Methotrexate by PD-Rx Pharmaceuticals, Inc. METHOTREXATE tablet
Methotrexate by
Drug Labeling and Warnings
Methotrexate by is a Prescription medication manufactured, distributed, or labeled by PD-Rx Pharmaceuticals, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
BOXED WARNING
(What is this?)
WARNINGS
METHOTREXATE SHOULD BE USED ONLY BY PHYSICIANS WHOSE KNOWLEDGE AND EXPERIENCE INCLUDE THE USE OF ANTIMETABOLITE THERAPY BECAUSE OF THE POSSIBILITY OF SERIOUS TOXIC REACTIONS (WHICH CAN BE FATAL):
METHOTREXATE SHOULD BE USED ONLY IN LIFE THREATENING NEOPLASTIC DISEASES, OR IN PATIENTS WITH PSORIASIS OR RHEUMATOID ARTHRITIS WITH SEVERE, RECALCITRANT, DISABLING DISEASE WHICH IS NOT ADEQUATELY RESPONSIVE TO OTHER FORMS OF THERAPY.
DEATHS HAVE BEEN REPORTED WITH THE USE OF METHOTREXATE IN THE TREATMENT OF MALIGNANCY, PSORIASIS, AND RHEUMATOID ARTHRITIS.
PATIENTS SHOULD BE CLOSELY MONITORED FOR BONE MARROW, LIVER, LUNG AND KIDNEY TOXICITIES. (See PRECAUTIONS.)
PATIENTS SHOULD BE INFORMED BY THEIR PHYSICIAN OF THE RISKS INVOLVED AND BE UNDER A PHYSICIAN’S CARE THROUGHOUT THERAPY.
- Methotrexate has been reported to cause fetal death and/or congenital anomalies. Therefore, it is not recommended for women of childbearing potential unless there is clear medical evidence that the benefits can be expected to outweigh the considered risks. Pregnant women with psoriasis or rheumatoid arthritis should not receive methotrexate. (See CONTRAINDICATIONS.)
- Methotrexate elimination is reduced in patients with impaired renal function, ascites, or pleural effusions. Such patients require especially careful monitoring for toxicity, and require dose reduction or, in some cases, discontinuation of methotrexate administration.
- Unexpectedly severe (sometimes fatal) bone marrow suppression, aplastic anemia, and gastrointestinal toxicity have been reported with concomitant administration of methotrexate (usually in high dosage) along with some non-steroidal anti-inflammatory drugs (NSAIDs). (See PRECAUTIONS, Drug Interactions.)
- Methotrexate causes hepatotoxicity, fibrosis and cirrhosis, but generally only after prolonged use. Acutely, liver enzyme elevations are frequently seen. These are usually transient and asymptomatic, and also do not appear predictive of subsequent hepatic disease. Liver biopsy after sustained use often shows histologic changes, and fibrosis and cirrhosis have been reported; these latter lesions may not be preceded by symptoms or abnormal liver function tests in the psoriasis population. For this reason, periodic liver biopsies are usually recommended for psoriatic patients who are under long-term treatment. Persistent abnormalities in liver function tests may precede appearance of fibrosis or cirrhosis in the rheumatoid arthritis population. (See PRECAUTIONS, Organ System Toxicity, Hepatic.)
- Methotrexate-induced lung disease is a potentially dangerous lesion, which may occur acutely at any time during therapy and which has been reported at doses as low as 7.5 mg/week. It is not always fully reversible. Pulmonary symptoms (especially a dry, nonproductive cough) may require interruption of treatment and careful investigation.
- Diarrhea and ulcerative stomatitis require interruption of therapy; otherwise, hemorrhagic enteritis and death from intestinal perforation may occur.
- Malignant lymphomas, which may regress following withdrawal of methotrexate, may occur in patients receiving low-dose methotrexate and, thus, may not require cytotoxic treatment. Discontinue methotrexate first and, if the lymphoma does not regress, appropriate treatment should be instituted.
- Like other cytotoxic drugs, methotrexate may induce “tumor lysis syndrome” in patients with rapidly growing tumors. Appropriate supportive and pharmacologic measures may prevent or alleviate this complication.
- Severe, occasionally fatal, skin reactions have been reported following single or multiple doses of methotrexate. Reactions have occurred within days of oral, intramuscular, intravenous, or intrathecal methotrexate administration. Recovery has been reported with discontinuation of therapy. (See PRECAUTIONS, Organ System Toxicity, Skin).
- Potentially fatal opportunistic infections, especially Pneumocystis carinii pneumonia, may occur with methotrexate therapy.
- Methotrexate given concomitantly with radiotherapy may increase the risk of soft tissue necrosis and osteonecrosis.
-
DESCRIPTION
Methotrexate, USP (formerly Amethopterin) is an antimetabolite used in the treatment of certain neoplastic diseases, severe psoriasis, and adult rheumatoid arthritis. Chemically methotrexate is N-[4-[[(2,4-diamino-6-pteridinyl)methyl]methylamino]benzoyl]-L-glutamic acid. The structural formula is:
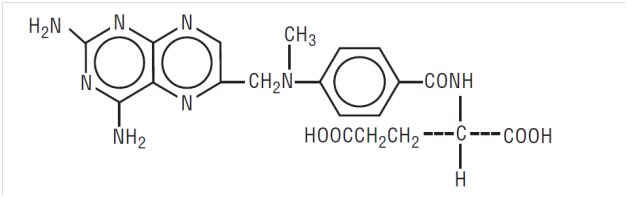
C 20H 22N 8O 5 M.W. 454.45
Methotrexate Tablets USP for oral administration, are available in bottles of 36 and 100. Methotrexate Tablets USP contain an amount of methotrexate sodium equivalent to 2.5 mg of methotrexate, USP.
Inactive Ingredients: Hydroxypropyl methylcellulose, lactose anhydrous, magnesium stearate, microcrystalline cellulose, polyethylene glycol, pregelatinized corn starch, propylene glycol, sodium carbonate monohydrate and talc.
-
CLINICAL PHARMACOLOGY
Methotrexate inhibits dihydrofolic acid reductase. Dihydrofolates must be reduced to tetrahydrofolates by this enzyme before they can be utilized as carriers of one-carbon groups in the synthesis of purine nucleotides and thymidylate. Therefore, methotrexate interferes with DNA synthesis, repair, and cellular replication. Actively proliferating tissues such as malignant cells, bone marrow, fetal cells, buccal and intestinal mucosa, and cells of the urinary bladder are in general more sensitive to this effect of methotrexate. When cellular proliferation in malignant tissues is greater than in most normal tissues, methotrexate may impair malignant growth without irreversible damage to normal tissues.
The mechanism of action in rheumatoid arthritis is unknown; it may affect immune function. Two reports describe in vitro methotrexate inhibition of DNA precursor uptake by stimulated mononuclear cells, and another describes in animal polyarthritis partial correction by methotrexate of spleen cell hyporesponsiveness and suppressed IL 2 production. Other laboratories, however, have been unable to demonstrate similar effects. Clarification of methotrexate’s effect on immune activity and its relation to rheumatoid immunopathogenesis await further studies.
In patients with rheumatoid arthritis, effects of methotrexate on articular swelling and tenderness can be seen as early as 3 to 6 weeks. Although methotrexate clearly ameliorates symptoms of inflammation (pain, swelling, stiffness), there is no evidence that it induces remission of rheumatoid arthritis nor has a beneficial effect been demonstrated on bone erosions and other radiologic changes which result in impaired joint use, functional disability, and deformity.
Most studies of methotrexate in patients with rheumatoid arthritis are relatively short term (3 to 6 months). Limited data from long-term studies indicate that an initial clinical improvement is maintained for at least two years with continued therapy.
In psoriasis, the rate of production of epithelial cells in the skin is greatly increased over normal skin. This differential in proliferation rates is the basis for the use of methotrexate to control the psoriatic process.
In a 6-month, double-blind, placebo-controlled trial of 127 pediatric patients with juvenile rheumatoid arthritis (JRA) (mean age, 10.1 years; age range 2.5 to 18 years, mean duration of disease, 5.1 years) on background non-steroidal anti-inflammatory drugs (NSAIDs) and/or prednisone, methotrexate given weekly at an oral dose of 10 mg/m 2 provided significant clinical improvement compared to placebo as measured by either the physician’s global assessment, or by a patient composite (25% reduction in the articular-severity score plus improvement in parent and physician global assessments of disease activity.) Over two-thirds of the patients in this trial had polyarticular-course JRA, and the numerically greatest response was seen in this subgroup treated with 10 mg/m 2/wk methotrexate. The overwhelming majority of the remaining patients had systemic-course JRA. All patients were unresponsive to NSAIDs; approximately one-third were using low dose corticosteroids. Weekly methotrexate at a dose of 5 mg/m 2 was not significantly more effective than placebo in this trial.
Pharmacokinetics
Absorption - In adults, oral absorption appears to be dose dependent. Peak serum levels are reached within one to two hours. At doses of 30 mg/m 2 or less, methotrexate is generally well absorbed with a mean bioavailability of about 60%. The absorption of doses greater than 80 mg/m 2 is significantly less, possibly due to a saturation effect.
In leukemic pediatric patients, oral absorption of methotrexate also appears to be dose dependent and has been reported to vary widely (23% to 95%). A twenty fold difference between highest and lowest peak levels (C max: 0.11 to 2.3 micromolar after a 20 mg/m 2 dose) has been reported. Significant interindividual variability has also been noted in time to peak concentration (T max: 0.67 to 4 hrs after a 15 mg/m 2 dose) and fraction of dose absorbed. The absorption of doses greater than 40 mg/m 2 has been reported to be significantly less than that of lower doses. Food has been shown to delay absorption and reduce peak concentration. Methotrexate is generally completely absorbed from parenteral routes of injection. After intramuscular injection, peak serum concentrations occur in 30 to 60 minutes. As in leukemic pediatric patients, a wide interindividual variability in the plasma concentrations of methotrexate has been reported in pediatric patients with JRA. Following oral administration of methotrexate in doses of 6.4 to 11.2 mg/m 2/week in pediatric patients with JRA, mean serum concentrations were 0.59 micromolar (range, 0.03 to 1.40) at 1 hour, 0.44 micromolar (range, 0.01 to 1.00) at 2 hours, and 0.29 micromolar (range, 0.06 to 0.58) at 3 hours. In pediatric patients receiving methotrexate for acute lymphocytic leukemia (6.3 to 30 mg/m 2), or for JRA (3.75 to 26.2 mg/m 2), the terminal half-life has been reported to range from 0.7 to 5.8 hours or 0.9 to 2.3 hours, respectively.
Distribution - After intravenous administration, the initial volume of distribution is approximately 0.18 L/kg (18% of body weight) and steady-state volume of distribution is approximately 0.4 to 0.8 L/kg (40% to 80% of body weight). Methotrexate competes with reduced folates for active transport across cell membranes by means of a single carrier-mediated active transport process. At serum concentrations greater than 100 micromolar, passive diffusion becomes a major pathway by which effective intracellular concentrations can be achieved. Methotrexate in serum is approximately 50% protein bound. Laboratory studies demonstrate that it may be displaced from plasma albumin by various compounds including sulfonamides, salicylates, tetracyclines, chloramphenicol, and phenytoin.
Methotrexate does not penetrate the blood-cerebrospinal fluid barrier in therapeutic amounts when given orally or parenterally. High CSF concentrations of the drug may be attained by intrathecal administration.
In dogs, synovial fluid concentrations after oral dosing were higher in inflamed than uninflamed joints. Although salicylates did not interfere with this penetration, prior prednisone treatment reduced penetration into inflamed joints to the level of normal joints.
Metabolism - After absorption, methotrexate undergoes hepatic and intracellular metabolism to polyglutamated forms which can be converted back to methotrexate by hydrolase enzymes. These polyglutamates act as inhibitors of dihydrofolate reductase and thymidylate synthetase. Small amounts of methotrexate polyglutamates may remain in tissues for extended periods. The retention and prolonged drug action of these active metabolites vary among different cells, tissues and tumors. A small amount of metabolism to 7-hydroxymethotrexate may occur at doses commonly prescribed. Accumulation of this metabolite may become significant at the high doses used in osteogenic sarcoma. The aqueous solubility of 7-hydroxymethotrexate is 3 to 5 fold lower than the parent compound. Methotrexate is partially metabolized by intestinal flora after oral administration.
Half-Life - The terminal half-life reported for methotrexate is approximately three to ten hours for patients receiving treatment for psoriasis, or rheumatoid arthritis or low dose antineoplastic therapy (less than 30 mg/m 2). For patients receiving high doses of methotrexate, the terminal half-life is eight to 15 hours.
Excretion - Renal excretion is the primary route of elimination and is dependent upon dosage and route of administration. With IV administration, 80% to 90% of the administered dose is excreted unchanged in the urine within 24 hours. There is limited biliary excretion amounting to 10% or less of the administered dose. Enterohepatic recirculation of methotrexate has been proposed.
Renal excretion occurs by glomerular filtration and active tubular secretion. Nonlinear elimination due to saturation of renal tubular reabsorption has been observed in psoriatic patients at doses between 7.5 and 30 mg. Impaired renal function, as well as concurrent use of drugs such as weak organic acids that also undergo tubular secretion, can markedly increase methotrexate serum levels. Excellent correlation has been reported between methotrexate clearance and endogenous creatinine clearance.
Methotrexate clearance rates vary widely and are generally decreased at higher doses. Delayed drug clearance has been identified as one of the major factors responsible for methotrexate toxicity. It has been postulated that the toxicity of methotrexate for normal tissues is more dependent upon the duration of exposure to the drug rather than the peak level achieved. When a patient has delayed drug elimination due to compromised renal function, a third space effusion, or other causes, methotrexate serum concentrations may remain elevated for prolonged periods.
The potential for toxicity from high dose regimens or delayed excretion is reduced by the administration of leucovorin calcium during the final phase of methotrexate plasma elimination. Pharmacokinetic monitoring of methotrexate serum concentrations may help identify those patients at high risk for methotrexate toxicity and aid in proper adjustment of leucovorin dosing. Guidelines for monitoring serum methotrexate levels, and for adjustment of leucovorin dosing to reduce the risk of methotrexate toxicity, are provided below in DOSAGE AND ADMINISTRATION.
Methotrexate has been detected in human breast milk. The highest breast milk to plasma concentration ratio reached was 0.08:1.
-
INDICATIONS AND USAGE
Neoplastic Diseases
Methotrexate is indicated in the treatment of gestational choriocarcinoma, chorioadenoma destruens and hydatidiform mole.
Methotrexate is used in maintenance therapy in combination with other chemotherapeutic agents.
Methotrexate is used alone or in combination with other anticancer agents in the treatment of breast cancer, epidermoid cancers of the head and neck, advanced mycosis fungoides (cutaneous T cell lymphoma), and lung cancer, particularly squamous cell and small cell types. Methotrexate is also used in combination with other chemotherapeutic agents in the treatment of advanced stage non-Hodgkin’s lymphomas.
Psoriasis
Methotrexate is indicated in the symptomatic control of severe, recalcitrant, disabling psoriasis that is not adequately responsive to other forms of therapy, but only when the diagnosis has been established, as by biopsy and/or after dermatologic consultation. It is important to ensure that a psoriasis “flare” is not due to an undiagnosed concomitant disease affecting immune responses.
Rheumatoid Arthritis including Polyarticular-Course Juvenile Rheumatoid Arthritis
Methotrexate is indicated in the management of selected adults with severe, active, rheumatoid arthritis (ACR criteria), or children with active polyarticular-course juvenile rheumatoid arthritis, who have had an insufficient therapeutic response to, or are intolerant of, an adequate trial of first-line therapy including full dose non-steroidal anti-inflammatory agents (NSAIDs).
Aspirin, NSAIDs, and/or low dose steroids may be continued, although the possibility of increased toxicity with concomitant use of NSAIDs including salicylates has not been fully explored. (See PRECAUTIONS, Drug Interactions.) Steroids may be reduced gradually in patients who respond to methotrexate. Combined use of methotrexate with gold, penicillamine, hydroxychloroquine, sulfasalazine, or cytotoxic agents, has not been studied and may increase the incidence of adverse effects. Rest and physiotherapy as indicated should be continued.
-
CONTRAINDICATIONS
Methotrexate can cause fetal death or teratogenic effects when administered to a pregnant woman. Methotrexate is contraindicated in pregnant women with psoriasis or rheumatoid arthritis and should be used in the treatment of neoplastic diseases only when the potential benefit outweighs the risk to the fetus. Women of childbearing potential should not be started on methotrexate until pregnancy is excluded and should be fully counseled on the serious risk to the fetus (See PRECAUTIONS) should they become pregnant while undergoing treatment. Pregnancy should be avoided if either partner is receiving methotrexate; during and for a minimum of three months after therapy for male patients, and during and for at least one ovulatory cycle after therapy for female patients. (See Boxed WARNINGS.)
Because of the potential for serious adverse reactions from methotrexate in breast fed infants, it is contraindicated in nursing mothers.
Patients with psoriasis or rheumatoid arthritis with alcoholism, alcoholic liver disease or other chronic liver disease should not receive methotrexate.
Patients with psoriasis or rheumatoid arthritis who have overt or laboratory evidence of immunodeficiency syndromes should not receive methotrexate.
Patients with psoriasis or rheumatoid arthritis who have preexisting blood dyscrasias, such as bone marrow hypoplasia, leukopenia, thrombocytopenia or significant anemia, should not receive methotrexate.
Patients with a known hypersensitivity to methotrexate should not receive the drug.
- WARNINGS - SEE BOXED WARNINGS.
-
PRECAUTIONS
General
Methotrexate has the potential for serious toxicity. (See Boxed WARNINGS.) Toxic effects may be related in frequency and severity to dose or frequency of administration but have been seen at all doses. Because they can occur at any time during therapy, it is necessary to follow patients on methotrexate closely. Most adverse reactions are reversible if detected early. When such reactions do occur, the drug should be reduced in dosage or discontinued and appropriate corrective measures should be taken. If necessary, this could include the use of leucovorin calcium and/or acute, intermittent hemodialysis with a high-flux dialyzer. (See OVERDOSAGE.) If methotrexate therapy is reinstituted, it should be carried out with caution, with adequate consideration of further need for the drug and with increased alertness as to possible recurrence of toxicity.
The clinical pharmacology of methotrexate has not been well studied in older individuals. Due to diminished hepatic and renal function as well as decreased folate stores in this population, relatively low doses should be considered, and these patients should be closely monitored for early signs of toxicity.
Information for Patients
Patients should be informed of the early signs and symptoms of toxicity, of the need to see their physician promptly if they occur, and the need for close follow-up, including periodic laboratory tests to monitor toxicity.
Both the physician and pharmacist should emphasize to the patient that the recommended dose is taken weekly in rheumatoid arthritis and psoriasis, and that mistaken daily use of the recommended dose has led to fatal toxicity. Patients should be encouraged to read the Patient Instructions sheet within the Dose Pack. Prescriptions should not be written or refilled on a PRN basis.
Patients should be informed of the potential benefit and risk in the use of methotrexate. The risk of effects on reproduction should be discussed with both male and female patients taking methotrexate.
Laboratory Tests
Patients undergoing methotrexate therapy should be closely monitored so that toxic effects are detected promptly. Baseline assessment should include a complete blood count with differential and platelet counts, hepatic enzymes, renal function tests, and a chest X-ray. During therapy of rheumatoid arthritis and psoriasis, monitoring of these parameters is recommended: hematology at least monthly, renal function and liver function every 1 to 2 months. More frequent monitoring is usually indicated during antineoplastic therapy. During initial or changing doses, or during periods of increased risk of elevated methotrexate blood levels (e.g., dehydration), more frequent monitoring may also be indicated.
Transient liver function test abnormalities are observed frequently after methotrexate administration and are usually not cause for modification of methotrexate therapy. Persistent liver function test abnormalities, and/or depression of serum albumin may be indicators of serious liver toxicity and require evaluation. (See PRECAUTIONS, Organ System Toxicity, Hepatic.)
A relationship between abnormal liver function tests and fibrosis or cirrhosis of the liver has not been established for patients with psoriasis. Persistent abnormalities in liver function tests may precede appearance of fibrosis or cirrhosis in the rheumatoid arthritis population.
Pulmonary function tests may be useful if methotrexate-induced lung disease is suspected, especially if baseline measurements are available.
Drug Interactions
Concomitant administration of some NSAIDs with high dose methotrexate therapy has been reported to elevate and prolong serum methotrexate levels, resulting in deaths from severe hematologic and gastrointestinal toxicity.
Caution should be used when NSAIDs and salicylates are administered concomitantly with lower doses of methotrexate. These drugs have been reported to reduce the tubular secretion of methotrexate in an animal model and may enhance its toxicity.
Despite the potential interactions, studies of methotrexate in patients with rheumatoid arthritis have usually included concurrent use of constant dosage regimens of NSAIDs, without apparent problems. It should be appreciated, however, that the doses used in rheumatoid arthritis (7.5 to 20 mg/wk) are somewhat lower than those used in psoriasis and that larger doses could lead to unexpected toxicity.
Methotrexate is partially bound to serum albumin, and toxicity may be increased because of displacement by certain drugs, such as salicylates, phenylbutazone, phenytoin, and sulfonamides. Renal tubular transport is also diminished by probenecid; use of methotrexate with this drug should be carefully monitored.
Oral antibiotics such as tetracycline, chloramphenicol, and nonabsorbable broad spectrum antibiotics, may decrease intestinal absorption of methotrexate or interfere with the enterohepatic circulation by inhibiting bowel flora and suppressing metabolism of the drug by bacteria.
Penicillins may reduce the renal clearance of methotrexate; increased serum concentrations of methotrexate with concomitant hematologic and gastrointestinal toxicity have been observed with methotrexate. Use of methotrexate with penicillins should be carefully monitored.
The potential for increased hepatotoxicity when methotrexate is administered with other hepatotoxic agents has not been evaluated. However, hepatotoxicity has been reported in such cases. Therefore, patients receiving concomitant therapy with methotrexate and other potential hepatotoxins (e.g., azathioprine, retinoids, sulfa-salazine) should be closely monitored for possible increased risk of hepatotoxicity.
Methotrexate may decrease the clearance of theophylline; theophylline levels should be monitored when used concurrently with methotrexate.
Certain side effects such as mouth sores may be reduced by folate supplementation with methotrexate.
Trimethoprim/sulfa-methoxazole has been reported rarely to increase bone marrow suppression in patients receiving methotrexate, probably by an additive antifolate effect.
The use of nitrous oxide anesthesia potentiates the effect of methotrexate on folate-dependent metabolic pathways, resulting in the potential for increased toxicity such as stomatitis, myelosuppression, and neurotoxicity. Avoid concomitant nitrous oxide anesthesia in patients receiving methotrexate. Use caution when administering methotrexate after a recent history of nitrous oxide administration.
Carcinogenesis, Mutagenesis, and Impairment of Fertility
No controlled human data exist regarding the risk of neoplasia with methotrexate. Methotrexate has been evaluated in a number of animal studies for carcinogenic potential with inconclusive results. Although there is evidence that methotrexate causes chromosomal damage to animal somatic cells and human bone marrow cells, the clinical significance remains uncertain. Non-Hodgkin’s lymphoma and other tumors have been reported in patients receiving low-dose oral methotrexate. However, there have been instances of malignant lymphoma arising during treatment with low-dose oral methotrexate, which have regressed completely following withdrawal of methotrexate, without requiring active anti-lymphoma treatment. Benefits should be weighed against the potential risks before using methotrexate alone or in combination with other drugs, especially in pediatric patients or young adults. Methotrexate causes embryotoxicity, abortion, and fetal defects in humans. It has also been reported to cause impairment of fertility, oligospermia and menstrual dysfunction in humans, during and for a short period after cessation of therapy.
Pregnancy
Psoriasis and rheumatoid arthritis: Methotrexate is in Pregnancy Category X. See CONTRAINDICATIONS.
Pediatric Use
Safety and effectiveness in pediatric patients have been established only in cancer chemotherapy and in polyarticular-course juvenile rheumatoid arthritis.
Published clinical studies evaluating the use of methotrexate in children and adolescents (i.e., patients 2 to 16 years of age) with JRA demonstrated safety comparable to that observed in adults with rheumatoid arthritis. (See CLINICAL PHARMACOLOGY, ADVERSE REACTIONS and DOSAGE AND ADMINISTRATION.)
Geriatric Use
Clinical studies of methotrexate did not include sufficient numbers of subjects age 65 and over to determine whether they respond differently from younger subjects. In general, dose selection for an elderly patient should be cautious reflecting the greater frequency of decreased hepatic and renal function, decreased folate stores, concomitant disease or other drug therapy (i.e. that interfere with renal function, methotrexate or folate metabolism) in this population (See PRECAUTIONS, Drug Interactions.) Since decline in renal function may be associated with increases in adverse events and serum creatinine measurements may over estimate renal function in the elderly, more accurate methods (i.e., creatine clearance) should be considered. Serum methotrexate levels may also be helpful. Elderly patients should be closely monitored for early signs of hepatic, bone marrow and renal toxicity. In chronic use situations, certain toxicities may be reduced by folate supplementation. Post-marketing experience suggests that the occurrence of bone marrow suppression, thrombocytopenia, and pneumonitis may increase with age. See Boxed WARNINGS and ADVERSE REACTIONS.
Organ System Toxicity
Gastrointestinal: If vomiting, diarrhea, or stomatitis occur, which may result in dehydration, methotrexate should be discontinued until recovery occurs. Methotrexate should be used with extreme caution in the presence of peptic ulcer disease or ulcerative colitis.
Hematologic: Methotrexate can suppress hematopoiesis and cause anemia, aplastic anemia, pancytopenia, leukopenia, neutropenia, and/or thrombocytopenia. In patients with malignancy and preexisting hematopoietic impairment, the drug should be used with caution, if at all. In controlled clinical trials in rheumatoid arthritis (n=128), leukopenia (WBC <3000/mm 3) was seen in 2 patients, thrombocytopenia (platelets <100,000/mm 3) in 6 patients, and pancytopenia in 2 patients.
In psoriasis and rheumatoid arthritis, methotrexate should be stopped immediately if there is a significant drop in blood counts. In the treatment of neoplastic diseases, methotrexate should be continued only if the potential benefit warrants the risk of severe myelosuppression. Patients with profound granulocytopenia and fever should be evaluated immediately and usually require parenteral broad-spectrum antibiotic therapy.
Hepatic: Methotrexate has the potential for acute (elevated transaminases) and chronic (fibrosis and cirrhosis) hepatotoxicity. Chronic toxicity is potentially fatal; it generally has occurred after prolonged use (generally two years or more) and after a total dose of at least 1.5 grams. In studies in psoriatic patients, hepatotoxicity appeared to be a function of total cumulative dose and appeared to be enhanced by alcoholism, obesity, diabetes and advanced age. An accurate incidence rate has not been determined; the rate of progression and reversibility of lesions is not known. Special caution is indicated in the presence of preexisting liver damage or impaired hepatic function.
In psoriasis, liver function tests, including serum albumin, should be performed periodically prior to dosing but are often normal in the face of developing fibrosis or cirrhosis. These lesions may be detectable only by biopsy. The usual recommendation is to obtain a liver biopsy at 1) pretherapy or shortly after initiation of therapy (2 to 4 months), 2) a total cumulative dose of 1.5 grams, and 3) after each additional 1.0 to 1.5 grams. Moderate fibrosis or any cirrhosis normally leads to discontinuation of the drug; mild fibrosis normally suggests a repeat biopsy in 6 months. Milder histologic findings such as fatty change and low grade portal inflammation are relatively common pretherapy. Although these mild changes are usually not a reason to avoid or discontinue methotrexate therapy, the drug should be used with caution.
In rheumatoid arthritis, age at first use of methotrexate and duration of therapy have been reported as risk factors for hepatotoxicity; other risk factors, similar to those observed in psoriasis, may be present in rheumatoid arthritis but have not been confirmed to date. Persistent abnormalities in liver function tests may precede appearance of fibrosis or cirrhosis in this population. There is a combined reported experience in 217 rheumatoid arthritis patients with liver biopsies both before and during treatment (after a cumulative dose of at least 1.5 g) and in 714 patients with a biopsy only during treatment. There are 64 (7%) cases of fibrosis and 1 (0.1%) case of cirrhosis. Of the 64 cases of fibrosis, 60 were deemed mild. The reticulin stain is more sensitive for early fibrosis and its use may increase these figures. It is unknown whether even longer use will increase these risks.
Liver function tests should be performed at baseline and at 4 to 8 week intervals in patients receiving methotrexate for rheumatoid arthritis. Pretreatment liver biopsy should be performed for patients with a history of excessive alcohol consumption, persistently abnormal baseline liver function test values or chronic hepatitis B or C infection. During therapy, liver biopsy should be performed if there are persistent liver function test abnormalities or there is a decrease in serum albumin below the normal range (in the setting of well controlled rheumatoid arthritis).
If the results of a liver biopsy show mild changes (Roenigk grades I, II, IIIa), methotrexate may be continued and the patient monitored as per recommendations listed above. Methotrexate should be discontinued in any patient who displays persistently abnormal liver function tests and refuses liver biopsy or in any patient whose liver biopsy shows moderate to severe changes (Roenigk grade IIIb or IV).
Infection or Immunologic States: Methotrexate should be used with extreme caution in the presence of active infection, and is usually contraindicated in patients with overt or laboratory evidence of immunodeficiency syndromes. Immunization may be ineffective when given during methotrexate therapy. Immunization with live virus vaccines is generally not recommended. There have been reports of disseminated vaccinia infections after smallpox immunization in patients receiving methotrexate therapy.
Hypogammaglobulinemia has been reported rarely.
Potentially fatal opportunistic infections, especially Pneumocystis carinii pneumonia, may occur with methotrexate therapy. When a patient presents with pulmonary symptoms, the possibility of Pneumocystis carinii pneumonia should be considered.
Pulmonary: Pulmonary symptoms (especially a dry nonproductive cough) or a nonspecific pneumonitis occurring during methotrexate therapy may be indicative of a potentially dangerous lesion and require interruption of treatment and careful investigation. Although clinically variable, the typical patient with methotrexate induced lung disease presents with fever, cough, dyspnea, hypoxemia, and an infiltrate on chest X-ray; infection (including pneumonia) needs to be excluded. This lesion can occur at all dosages.
Renal: Methotrexate may cause renal damage that may lead to acute renal failure. Nephrotoxicity is due primarily to the precipitation of methotrexate and 7-hydroxymethotrexate in the renal tubules. Close attention to renal function including adequate hydration, urine alkalinization and measurement of serum methotrexate and creatinine levels are essential for safe administration.
Skin: Severe, occasionally fatal, dermatologic reactions, including toxic epidermal necrolysis, Stevens-Johnson syndrome, exfoliative dermatitis, skin necrosis, and erythema multiforme, have been reported in children and adults, within days of oral, intramuscular, intravenous, or intrathecal methotrexate administration. Reactions were noted after single or multiple, low, intermediate or high doses of methotrexate in patients with neoplastic and non-neoplastic diseases.
Other Precautions: Methotrexate should be used with extreme caution in the presence of debility.
Methotrexate exits slowly from third space compartments (e.g., pleural effusions or ascites). This results in a prolonged terminal plasma half-life and unexpected toxicity. In patients with significant third space accumulations, it is advisable to evacuate the fluid before treatment and to monitor plasma methotrexate levels.
Lesions of psoriasis may be aggravated by concomitant exposure to ultraviolet radiation. Radiation dermatitis and sunburn may be “recalled” by the use of methotrexate.
-
ADVERSE REACTIONS
IN GENERAL, THE INCIDENCE AND SEVERITY OF ACUTE SIDE EFFECTS ARE RELATED TO DOSE AND FREQUENCY OF ADMINISTRATION. THE MOST SERIOUS REACTIONS ARE DISCUSSED ABOVE UNDER ORGAN SYSTEM TOXICITY IN THE PRECAUTION SECTION. THAT SECTION SHOULD ALSO BE CONSULTED WHEN LOOKING FOR INFORMATION ABOUT ADVERSE REACTIONS WITH METHOTREXATE.
The most frequently reported adverse reactions include ulcerative stomatitis, leukopenia, nausea, and abdominal distress. Other frequently reported adverse effects are malaise, undue fatigue, chills and fever, dizziness and decreased resistance to infection.
Other adverse reactions that have been reported with methotrexate are listed below by organ system. In the oncology setting, concomitant treatment and the underlying disease make specific attribution of a reaction to methotrexate difficult.
Alimentary System: gingivitis, pharyngitis, stomatitis, anorexia, nausea, vomiting, diarrhea, hematemesis, melena, gastrointestinal ulceration and bleeding, enteritis, pancreatitis.
Blood and Lymphatic System Disorders: suppressed hematopoiesis causing anemia, aplastic anemia, pancytopenia, leukopenia, neutropenia and/or thrombocytopenia, lymphadenopathy and lymphoproliferative disorders (including reversible). Hypogammaglobulinemia has been reported rarely.
Cardiovascular: pericarditis, pericardial effusion, hypotension, and thromboembolic events (including arterial thrombosis, cerebral thrombosis, deep vein thrombosis, retinal vein thrombosis, thrombophlebitis, and pulmonary embolus).
Central Nervous System: headaches, drowsiness, blurred vision, transient blindness, speech impairment including dysarthria and aphasia, hemiparesis, paresis and convulsions have also occurred following administration of methotrexate. Following low doses, there have been occasional reports of transient subtle cognitive dysfunction, mood alteration, unusual cranial sensations, leukoencephalopathy, or encephalopathy.
Hepatobiliary: disorders, hepatotoxicity, acute hepatitis, chronic fibrosis and cirrhosis, decrease in serum albumin, liver enzyme elevations.
Infection: There have been case reports of sometimes fatal opportunistic infections in patients receiving methotrexate therapy for neoplastic and non-neoplastic diseases. Pneumocystis carinii pneumonia was the most common opportunistic infection. There have also been reports of infections, pneumonia, sepsis, nocardiosis, histoplasmosis, cryptococcosis, herpes zoster, H. simplex hepatitis, and disseminated H. simplex.
Musculoskeletal System: stress fracture.
Ophthalmic: conjunctivitis, serious visual changes of unknown etiology.
Pulmonary System: respiratory fibrosis, respiratory failure, interstitial pneumonitis; deaths have been reported, and chronic interstitial obstructive pulmonary disease has occasionally occurred.
Skin: erythematous rashes, pruritus, urticaria, photosensitivity, pigmentary changes, alopecia, ecchymosis, telangiectasia, acne, furunculosis, erythema multiforme, toxic epidermal necrolysis, Stevens-Johnson syndrome, skin necrosis, skin ulceration, and exfoliative dermatitis.
Urogenital System: severe nephropathy or renal failure, azotemia, cystitis, hematuria; defective oogenesis or spermatogenesis, transient oligospermia, menstrual dysfunction, vaginal discharge, and gynecomastia; infertility, abortion, fetal defects.
Other rarer reactions related to or attributed to the use of methotrexate such as nodulosis, vasculitis, arthralgia/myalgia, loss of libido/impotence, diabetes, osteoporosis, sudden death, reversible lymphomas, tumor lysis syndrome, soft tissue necrosis and osteonecrosis. Anaphylactoid reactions have been reported.
Adverse Reactions in Double-Blind Rheumatoid Arthritis Studies
The approximate incidences of methotrexate attributed (i.e., placebo rate subtracted) adverse reactions in 12 to 18 week double-blind studies of patients (n=128) with rheumatoid arthritis treated with low-dose oral (7.5 to 15 mg/week) pulse methotrexate, are listed below. Virtually all of these patients were on concomitant nonsteroidal anti-inflammatory drugs and some were also taking low dosages of corticosteroids. Hepatic histology was not examined in these short-term studies. (See PRECAUTIONS.)
Incidence greater than 10%:Elevated liver function tests 15%, nausea/vomiting 10%.
Incidence 3% to 10%:Stomatitis, thrombocytopenia, (platelet count less than 100,000/mm 3).
Incidence 1% to 3%:Rash/pruritus/dermatitis, diarrhea, alopecia, leukopenia (WBC less than 3000/mm 3), pancytopenia, dizziness.
Two other controlled trials of patients (n=680) with Rheumatoid Arthritis on 7.5 mg to 15 mg/wk oral doses showed an incidence of interstitial pneumonitis of 1%. (See PRECAUTIONS.)
Other less common reactions included decreased hematocrit, headache, upper respiratory infection, anorexia, arthralgias, chest pain, coughing, dysuria, eye discomfort, epistaxis, fever, infection, sweating, tinnitus, and vaginal discharge.
Adverse Reactions in Psoriasis
There are no recent placebo-controlled trials in patients with psoriasis. There are two literature reports (Roenigk, 1969 and Nyfors, 1978) describing large series (n=204, 248) of psoriasis patients treated with methotrexate. Dosages ranged up to 25 mg per week and treatment was administered for up to four years. With the exception of alopecia, photosensitivity, and “burning of skin lesions” (each 3% to 10%), the adverse reaction rates in these reports were very similar to those in the rheumatoid arthritis studies. Rarely, painful plaque erosions may appear.
Adverse Reactions in JRA Studies
The approximate incidences of adverse reactions reported in pediatric patients with JRA treated with oral, weekly doses of methotrexate (5 to 20 mg/m 2/wk or 0.1 to 0.65 mg/kg/wk) were as follows (virtually all patients were receiving concomitant nonsteroidal anti-inflammatory drugs, and some also were taking low doses of cortico-steroids): elevated liver function tests, 14%; gastrointestinal reactions (e.g., nausea, vomiting, diarrhea), 11%; stomatitis, 2%; leukopenia, 2%; headache, 1.2%; alopecia, 0.5%; dizziness, 0.2%; and rash, 0.2%. Although there is experience with dosing up to 30 mg/m 2/wk in JRA, the published data for doses above 20 mg/m 2/wk are too limited to provide reliable estimates of adverse reaction rates.
-
OVERDOSAGE
Leucovorin is indicated to diminish the toxicity and counteract the effect of inadvertently administered overdosages of methotrexate. Leucovorin administration should begin as promptly as possible. As the time interval between methotrexate administration and leucovorin initiation increases, the effectiveness of leucovorin in counteracting toxicity decreases. Monitoring of the serum methotrexate concentration is essential in determining the optimal dose and duration of treatment with leucovorin.
In cases of massive overdosage, hydration and urinary alkalinization may be necessary to prevent the precipitation of methotrexate and/or its metabolites in the renal tubules. Generally speaking, neither hemodialysis nor peritoneal dialysis has been shown to improve methotrexate elimination. However, effective clearance of methotrexate has been reported with acute, intermittent hemodialysis using a high-flux dialyzer (Wall, SM et al: Am J Kidney Dis28(6): 846-854, 1996).
In postmarketing experience, overdose with methotrexate has generally occurred with oral and intrathecal administration, although intravenous and intramuscular overdose have also been reported.
Reports of oral overdose often indicate accidental daily administration instead of weekly (single or divided doses). Symptoms commonly reported following oral overdose include those symptoms and signs reported at pharmacologic doses, particularly hematologic and gastrointestinal reaction. For example, leukopenia, thrombocytopenia, anemia, pancytopenia, bone marrow suppression, mucositis, stomatitis, oral ulceration, nausea, vomiting, gastrointestinal ulceration, gastrointestinal bleeding. In some cases, no symptoms were reported. There have been reports of death following overdose. In these cases, events such as sepsis or septic shock, renal failure, and aplastic anemia were also reported.
-
DOSAGE AND ADMINISTRATION
Neoplastic Diseases
Oral administration in tablet form is often preferred when low doses are being administered since absorption is rapid and effective serum levels are obtained.
Choriocarcinoma and similar trophoblastic diseases: Methotrexate is administered orally or intramuscularly in doses of 15 to 30 mg daily for a five-day course. Such courses are usually repeated for 3 to 5 times as required, with rest periods of one or more weeks interposed between courses, until any manifesting toxic symptoms subside. The effectiveness of therapy is ordinarily evaluated by 24 hour quantitative analysis of urinary chorionic gonadotropin (hCG), which should return to normal or less than 50 IU/24 hr usually after the third or fourth course and usually be followed by a complete resolution of measurable lesions in 4 to 6 weeks. One to two courses of methotrexate after normalization of hCG is usually recommended. Before each course of the drug careful clinical assessment is essential. Cyclic combination therapy of methotrexate with other antitumor drugs has been reported as being useful.
Since hydatidiform mole may precede choriocarcinoma, prophylactic chemotherapy with methotrexate has been recommended.
Chorioadenoma destruens is considered to be an invasive form of hydatidiform mole. Methotrexate is administered in these disease states in doses similar to those recommended for choriocarcinoma.
Leukemia: Acute lymphoblastic leukemia in pediatric patients and young adolescents is the most responsive to present day chemotherapy. In young adults and older patients, clinical remission is more difficult to obtain and early relapse is more common.
Methotrexate alone or in combination with steroids was used initially for induction of remission in acute lymphoblastic leukemias. More recently corticosteroid therapy, in combination with other antileukemic drugs or in cyclic combinations with methotrexate included, has appeared to produce rapid and effective remissions. When used for induction, methotrexate in doses of 3.3 mg/m 2 in combination with 60 mg/m 2 of prednisone, given daily, produced remissions in 50% of patients treated, usually within a period of 4 to 6 weeks. Methotrexate in combination with other agents appears to be the drug of choice for securing maintenance of drug-induced remissions. When remission is achieved and supportive care has produced general clinical improvement, maintenance therapy is initiated, as follows: Methotrexate is administered 2 times weekly either by mouth or intramuscularly in total weekly doses of 30 mg/m 2. It has also been given in doses of 2.5 mg/kg intravenously every 14 days. If and when relapse does occur, reinduction of remission can again usually be obtained by repeating the initial induction regimen.
A variety of combination chemotherapy regimens have been used for both induction and maintenance therapy in acute lymphoblastic leukemia. The physician should be familiar with the new advances in antileukemic therapy.
Lymphomas: In Burkitt’s tumor, Stages I-II, methotrexate has produced prolonged remissions in some cases. Recommended dosage is 10 to 25 mg/day orally for 4 to 8 days. In Stage III, methotrexate is commonly given concomitantly with other anti-tumor agents. Treatment in all stages usually consists of several courses of the drug interposed with 7 to 10 day rest periods. Lymphosarcomas in Stage III may respond to combined drug therapy with methotrexate given in doses of 0.625 to 2.5 mg/kg daily.
Mycosis Fungoides (cutaneous T cell lymphoma): Therapy with methotrexate as a single agent appears to produce clinical responses in up to 50% of patients treated. Dosage in early stages is usually 5 to 50 mg once weekly. Dose reduction or cessation is guided by patient response and hematologic monitoring. Methotrexate has also been administered twice weekly in doses ranging from 15 to 37.5 mg in patients who have responded poorly to weekly therapy.
Psoriasis, Rheumatoid Arthritis, and Juvenile Rheumatoid Arthritis
Adult Rheumatoid Arthritis: Recommended Starting Dosage Schedules
1. Single oral doses of 7.5 mg once weekly.
2. Divided oral dosages of 2.5 mg at 12 hour intervals for 3 doses given as a course once weekly.
Polyarticular-Course Juvenile Rheumatoid Arthritis: The recommended starting dose is 10 mg/m 2 given once weekly.
For either adult RA or polyarticular-course JRA dosages may be adjusted gradually to achieve an optimal response. Limited experience shows a significant increase in the incidence and severity of serious toxic reactions, especially bone marrow suppression, at doses greater than 20 mg/wk in adults. Although there is experience with doses up to 30 mg/m 2/wk in children, there are too few published data to assess how doses over 20 mg/m 2/wk might affect the risk of serious toxicity in children. Experience does suggest, however, that children receiving 20 to 30 mg/m 2/wk (0.65 to 1.0 mg/kg/wk) may have better absorption and fewer gastrointestinal side effects if methotrexate is administered either intramuscularly or subcutaneously.
Therapeutic response usually begins within 3 to 6 weeks and the patient may continue to improve for another 12 weeks or more.
The optimal duration of therapy is unknown. Limited data available from long-term studies in adults indicate that the initial clinical improvement is maintained for at least two years with continued therapy. When methotrexate is discontinued, the arthritis usually worsens within 3 to 6 weeks.
The patient should be fully informed of the risks involved and should be under constant supervision of the physician. (See Information for Patients under PRECAUTIONS.) Assessment of hematologic, hepatic, renal, and pulmonary function should be made by history, physical examination, and laboratory tests before beginning, periodically during, and before reinstituting methotrexate therapy. (See PRECAUTIONS.) Appropriate steps should be taken to avoid conception during methotrexate therapy. (See PRECAUTIONS and CONTRAINDICATIONS.)
All schedules should be continually tailored to the individual patient. An initial test dose may be given prior to the regular dosing schedule to detect any extreme sensitivity to adverse effects. (See ADVERSE REACTIONS.) Maximal myelosuppression usually occurs in seven to ten days.
Psoriasis: Recommended Starting Dose Schedules
1. Weekly single oral, IM or IV dose schedule: 10 to 25 mg per week until adequate response is achieved.
2. Divided oral dose schedule: 2.5 mg at 12-hour intervals for three doses.
Dosages in each schedule may be gradually adjusted to achieve optimal clinical response; 30 mg/week should not ordinarily be exceeded.
Once optimal clinical response has been achieved, each dosage schedule should be reduced to the lowest possible amount of drug and to the longest possible rest period. The use of methotrexate may permit the return to conventional topical therapy, which should be encouraged.
- HANDLING AND DISPOSAL
-
HOW SUPPLIED
Methotrexate Tablets USP contain an amount of methotrexate sodium equivalent to 2.5 mg of methotrexate, USP.
2.5 mg: Yellow, oval-shaped, scored tablet debossed with stylized b/572 on one side. Available in bottles of 30 tablets (NDC: 43063-439-30).
Dispense in a tight, light-resistant container as defined in the USP, with a child-resistant closure (as required).
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature].
Protect from light.
KEEP THIS AND ALL MEDICATIONS OUT OF THE REACH OF CHILDREN.
-
REFERENCES
1. Controlling occupational exposure to hazardous drugs (OSHA Work-Practice Guidelines). Am J Health Syst Pharm 1996; 53: 1669-1685.
2. National Study Commission on Cytotoxic Exposure - Recommendations for Handling Cytotoxic Agents. Available from Louis P. Jeffrey, Sc D, Chairman, National Study Commission on Cytotoxic Exposure, Massachusetts College of Pharmacy and Allied Health Sciences, 179 Longwood Avenue, Boston, Massachusetts 02115.
3. Clinical Oncological Society of Australia: Guidelines and recommendations for safe handling of antineoplastic agents. Med J Australia 1983; 1:426-428.
4. Jones RB, et al. Safe handling of chemotherapeutic agents: A report from the Mount Sinai Medical Center. CA - A Cancer Journal for Clinicians Sept/Oct 1983; 258-263.
5. American Society of Hospital Pharmacists technical assistance bulletin on handling cytotoxic and hazardous drugs. Am J Hosp Pharm 1990; 47:1033-1049.
Rev. C 3/2018
-
Patient Information
Methotrexate (meth” oh trex’ ate) Tablets USP
Read the Patient Instructions that come with methotrexate before you start using it and each time you get a refill. There may be new information. This leaflet does not take the place of talking with your doctor about your condition.
What is the most important information I should know about methotrexate?
- Methotrexate can cause serious side effects that may be life threatening (see "What are the possible or reasonably likely side effects of methotrexate?"). Most side effects can be found by medical tests before they become serious. Your doctor may do regular tests to check how methotrexate is affecting your body. It is important that you stay under a doctor's care while taking methotrexate. Call your doctor right away to report any side effects or symptoms you get.
- Methotrexate can cause birth defects or death of an unborn child. Therefore, if you are pregnant or your sexual partner is pregnant, or plans to become pregnant, do not take methotrexate. Neither you nor your partner should become pregnant while taking methotrexate. Women should wait at least 1 menstrual cycle after stopping treatment with methotrexate before getting pregnant. Men should wait at least 3 months after stopping treatment with methotrexate before getting their partner pregnant. Women who can become pregnant should have a pregnancy test before starting methotrexate. During treatment with methotrexate men whose partners and women who are able to get pregnant should use effective birth control.
What is methotrexate?
Methotrexate is a prescription medicine used in treating certain cancers, severe rheumatoid arthritis including polyarticular juvenile rheumatoid arthritis, and severe psoriasis.
Who should not take methotrexate?
Do not take methotrexate if:
- you are pregnant or planning to become pregnant. Methotrexate can cause birth defects or death to your unborn child. See ‘What is the most important information I should know about methotrexate?”
- you are breastfeeding. Methotrexate can harm your baby. You will need to decide either to breastfeed or to take methotrexate, but not both.
- you have any conditions that weaken your immune system (immunodeficiency conditions).
- your bone marrow does not make enough blood cells, or if you have low white blood cell counts, low platelet counts, or serious anemia.
- you drink alcohol or have liver problems from alcohol abuse.
- you have chronic liver disease.
- you are allergic to methotrexate or any of the ingredients in methotrexate. See the end of this leaflet for a complete list of ingredients in methotrexate.
Before using methotrexate tell your doctor:
- about all your medical problems including if you:
- have kidney problems or are getting dialysis treatments
- have liver problems
- have fluid in your stomach area (ascites)
- have lung problems or fluid in your lungs (pleural effusion)
- about all the medicines you take including prescription and nonprescription medicines, vitamins and herbal supplements. Methotrexate and certain other medicines can affect each other and cause serious side effects. Do not start or change any medicines unless you have talked to your doctor and your doctor has told you it is safe. Know all the medicines that you take and keep a list of them with you at all times to show doctors and pharmacists.
How should I take methotrexate?
- Take methotrexate exactly as prescribed by your doctor. Your dose of methotrexate and when you take it will depend on the condition that is being treated. Do not take more methotrexate than prescribed. Do not change your dose of methotrexate unless your doctor has told you to. For treatment of severe psoriasis, and severe rheumatoid arthritis including juvenile rheumatoid arthritis, methotrexate should be taken weekly, not every day. This weekly dose is taken either at one time or in several doses.
- If you miss a dose of methotrexate call your doctor to ask if you should take the dose or not.
- If you take too much methotrexate call your doctor or go to your nearest emergency room right away. You will need to take a medicine called an antidote as soon as possible.
- Call your doctor right away for further instructions if you get dehydrated (lose a large amount of body fluids). This can happen if you are sick and have a fever, vomiting, or diarrhea. Dehydration can also happen when you sweat a lot with activities or exercise and don't drink enough fluids.
- Stop taking methotrexate if you get diarrhea, or if you get sores in your mouth. Call your doctor right away. If you keep taking methotrexate with these symptoms, you could get serious bleeding or tearing of your digestive tract.
- Your doctor should do regular tests to monitor how methotrexate is affecting your body. Check with your doctor after having any blood tests before taking methotrexate again. Your doctor will tell you if it is safe to take more methotrexate.
- Certain side effects such as mouth sores may be reduced by folate supplementation with methotrexate.
What should I avoid while taking methotrexate?
Do not:
- get pregnant or try to become pregnant. See “What is the most important information I should know about methotrexate?”
- breastfeed. See "Who should not take methotrexate?"
- drink alcohol. Alcohol drinks, including beer and wine, may increase some of the side effects with methotrexate, including the chance of liver damage.
- take certain live virus vaccines.
What are the possible or reasonably likely side effects of methotrexate?
Methotrexate can cause serious and life-threatening problems including (see “What is the most important information I should know about methotrexate”?):
- birth defects and death of an unborn child
- serious anemia, lower white cells, red cells, and platelets in your blood
- liver damage
- kidney damage
- lung disease
- cancer of the lymph system (lymphoma)
- severe skin reactions and rashes
- opportunistic infections such as Pneumocystis carinii pneumonia
- soft tissue and bone damage if you are getting radiation therapy at the same time you are taking methotrexate
The most common side effects of methotrexate include:
- mouth sores
- low white blood cells
- nausea, upset stomach
- feeling poorly
- tiredness, chills, fever, dizziness
- higher chance for getting infections
- diarrhea
- vomiting
- hair loss
- easy bruising
Stop taking methotrexate and call your doctor right away if you get diarrhea, mouth sores, a fever, dehydration, cough, bleeding, shortness of breath, any signs of infection, or a skin rash. If you have any questions about these or other side effects, talk to your doctor. These are not all the side effects of methotrexate. Ask your doctor or pharmacist for more information.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store methotrexate?
- Store methotrexate at room temperature between 68° to 77°F (20° to 25°C).
- Keep methotrexate away from light.
- Keep methotrexate and all medicines out of the reach of children.
General information about methotrexate tablets
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use methotrexate for a condition for which it was not prescribed. Do not give methotrexate to other people, even if they have the same symptoms that you have. It may harm them.
This leaflet summaries the most important information about methotrexate. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about methotrexate that is written for healthcare professionals.
For additional information, please contact TEVA PHARMACEUTICALS USA, INC. at our toll-free number: 1-888-838-2872.
What are the ingredients in methotrexate?
Active Ingredient: methotrexate sodium
Inactive Ingredients: hydroxypropyl methylcellulose, lactose anhydrous, magnesium stearate, microcrystalline cellulose, polyethylene glycol, pregelatinized corn starch, propylene glycol, sodium carbonate monohydrate and talc.
TEVA PHARMACEUTICALS USA, INC.
North Wales, PA 19454This Patient Information has been approved by the U.S. Food and Drug Administration. Iss. 2/2016
- Package/Label Display Panel
-
INGREDIENTS AND APPEARANCE
METHOTREXATE
methotrexate tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 43063-439(NDC:0555-0572) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength METHOTREXATE SODIUM (UNII: 3IG1E710ZN) (METHOTREXATE - UNII:YL5FZ2Y5U1) METHOTREXATE 2.5 mg Inactive Ingredients Ingredient Name Strength HYPROMELLOSES (UNII: 3NXW29V3WO) ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) STARCH, CORN (UNII: O8232NY3SJ) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) SODIUM CARBONATE MONOHYDRATE (UNII: 2A1Q1Q3557) TALC (UNII: 7SEV7J4R1U) Product Characteristics Color yellow Score 2 pieces Shape OVAL Size 8mm Flavor Imprint Code b;572 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 43063-439-30 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 08/22/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA081099 11/01/1990 Labeler - PD-Rx Pharmaceuticals, Inc. (156893695) Registrant - PD-Rx Pharmaceuticals, Inc. (156893695) Establishment Name Address ID/FEI Business Operations PD-Rx Pharmaceuticals, Inc. 156893695 repack(43063-439)

© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.