PACERONE- amiodarone hydrochloride tablet
Pacerone by
Drug Labeling and Warnings
Pacerone by is a Prescription medication manufactured, distributed, or labeled by Upsher-Smith Laboratories, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PACERONE® TABLETS safely and effectively. See full prescribing information for PACERONE® TABLETS.
PACERONE® (amiodarone hydrochloride) tablets, for oral use
Initial U.S. Approval: 1985WARNING: PULMONARY, HEPATIC, and CARDIAC TOXICITY
See full prescribing information for complete boxed warning.
- Reserve Pacerone® for patients with the indicated life-threatening arrhythmias because its use is accompanied by substantial toxicity, some also life-threatening. Utilize alternative agents first. (1)
- Pacerone®’s life-threatening toxicities include pulmonary (5.2), hepatic (5.3), and proarrhythmic (5.4).
- Initiate under hospital or specialist supervision. (5)
INDICATIONS AND USAGE
Pacerone® is an antiarrhythmic indicated for:
- Recurrent ventricular fibrillation. (1)
- Recurrent hemodynamically unstable ventricular tachycardia. (1)
DOSAGE AND ADMINISTRATION
Initiate treatment with a loading doses of 800 to 1600 mg/day until initial therapeutic response occurs (usually 1 to 3 weeks). Once adequate arrhythmia control is achieved, or if side effects become prominent, reduce Pacerone® dose to 600 to 800 mg/day for one month and then to the maintenance dose, usually 400 mg/day. (2)
DOSAGE FORMS AND STRENGTHS
Tablets, 400 mg. (3)
CONTRAINDICATIONS
Pacerone® is contraindicated in patients with (4):
- Cardiogenic shock.
- Sick sinus syndrome, second- or third-degree AV block, bradycardia leading to syncope without a functioning pacemaker.
- Known hypersensitivity to the drug or any of its components.
WARNINGS AND PRECAUTIONS
- Persistence of Adverse Effects: Adverse reactions and drug interaction can persist for several weeks following discontinuation. (5.1)
- Impaired Vision: Corneal microdeposits (common; reversible), optic neuropathy/neuritis (rare; may lead to blindness). (5.5)
- Thyroid Abnormalities: Hyperthyroidism or hypothyroidism. (5.6)
ADVERSE REACTIONS
The most common reactions (>1%) leading to discontinuation of Pacerone® include pulmonary toxicity, paroxysmal ventricular tachycardia, congestive heart failure, and elevation of liver enzymes. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Upsher-Smith Laboratories, LLC at 1-855-899-9180 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Avoid coadministration of amiodarone with other antiarrhythmics and drugs known to prolong the QT interval. (7)
- Amiodarone is a substrate for CYP3A and CYP2C8, so inhibitors and inducers affect amiodarone exposure. (7)
- Amiodarone inhibits P-glycoprotein and CYP1A2, CYP2C9, CYP2D6, and CYP3A, increasing exposure to other drugs. (7)
USE IN SPECIFIC POPULATIONS
- Pregnancy: May cause fetal harm. (8.1)
- Lactation: Breastfeeding not recommended. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 11/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: PULMONARY, HEPATIC and CARDIAC TOXICITY
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Persistence of Adverse Effects
5.2 Pulmonary Toxicity
5.3 Hepatic Injury
5.4 Worsened Arrhythmia
5.5 Visual Impairment and Loss of Vision
5.6 Thyroid Abnormalities
5.7 Bradycardia
5.8 Implantable Cardiac Devices
5.9 Fetal Toxicity
5.10 Peripheral Neuropathy
5.11 Photosensitivity and Skin Discoloration
5.12 Surgery
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: PULMONARY, HEPATIC and CARDIAC TOXICITY
- Pacerone® is intended for use only in patients with the indicated life-threatening arrhythmias because its use is accompanied by substantial toxicity [see Indications and Usage (1)].
- Pacerone® can cause pulmonary toxicity (hypersensitivity pneumonitis or interstitial/alveolar pneumonitis) that has resulted in clinically manifest disease at rates as high as 17% in some series of patients. Pulmonary toxicity has been fatal about 10% of the time. Obtain a baseline chest X-ray and pulmonary-function tests, including diffusion capacity, when Pacerone® therapy is initiated. Repeat history, physical exam, and chest X-ray every 3 to 6 months [see Warnings andPrecautions 5.2)].
- Pacerone® can cause hepatoxicity, which can be fatal. Obtain baseline and periodic liver transaminases and discontinue or reduce dose if the increase exceeds three times normal, or doubles in a patient with an elevated baseline. Discontinue Pacerone® if the patient experiences signs or symptoms of clinical liver injury [see Warnings andPrecautions (5.3)].
- Pacerone® can exacerbate arrhythmias. Initiate Pacerone® in a clinical setting where continuous electrocardiograms and cardiac resuscitation are available [see Warnings and Precautions (5.4)].
-
1 INDICATIONS AND USAGE
Pacerone® is indicated for the treatment of documented, life-threatening recurrent ventricular fibrillation and life-threatening recurrent hemodynamically unstable tachycardia in adults who have not responded to adequate doses of other available antiarrhythmics or when alternative agents cannot be tolerated.
-
2 DOSAGE AND ADMINISTRATION
Dosage must be individualized based on severity of arrhythmia and response. Use the lowest effective dose. Obtain baseline chest x-ray, pulmonary function tests, thyroid function tests, and liver aminotransferases. Correct hypokalemia, hypomagnesemia, and hypocalcemia before initiating treatment
Recommended Dosage:
Initiate treatment with a loading doses of 800 to 1600 mg/day until initial therapeutic response occurs (usually 1 to 3 weeks). Once adequate arrhythmia control is achieved, or if side effects become prominent, reduce Pacerone® dose to 600 to 800 mg/day for one month and then to the maintenance dose, usually 400 mg/day.
Administration:
Administer Pacerone® consistently with regard to meals [see Clinical Pharmacology (12.3)]. Administration of Pacerone® in divided doses with meals is suggested for total daily doses of 1000 mg or higher, or when gastrointestinal intolerance occurs.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Persistence of Adverse Effects
Because of the long half-life of amiodarone (15 to 142 days) and its active metabolite desethylamiodarone (14 to 75 days), adverse reactions and drug interactions can persist for several weeks following amiodarone discontinuation [see Clinical Pharmacology(12.3)].
5.2 Pulmonary Toxicity
Pacerone® may cause a clinical syndrome of cough and progressive dyspnea accompanied by functional, radiographic, gallium-scan, and pathological data consistent with pulmonary toxicity. Pulmonary toxicity secondary to Pacerone® may result from either indirect or direct toxicity as represented by hypersensitivity pneumonitis (including eosinophilic pneumonia) or interstitial/alveolar pneumonitis, respectively. Rates of pulmonary toxicity have been reported to be as high as 17% and is fatal in about 10% of cases. Obtain a baseline chest X-ray and pulmonary-function tests, including diffusion capacity, when Pacerone® therapy is initiated. Repeat history, physical exam, and chest X-ray every 3 to 6 months or if symptoms occur. Consider alternative antiarrhythmic therapy if the patient experiences signs or symptoms of pulmonary toxicity. Prednisone 40 to 60 mg/day tapered over several weeks may be helpful in treating pulmonary toxicity.
Adult Respiratory Distress Syndrome (ARDS)
Postoperatively, occurrences of ARDS have been reported in patients receiving Pacerone® therapy who have undergone either cardiac or noncardiac surgery. Although patients usually respond well to vigorous respiratory therapy, in rare instances the outcome has been fatal.
5.3 Hepatic Injury
Asymptomatic elevations of hepatic enzyme levels are seen frequently, but Pacerone® can cause life-threatening hepatic injury. Histology has resembled that of alcoholic hepatitis or cirrhosis. Obtain baseline and periodic liver transaminases. If transaminases exceed three times normal, or doubles in a patient with an elevated baseline, discontinue or reduce dose of Pacerone®, obtain follow-up tests and treat appropriately.
5.4 Worsened Arrhythmia
Pacerone® can exacerbate the presenting arrhythmia in about 2 to 5% of patients or cause new ventricular fibrillation, incessant ventricular tachycardia, increased resistance to cardioversion, and polymorphic ventricular tachycardia associated with QTc prolongation (Torsade de Pointes [TdP]).
Correct hypokalemia, hypomagnesemia, and hypocalcemia before initiating treatment with Pacerone®, as these disorders can exaggerate the degree of QTc prolongation and increase the potential for TdP. Give special attention to electrolyte and acid-base balance in patients experiencing severe or prolonged diarrhea or receiving drugs affecting electrolyte levels, such as diuretics, laxatives, systemic corticosteroids, or amphotericin B.
5.5 Visual Impairment and Loss of Vision
Optic Neuropathy and Optic Neuritis
Cases of optic neuropathy and optic neuritis, usually resulting in visual impairment and sometimes permanent blindness, have been reported in patients treated with amiodarone and may occur at any time during therapy. If symptoms of visual impairment appear, such as changes in visual acuity and decreases in peripheral vision, consider discontinuing Pacerone® and promptly refer for ophthalmic examination. Regular ophthalmic examination, including funduscopy and slit-lamp examination, is recommended during administration of Pacerone®[see Adverse Reactions (6.1)].
Corneal Microdeposits
Corneal microdeposits appear in the majority of adults treated with Pacerone®. They are usually discernible only by slit-lamp examination, but give rise to symptoms such as visual halos or blurred vision in as many as 10% of patients. Corneal microdeposits are reversible upon reduction of dose or termination of treatment. Asymptomatic microdeposits alone are not a reason to reduce dose or discontinue treatment [see Adverse Reactions (6.1)].
5.6 Thyroid Abnormalities
Pacerone® inhibits peripheral conversion of thyroxine (T4) to triiodothyronine (T3) and may cause increased thyroxine levels, decreased T3 levels, and increased levels of inactive reverse T3 (rT3) in clinically euthyroid patients. Pacerone® can cause either hypothyroidism (reported in up to 10% of patients) or hyperthyroidism (occurring in about 2% of patients). Monitor thyroid function prior to treatment and periodically thereafter, particularly in elderly patients, and in any patient with a history of thyroid nodules, goiter, or other thyroid dysfunction.
Hyperthyroidism may induce arrhythmia breakthrough. If any new signs of arrhythmia appear, the possibility of hyperthyroidism should be considered. Antithyroid drugs, β-adrenergic blockers, temporary corticosteroid therapy may be necessary to treat the symptoms of hyperthyroidism. The action of antithyroid drugs may be delayed in amiodarone-induced thyrotoxicosis because of substantial quantities of preformed thyroid hormones stored in the gland. Radioactive iodine therapy is contraindicated because of the low radioiodine uptake associated with amiodarone-induced hyperthyroidism. Pacerone®-induced hyperthyroidism may be followed by a transient period of hypothyroidism.
Hypothyrodism may be primary or subsequent to resolution of preceding amiodarone-induced hyperthyroidism. Severe hypothyroidism and myxedema coma, sometimes fatal, have been reported in association with amiodarone therapy. In some clinically hypothyroid amiodarone-treated patients, free thyroxine index values may be normal. Manage hypothyroidism by reducing the dose of or discontinuing Pacerone® and thyroid hormone supplementation.
5.7 Bradycardia
Pacerone® causes symptomatic bradycardia or sinus arrest with suppression of escape foci in 2 to 4% of patients. The risk is increased by electrolytic disorders or use of concomitant antiarrhythmics or negative chronotropes [see Drug Interactions (7)]. Bradycardia may require a pacemaker for rate control.
Postmarketing cases of symptomatic bradycardia, some requiring pacemaker insertion and at least one fatal, have been reported when ledipasvir/sofosbuvir or sofosbuvir with simeprevir were initiated in patients on amiodarone. Bradycardia generally occurred within hours to days, but in some cases presented up to 2 weeks after initiating antiviral treatment. Bradycardia generally resolved after discontinuation of antiviral treatment. The mechanism for this effect is unknown. Monitor heart rate in patients taking or recently discontinuing amiodarone when starting antiviral treatment [see Drug Interactions (7)].
5.8 Implantable Cardiac Devices
In patients with implanted defibrillators or pacemakers, chronic administration of antiarrhythmic drugs may affect pacing or defibrillation thresholds. Therefore, at the inception of and during amiodarone treatment, pacing and defibrillation thresholds should be assessed.
5.9 Fetal Toxicity
Pacerone® may cause fetal harm when administered to a pregnant woman. Fetal exposure may increase the potential for cardiac, thyroid, neurodevelopmental, neurological, and growth effects in neonate [see Use in Specific Populations (8.1)].
5.10 Peripheral Neuropathy
Chronic administration of Pacerone® may lead to peripheral neuropathy, which may not resolve when Pacerone® is discontinued.
5.11 Photosensitivity and Skin Discoloration
Pacerone® induces photosensitization in about 10% of patients; some protection may be afforded sun-barrier creams or protective clothing. During long-term treatment, a blue-gray discoloration of the exposed skin may occur. The risk may be increased in patients of fair complexion or those with excessive sun exposure. Some reversal of discoloration may occur upon drug discontinuation.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described in more detail in other sections of the prescribing information:
- Pulmonary Toxicity [see Warnings and Precautions (5.2)]
- Hepatic Injury [see Warnings and Precautions (5.3)]
- Worsened Arrhythmia [see Warnings and Precautions (5.4)]
- Visual Impairment and Loss of Vision [see Warnings and Precautions (5.5)]
- Thyroid Abnormalities [see Warnings and Precautions (5.6)]
- Bradycardia [see Warnings and Precautions (5.7)]
- Peripheral Neuropathy [see Warnings and Precautions (5.10)]
- Photosensitivity and Skin Discoloration [see Warnings and Precautions (5.11)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
At the usual maintenance dose (400 mg/day) and above, Pacerone® causes adverse reactions in about three-fourths of all patients, resulting in discontinuation in 7 to 18%.
In surveys of almost 5,000 patients treated in open U.S. studies and in published reports of treatment with Pacerone®, the adverse reactions most frequently requiring discontinuation of Pacerone® included pulmonary infiltrates or fibrosis, paroxysmal ventricular tachycardia, congestive heart failure, and elevation of liver enzymes. Other symptoms causing discontinuations less often included visual disturbances, photosensitivity, blue skin discoloration, hyperthyroidism, and hypothyroidism.
The following side-effect rates are based on a retrospective study of 241 patients treated for 2 to 1,515 days (mean 441.3 days):
Thyroid
Common
Hypothyroidism, hyperthyroidism.
Cardiovascular
Common
Congestive heart failure, cardiac arrhythmias, SA node dysfunction.
Gastrointestinal
Very common
Nausea, vomiting.
Common
Constipation, anorexia, abdominal pain.
Dermatologic
Common
Solar dermatitis/photosensitivity.
Neurologic
Common
Malaise and fatigue, tremor/abnormal involuntary movements, lack of coordination, abnormal gait/ataxia, dizziness, paresthesias, decreased libido, insomnia, headache, sleep disturbances.
Ophthalmic
Common
Visual disturbances.
Hepatic
Common
Abnormal liver-function tests, nonspecific hepatic disorders.
Respiratory
Common
Pulmonary inflammation or fibrosis.
Other
Common
Flushing, abnormal taste and smell, edema, abnormal salivation, coagulation abnormalities.
Uncommon
Blue skin discoloration, rash, spontaneous ecchymosis, alopecia, hypotension, and cardiac conduction abnormalities.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of Pacerone®. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hematologic
Hemolytic anemia, aplastic anemia, pancytopenia, neutropenia, thrombocytopenia, agranulocytosis, granuloma.
Immune
Anaphylactic/anaphylactoid reaction (including shock), angioedema.
Neurologic
Pseudotumor cerebri, parkinsonian symptoms such as akinesia and bradykinesia (sometimes reversible with discontinuation of therapy), demyelinating polyneuropathy.
Psychiatric
Hallucination, confusional state, disorientation, delirium.
Cardiac
Hypotension (sometimes fatal), sinus arrest.
Respiratory
Eosinophilic pneumonia, acute respiratory distress syndrome in the post-operative setting, bronchospasm, bronchiolitis obliterans organizing pneumonia, pulmonary alveolar hemorrhage, pleural effusion, pleuritis.
Gastrointestinal
Pancreatitis, acute pancreatitis.
Hepatic
Hepatitis, cholestatic hepatitis, cirrhosis.
Skin and Subcutaneous Tissue Disorders
Urticaria, toxic epidermal necrolysis (sometimes fatal), erythema multiforme, Stevens-Johnson syndrome, exfoliative dermatitis, bullous dermatitis, drug rash with eosinophilia and systemic symptoms (DRESS), eczema, pruritus, skin cancer, lupus-like syndrome.
Musculoskeletal
Myopathy, muscle weakness, rhabdomyolysis.
Renal
Renal impairment, renal insufficiency, acute renal failure.
Reproductive
Epididymitis, impotence.
Body as a whole
Fever, dry mouth.
Endocrine and metabolic
Thyroid nodules/ thyroid cancer, syndrome of inappropriate antidiuretic hormone secretion (SIADH).
Vascular
Vasculitis.
-
7 DRUG INTERACTIONS
Because of amiodarone’s long half-life, expect drug interactions to persist for weeks to months after discontinuation of amiodarone.
Drug interactions with amiodarone are described in Table 1 below.
Table 1: Amiodarone Drug Interactions Concomitant Drug
Class/Name
Examples
Clinical Comment
Pharmacodynamic Interactions
QT Prolonging Drugs
class I and III antiarrhythmics, lithium, certain phenothiazines, tricyclic antidepressants, certain
fluoroquinolone and macrolide antibiotics, azole antifungals, halogenated inhalation anesthetic agents
Increased risk of Torsade de Pointes.
Avoid concomitant use.
Negative Chronotropes
digoxin, beta blockers, verapamil, diltiazem, clonidine, ivabradine
Potentiates the electrophysiologic and hemodynamic effects of amiodarone, resulting in bradycardia, sinus arrest, and AV block. Monitor heart rate.
Pharmacokinetic Interactions
CYP450 Inhibitors
grapefruit juice, certain fluoroquinolone and macrolide antibiotics, azole antifungals,
cimetidine, certain protease inhibitors
Increased exposure of amiodarone. Avoid concomitant use.
CYP450 Inducers
St. John’s Wort
Reduced amiodarone serum levels.
Cyclosporine
Increased plasma levels of cyclosporine have been reported resulting in elevated creatinine, despite reduction of cyclosporine dose. Monitor cyclosporine drug levels and renal function with concomitant use.
Cholestyramine
Reduced amiodarone serum levels.
Antiarrhythmics
quinidine, procainamide, flecainide
Reserve concomitant use for patients who are unresponsive to a single agent. Antiarrhythmic metabolism inhibited by amiodarone. Initiate antiarrhythmic at a lower than usual dose and monitor patient carefully. Reduce dose levels of previously administered antiarrhythmic by 30 to 50% for several days after transitioning to oral amiodarone. Evaluate continued need for antiarrhythmic.
Digoxin
Increased digoxin concentration. Reduce digoxin by half or discontinue. If continued, monitor for evidence of toxicity.
HMG-CoA Reductase
Inhibitors
simvastatin, lovastatin, atorvastatin
Increased plasma concentration of HMGCoA reductase inhibitor.
Limit the dose of lovastatin to 40 mg.
Limit the coadministered dose of simvastatin to 20 mg.
Lower starting dose of other CYP3A4 substrates may be required.
Warfarin
Potentiates anticoagulant response and can result in serious or fatal bleeding. Coadministration increases prothrombin time by 100% after 3 to 4 days. Reduce warfarin dose by one-third to one-half and monitor prothrombin times.
Phenytoin
Increased steady-state levels of phenytoin. Monitor phenytoin levels.
Hepatitis C Direct
Acting Antiviral
sofosbuvir
Cases of symptomatic bradyarrhythmia requiring pacemaker insertion have been reported in patients on oral maintenance amiodarone who initiated therapy with sofosbuvir.
CYP3A Substrate
lidocaine
Sinus bradycardia has been reported with oral amiodarone in combination with lidocaine given for local anesthesia. Monitor heart rate. A lower starting dose of lidocaine may be required.
CYP3A Substrate
fentanyl
Fentanyl in combination with amiodarone may cause hypotension, bradycardia, and decreased cardiac output.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from postmarketing reports and published case series indicate that amiodarone use in pregnant women may increase the risk for fetal adverse effects including neonatal hypo- and hyperthyroidism, neonatal bradycardia, neurodevelopmental abnormalities, preterm birth and fetal growth restriction. Amiodarone and its metabolite, desethylamiodarone (DEA), cross the placenta. Untreated underlying arrhythmias, including ventricular arrhythmias, during pregnancy pose a risk to the mother and fetus (see ClinicalConsiderations). In animal studies, administration of amiodarone to rabbits, rats, and mice during organogenesis resulted in embryofetal toxicity at doses less than the maximum recommended human maintenance dose (see Data). Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and or Embryo/Fetal Risk
The incidence of ventricular tachycardia is increased and may be more symptomatic during pregnancy. Ventricular arrhythmias most often occur in pregnant women with underlying cardiomyopathy, congenital heart disease, valvular heart disease, or mitral valve prolapse. Most tachycardia episodes are initiated by ectopic beats and the occurrence of arrhythmia episodes may therefore, increase during pregnancy due to the increased propensity to ectopic activity. Breakthrough arrhythmias may also occur during pregnancy, as therapeutic treatment levels may be difficult to maintain due to the increased volume of distribution and increased drug metabolism inherent in the pregnant state.
Fetal/Neonatal Adverse Reactions
Amiodarone and its metabolite have been shown to cross the placenta. Adverse fetal effects associated with maternal amiodarone use during pregnancy may include neonatal bradycardia, QT prolongation, and periodic ventricular extrasystoles, neonatal hypothyroidism (with or without goiter) detected antenatally or in the newborn and reported even after a few days of exposure, neonatal hyperthyroxinemia, neurodevelopmental abnormalities independent of thyroid function, including speech delay and difficulties with written language and arithmetic, delayed motor development, and ataxia, jerk nystagmus with synchronous head titubation, fetal growth restriction, and premature birth. Monitor the newborn for signs and symptoms of thyroid disorder and cardiac arrhythmias.
Labor and Delivery
Risk of arrhythmias may increase during labor and delivery. Patients treated with Pacerone® should be monitored continuously during labor and delivery [see Warnings and Precautions (5.4)].
Data
Animal Data
In pregnant rats and rabbits during the period of organogenesis, amiodarone HCl in doses of 25 mg/kg/day (approximately 0.4 and 0.9 times, respectively, the maximum recommended human maintenance dose*) had no adverse effects on the fetus. In the rabbit, 75 mg/kg/day (approximately 2.7 times the maximum recommended human maintenance dose*) caused abortions in greater than 90% of the animals. In the rat, doses of 50 mg/kg/day or more were associated with slight displacement of the testes and an increased incidence of incomplete ossification of some skull and digital bones; at 100 mg/kg/day or more, fetal body weights were reduced; at 200 mg/kg/day, there was an increased incidence of fetal resorption. (These doses in the rat are approximately 0.8, 1.6 and 3.2 times the maximum recommended human maintenance dose*) Adverse effects on fetal growth and survival also were noted in one of two strains of mice at a dose of 5 mg/kg/day (approximately 0.04 times the maximum recommended human maintenance dose*).
*600 mg in a 60 kg patient (doses compared on a body surface area basis)
8.2 Lactation
Risk Summary
Amiodarone and one of its major metabolites, DEA, are present in breastmilk at between 3.5% and 45% of the maternal weight adjusted dosage of amiodarone. There are cases of hypothyroidism and bradycardia in breastfed infants, although it is unclear if these effects are due to amiodarone exposure in breastmilk. Breastfeeding is not recommended during treatment with Pacerone®[seeWarnings and Precautions (5.6, 5.7)].
8.3 Females and Males of Reproductive Potential
Infertility
Based on animal fertility studies, Pacerone® may reduce female and male fertility. It is not known if this effect is reversible. [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of Pacerone® (amiodarone hydrochloride) in pediatric patients have not been established.
8.5 Geriatric Use
Normal subjects over 65 years of age show lower clearances and increased drug half-life than younger subjects [see Clinical Pharmacology (12.3)]. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosingrange, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drugtherapy.
-
10 OVERDOSAGE
There have been cases, some fatal, of Pacerone® overdose.
Monitor the patient’s cardiac rhythm and blood pressure, and, if bradycardia ensues, a β-adrenergic agonist or a pacemaker may be used. Treat hypotension with inadequate tissue perfusion with positive inotropic and vasopressor agents. Neither Pacerone® nor its metabolite is dialyzable.
-
11 DESCRIPTION
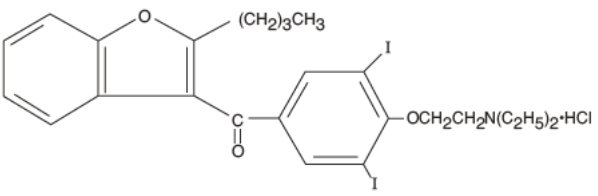
Pacerone® (amiodarone hydrochloride) tablets are a member of a class of antiarrhythmic drugs with predominantly Class III (Vaughan Williams’ classification) effects, available for oral administration as light yellow, scored tablets. Each tablet for oral administration contains 400 mg of amiodarone hydrochloride. In addition, each tablet contains the following inactive ingredients: colloidal silicon dioxide, corn starch, lactose monohydrate, magnesium stearate, povidone, and D&C yellow No. 10 aluminum lake. Amiodarone hydrochloride, the active ingredient in Pacerone® Tablets, is a benzofuran derivative: 2-butyl-3-benzofuranyl 4-[2-(diethylamino)-ethoxy]-3,5-diiodophenyl ketone hydrochloride.
The structural formula is as follows:
C25H29I2NO3HCl Molecular Weight: 681.8
Amiodarone hydrochloride is a white to cream-colored crystalline powder. It is slightly soluble in water, soluble in alcohol, and freely soluble in chloroform. It contains 37.3% iodine by weight.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Amiodarone is considered a class III antiarrhythmic drug, but it possesses electrophysiologic characteristics of all four Vaughan Williams classes. Like class I drugs, amiodarone blocks sodium channels at rapid pacing frequencies, and like class II drugs, amiodarone exerts a noncompetitive antisympathetic action. One of its main effects, with prolonged administration, is to lengthen the cardiac action potential, a class III effect. The negative chronotropic effect of amiodarone in nodal tissues is similar to the effect of class IV drugs. In addition to blocking sodium channels, amiodarone blocks myocardial potassium channels, which contributes to slowing of conduction and prolongation of refractoriness. The antisympathetic action and the block of calcium and potassium channels are responsible for the negative dromotropic effects on the sinus node and for the slowing of conduction and prolongation of refractoriness in the atrioventricular (AV) node. Its vasodilatory action can decrease cardiac workload and consequently myocardial oxygen consumption.
Pacerone® prolongs the duration of the action potential of all cardiac fibers while causing minimal reduction of dV/dt (maximal upstroke velocity of the action potential). The refractory period is prolonged in all cardiac tissues. Pacerone® increases the cardiac refractory period without influencing resting membrane potential, except in automatic cells where the slope of the prepotential is reduced, generally reducing automaticity. These electrophysiologic effects are reflected in a decreased sinus rate of 15 to 20%, increased PR and QT intervals of about 10%, the development of U-waves, and changes in T-wave contour. These changes should not require discontinuation of Pacerone® as they are evidence of its pharmacological action, although Pacerone® can cause marked sinus bradycardia or sinus arrest and heart block [see Warnings and Precautions (5.4)].
Hemodynamics
In animal studies and after intravenous administration in man, Pacerone® relaxes vascular smooth muscle, reduces peripheral vascular resistance (afterload), and slightly increases cardiac index. After oral dosing, however, Pacerone® produces no significant change in left ventricular ejection fraction (LVEF), even in patients with depressed LVEF. After acute intravenous dosing in man, Pacerone® may have a mild negative inotropic effect.
12.2 Pharmacodynamics
There is no well-established relationship between plasma concentration and effectiveness, but it does appear that concentrations much below 1 mg/L are often ineffective and that levels above 2.5 mg/L are generally not needed. Plasma-concentration measurements can be used to identify patients whose levels are unusually low, and who might benefit from a dose increase, or unusually high, and who might have dosage reduction in the hope of minimizing side effects.
Effects on abnormal rhythms are not seen before 2 to 3 days and usually require 1 to 3 weeks, even when a loading dose is used. There may be a continued increase in effect for longer periods still. There is evidence that the time to effect is shorter when a loading-dose regimen is used.
Consistent with the slow rate of elimination, antiarrhythmic effects persist for weeks or months after Pacerone® is discontinued, but the time of recurrence is variable and unpredictable. In general, when the drug is resumed after recurrence of the arrhythmia, control is established relatively rapidly compared to the initial response, presumably because tissue stores were not wholly depleted.
12.3 Pharmacokinetics
Absorption
Following oral administration in humans, Pacerone® is slowly and variably absorbed. The bioavailability of Pacerone® is approximately 50%. Maximum plasma concentrations are attained 3 to 7 hours after a single dose. Plasma concentrations with chronic dosing at 100 to 600 mg/day are approximately dose proportional, with a mean 0.5 mg/L increase for each 100 mg/day. These means, however, include considerable individual variability.
Food increases the rate and extent of absorption of Pacerone®. The effects of food upon the bioavailability of Pacerone® have been studied in 30 healthy subjects who received a single 600-mg dose immediately after consuming a high-fat meal and following an overnight fast. The area under the plasma concentration-time curve (AUC) and the peak plasma concentration (Cmax) of amiodarone increased by 2.3 (range 1.7 to 3.6) and 3.8 (range 2.7 to 4.4) times, respectively, in the presence of food. Food also increased the rate of absorption of amiodarone, decreasing the time to peak plasma concentration (Tmax) by 37%. The mean AUC and mean Cmax of the major metabolite of amiodarone, DEA increased by 55% (range 58 to 101%) and 32% (range 4 to 84%), respectively, but there was no change in the Tmax in the presence of food.
Distribution
Pacerone® is highly protein-bound (approximately 96%). Pacerone® has a very large but variable volume of distribution, averaging about 60 L/kg, because of extensive accumulation in various sites, especially adipose tissue and highly perfused organs, such as the liver, lung, and spleen.
One major metabolite of Pacerone®, DEA, has been identified in man; it accumulates to an even greater extent in almost all tissues. No data are available on the activity of DEA in humans, but in animals, it has significant electrophysiologic and antiarrhythmic effects generally similar to amiodarone itself. DEA’s precise role and contribution to the antiarrhythmic activity of oral amiodarone are not certain. The development of maximal ventricular class III effects after oral Pacerone® administration in humans correlates more closely with DEA accumulation over time than with amiodarone accumulation.
Elimination
Following single dose administration in 12 healthy subjects, Pacerone® exhibited multi-compartmental pharmacokinetics with a mean apparent plasma terminal elimination half-life of 58 days (range 15 to 142 days) for amiodarone and 36 days (range 14 to 75 days) for the active metabolite (DEA). In patients, following discontinuation of chronic oral therapy, Pacerone® has been shown to have a biphasic elimination with an initial 50% reduction of plasma levels after 2.5 to 10 days. A much slower terminal plasma-elimination phase shows a half-life of the parent compound ranging from 26 to 107 days, with a mean of approximately 53 days and most patients in the 40- to 55-day range. In the absence of a loading-dose period, steady-state plasma concentrations, at constant oral dosing, would therefore be reached between 130 and 535 days, with an average of 265 days. For the metabolite, the mean plasma-elimination half-life was approximately 61 days. These data probably reflect an initial elimination of drug from well-perfused tissue (the 2.5- to 10-day half-life phase), followed by a terminal phase representing extremely slow elimination from poorly perfused tissue compartments such as fat.
The considerable inter-subject variation in both phases of elimination, as well as uncertainty as to what compartment is critical to drug effect, requires attention to individual responses once arrhythmia control is achieved with loading doses because the correct maintenance dose is determined, in part, by the elimination rates. Individualize maintenance doses of Pacerone®[see Dosage andAdministration (2)].
Metabolism
Amiodarone is metabolized to DEA by the cytochrome P450 (CYP) enzyme group, specifically CYP3A and CYP2C8. The CYP3A isoenzyme is present in both the liver and intestines. In vitro, amiodarone and DEA exhibit a potential to inhibit CYP2C9, CYP2C19, CYP2D6, CYP3A, CYP2A6, CYP2B6 and CYP2C8. Amiodarone and DEA have also a potential to inhibit some transporters such as P-glycoprotein and organic cation transporter (OCT2).
Excretion
Amiodarone is eliminated primarily by hepatic metabolism and biliary excretion and there is negligible excretion of amiodarone or DEA in urine. Neither amiodarone nor DEA is dialyzable.
Specific Populations
Effect of Age
Normal subjects over 65 years of age show lower clearances (about 100 mL/hr/kg) than younger subjects (about 150 mL/hr/kg) and an increase in t½ from about 20 to 47 days.
Renal Impairment
Renal impairment does not influence the pharmacokinetics of amiodarone or DEA.
Hepatic Impairment
After a single dose of intravenous amiodarone to cirrhotic patients, significantly lower Cmax and average concentration values are seen for DEA, but mean amiodarone levels are unchanged.
Cardiac Disease
In patients with severe left ventricular dysfunction, the pharmacokinetics of amiodarone are not significantly altered but the terminal elimination t½ of DEA is prolonged.
Although no dosage adjustment for patients with renal, hepatic, or cardiac abnormalities has been defined during chronic treatment with oral amiodarone, close clinical monitoring is prudent for elderly patients and those with severe left ventricular dysfunction.
Drug Interactions
Effects of Other Agents on Amiodarone
Grapefruit juice
Grapefruit juice given to healthy volunteers increased amiodarone AUC by 50% and Cmax by 84%, and decreased DEA to unquantifiable concentrations.
Cimetidine
Inhibits CYP3A and can increase serum amiodarone levels.
Cholestyramine
Reduces enterohepatic circulation of amiodarone thereby increasing its elimination. This results in reduced amiodarone serum levels and half-life.
Effects of Amiodarone on Agents
CYP3A Substrate
Amiodarone taken concomitantly with quinidine increases the quinidine serum concentration by 33% after two days. Amiodarone taken concomitantly with procainamide for less than seven days increases plasma concentrations of procainamide and n-acetyl procainamide by 55% and 33%, respectively.
Loratadine
A non-sedating antihistaminic, is metabolized primarily by CYP3A and its metabolism can be inhibited by amiodarone.
Metabolism of lidocaine can be inhibited by amiodarone.
Cyclophosphamide
A prodrug, metabolized by CYP450 including CYP3A to an active metabolite. The metabolism of cyclophosphamide may be inhibited by amiodarone.
Clopidogrel
An inactive thienopyridine prodrug, is metabolized in the liver by CYP3A to an active metabolite. A potential interaction between clopidogrel and amiodarone resulting in ineffective inhibition of platelet aggregation has been reported.
Macrolide/Ketolide Antibiotics
Amiodarone can inhibit the metabolism of macrolide/ketolide antibiotics (except for azithromycin) and systemic azole antifungal drugs.
P-Glycoprotein Substrates:
Amiodarone taken concomitantly with digoxin increases the serum digoxin concentration by 70% after one day.
Dabigatran Etexilate
When taken concomitantly with oral amiodarone can result in elevated serum concentration of dabigatran.
Dextromethorphan
A substrate for both CYP2D6 and CYP3A. Amiodarone inhibits CYP2D6. Chronic (> 2 weeks) oral amiodarone administration impairs metabolism of dextromethorphan can lead to increased serum concentrations.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Amiodarone HCl was associated with a statistically significant, dose-related increase in the incidence of thyroid tumors (follicular adenoma and/or carcinoma) in rats. The incidence of thyroid tumors was greater than control at the lowest dose level tested, i.e., 5 mg/kg/day (approximately 0.08 times the maximum recommended human maintenance dose*).
Mutagenicity studies (Ames, micronucleus, and lysogenic tests) with Pacerone® were negative.
In a study in which amiodarone HCl was administered to male and female rats, beginning 9 weeks prior to mating, reduced fertility was observed at a dose level of 90 mg/kg/day (approximately 1.4 times the maximum recommended human maintenance dose*).
*600 mg in a 60 kg patient (dose compared on a body surface area basis)
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Pacerone® (Amiodarone Hydrochloride) Tablets, 400 mg, are available in bottles of 30 tablets (NDC: 0245-0145-30), bottles of 100 tablets (NDC: 0245-0145-11) and bottles of 500 tablets (NDC: 0245-0145-15). The 400 mg tablets are light yellow, oval-shaped, scored, uncoated tablets, debossed with “P400” on the unscored side, and “01” to the left and “45” to the right of the score on the reverse side.
Store at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature].
Keep tightly closed. Protect from light. Dispense in a tight, light-resistant container as defined in the USP, with a child-resistant closure as required.
Keep out of the reach of children.
This product's label may have been revised after this insert was used in production. For further product information and current package insert, please visit www.upsher-smith.com or call 1-888-650-3789.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their prescriber of a known or suspected pregnancy [see Use in Specific Populations (8.1)].
Advise women that breastfeeding is not recommended during treatment with Pacerone®[see Use in Specific Populations (8.2)].
Advise patients to avoid grapefruit juice and St. John's Wort.
Advise patients to seek medical attention if they experience the signs and symptoms of pulmonary toxicity, worsening arrhythmia, bradycardia, visual impairment, or hypo- and hyperthyroidism.
Manufactured for
UPSHER-SMITH LABORATORIES, LLC.
Maple Grove, MN 55369
© 2018 Upsher-Smith Laboratories, LLC.
Rev. 11/18
-
MEDICATION GUIDE
Medication Guide
Pacerone® (PĀS ǝr ōn) Tablets
(Amiodarone HCl)
What is the most important information I should know about Pacerone® Tablets?
Pacerone® Tablets can cause serious side effects that can lead to death including:
- lung problems
- liver problems
- worsening of heartbeat problems
Call your healthcare provider or get medical help right away if you have any of the following symptoms during treatment with Pacerone® Tablets:
- trouble breathing, wheezing, shortness of breath, coughing chest pain, spitting up of blood, or fever
- nausea or vomiting, brown or dark-colored urine, feel more tired than usual, yellowing of your skin or the whites of your eyes (jaundice), or right upper stomach-area pain
- heart pounding, skipping a beat, beating fast or slowly, feel light-headed, or if you faint
- vision problems, including blurred vision, see halos, or your eyes become sensitive to light. You should have regular eye exams before and during treatment with Pacerone® Tablets.
Pacerone® Tablets should be started in a hospital so that your medical condition can be carefully monitored.
Pacerone® Tablets should only be used to treat people who have been diagnosed with life-threatening heartbeat problems called ventricular arrhythmias, when other treatments did not work or you cannot tolerate them.
Pacerone® Tablets can cause other serious side effects. See “What are the possible side effects of Pacerone® Tablets?”
If you get serious side effects during treatment you may need to stop Pacerone® Tablets, have your dose changed, or get medical treatment. Talk with your healthcare provider before you stop taking Pacerone® Tablets.
You may still have side effects after stopping Pacerone® Tablets because the medicine stays in your body for months after treatment is stopped.
You should have regular check-ups, blood tests, chest x-rays before and during treatment with Pacerone® Tablets to check for serious side effects. You should also have lung function tests before starting treatment with Pacerone® Tablets.
What are Pacerone® Tablets?
Pacerone® Tablets are a prescription medicine used to treat people who have been diagnosed with life-threatening heartbeat problems called ventricular arrhythmias, when other treatments did not work or you cannot tolerate them.
It is not known if Pacerone® Tablets are safe and effective in children.
Who should not take Pacerone® Tablets?
Do not take Pacerone® Tablets if you:
- have a serious heart problem called cardiogenic shock
- have certain types of the heart condition called heart block, with or without a slow heart rate
- have a slow heart rate with dizziness or lightheadedness, and you do not have an implanted pacemaker
- are allergic to amiodarone, iodine, or any of the other ingredients in Pacerone® Tablets. See the end of this Medication Guide for a complete list of ingredients in Pacerone® Tablets.
Before taking Pacerone® Tablets tell your healthcare provider about all of your medical conditions including if you:
- have lung or breathing problems
- have liver problems
- have or had thyroid problems
- have a slow heart rate or blood pressure problems
- have diarrhea or have had diarrhea for a long period of time
- have been told that you have low levels of potassium, magnesium, or calcium in your blood
- have an implanted pacemaker or defribrillator
- if you plan to have surgery with general anesthesia
- are pregnant or plan to become pregnant. Amiodarone hydrochloride may harm your unborn baby. Tell your healthcare provider right away if you become pregnant during treatment with Pacerone® Tablets. Amiodarone hydrochloride can stay in your body for months after treatment is stopped.
- are breastfeeding or plan to breastfeed. Amiodarone hydrochloride can pass into your breast milk and may harm your baby. You should not breast feed while taking Pacerone® Tablets. Amiodarone hydrochloride can stay in your body for months after treatment is stopped. Talk to your healthcare provider about the best way to feed your baby during this time.
Tell your healthcare provider about all the medicines you take including prescription and over-the-counter medicines, vitamins and herbal supplements. Pacerone® Tablets and certain other medicines can affect with each other and cause serious side effects. You can ask your pharmacist for a list of medicines that interact with Pacerone® Tablets.
How should I take Pacerone® Tablets?
- When you are discharged from the hospital, take Pacerone® Tablets exactly as your doctor tells you to take them.
- Your healthcare provider will tell you how many Pacerone® Tablets to take and when to take them.
- Your healthcare provider may change your dose of Pacerone® Tablets as needed if your heart rhythm is controlled, or if you have certain side effects. Your healthcare provider should monitor you carefully when your dose of Pacerone® Tablets is being changed.
- Take your dose of Pacerone® Tablets the same way each time, either with or without food.
- If you take too many Pacerone® Tablets, call your healthcare provider or go to the nearest hospital emergency room right away. If you miss a dose, wait and take your next dose at your regular time. Do not take two doses at the same. Continue with your next regularly scheduled dose.
What should I avoid while taking Pacerone® Tablets?
- Avoid drinking grapefruit juice during treatment with Pacerone® Tablets. Drinking grapefruit juice with Pacerone® Tablets may increase the amount of Pacerone® Tablets in your blood, and this may lead to side effects.
- Pacerone® Tablets can make your skin sensitive to sunlight. You could get severe sunburn. Use sunscreen and wear a hat and clothes that cover your skin to help protect you if you must be in sunlight. Talk to your healthcare provider if you get a sunburn. See “Skin problems” in the Medication Guide section “What are the possible side effects of Pacerone® Tablets?” below.
What are the possible side effects of Pacerone® Tablets?
Pacerone® Tablets can cause serious side effects, including:
- See “What is the most important information I should know about Pacerone® Tablets?”
- Nerve problems. Pacerone® Tablets can cause nerve problems. Call your healthcare provider if you develop symptoms of nerve problems, including: a feeling of “pins and needles” or numbness in your hands, legs, or feet, muscle weakness, uncontrolled movements, poor coordination, or trouble walking.
- Skin problems. Pacerone® Tablets can cause your skin to be more sensitive to the sun or turn a bluish-gray color. People who have fair skin or people who have a lot of sun exposure may be more at risk for these skin problems. Some of the bluish-gray skin color may return to normal after stopping Pacerone® Tablets.
- Thyroid problems. Pacerone® Tablets can cause you to have either decreased thyroid function (hypothyroidism), which can sometimes be severe, or an overactive thyroid (hyperthyroidism), which can be severe.
- ○ If you develop decreased thyroid function during treatment with Pacerone® Tablets, your healthcare provider may need to reduce your dose or stop your treatment with Pacerone® Tablets, and possibly prescribe medicine to replace your thyroid hormone.
- ○ An overactive thyroid can cause you to produce too much thyroid hormone. You can have abnormal heartbeats even while you are receiving Pacerone® Tablets. Your healthcare provider may prescribe certain medicines to treat your overactive thyroid. Call your healthcare provider if you get any abnormal heart beats during treatment with Pacerone® Tablets. This may mean that you have an overactive thyroid.
- ○ Your healthcare provider should do tests to check your thyroid function before you start and during treatment with Pacerone® Tablets.
- ○
Call your healthcare provider if you develop any of the following symptoms of a thyroid problem during treatment with Pacerone® Tablets:
- weakness
- weight loss or weight gain
- heat or cold intolerance
- hair thinning
- sweating
- changes in your menstrual periods
- swelling of your neck (goiter)
- nervousness
- irritability
- restlessness
- decreased concentration
- feeling depressed (in the elderly)
- tremor
The most common side effects of Pacerone® Tablets include:
- lung problems
- heartbeat problems
- heart problems
- liver problems
Pacerone® Tablets may affect fertility in males and females. It is not known if the effects are reversible. Talk to your healthcare provider if you have concerns about fertility.
These are not all the possible side effects of Pacerone® Tablets. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store Pacerone® Tablets?
- Store Pacerone® Tablets at room temperature between 68° to 77°F (20° to 25°C).
- Keep Pacerone® Tablets in a tightly closed container and protect from light.
Keep Pacerone® Tablets and all medicines out of the reach of children.
General information about the safe and effective use of Pacerone® Tablets
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use
Pacerone® Tablets for a condition for which it was not prescribed. Do not give Pacerone® Tablets to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about Pacerone® Tablets that is written for health professionals.
What are the ingredients in Pacerone® Tablets?
Active Ingredient: amiodarone hydrochloride, 400 mg
Inactive Ingredients: colloidal silicon dioxide, corn starch, lactose monohydrate, magnesium stearate, povidone and D&C yellow No. 10 aluminum lake.
Manufactured for
UPSHER-SMITH LABORATORIES, LLC.
Maple Grove, MN 55369
OS7841
MF0145REV11/18
© 2018 Upsher-Smith Laboratories, LLC.
Pacerone is a registered trademark of Upsher-Smith Laboratories, LLC.
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised 11/18
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC: 0245-0145-30
Pacerone®
(Amiodarone HCl) Tablets
400 mg
PHARMACIST: Dispense the Medication Guide provided separately to each patient.
30 Tablets
Rx only
UPSHER-SMITH
-
INGREDIENTS AND APPEARANCE
PACERONE
amiodarone hydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0245-0145 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Amiodarone Hydrochloride (UNII: 976728SY6Z) (Amiodarone - UNII:N3RQ532IUT) Amiodarone Hydrochloride 400 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) STARCH, CORN (UNII: O8232NY3SJ) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) D&C YELLOW NO. 10 (UNII: 35SW5USQ3G) Product Characteristics Color YELLOW (light yellow) Score 2 pieces Shape OVAL Size 16mm Flavor Imprint Code P400;01;45 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0245-0145-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 06/30/2000 09/30/2023 2 NDC: 0245-0145-11 100 in 1 BOTTLE; Type 0: Not a Combination Product 06/30/2000 12/31/2005 3 NDC: 0245-0145-15 500 in 1 BOTTLE; Type 0: Not a Combination Product 06/30/2000 09/30/2023 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA075315 06/30/2000 09/30/2023 Labeler - Upsher-Smith Laboratories, Inc. (047251004)

Trademark Results [Pacerone]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 PACERONE 75028678 2127660 Live/Registered |
UPSHER-SMITH LABORATORIES, LLC 1995-12-06 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.