reach of children and destroy any unused medication appropriately [see Patient Counseling Information (17)].
5.5 Neonatal Opioid Withdrawal Syndrome
Neonatal opioid withdrawal syndrome (NOWS) is an expected and treatable outcome of prolonged use of opioids during pregnancy, whether that use is medically-authorized or illicit. Unlike opioid withdrawal syndrome in adults, NOWS may be life-threatening if not recognized and treated in the neonate. Healthcare professionals should observe newborns for signs of NOWS and manage accordingly [see Use in Specific Populations (8.1)].
Advise pregnant women receiving opioid addiction treatment with buprenorphine and naloxone sublingual tablets of the risk of neonatal opioid withdrawal syndrome and ensure that appropriate treatment will be available [see Use in Specific Populations (8.1)]. This risk must be balanced against the risk of untreated opioid addiction which often results in continued or relapsing illicit opioid use and is associated with poor pregnancy outcomes. Therefore, prescribers should discuss the importance and benefits of management of opioid addiction throughout pregnancy.
5.6 Adrenal Insufficiency
Cases of adrenal insufficiency have been reported with opioid use, more often following greater than one month of use. Presentation of adrenal insufficiency may include non-specific symptoms and signs including nausea, vomiting, anorexia, fatigue, weakness, dizziness, and low blood pressure. If adrenal insufficiency is suspected, confirm the diagnosis with diagnostic testing as soon as possible. If adrenal insufficiency is diagnosed, treat with physiologic replacement doses of corticosteroids. Wean the patient off of the opioid to allow adrenal function to recover and continue corticosteroid treatment until adrenal function recovers. Other opioids may be tried as some cases reported use of a different opioid without recurrence of adrenal insufficiency. The information available does not identify any particular opioids as being more likely to be associated with adrenal insufficiency.
5.7 Risk of Opioid Withdrawal with Abrupt Discontinuation
Buprenorphine is a partial agonist at the mu-opioid receptor and chronic administration produces physical dependence of the opioid-type, characterized by withdrawal signs and symptoms upon abrupt discontinuation or rapid taper. The withdrawal syndrome is typically milder than seen with full agonists and may be delayed in onset [see Drug Abuse and Dependence (9.3)]. When discontinuing buprenorphine and naloxone sublingual tablets, gradually taper the dosage [see Dosage and Administration (2.7)].
5.8 Risk of Hepatitis, Hepatic Events
Cases of cytolytic hepatitis and hepatitis with jaundice have been observed in individuals receiving buprenorphine in clinical trials and through post-marketing adverse event reports. The spectrum of abnormalities ranges from transient asymptomatic elevations in hepatic transaminases to case reports of death, hepatic failure, hepatic necrosis, hepatorenal syndrome, and hepatic encephalopathy. In many cases, the presence of pre-existing liver enzyme abnormalities, infection with hepatitis B or hepatitis C virus, concomitant usage of other potentially hepatotoxic drugs, and ongoing injecting drug use may have played a causative or contributory role. In other cases, insufficient data were available to determine the etiology of the abnormality. Withdrawal of buprenorphine has resulted in amelioration of acute hepatitis in some cases; however, in other cases no dose reduction was necessary. The possibility exists that buprenorphine had a causative or contributory role in the development of the hepatic abnormality in some cases. Liver function tests, prior to initiation of treatment is recommended to establish a baseline. Periodic monitoring of liver function during treatment is also recommended. A biological and etiological evaluation is recommended when a hepatic event is suspected. Depending on the case, buprenorphine and naloxone sublingual tablets may need to be carefully discontinued to prevent withdrawal signs and symptoms and a return by the patient to illicit drug use, and strict monitoring of the patient should be initiated.
5.9 Hypersensitivity Reactions
Cases of hypersensitivity to buprenorphine and naloxone containing products have been reported both in clinical trials and in the post-marketing experience. Cases of bronchospasm, angioneurotic edema, and anaphylactic shock have been reported. The most common signs and symptoms include rashes, hives, and pruritus. A history of hypersensitivity to buprenorphine or naloxone is a contraindication to the use of buprenorphine and naloxone sublingual tablets.
5.10 Precipitation of Opioid Withdrawal Signs and Symptoms
Because it contains naloxone, buprenorphine and naloxone sublingual tablet is highly likely to produce marked and intense withdrawal signs and symptoms if misused parenterally by individuals dependent on full opioid agonists such as heroin, morphine, or methadone. Because of the partial agonist properties of buprenorphine, buprenorphine and naloxone sublingual tablets may precipitate opioid withdrawal signs and symptoms in such persons if administered sublingually before the agonist effects of the opioid have subsided.
5.11 Risk of Overdose in Opioid Naïve Patients
There have been reported deaths of opioid naive individuals who received a 2 mg dose of buprenorphine as a sublingual tablet for analgesia. Buprenorphine and naloxone sublingual tablets are not appropriate as an analgesic.
5.12 Use in Patients with Impaired Hepatic Function
Buprenorphine/naloxone products are not recommended in patients with severe hepatic impairment and may not be appropriate for patients with moderate hepatic impairment. The doses of buprenorphine and naloxone in this fixed-dose combination product cannot be individually titrated, and hepatic impairment results in a reduced clearance of naloxone to a much greater extent than buprenorphine. Therefore, patients with severe hepatic impairment will be exposed to substantially higher levels of naloxone than patients with normal hepatic function. This may result in an increased risk of precipitated withdrawal at the beginning of treatment (induction) and may interfere with buprenorphine’s efficacy throughout treatment. In patients with moderate hepatic impairment, the differential reduction of naloxone clearance compared to buprenorphine clearance is not as great as in subjects with severe hepatic impairment. However, buprenorphine/naloxone products are not recommended for initiation of treatment (induction) in patients with moderate hepatic impairment due to the increased risk of precipitated withdrawal. Buprenorphine/naloxone products may be used with caution for maintenance treatment in patients with moderate hepatic impairment who have initiated treatment on a buprenorphine product without naloxone. However, patients should be carefully monitored and consideration given to the possibility of naloxone interfering with buprenorphine’s efficacy [see Use in Specific Populations (8.6)].
5.13 Impairment of Ability to Drive or Operate Machinery
Buprenorphine and naloxone sublingual tablets may impair the mental or physical abilities required for the performance of potentially dangerous tasks such as driving a car or operating machinery, especially during treatment induction and dose adjustment. Caution patients about driving or operating hazardous machinery until they are reasonably certain that buprenorphine and naloxone sublingual tablet therapy does not adversely affect his or her ability to engage in such activities.
5.14 Orthostatic Hypotension
Like other opioids, buprenorphine and naloxone sublingual tablets may produce orthostatic hypotension in ambulatory patients.
5.15 Elevation of Cerebrospinal Fluid Pressure
Buprenorphine, like other opioids, may elevate cerebrospinal fluid pressure and should be used with caution in patients with head injury, intracranial lesions, and other circumstances when cerebrospinal pressure may be increased. Buprenorphine can produce miosis and changes in the level of consciousness that may interfere with patient evaluation.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the labeling:
- Addiction, Abuse, and Misuse [see Warnings and Precautions (5.1)]
- Respiratory and CNS Depression [see Warnings and Precautions (5.2, 5.3)]
- Neonatal Opioid Withdrawal Syndrome [see Warnings and Precautions (5.5)]
- Adrenal Insufficiency [see Warnings and Precautions (5.6)]
- Opioid Withdrawal [see Warnings and Precautions (5.7, 5.10)]
- Hepatitis, Hepatic Events [see Warnings and Precautions (5.8)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.9)]
- Orthostatic Hypotension [see Warnings and Precautions (5.14)]
- Elevation of Cerebrospinal Fluid Pressure [see Warnings and Precautions (5.15)]
- Elevation of Intracholedochal Pressure [see Warnings and Precautions (5.16)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of buprenorphine and naloxone sublingual tablets was evaluated in 497 opioid-dependent subjects. The prospective evaluation of buprenorphine and naloxone sublingual tablets was supported by clinical trials using buprenorphine tablets without naloxone and other trials using buprenorphine sublingual solutions. In total, safety data were available from 3214 opioid-dependent subjects exposed to buprenorphine at doses in the range used in treatment of opioid addiction.
Few differences in adverse event profile were noted between buprenorphine and naloxone sublingual tablets and buprenorphine sublingual tablets or buprenorphine administered as a sublingual solution.
The following adverse events were reported to occur by at least 5% of patients in a 4-week study (Table 1).
Table 1. Adverse Events ≥5% by Body System and Treatment Group in a 4-week Study N (%) N (%) Body System / Adverse Event (COSTART Terminology) Buprenorphine and Naloxone Sublingual Tablets
16 mg/day
N=107Placebo
N=107Body as a Whole Asthenia 7 (6.5%) 7 (6.5%) Chills 8 (7.5%) 8 (7.5%) Headache 39 (36.4%) 24 (22.4%) Infection 6 (5.6%) 7 (6.5%) Pain 24 (22.4%) 20 (18.7%) Pain Abdomen 12 (11.2%) 7 (6.5%) Pain Back 4 (3.7%) 12 (11.2%) Withdrawal Syndrome 27 (25.2%) 40 (37.4%) Cardiovascular System Vasodilation 10 (9.3%) 7 (6.5%) Digestive System Constipation 13 (12.1%) 3 (2.8%) Diarrhea 4 (3.7%) 16 (15.0%) Nausea 16 (15.0%) 12 (11.2%) Vomiting 8 (7.5%) 5 (4.7%) Nervous System Insomnia 15 (14.0%) 17 (15.9%) Respiratory System Rhinitis 5 (4.7%) 14 (13.1%) Skin and Appendages Sweating 15 (14.0%) 11 (10.3%) The adverse event profile of buprenorphine was also characterized in the dose-controlled study of buprenorphine solution, over a range of doses in four months of treatment. Table 2 shows adverse events reported by at least 5% of subjects in any dose group in the dose-controlled study.
Table 2. Adverse Events (≥ 5%) by Body System and Treatment Group in a 16-week Study *Sublingual solution. Doses in this table cannot necessarily be delivered in tablet form, but for comparison purposes:
"Very low" dose (1 mg solution) would be less than a tablet dose of 2 mg
"Low" dose (4 mg solution) approximates a 6 mg tablet dose
"Moderate" dose (8 mg solution) approximates a 12 mg tablet dose
"High" dose (16 mg solution) approximates a 24 mg tablet doseBody System/ Adverse Event (COSTART Terminology) Buprenorphine Dose* Very Low*
(N=184)Low*
(N=180)Moderate*
(N=186)High*
(N=181)Total*
(N=731)N (%) N (%) N (%) N (%) N (%) Body as a Whole Abscess 9 (5%) 2(1%) 3 (2%) 2 (1%) 16 (2%) Asthenia 26 (14%) 28 (16%) 26 (14%) 24 (13%) 104 (14%) Chills 11 (6%) 12 (7%) 9 (5%) 10 (6%) 42 (6%) Fever 7 (4%) 2 (1%) 2 (1%) 10 (6%) 21 (3%) Flu Syndrome 4 (2%) 13 (7%) 19 (10%) 8 (4%) 44 (6%) Headache 51 (28%) 62 (34%) 54 (29%) 53 (29%) 220 (30%) Infection 32 (17%) 39 (22%) 38 (20%) 40 (22%) 149 (20%) Injury Accidental 5 (3%) 10 (6%) 5 (3%) 5 (3%) 25 (3%) Pain 47 (26%) 37 (21%) 49 (26%) 44 (24%) 177 (24%) Pain Back 18 (10%) 29 (16%) 28 (15%) 27 (15%) 102 (14%) Withdrawal Syndrome 45 (24%) 40 (22%) 41 (22%) 36 (20%) 162 (22%) Digestive System Constipation 10 (5%) 23 (13%) 23 (12%) 26 (14%) 82 (11%) Diarrhea 19 (10%) 8 (4%) 9 (5%) 4 (2%) 40 (5%) Dyspepsia 6 (3%) 10 (6%) 4 (2%) 4 (2%) 24 (3%) Nausea 12 (7%) 22 (12%) 23 (12%) 18 (10%) 75 (10%) Vomiting 8 (4%) 6 (3%) 10 (5%) 14 (8%) 38 (5%) Nervous System Anxiety 22 (12%) 24 (13%) 20 (11%) 25 (14%) 91 (12%) Depression 24 (13%) 16 (9%) 25 (13%) 18 (10%) 83 (11%) Dizziness 4 (2%) 9 (5%) 7 (4%) 11 (6%) 31 (4%) Insomnia 42 (23%) 50 (28%) 43 (23%) 51 (28%) 186 (25%) Nervousness 12 (7%) 11 (6%) 10 (5%) 13 (7%) 46 (6%) Somnolence 5 (3%) 13 (7%) 9 (5%) 11 (6%) 38 (5%) Respiratory System Cough Increase 5 (3%) 11 (6%) 6 (3%) 4 (2%) 26 (4%) Pharyngitis 6 (3%) 7 (4%) 6 (3%) 9 (5%) 28 (4%) Rhinitis 27 (15%) 16 (9%) 15 (8%) 21 (12%) 79 (11%) Skin and Appendages Sweat 23 (13%) 21 (12%) 20 (11%) 23 (12%) 87 (12%) Special Senses Runny Eyes 13 (7%) 9 (5%) 6 (3%) 6 (3%) 34 (5%) 6.2 Post-marketing Experience
The following adverse reactions have been identified during post approval use of buprenorphine/naloxone. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The most frequently reported post-marketing adverse event not observed in clinical trials was peripheral edema.
Serotonin syndrome: Cases of serotonin syndrome, a potentially life-threatening condition, have been reported during concomitant use of opioids with serotonergic drugs.
Adrenal insufficiency: Cases of adrenal insufficiency have been reported with opioid use, more often following greater than one month of use.
Anaphylaxis: Anaphylaxis has been reported with ingredients contained in buprenorphine and naloxone sublingual tablets.
Androgen deficiency: Cases of androgen deficiency have occurred with chronic use of opioids [see Clinical Pharmacology (12.2)].
Local reactions: Glossodynia, glossitis, oral mucosal erythema, oral hypoesthesia, and stomatitis
-
7 DRUG
INTERACTIONS
Table 3 Includes clinically significant drug interactions with buprenorphine and naloxone sublingual tablets
Table 3. Clinically Significant Drug Interactions Benzodiazepines or other Central Nervous System (CNS) Depressants Clinical Impact: Due to additive pharmacologic effects, the concomitant use of benzodiazepines or other CNS depressants, including alcohol, increases the risk of respiratory depression, profound sedation, coma, and death. Intervention: Cessation of benzodiazepines or other CNS depressants is preferred in most cases of concomitant use. In some cases, monitoring in a higher level of care for taper may be appropriate. In others, gradually tapering a patient off of a prescribed benzodiazepine or other CNS depressant or decreasing to the lowest effective dose may be appropriate.
Before co-prescribing benzodiazepines for anxiety or insomnia, ensure that patients are appropriately diagnosed and consider alternative medications and non-pharmacologic treatments. [see Warnings and Precautions (5.2, 5.3)].Examples: Alcohol, non-benzodiazepine sedatives/hypnotics, anxiolytics, tranquilizers, muscle relaxants, general anesthetics, antipsychotics, and other opioids. Inhibitors of CYP3A4 Clinical Impact: The concomitant use of buprenorphine and CYP3A4 inhibitors can increase the plasma concentration of buprenorphine, resulting in increased or prolonged opioid effects, particularly when an inhibitor is added after a stable dose of buprenorphine and naloxone sublingual tablet is achieved.
After stopping a CYP3A4 inhibitor, as the effects of the inhibitor decline, the buprenorphine plasma concentration will decrease [see Clinical Pharmacology (12.3)], potentially resulting in decreased opioid efficacy or a withdrawal syndrome in patients who had developed physical dependence to buprenorphine.Intervention: If concomitant use is necessary, consider dosage reduction of buprenorphine and naloxone sublingual tablets until stable drug effects are achieved. Monitor patients for respiratory depression and sedation at frequent intervals.
If a CYP3A4 inhibitor is discontinued, consider increasing the buprenorphine and naloxone sublingual tablets dosage until stable drug effects are achieved. Monitor for signs of opioid withdrawal.Examples: Macrolide antibiotics (e.g., erythromycin), azole-antifungal agents (e.g. ketoconazole), protease inhibitors (e.g., ritonavir) CYP3A4 Inducers Clinical Impact: The concomitant use of buprenorphine and CYP3A4 inducers can decrease the plasma concentration of buprenorphine [see Clinical Pharmacology (12.3)], potentially resulting in decreased efficacy or onset of a withdrawal syndrome in patients who have developed physical dependence to buprenorphine.
After stopping a CYP3A4 inducer, as the effects of the inducer decline, the buprenorphine plasma concentration will increase [see Clinical Pharmacology (12.3)], which could increase or prolong both therapeutic effects and adverse reactions and may cause serious respiratory depression.Intervention: If concomitant use is necessary, consider increasing the buprenorphine and naloxone sublingual tablets dosage until stable drug effects are achieved. Monitor for signs of opioid withdrawal.
If a CYP3A4 inducer is discontinued, consider buprenorphine and naloxone sublingual tablets dosage reduction and monitor for signs of respiratory depression.Examples: Rifampin, carbamazepine, phenytoin Antiretrovirals: Non-nucleoside reverse transcriptase inhibitors (NNRTIs) Clinical Impact: Non-nucleoside reverse transcriptase inhibitors (NNRTIs) are metabolized principally by CYP3A4. Efavirenz, nevirapine, and etravirine are known CYP3A inducers, whereas delavirdine is a CYP3A inhibitor. Significant pharmacokinetic interactions between NNRTIs (e.g., efavirenz and delavirdine) and buprenorphine have been shown in clinical studies, but these pharmacokinetic interactions did not result in any significant pharmacodynamic effects. Intervention: Patients who are on chronic buprenorphine and naloxone sublingual tablets treatment should have their dose monitored if NNRTIs are added to their treatment regimen. Examples: efavirenz, nevirapine, etravirine, delavirdine Antiretrovirals: Protease inhibitors (PIs) Clinical Impact: Studies have shown some antiretroviral protease inhibitors (PIs) with CYP3A4 inhibitory activity (nelfinavir, lopinavir/ritonavir, ritonavir) have little effect on buprenorphine pharmacokinetic and no significant pharmacodynamic effects. Other PIs with CYP3A4 inhibitory activity (atazanavir and atazanavir/ritonavir) resulted in elevated levels of buprenorphine and norbuprenorphine, and patients in one study reported increased sedation. Symptoms of opioid excess have been found in post-marketing reports of patients receiving buprenorphine and atazanavir with and without ritonavir concomitantly. Intervention: Monitor patients taking buprenorphine and naloxone sublingual tablets and atazanavir with and without ritonavir, and reduce dose of buprenorphine and naloxone sublingual tablets if warranted. Examples: atazanavir, ritonavir Antiretrovirals: Nucleoside reverse transcriptase inhibitors (NRTIs) Clinical Impact: Nucleoside reverse transcriptase inhibitors (NRTIs) do not appear to induce or inhibit the P450 enzyme pathway, thus no interactions with buprenorphine are expected. Intervention: None Serotonergic Drugs Clinical Impact: The concomitant use of opioids with other drugs that affect the serotonergic neurotransmitter system has resulted in serotonin syndrome. Intervention: If concomitant use is warranted, carefully observe the patient, particularly during treatment initiation and dose adjustment. Discontinue buprenorphine and naloxone sublingual tablets if serotonin syndrome is suspected. Examples: Selective serotonin reuptake inhibitors, serotonin and norepinephrine reuptake inhibitors, tricyclic antidepressants (TCAs), triptans, 5-HT3 receptor antagonists, drugs that affect the serotonin neurotransmitter system (e.g., mirtazapine, trazodone, tramadol), certain muscle relaxants (i.e., cyclobenzaprine, metaxalone), monoamine oxidase (MAO) inhibitors (those intended to treat psychiatric disorders and also others, such as linezolid and intravenous methylene blue). Monoamine Oxidase Inhibitors (MAOIs) Clinical Impact: MAOI interactions with opioids may manifest as serotonin syndrome or opioid toxicity (e.g., respiratory depression, coma). Intervention: The use of buprenorphine and naloxone sublingual tablets is not recommended for patients taking MAOIs or within 14 days of stopping such treatment. Examples: phenelzine, tranylcypromine, linezolid Muscle Relaxants Clinical Impact: Buprenorphine may enhance the neuromuscular blocking action of skeletal muscle relaxants and produce an increased degree of respiratory depression. Intervention: Monitor patients receiving muscle relaxants and buprenorphine and naloxone sublingual tablets for signs of respiratory depression that may be greater than otherwise expected and decrease the dosage of buprenorphine and naloxone sublingual tablets and/or the muscle relaxant as necessary. Diuretics Clinical Impact: Opioids can reduce the efficacy of diuretics by inducing the release of antidiuretic hormone. Intervention: Monitor patients for signs of diminished diuresis and/or effects on blood pressure and increase the dosage of the diuretic as needed. Anticholinergic Drugs Clinical Impact: The concomitant use of anticholinergic drugs may increase the risk of urinary retention and/or severe constipation, which may lead to paralytic ileus. Intervention: Monitor patients for signs of urinary retention or reduced gastric motility when buprenorphine and naloxone sublingual tablets are used concomitantly with anticholinergic drugs. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
The data on use of buprenorphine, one of the active ingredients in buprenorphine and naloxone sublingual tablets, in pregnancy, are limited; however, these data do not indicate an increased risk of major malformations specifically due to buprenorphine exposure. There are limited data from randomized clinical trials in women maintained on buprenorphine that were not designed appropriately to assess the risk of major malformations [see Data]. Observational studies have reported on congenital malformations among buprenorphine-exposed pregnancies, but were also not designed appropriately to assess the risk of congenital malformations specifically due to buprenorphine exposure [see Data]. The extremely limited data on sublingual naloxone exposure in pregnancy are not sufficient to evaluate a drug-associated risk.
Reproductive and developmental studies in rats and rabbits identified adverse events at clinically relevant and higher doses. Embryo-fetal death was also observed in both rats and rabbits administered buprenorphine during the period of organogenesis at doses approximately 6 and 0.3 times, respectively, the human sublingual dose of 16 mg/day of buprenorphine. Pre-and post-natal development studies in rats demonstrated increased neonatal deaths at 0.3 times and above and dystocia at approximately 3 times the human sublingual dose of 16 mg/day of buprenorphine. No clear teratogenic effects were seen when buprenorphine was administered during organogenesis with a range of doses equivalent to or greater than the human sublingual dose of 16 mg/day of buprenorphine. However, increases in skeletal abnormalities were noted in rats and rabbits administered buprenorphine daily during organogenesis at doses approximately 0.6 times and approximately equal to the human sublingual dose of 16 mg/day of buprenorphine, respectively. In a few studies, some events such as acephalus and omphalocele were also observed but these findings were not clearly treatment-related [see Data]. Based on animal data, advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Disease-associated maternal and embryo-fetal risk
Untreated opioid addiction in pregnancy is associated with adverse obstetrical outcomes such as low birth weight, preterm birth, and fetal death. In addition, untreated opioid addiction often results in continued or relapsing illicit opioid use.
Dose Adjustment during Pregnancy and the Postpartum Period
Dosage adjustments of buprenorphine may be required during pregnancy, even if the patient was maintained on a stable dose prior to pregnancy. Withdrawal signs and symptoms should be monitored closely and the dose adjusted as necessary.
Fetal/neonatal adverse reactions
Neonatal opioid withdrawal syndrome may occur in newborn infants of mothers who are receiving treatment with buprenorphine and naloxone sublingual tablets.
Neonatal opioid withdrawal syndrome presents as irritability, hyperactivity and abnormal sleep pattern, high pitched cry, tremor, vomiting, diarrhea, and/or failure to gain weight. Signs of neonatal withdrawal usually occur in the first days after birth. The duration and severity of neonatal opioid withdrawal syndrome may vary. Observe newborns for signs of neonatal opioid withdrawal syndrome and manage accordingly [see Warnings and Precautions (5.5)].
Opioid-dependent women on buprenorphine maintenance therapy may require additional analgesia during labor.
Studies have been conducted to evaluate neonatal outcomes in women exposed to buprenorphine during pregnancy. Limited data from trials, observational studies, case series, and case reports on buprenorphine use in pregnancy do not indicate an increased risk of major malformations specifically due to buprenorphine. Several factors may complicate the interpretation of investigations of the children of women who take buprenorphine during pregnancy, including maternal use of illicit drugs, late presentation for prenatal care, infection, poor compliance, poor nutrition, and psychosocial circumstances. Interpretation of data is complicated further by the lack of information on untreated opioid-dependent pregnant women, who would be the most appropriate group for comparison. Rather, women on another form of opioid medication-assisted treatment, or women in the general population are generally used as the comparison group. However, women in these comparison groups may be different from women prescribed buprenorphine-containing products with respect to maternal factors that may lead to poor pregnancy outcomes.
In a multicenter, double-blind, randomized, controlled trial [Maternal Opioid Treatment: Human Experimental Research (MOTHER)] designed primarily to assess neonatal opioid withdrawal effects, opioid-dependent pregnant women were randomized to buprenorphine (n=86) or methadone (n=89) treatment, with enrollment at an average gestational age of 18.7 weeks in both groups. A total of 28 of the 86 women in the buprenorphine group (33%) and 16 of the 89 women in the methadone group (18%) discontinued treatment before the end of pregnancy.
Among women who remained in treatment until delivery, there was no difference between buprenorphine-treated and methadone-treated groups in the number of neonates requiring NOWS treatment or in the peak severity of NOWS. Buprenorphine-exposed neonates required less morphine (mean total dose, 1.1 mg vs. 10.4 mg), had shorter hospital stays (10.0 days vs. 17.5 days), and shorter duration of treatment for NOWS (4.1 days vs. 9.9 days) compared to the methadone-exposed group. There were no differences between groups in other primary outcomes (neonatal head circumference) or secondary outcomes (weight and length at birth, preterm birth, gestational age at delivery, and 1-minute and 5-minute Apgar scores), or in the rates of maternal or neonatal adverse events. The outcomes among mothers who discontinued treatment before delivery and may have relapsed to illicit opioid use are not known. Because of the imbalance in discontinuation rates between the buprenorphine and methadone groups, the study findings are difficult to interpret.
The exposure margins listed below are based on body surface area comparisons (mg/m2) to the human sublingual dose of 16 mg buprenorphine via buprenorphine and naloxone sublingual tablets.
Effects on embryo-fetal development were studied in Sprague-Dawley rats and Russian white rabbits following oral (1:1) and intramuscular (IM) (3:2) administration of mixtures of buprenorphine and naloxone during the period of organogenesis. Following oral administration to rats, no teratogenic effects were observed at buprenorphine doses up to 250 mg/kg/day (estimated exposure approximately 150 times the human sublingual dose of 16 mg) in the presence of maternal toxicity (mortality). Following oral administration to rabbits, no teratogenic effects were observed at buprenorphine doses up to 40 mg/kg/day (estimated exposure approximately 50 times the human sublingual dose of 16 mg) in the absence of clear maternal toxicity. No definitive drug-related teratogenic effects were observed in rats and rabbits at IM doses up to 30 mg/kg/day (estimated exposure approximately 20 times and 35 times, respectively, the human sublingual dose of 16 mg). Maternal toxicity resulting in mortality was noted in these studies in both rats and rabbits. Acephalus was observed in one rabbit fetus from the low-dose group and omphalocele was observed in two rabbit fetuses from the same litter in the mid-dose group; no findings were observed in fetuses from the high-dose group. Maternal toxicity was seen in the high-dose group but not at the lower doses where the findings were observed. Following oral administration of buprenorphine to rats, dose-related post-implantation losses, evidenced by increases in the numbers of early resorptions with consequent reductions in the numbers of fetuses, were observed at doses of 10 mg/kg/day or greater (estimated exposure approximately 6 times the human sublingual dose of 16 mg). In the rabbit, increased post-implantation losses occurred at an oral dose of 40 mg/kg/day. Following IM administration in the rat and the rabbit, post-implantation losses, as evidenced by decreases in live fetuses and increases in resorptions, occurred at 30 mg/kg/day.
Buprenorphine was not teratogenic in rats or rabbits after IM or subcutaneous (SC) doses up to 5 mg/kg/day (estimated exposure was approximately 3 and 6 times, respectively, the human sublingual dose of 16 mg), after IV doses up to 0.8 mg/kg/day (estimated exposure was approximately 0.5 times and equal to, respectively, the human sublingual dose of 16 mg), or after oral doses up to 160 mg/kg/day in rats (estimated exposure was approximately 95 times the human sublingual dose of 16 mg) and 25 mg/kg/day in rabbits (estimated exposure was approximately 30 times the human sublingual dose of 16 mg). Significant increases in skeletal abnormalities (e.g., extra thoracic vertebra or thoraco-lumbar ribs) were noted in rats after SC administration of 1 mg/kg/day and up (estimated exposure was approximately 0.6 times the human sublingual dose of 16 mg), but were not observed at oral doses up to 160 mg/kg/day. Increases in skeletal abnormalities in rabbits after IM administration of 5 mg/kg/day (estimated exposure was approximately 6 times the human sublingual dose of 16 mg) in the absence of maternal toxicity or oral administration of 1 mg/kg/day or greater (estimated exposure was approximately equal to the human sublingual dose of 16 mg) were not statistically significant.
In rabbits, buprenorphine produced statistically significant pre-implantation losses at oral doses of 1 mg/kg/day or greater and post-implantation losses that were statistically significant at IV doses of 0.2 mg/kg/day or greater (estimated exposure approximately 0.3 times the human sublingual dose of 16 mg). No maternal toxicity was noted at doses causing post-implantation loss in this study.
Dystocia was noted in pregnant rats treated intramuscularly with buprenorphine from Gestation Day 14 through Lactation Day 21 at 5 mg/kg/day (approximately 3 times the human sublingual dose of 16 mg). Fertility, and pre- and postnatal development studies with buprenorphine in rats indicated increases in neonatal mortality after oral doses of 0.8 mg/kg/day and up (approximately 0.5 times the human sublingual dose of 16 mg), after IM doses of 0.5 mg/kg/day and up (approximately 0.3 times the human sublingual dose of 16 mg), and after SC doses of 0.1 mg/kg/day and up (approximately 0.06 times the human sublingual dose of 16 mg). An apparent lack of milk production during these studies likely contributed to the decreased pup viability and lactation indices. Delays in the occurrence of righting reflex and startle response were noted in rat pups at an oral dose of 80 mg/kg/day (approximately 50 times the human sublingual dose of 16 mg).
8.2 Lactation
Based on two studies in 13 lactating women maintained on buprenorphine treatment, buprenorphine and its metabolite norbuprenorphine were present in low levels in human milk and available data have not shown adverse reactions in breastfed infants. There are no data on the combination product buprenorphine/naloxone in breastfeeding, however oral absorption of naloxone is limited. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for buprenorphine and naloxone sublingual tablets and any potential adverse effects on the breastfed child from the drug or from the underlying maternal condition.
Advise breastfeeding women taking buprenorphine products to monitor the infant for increased drowsiness and breathing difficulties.
Data were consistent from two studies (N=13) of breastfeeding infants whose mothers were maintained on sublingual doses of buprenorphine ranging from 2.4 to 24 mg/day, showing that the infants were exposed to less than 1% of the maternal daily dose.
In a study of six lactating women who were taking a median sublingual buprenorphine dose of 0.29 mg/kg/day 5 to 8 days after delivery, breast milk provided a median infant dose of 0.42 mcg/kg/day of buprenorphine and 0.33 mcg/kg/day of norbuprenorphine, equal to 0.2% and 0.12%, respectively, of the maternal weight-adjusted dose (relative dose/kg (%) of norbuprenorphine was calculated from the assumption that buprenorphine and norbuprenorphine are equipotent).
Data from a study of seven lactating women who were taking a median sublingual buprenorphine dose of 7 mg/day an average of 1.12 months after delivery indicated that the mean milk concentrations (Cavg) of buprenorphine and norbuprenorphine were 3.65 mcg/L and 1.94 mcg/L, respectively. Based on the study data, and assuming milk consumption of 150 mL/kg/day, an exclusively breastfed infant would receive an estimated mean absolute infant dose (AID) of 0.55 mcg/kg/day of buprenorphine and 0.29 mcg/kg/day of norbuprenorphine, or a mean relative infant dose (RID) of 0.38% and 0.18%, respectively, of the maternal weight-adjusted dose.
8.3 Females and Males of Reproductive Potential
Chronic use of opioids may cause reduced fertility in females and males of reproductive potential. It is not known whether these effects on fertility are reversible [see Adverse Reactions (6.2), Clinical Pharmacology (12.2), Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of buprenorphine and naloxone sublingual tablets have not been established in pediatric patients. This product is not appropriate for the treatment of neonatal abstinence syndrome in neonates because it contains naloxone, an opioid antagonist.
8.5 Geriatric Use
Clinical studies of buprenorphine and naloxone sublingual tablets, buprenorphine and naloxone sublingual film, or buprenorphine sublingual tablets did not include sufficient numbers of subjects aged 65 and over to determine whether they responded differently than younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. Due to possible decreased hepatic, renal, or cardiac function and of concomitant disease or other drug therapy in geriatric patients, the decision to prescribe buprenorphine and naloxone sublingual tablets should be made cautiously in individuals 65 years of age or older and these patients should be monitored for signs and symptoms of toxicity or overdose.
8.6 Hepatic Impairment
The effect of hepatic impairment on the pharmacokinetics of buprenorphine and naloxone has been evaluated in a pharmacokinetic study. Both drugs are extensively metabolized in the liver. While no clinically significant changes have been observed in subjects with mild hepatic impairment; the plasma levels have been shown to be higher and half-life values have been shown to be longer for both buprenorphine and naloxone in subjects with moderate and severe hepatic impairment. The magnitude of the effects on naloxone are greater than that on buprenorphine in both moderately and severely impaired subjects. The difference in magnitude of the effects on naloxone and buprenorphine are greater in subjects with severe hepatic impairment than in subjects with moderate hepatic impairment, and therefore the clinical impact of these effects is likely to be greater in patients with severe hepatic impairment than in patients with moderate hepatic impairment. Buprenorphine/naloxone products should be avoided in patients with severe hepatic impairment and may not be appropriate for patients with moderate hepatic impairment [see Warnings and Precautions (5.12), Clinical Pharmacology (12.3)].
-
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
Buprenorphine and naloxone sublingual tablets contain buprenorphine, a Schedule III controlled substance under the Controlled Substances Act.
Under the Drug Addiction Treatment Act (DATA) codified at 21 U.S.C. 823(g), prescription use of this product in the treatment of opioid dependence is limited to healthcare providers who meet certain qualifying requirements, and who have notified the Secretary of Health and Human Services (HHS) of their intent to prescribe this product for the treatment of opioid dependence and have been assigned a unique identification number that must be included on every prescription.
9.2 Abuse
Buprenorphine, like morphine and other opioids, has the potential for being abused and is subject to criminal diversion. This should be considered when prescribing or dispensing buprenorphine in situations when the clinician is concerned about an increased risk of misuse, abuse, or diversion. Healthcare professionals should contact their state professional licensing board or state controlled substances authority for information on how to prevent and detect abuse or diversion of this product.
Patients who continue to misuse, abuse, or divert buprenorphine products or other opioids should be provided with, or referred to, more intensive and structured treatment.
Abuse of buprenorphine poses a risk of overdose and death. This risk is increased with the abuse of buprenorphine and alcohol and other substances, especially benzodiazepines.
The healthcare provider may be able to more easily detect misuse or diversion by maintaining records of medication prescribed including date, dose, quantity, frequency of refills, and renewal requests of medication prescribed.
Proper assessment of the patient, proper prescribing practices, periodic re-evaluation of therapy, and proper handling and storage of the medication are appropriate measures that help to limit abuse of opioid drugs.
9.3 Dependence
Buprenorphine is a partial agonist at the mu-opioid receptor and chronic administration produces physical dependence of the opioid-type, characterized by moderate withdrawal signs and symptoms upon abrupt discontinuation or rapid taper. The withdrawal syndrome is typically milder than seen with full agonists and may be delayed in onset [see Warnings and Precautions (5.7)].
Neonatal opioid withdrawal syndrome (NOWS) is an expected and treatable outcome of prolonged use of opioids during pregnancy [see Warnings and Precautions (5.5)].
-
10 OVERDOSAGE
The manifestations of acute overdose include pinpoint pupils, sedation, hypotension, respiratory depression, and death.
In the event of overdose, the respiratory and cardiac status of the patient should be monitored carefully. When respiratory or cardiac functions are depressed, primary attention should be given to the re-establishment of adequate respiratory exchange through provision of a patent airway and institution of assisted or controlled ventilation. Oxygen, IV fluids, vasopressors, and other supportive measures should be employed as indicated.
In the case of overdose, the primary management should be the re-establishment of adequate ventilation with mechanical assistance of respiration, if required. Naloxone may be of value for the management of buprenorphine overdose. Higher than normal doses and repeated administration may be necessary. The long duration of action of buprenorphine and naloxone sublingual tablets should be taken into consideration when determining the length of treatment and medical surveillance needed to reverse the effects of an overdose. Insufficient duration of monitoring may put patients at risk.
-
11 DESCRIPTION
Buprenorphine and naloxone sublingual tablet is an orange, round flat-faced beveled edge tablet, debossed with an alphanumeric word identifying the product strength. It contains buprenorphine HCl, a partial agonist at the mu-opioid receptor, and naloxone HCl dihydrate, an opioid receptor antagonist, at a ratio of 4:1 (ratio of free bases). It is intended for sublingual administration and is available in two dosage strengths, 2 mg buprenorphine with 0.5 mg naloxone and 8 mg buprenorphine with 2 mg naloxone. Each sublingual tablet also contains the following inactive ingredients: lactose monohydrate, povidone K29/32, acesulfame potassium, FD&C Yellow No.6 aluminum lake, natural lemon flavor 717297 (corn syrup solids, maltodextrin, modified starch, natural flavorings, tocopherol), citric acid anhydrous, trisodium citrate dihydrate, corn starch, mannitol, and magnesium stearate.
Chemically, buprenorphine HCl is (2S)-2-[17-Cyclopropylmethyl-4,5α-epoxy-3-hydroxy-6-methoxy-6α,14-ethano-14α-morphinan-7α-yl]-3,3-dimethylbutan-2-ol hydrochloride. It has the following chemical structure:

Buprenorphine HCl has the molecular formula C29H41NO4 HCl and the molecular weight is 504.10. It is a white or off-white crystalline powder, sparingly soluble in water, freely soluble in methanol, soluble in alcohol, and practically insoluble in cyclohexane.
Chemically, naloxone HCl dihydrate is 17-Allyl-4, 5 α -epoxy-3, 14-dihydroxymorphinan-6-one hydrochloride dihydrate. It has the following chemical structure:
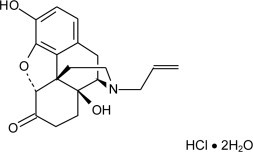
Naloxone hydrochloride dihydrate has the molecular formula C19H21NO4 HCl 2H20 and the molecular weight is 399.87. It is a white to slightly off-white powder and is freely soluble in water, soluble in alcohol, and practically insoluble in toluene and ether.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Buprenorphine and naloxone sublingual tablet contains buprenorphine and naloxone. Buprenorphine is a partial agonist at the mu-opioid receptor and an antagonist at the kappa-opioid receptor. Naloxone is an opioid antagonist and produces opioid withdrawal signs and symptoms in individuals physically dependent on full opioid agonists when administered parenterally.
12.2 Pharmacodynamics
Comparisons of buprenorphine to full opioid agonists such as methadone and hydromorphone suggest that sublingual buprenorphine produces typical opioid agonist effects which are limited by a ceiling effect.
In opioid-experienced subjects who were not physically dependent, acute sublingual doses of buprenorphine/naloxone tablets produced opioid agonist effects which reached a maximum between doses of 8/2 mg and 16/4 mg buprenorphine/naloxone.
Opioid agonist ceiling-effects were also observed in a double-blind, parallel-group, dose-ranging comparison of single doses of buprenorphine sublingual solution (1, 2, 4, 8, 16, or 32 mg), placebo, and a full agonist control at various doses. The treatments were given in ascending dose order at intervals of at least one week to 16 opioid-experienced subjects who were not physically dependent. Both active drugs produced typical opioid agonist effects. For all measures for which the drugs produced an effect, buprenorphine produced a dose-related response. However, in each case, there was a dose that produced no further effect. In contrast, the highest dose of the full agonist control always produced the greatest effects. Agonist objective rating scores remained elevated for the higher doses of buprenorphine (8 to 32 mg) longer than for the lower doses and did not return to baseline until 48 hours after drug administration. The onset of effects appeared more rapidly with buprenorphine than with the full agonist control, with most doses nearing peak effect after 100 minutes for buprenorphine compared to 150 minutes for the full agonist control.
Buprenorphine in IV (2, 4, 8, 12 and 16 mg) and sublingual (12 mg) doses have been administered to opioid-experienced subjects who were not physically dependent to examine cardiovascular, respiratory, and subjective effects at doses comparable to those used for treatment of opioid dependence. Compared to placebo, there were no statistically significant differences among any of the treatment conditions for blood pressure, heart rate, respiratory rate, O2 saturation, or skin temperature across time. Systolic BP was higher in the 8 mg group than placebo (3-hour AUC values). Minimum and maximum effects were similar across all treatments. Subjects remained responsive to low voice and responded to computer prompts. Some subjects showed irritability, but no other changes were observed.
The respiratory effects of sublingual buprenorphine were compared with the effects of methadone in a double-blind, parallel-group, dose-ranging comparison of single doses of buprenorphine sublingual solution (1, 2, 4, 8, 16, or 32 mg) and oral methadone (15, 30, 45, or 60 mg) in non-dependent, opioid-experienced volunteers. In this study, hypoventilation not requiring medical intervention was reported more frequently after buprenorphine doses of 4 mg and higher than after methadone. Both drugs decreased O2 saturation to the same degree.
Physiologic and subjective effects following acute sublingual administration of buprenorphine tablets and buprenorphine/naloxone tablets were similar at equivalent dose levels of buprenorphine. Naloxone had no clinically significant effect when administered by the sublingual route, although blood levels of the drug were measurable. Buprenorphine/naloxone, when administered sublingually to an opioid-dependent cohort, was recognized as an opioid agonist, whereas when administered intramuscularly, combinations of buprenorphine with naloxone produced opioid antagonist actions similar to naloxone. This finding suggests that the naloxone in buprenorphine/naloxone tablets may deter injection of buprenorphine/naloxone tablets by persons with active substantial heroin or other full mu-opioid dependence. However, clinicians should be aware that some opioid-dependent persons, particularly those with a low level of full mu-opioid physical dependence or those whose opioid physical dependence is predominantly to buprenorphine, abuse buprenorphine/naloxone combinations by the intravenous or intranasal route. In methadone-maintained patients and heroin-dependent subjects, IV administration of buprenorphine/naloxone combinations precipitated opioid withdrawal signs and symptoms and was perceived as unpleasant and dysphoric. In morphine-stabilized subjects, intravenously administered combinations of buprenorphine with naloxone produced opioid antagonist and withdrawal signs and symptoms that were ratio-dependent; the most intense withdrawal signs and symptoms were produced by 2:1 and 4:1 ratios, less intense by an 8:1 ratio.
Effects on the Endocrine System
Opioids inhibit the secretion of adrenocorticotropic hormone (ACTH), cortisol, and luteinizing hormone (LH) in humans [see Adverse Reactions (6.2)]. They also stimulate prolactin, growth hormone (GH) secretion, and pancreatic secretion of insulin and glucagon.
Chronic use of opioids may influence the hypothalamic-pituitary-gonadal axis, leading to androgen deficiency that may manifest as low libido, impotence, erectile dysfunction, amenorrhea, or infertility. The causal role of opioids in the clinical syndrome of hypogonadism is unknown because the various medical, physical, lifestyle, and psychological stressors that may influence gonadal hormone levels have not been adequately controlled for in studies conducted to date. Patients presenting with symptoms of androgen deficiency should undergo laboratory evaluation.
12.3 Pharmacokinetics
Plasma levels of buprenorphine and naloxone increased with the sublingual dose of buprenorphine and naloxone sublingual tablets (Table 4). There was wide inter-patient variability in the sublingual absorption of buprenorphine and naloxone, but within subjects the variability was low. Both Cmax and AUC of buprenorphine increased in a linear fashion with the increase in dose (in the range of 4 to 16 mg), although the increase was not directly dose-proportional.
Naloxone did not affect the pharmacokinetics of buprenorphine and both buprenorphine and naloxone sublingual tablets. At the three naloxone doses of 1, 2, and 4 mg, levels above the limit of quantitation (0.05 ng/mL) were not detected beyond 2 hours in seven of eight subjects. In one individual, at the 4 mg dose, the last measurable concentration was at 8 hours. Within each subject (for most of the subjects), across the doses there was a trend toward an increase in naloxone concentrations with increase in dose. Mean peak naloxone levels ranged from 0.11 to 0.28 ng/mL in the dose range of 1 to 4 mg.
Table 4. Pharmacokinetic parameters (Mean ± SD) of buprenorphine, norbuprenorphine, and naloxone following buprenorphine and naloxone sublingual tablets administration *Tmax is reported as median value with range PK Parameter Buprenorphine and Naloxone Sublingual Tablets Dose (mg) 2 mg/0.5 mg 8 mg/2 mg Buprenorphine Cmax (ng/mL) 0.780 ± 0.323 2.58 ± 1.10 Tmax (hr)* 1.50 (0.75-3.00) 1.50 (0.50-3.03) AUCinf (ng.hr/mL) 7.651 ± 2.650 25.31 ± 9.500 t½ (hr) 30.75 ± 15.04 31.94 ± 15.27 Norbuprenorphine Cmax (ng/mL) 0.293 ± 0.129 1.35 ± 0.977 Tmax (hr)* 1.25 (0.50-8.00) 1.25 (0.75-12.00) AUCinf (ng.hr/mL) 13.59 ± 4.887 52.84 ± 31.15 t½ (hr) 45.84 ± 15.85 44.76 ± 28.74 Naloxone Cmax (pg/mL) 51.3 ± 21.1 135 ± 57.3 Tmax (hr)* 0.75 (0.30-1.50) 0.75 (0.50-1.25) AUCinf (pg.hr/mL) 124.2 ± 52.49 374.6 ± 132.8 t½ (hr) 5.15 ± 5.28 7.65 ± 3. 99 Buprenorphine is approximately 96% protein bound, primarily to alpha and beta globulin.
Naloxone is approximately 45% protein bound, primarily to albumin.
Buprenorphine undergoes both N-dealkylation to norbuprenorphine and glucuronidation. The N-dealkylation pathway is mediated primarily by the CYP3A4. Norbuprenorphine, the major metabolite, can further undergo glucuronidation. Norbuprenorphine has been found to bind opioid receptors in vitro; however, it is not known whether norbuprenorphine contributes to the overall effect of buprenorphine and naloxone sublingual tablets. Naloxone undergoes direct glucuronidation to naloxone-3-glucuronide as well as N-dealkylation, and reduction of the 6-oxo group.
A mass balance study of buprenorphine showed complete recovery of radiolabel in urine (30%) and feces (69%) collected up to 11 days after dosing. Almost all of the dose was accounted for in terms of buprenorphine, norbuprenorphine, and two unidentified buprenorphine metabolites. In urine, most of buprenorphine and norbuprenorphine was conjugated (buprenorphine, 1% free and 9.4% conjugated; norbuprenorphine, 2.7% free and 11% conjugated). In feces, almost all of the buprenorphine and norbuprenorphine were free (buprenorphine, 33% free and 5% conjugated; norbuprenorphine, 21% free and 2% conjugated). When buprenorphine and naloxone sublingual tablets are administered sublingually, buprenorphine has a mean elimination half-life ranging from 24 to 42 hours and naloxone has a mean elimination half-life ranging from 2 to 12 hours.
CYP3A4 Inhibitors and Inducers
Buprenorphine has been found to be a CYP2D6 and CYP3A4 inhibitor and its major metabolite, norbuprenorphine, has been found to be a moderate CYP2D6 inhibitor in in-vitro studies employing human liver microsomes. However, the relatively low plasma concentrations of buprenorphine and norbuprenorphine resulting from therapeutic doses are not expected to raise significant drug-drug interaction concerns [see Drug Interactions (7)] .
In a pharmacokinetic study, the disposition of buprenorphine and naloxone were determined after administering a 2.0/0.5 mg buprenorphine and naloxone sublingual tablet in subjects with varied degrees of hepatic impairment as indicated by Child-Pugh criteria. The disposition of buprenorphine and naloxone in patients with hepatic impairment were compared to disposition in subjects with normal hepatic function.
In subjects with mild hepatic impairment, the changes in mean Cmax, AUC0-last, and half-life values of both buprenorphine and naloxone were not clinically significant. No dosing adjustment is needed in patients with mild hepatic impairment.
For subjects with moderate and severe hepatic impairment, mean Cmax, AUC0-last, and half-life values of both buprenorphine and naloxone were increased; the effects on naloxone are greater than that on buprenorphine (Table 5).
Table 5. Changes in Pharmacokinetic Parameters in Subjects with Moderate and Severe Hepatic Impairment Hepatic Impairment PK Parameters Increase in buprenorphine compared to healthy subjects Increase in naloxone compared to healthy subjects Moderate Cmax 8% 170% AUC0-last 64% 218% Half-life 35% 165% Severe Cmax 72% 1030% AUC0-last 181% 1302% Half-life 57% 122% The difference in magnitude of the effects on naloxone and buprenorphine are greater in subjects with severe hepatic impairment than subjects with moderate hepatic impairment [see Warnings and Precautions (5.12), Use in Specific Populations (8.6)].
In subjects with HCV infection but no sign of hepatic impairment, the changes in the mean Cmax, AUC0-last, and half-life values of buprenorphine and naloxone were not clinically significant in comparison to healthy subjects without HCV infection.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
A carcinogenicity study of buprenorphine/naloxone (4:1 ratio of the free bases) was performed in Alderley Park rats. Buprenorphine/naloxone was administered in the diet at doses of approximately 7, 31, and 123 mg/kg/day for 104 weeks (estimated exposure was approximately 4, 18, and 44 times the recommended human sublingual dose of 16/4 mg buprenorphine/naloxone based on buprenorphine AUC comparisons). A statistically significant increase in Leydig cell adenomas was observed in all dose groups. No other drug-related tumors were noted.
Carcinogenicity studies of buprenorphine were conducted in Sprague-Dawley rats and CD-1 mice. Buprenorphine was administered in the diet to rats at doses of 0.6, 5.5, and 56 mg/kg/day (estimated exposure was approximately 0.4, 3, and 35 times the recommended human daily sublingual dose of 16 mg on a mg/m2 basis) for 27 months. As in the buprenorphine/naloxone carcinogenicity study in rat, statistically significant dose-related increases in Leydig cell tumors occurred. In an 86-week study in CD-1 mice, buprenorphine was not carcinogenic at dietary doses up to 100 mg/kg/day (estimated exposure was approximately 30 times the recommended human daily sublingual dose of 16 mg on a mg/m2 basis).
The 4:1 combination of buprenorphine and naloxone was not mutagenic in a bacterial mutation assay (Ames test) using four strains of S. typhimurium and two strains of E. coli. The combination was not clastogenic in an in vitro cytogenetic assay in human lymphocytes or in an IV micronucleus test in the rat.
Buprenorphine was studied in a series of tests utilizing gene, chromosome, and DNA interactions in both prokaryotic and eukaryotic systems. Results were negative in yeast (S. cerevisiae) for recombinant, gene convertant, or forward mutations; negative in Bacillus subtilis “rec” assay, negative for clastogenicity in CHO cells, Chinese hamster bone marrow and spermatogonia cells, and negative in the mouse lymphoma L5178Y assay.
Results were equivocal in the Ames test: negative in studies in two laboratories, but positive for frame shift mutation at a high dose (5 mg/plate) in a third study. Results were positive in the Green-Tweets (E. coli) survival test, positive in a DNA synthesis inhibition test with testicular tissue from mice, for both in vivo and in vitro incorporation of [3H]thymidine, and positive in unscheduled DNA synthesis test using testicular cells from mice.
Dietary administration of buprenorphine in the rat at dose levels of 500 ppm or greater (equivalent to approximately 47 mg/kg/day or greater; estimated exposure approximately 28 times the recommended human daily sublingual dose of 16 mg on a mg/m2 basis) produced a reduction in fertility demonstrated by reduced female conception rates. A dietary dose of 100 ppm (equivalent to approximately 10 mg/kg/day; estimated exposure approximately 6 times the recommended human daily sublingual dose of 16 mg on a mg/m2 basis) had no adverse effect on fertility.
-
14 CLINICAL STUDIES
Clinical data on the safety and efficacy of buprenorphine and naloxone sublingual tablets were derived from studies of buprenorphine sublingual tablet formulations, with and without naloxone, and from studies of sublingual administration of a more bioavailable ethanolic solution of buprenorphine.
Buprenorphine and naloxone sublingual tablets were studied in 575 patients, buprenorphine (without naloxone) tablets in 1834 patients and buprenorphine sublingual solutions in 2470 patients. A total of 1270 women received buprenorphine in those clinical trials. Dosing recommendations are based on data from one trial of both tablet formulations and two trials of the ethanolic solution. All trials used buprenorphine in conjunction with psychosocial counseling as part of a comprehensive addiction treatment program. There were no clinical studies conducted to assess the efficacy of buprenorphine as the only component of treatment.
In a double-blind placebo- and active-controlled study, 326 heroin-addicted subjects were randomly assigned to either buprenorphine and naloxone sublingual tablets, 16/4 mg per day; buprenorphine sublingual tablets, 16 mg per day; or placebo sublingual tablets. For subjects randomized to either active treatment, dosing began with one 8 mg buprenorphine sublingual tablet on Day 1, followed by 16 mg (two 8 mg tablets) of buprenorphine sublingual tablets on Day 2. On Day 3, those randomized to receive buprenorphine and naloxone sublingual tablets were switched to the combination tablet. Subjects randomized to placebo received one placebo tablet on Day 1 and two placebo tablets per day thereafter for four weeks. Subjects were seen daily in the clinic (Monday through Friday) for dosing and efficacy assessments. Take-home doses were provided for weekends. Subjects were instructed to hold the medication under the tongue for approximately 5 to 10 minutes until completely dissolved. Subjects received counseling regarding HIV infection and up to one hour of individualized counseling per week. The primary study comparison was to assess the efficacy of buprenorphine and naloxone sublingual tablets and buprenorphine sublingual tablets individually against placebo sublingual tablet. The percentage of thrice-weekly urine samples that were negative for non-study opioids was statistically higher for both buprenorphine and naloxone sublingual tablets and buprenorphine sublingual tablets than for placebo sublingual tablets.
In a double-blind, double-dummy, parallel-group study comparing buprenorphine ethanolic solution to a full agonist active control, 162 subjects were randomized to receive the ethanolic sublingual solution of buprenorphine at 8 mg/day (a dose which is roughly comparable to a dose of 12/3 mg per day of buprenorphine and naloxone sublingual tablets or 12 mg per day of buprenorphine sublingual tablets), or two relatively low doses of active control, one of which was low enough to serve as an alternative to placebo, during a 3 to 10 day induction phase, a 16-week maintenance phase and a 7-week detoxification phase. Buprenorphine was titrated to maintenance dose by Day 3; active control doses were titrated more gradually.
Maintenance dosing continued through Week 17, and then medications were tapered by approximately 20% to 30% per week over Weeks 18 through 24, with placebo dosing for the last two weeks. Subjects received individual and/or group counseling weekly.
Based on retention in treatment and the percentage of thrice-weekly urine samples negative for non-study opioids, buprenorphine was more effective than the low dose of the control, in keeping heroin addicts in treatment and in reducing their use of opioids while in treatment. The effectiveness of buprenorphine, 8 mg per day, was similar to that of the moderate active control dose but equivalence was not demonstrated.
In a dose-controlled, double-blind, parallel-group, 16-week study, 731 subjects were randomized to receive one of four doses of buprenorphine ethanolic solution: 1 mg, 4 mg, 8 mg, and 16 mg. Buprenorphine was titrated to maintenance doses over 1 to 4 days and continued for 16 weeks. Subjects received at least one session of AIDS education and additional counseling ranging from one hour per month to one hour per week, depending on site.
Based on retention in treatment and the percentage of thrice-weekly urine samples negative for non-study opioids, the three highest tested doses were superior to the 1 mg dose. Therefore, this study showed that a range of buprenorphine doses may be effective. The 1 mg dose of buprenorphine sublingual solution can be considered to be somewhat lower than a 2 mg tablet dose. The other doses used in the study encompass a range of tablet doses from approximately 6 mg to approximately 24 mg.
-
16 HOW
SUPPLIED/STORAGE AND HANDLING
Buprenorphine and Naloxone Sublingual Tablet is an orange, round flat faced beveled edge tablet debossed with an alphanumeric word identifying the product strength (RP on one side for each strength, and “n2” and “n8” on 2 mg and 8 mg tablets, respectively), supplied in white HDPE bottles:
- NDC: 42858-601-03 (buprenorphine 2 mg and naloxone 0.5mg/sublingual tablet; content expressed in terms of free base, equivalent to 2.16 mg buprenorphine hydrochloride USP and 0.61 mg naloxone hydrochloride dihydrate USP) – 30 tablets per bottle
- NDC: 42858-602-03 (buprenorphine 8 mg and naloxone 2 mg/sublingual tablet; content expressed in terms of free base, equivalent to 8.64 mg buprenorphine hydrochloride USP and 2.44 mg naloxone hydrochloride dihydrate USP) – 30 tablets per bottle
Store at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature].
Store Buprenorphine and Naloxone Sublingual Tablets securely and dispose of properly [see Patient Counseling Information (17)].
-
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Medication Guide)
Because of the risks associated with accidental ingestion, misuse, and abuse, advise patients to store buprenorphine and naloxone sublingual tablets securely, out of sight and reach of children, and in a location not accessible by others, including visitors to the home [see Warnings and Precautions (5.1, 5.4), Drug Abuse and Dependence (9.2)]. Inform patients that leaving buprenorphine and naloxone sublingual tablets unsecured can pose a deadly risk to others in the home.
Advise patients and caregivers that when medicines are no longer needed, they should be disposed of promptly. Expired, unwanted, or unused buprenorphine and naloxone sublingual tablets should be disposed of by flushing the unused medication down the toilet if a drug take-back option is not readily available. Inform patients that they can visit www.fda.gov/drugdisposal for a complete list of medicines recommended for disposal by flushing, as well as additional information on disposal of unused medicines.
Before initiating treatment with buprenorphine and naloxone sublingual tablets, explain the points listed below to caregivers and patients. Instruct patients to read the Medication Guide each time buprenorphine and naloxone sublingual tablets are dispensed because new information may be available.
- Buprenorphine and naloxone sublingual tablets must be administered whole. Advise patients not to cut, chew, or swallow buprenorphine and naloxone sublingual tablets.
- Inform patients and caregivers that potentially fatal additive effects may occur if buprenorphine and naloxone sublingual tablets is used with benzodiazepines or other CNS depressants, including alcohol. Counsel patients that such medications should not be used concomitantly unless supervised by a health care provider [see Warnings and Precautions (5.2, 5.3), Drug Interactions (7)].
- Advise patients that buprenorphine and naloxone sublingual tablets contain an opioid that can be a target for people who abuse prescription medications or street drugs, to keep their tablets in a safe place, and to protect them from theft.
- Instruct patients to keep buprenorphine and naloxone sublingual tablets in a secure place, out of the sight and reach of children. Accidental or deliberate ingestion by a child may cause respiratory depression that can result in death. Advise patients to seek medical attention immediately if a child is exposed to buprenorphine and naloxone sublingual tablets.
- Inform patients that opioids could cause a rare but potentially life-threatening condition resulting from concomitant administration of serotonergic drugs. Warn patients of the symptoms of serotonin syndrome and to seek medical attention right away if symptoms develop. Instruct patients to inform their healthcare providers if they are taking, or plan to take, serotonergic medications [see Drug Interactions (7)].
- Inform patients that opioids could cause adrenal insufficiency, a potentially life-threatening condition. Adrenal insufficiency may present with non-specific symptoms and signs such as nausea, vomiting, anorexia, fatigue, weakness, dizziness, and low blood pressure. Advise patients to seek medical attention if they experience a constellation of these symptoms [see Warnings and Precautions (5.6)].
- Advise patients never to give buprenorphine and naloxone sublingual tablets to anyone else, even if he or she has the same signs and symptoms. It may cause harm or death.
- Advise patients that selling or giving away this medication is against the law.
- Caution patients that buprenorphine and naloxone sublingual tablets may impair the mental or physical abilities required for the performance of potentially dangerous tasks such as driving or operating machinery. Caution should be taken especially during drug induction and dose adjustment and until individuals are reasonably certain that buprenorphine therapy does not adversely affect their ability to engage in such activities [see Warnings and Precautions (5.13)].
- Advise patients not to change the dosage of buprenorphine and naloxone sublingual tablets without consulting their healthcare provider.
- Advise patients that if they miss a dose of buprenorphine and naloxone sublingual tablets that they should take it as soon as they remember. If it is almost time for the next dose, they should skip the missed dose and take the next dose at the regular time.
- Advise patients to take buprenorphine and naloxone sublingual tablets once a day.
- Inform patients that buprenorphine and naloxone sublingual tablets can cause drug dependence and that withdrawal signs and symptoms may occur when the medication is discontinued.
- Advise patients seeking to discontinue treatment with buprenorphine for opioid dependence to work closely with their healthcare provider on a tapering schedule and inform them of the potential to relapse to illicit drug use associated with discontinuation of opioid agonist/partial agonist medication-assisted treatment.
- Advise patients that, like other opioids, buprenorphine and naloxone sublingual tablets may produce orthostatic hypotension in ambulatory individuals [see Warnings and Precautions (5.14)].
- Advise patients to inform their healthcare provider if any other prescription medications, over-the-counter medications, or herbal preparations are prescribed or currently being used [see Drug Interactions (7)].
- Advise women that if they are pregnant while being treated with buprenorphine and naloxone sublingual tablets, the baby may have signs of withdrawal at birth and that withdrawal is treatable [see Warnings and Precautions (5.5), Use in Specific Populations (8.1)].
- Advise women who are breastfeeding to monitor the infant for drowsiness and difficulty breathing [see Use in Specific Populations (8.2)].
- Inform patients that chronic use of opioids may cause reduced fertility. It is not known whether these effects on fertility are reversible [see Use in Specific Populations (8.3)].
- Advise patients to inform their family members that, in the event of emergency, the treating healthcare provider or emergency room staff should be informed that the patient is physically dependent on an opioid and that the patient is being treated with buprenorphine and naloxone sublingual tablets.
Manufactured by:
Purdue Pharma L.P.
Stamford, CT 06901 -
MEDICATION GUIDE
MEDICATION GUIDE
Buprenorphine (byoo pre Nor feen) and Naloxone (nah LOX own) Sublingual Tablets (CIII)IMPORTANT:
Keep buprenorphine and naloxone sublingual tablets in a secure place away from children. Accidental use by a child is a medical emergency and can result in death. If a child accidentally uses buprenorphine and naloxone sublingual tablets, get emergency help right away.Read this Medication Guide that comes with buprenorphine and naloxone sublingual tablets before you start taking it and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking to your doctor. Talk to your doctor or pharmacist if you have questions about buprenorphine and naloxone sublingual tablets. Share the important information in this Medication Guide with members of your household. What is the most important information I should know about buprenorphine and naloxone sublingual tablets? - Buprenorphine is a medicine in buprenorphine and naloxone sublingual tablets that can cause serious and life‐threatening problems, especially if you take or use certain other medicines or drugs. Call your healthcare provider right away or get emergency help if you:
- feel faint or dizzy
- have mental changes such as confusion
- have slower breathing than you normally have
- have severe sleepiness
- have blurred vision
- have problems with coordination
- have slurred speech
- cannot think well or clearly
- have slowed reflexes
- have a high body temperature
- feel agitated
- have stiff muscles
- have trouble walking
- Do not switch from buprenorphine and naloxone sublingual tablets to other medicines that contain buprenorphine without talking with your doctor. The amount of buprenorphine in a dose of buprenorphine and naloxone sublingual tablets is not the same as the amount of buprenorphine in other medicines that contain buprenorphine. Your doctor will prescribe a starting dose of buprenorphine and naloxone sublingual tablets that may be different than other buprenorphine containing medicines you may have been taking.
- Buprenorphine and naloxone sublingual tablets contain an
opioid that can cause physical dependence.
- Do not stop taking buprenorphine and naloxone sublingual tablets without talking to your doctor. You could become sick with uncomfortable withdrawal signs and symptoms because your body has become used to this medicine
- Physical dependence is not the same as drug addiction
- Buprenorphine and naloxone sublingual tablets are not for occasional or “as needed” use
- An overdose, and even death, can happen if you take benzodiazepines, sedatives, tranquilizers, antidepressants, or alcohol while using buprenorphine and naloxone sublingual tablets. Ask your doctor what you should do if you are taking one of these.
- Call a doctor or get emergency help right away if you:
- Feel sleepy and uncoordinated
- Have blurred vision
- Have slurred speech
- Cannot think well or clearly
- Have slowed reflexes and breathing
- Do not inject (“shoot‐up”) or snort buprenorphine and naloxone
sublingual tablets.
- Injecting buprenorphine and naloxone sublingual tablets may cause life‐threatening infections and other serious health problems.
- Crushing and/or dissolving buprenorphine and naloxone sublingual tablets and then injecting it (“shooting up”) could cause serious precipitated withdrawal (sudden, serious, withdrawal symptoms such as pain, cramps, vomiting and diarrhea) in people who are physically dependent on other opioids.
- Snorting buprenorphine and naloxone sublingual tablets could cause precipitated withdrawal. In an emergency, have family members tell emergency department staff that you are physically dependent on an opioid and are being treated with buprenorphine and naloxone sublingual tablets.
What is buprenorphine and naloxone sublingual tablets? - Buprenorphine and naloxone sublingual tablets is a prescription medicine used to treat adults who are addicted to (dependent on) opioid drugs (either prescription or illegal) as part of a complete treatment program that also includes counseling and behavioral therapy.
Buprenorphine and naloxone sublingual tablets is a controlled substance (CIII) because it contains buprenorphine, which can be a target for people who abuse prescription medicines or street drugs. Keep your buprenorphine and naloxone sublingual tablets in a safe place to protect it from theft. Never give your buprenorphine and naloxone sublingual tablets to anyone else; it can cause death or harm them. Selling or giving away this medicine is against the law. - It is not known if buprenorphine and naloxone sublingual tablets is safe or effective in children.
Who should not take buprenorphine and naloxone sublingual tablets? Do not take buprenorphine and naloxone sublingual tablets if you are allergic to buprenorphine or naloxone. What should I tell my doctor before taking buprenorphine and naloxone sublingual tablets? Buprenorphine and naloxone sublingual tablets may not be right for you. Before taking buprenorphine and naloxone sublingual tablets, tell your doctor if you: - Have liver or kidney problems
- Have trouble breathing or lung problems
- Have an enlarged prostate gland (men)
- Have a head injury or brain problem
- Have problems urinating
- Have a curve in your spine that affects your breathing
- Have gallbladder problems
- Have adrenal gland problems
- Have Addison’s disease
- Have low thyroid (hypothyroidism)
- Have a history of alcoholism
- Have mental problems such as hallucinations (seeing or hearing things that are not there)
- Have any other medical condition
- Are pregnant or plan to become pregnant. If you take buprenorphine and naloxone sublingual tablets while pregnant, your baby may have signs of opioid withdrawal at birth. Neonatal opioid withdrawal syndrome (NOWS) is an expected and treatable outcome of prolonged use of opioids during pregnancy. Talk to your doctor if you are pregnant or plan to become pregnant.
- Are breastfeeding or plan to breastfeed. Buprenorphine and naloxone can pass into your milk and may harm your baby. Talk to your doctor about the best way to feed your baby if you take buprenorphine and naloxone sublingual tablets. Monitor your baby for increased sleepiness and breathing problems.
Tell your doctor about all the medicines you take, including prescription and over‐the‐counter medicines, vitamins, and herbal supplements. Buprenorphine and naloxone sublingual tablets may affect the way other medicines work, and other medicines may affect how buprenorphine and naloxone sublingual tablets work. Some medicines may cause serious or life‐threatening medical problems when taken with buprenorphine and naloxone sublingual tablets. Sometimes the doses of certain medicines and buprenorphine and naloxone sublingual tablets may need to be changed if used together. Do not take any medicine while using buprenorphine and naloxone sublingual tablets until you have talked with your doctor. Your doctor will tell you if it is safe to take other medicines while you are taking buprenorphine and naloxone sublingual tablets. Be especially careful about taking other medicines that may make you sleepy, such as muscle relaxants, pain medicines, tranquilizers, antidepressant medicines, sleeping pills, anxiety medicines, or antihistamines. Know the medicines you take. Keep a list of them to show your doctor or pharmacist each time you get a new medicine. How should I take buprenorphine and naloxone sublingual tablets? - Always take buprenorphine and naloxone sublingual tablets exactly as your doctor tells you. Your doctor may change your dose after seeing how it affects you. Do not change your dose unless your doctor tells you to change it.
- Do not take buprenorphine and naloxone sublingual tablets more often than prescribed by your doctor.
- If you are prescribed a dose of 2 or more buprenorphine
and naloxone sublingual tablets at the same time:
- Ask your doctor for instructions on the right way to take buprenorphine and naloxone sublingual tablets
- Follow the same instructions every time you take a dose of buprenorphine and naloxone sublingual tablets
- Put the tablets under your tongue. Let them dissolve completely.
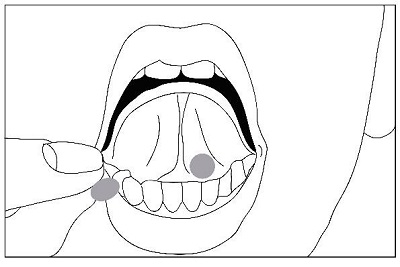
- While buprenorphine and naloxone sublingual tablet is dissolving, do not chew or swallow the tablet because the medicine will not work as well.
- Talking while the tablet is dissolving can affect how well the medicine in buprenorphine and naloxone sublingual tablets is absorbed.
- If you miss a dose of buprenorphine and naloxone sublingual tablets, take your medicine when you remember. If it is almost time for your next dose, skip the missed dose and take the next dose at your regular time. Do not take 2 doses at the same time unless your doctor tells you to. If you are not sure about your dosing, call your doctor.
- Do not stop taking buprenorphine and naloxone sublingual tablets suddenly. You could become sick and have withdrawal symptoms because your body has become used to the medicine. Physical dependence is not the same as drug addiction. Your doctor can tell you more about the differences between physical dependence and drug addiction. To have fewer withdrawal symptoms, ask your doctor how to stop using buprenorphine and naloxone sublingual tablets the right way.
- If you take too much buprenorphine and naloxone sublingual tablets or overdose, call Poison Control or get emergency medical help right away.
What should I avoid while taking buprenorphine and naloxone sublingual tablets? - Do not drive, operate heavy machinery, or perform any other dangerous activities until you know how this medication affects you. Buprenorphine can cause drowsiness and slow reaction times. This may happen more often in the first few weeks of treatment when your dose is being changed, but can also happen if you drink alcohol or take other sedative drugs when you take buprenorphine and naloxone sublingual tablets.
- You should not drink alcohol while using buprenorphine and naloxone sublingual tablets, as this can lead to loss of consciousness or even death.
What are the possible side effects of buprenorphine and naloxone sublingual tablets? Buprenorphine and naloxone sublingual tablets can cause serious side effects including: - See “What is the most important information I should know about buprenorphine and naloxone sublingual tablets?”
- Respiratory problems. You have a higher risk of death and coma if you take buprenorphine and naloxone sublingual tablets with other medicines, such as benzodiazepines.
- Sleepiness, dizziness, and problems with coordination
- Dependency or abuse
- Liver problems. Call your doctor right away if you notice any of these signs of liver problems: Your skin or the white part of your eyes turning yellow (jaundice), urine turning dark, stools turning light in color, you have less of an appetite, or you have stomach (abdominal) pain or nausea. Your doctor should do tests before you start taking and while you take buprenorphine and naloxone sublingual tablets.
- Allergic reaction. You may have a rash, hives, swelling of the face, wheezing, or a loss of blood pressure and consciousness. Call a doctor or get emergency help right away.
- Opioid withdrawal. This can include: shaking, sweating more than normal, feeling hot or cold more than normal, runny nose, watery eyes, goose bumps, diarrhea, vomiting, and muscle aches. Tell your doctor if you develop any of these symptoms.
- Decrease in blood pressure. You may feel dizzy if you get up too fast from sitting or lying down.
- Nausea
- Vomiting
- Drug withdrawal syndrome
- Headache
- Sweating
- Numb mouth
- Constipation
- Swollen and/or painful tongue
- The inside of your mouth is more red than normal
- Intoxication (feeling lightheaded or drunk)
- Disturbance in attention
- Irregular heart beat (palpitations)
- Decrease in sleep (insomnia)
- Blurred vision
- Back pain
- Fainting
- Dizziness
- Sleepiness
Tell your doctor about any side effect that bothers you or that does not go away. These are not all the possible side effects of buprenorphine and naloxone sublingual tablets. For more information, ask your doctor or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1‐800‐FDA‐1088. How should I store buprenorphine and naloxone sublingual tablets? - Store buprenorphine and naloxone sublingual tablets at room temperature between 68°F to77°F (20°C to 25°C).
- Keep buprenorphine and naloxone sublingual tablets in a safe place, out of the sight and reach of children.
- Dispose of unused buprenorphine and naloxone sublingual tablets as soon as you no longer need them.
- Dispose of expired, unwanted, or unused buprenorphine and naloxone sublingual tablets by promptly flushing down the toilet, if a drug take-back option is not readily available. Visit www.fda.gov/drugdisposal for additional information on disposal of unused medicines.
General information about the safe and effective use of buprenorphine and naloxone sublingual tablets. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not take buprenorphine and naloxone sublingual tablets for a condition for which it was not prescribed. Do not give buprenorphine and naloxone sublingual tablets to other people, even if they have the same symptoms you have. It may harm them and it is against the law. This Medication Guide summarizes the most important information about buprenorphine and naloxone sublingual tablets. If you would like more information, talk to your doctor or pharmacist. You can ask your doctor or pharmacist for information that is written for health professionals. For more information call Rhodes Pharmaceuticals L.P. at 1-888-827-0616. What are the ingredients in Buprenorphine and Naloxone Sublingual Tablets? Active Ingredients: buprenorphine and naloxone Inactive Ingredients: lactose monohydrate, povidone K29/32, acesulfame potassium, FD&C Yellow No.6 aluminum lake, natural lemon flavor 717297 (corn syrup solids, maltodextrin, modified starch, natural flavorings, tocopherol), citric acid anhydrous, trisodium citrate dihydrate, corn starch, mannitol, and magnesium stearate. Manufactured by:
Purdue Pharma L.P.
Stamford, CT 06901Marketed by:
Rhodes Pharmaceuticals L.P.
Coventry, RI 02816This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 01/2020 - PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
BUPRENORPHINE AND NALOXONE
buprenorphine and naloxone tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 42858-601 Route of Administration SUBLINGUAL DEA Schedule CIII Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BUPRENORPHINE HYDROCHLORIDE (UNII: 56W8MW3EN1) (BUPRENORPHINE - UNII:40D3SCR4GZ) BUPRENORPHINE 2 mg NALOXONE HYDROCHLORIDE DIHYDRATE (UNII: 5Q187997EE) (NALOXONE - UNII:36B82AMQ7N) NALOXONE 0.5 mg Inactive Ingredients Ingredient Name Strength Lactose monohydrate (UNII: EWQ57Q8I5X) Povidone K30 (UNII: U725QWY32X) Acesulfame Potassium (UNII: 23OV73Q5G9) Anhydrous Citric Acid (UNII: XF417D3PSL) Trisodium Citrate Dihydrate (UNII: B22547B95K) Mannitol (UNII: 3OWL53L36A) Magnesium Stearate (UNII: 70097M6I30) Corn syrup (UNII: 9G5L16BK6N) Maltodextrin (UNII: 7CVR7L4A2D) .Alpha.-Tocopherol (UNII: H4N855PNZ1) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) STARCH, CORN (UNII: O8232NY3SJ) Product Characteristics Color ORANGE Score no score Shape ROUND Size 6mm Flavor Imprint Code RP;n2 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 42858-601-03 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 04/13/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA205601 04/13/2020 BUPRENORPHINE AND NALOXONE
buprenorphine and naloxone tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 42858-602 Route of Administration SUBLINGUAL DEA Schedule CIII Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BUPRENORPHINE HYDROCHLORIDE (UNII: 56W8MW3EN1) (BUPRENORPHINE - UNII:40D3SCR4GZ) BUPRENORPHINE 8 mg NALOXONE HYDROCHLORIDE DIHYDRATE (UNII: 5Q187997EE) (NALOXONE - UNII:36B82AMQ7N) NALOXONE 2 mg Inactive Ingredients Ingredient Name Strength Lactose monohydrate (UNII: EWQ57Q8I5X) Povidone K30 (UNII: U725QWY32X) Acesulfame Potassium (UNII: 23OV73Q5G9) Anhydrous Citric Acid (UNII: XF417D3PSL) Trisodium Citrate Dihydrate (UNII: B22547B95K) Mannitol (UNII: 3OWL53L36A) Magnesium Stearate (UNII: 70097M6I30) Corn syrup (UNII: 9G5L16BK6N) Maltodextrin (UNII: 7CVR7L4A2D) .Alpha.-Tocopherol (UNII: H4N855PNZ1) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) STARCH, CORN (UNII: O8232NY3SJ) Product Characteristics Color ORANGE Score no score Shape ROUND Size 11mm Flavor Imprint Code RP;n8 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 42858-602-03 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 04/13/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA205601 04/13/2020 Labeler - Rhodes Parmaceuticals L.P. (831928986) Establishment Name Address ID/FEI Business Operations Purdue Pharmaceuticals L.P. 132080875 MANUFACTURE(42858-601, 42858-602) Establishment Name Address ID/FEI Business Operations Halo Pharmaceutical Inc. 829609168 MANUFACTURE(42858-601, 42858-602)


© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.