ADEMPAS- riociguat tablet, film coated
Adempas by
Drug Labeling and Warnings
Adempas by is a Prescription medication manufactured, distributed, or labeled by Bayer HealthCare Pharmaceuticals Inc., Sharp Corporation, Bayer AG. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ADEMPAS safely and effectively. See full prescribing information for ADEMPAS.
ADEMPAS (riociguat) tablets, for oral use
Initial U.S. Approval: 2013WARNING: EMBRYO-FETAL TOXICITY
See full prescribing information for complete boxed warning.
- Do not administer Adempas to a pregnant female because it may cause fetal harm. (4.1, 5.1, 8.1)
- Females of reproductive potential: Exclude pregnancy before start of treatment, monthly during treatment, and 1 month after treatment discontinuation. Prevent pregnancy during treatment and for one month after treatment discontinuation by use of effective forms of contraception. (2.3, 5.1,5.2, 8.6)
- For females, Adempas is available only through a restricted program called the Adempas REMS Program. (5.1,5.2)
INDICATIONS AND USAGE
Adempas is a soluble guanylate cyclase (sGC) stimulator indicated for the treatment of adults with:
- Persistent/recurrent Chronic Thromboembolic Pulmonary Hypertension (CTEPH) (WHO Group 4) after surgical treatment or inoperable CTEPH to improve exercise capacity and WHO functional class. (1.1)
- Pulmonary Arterial Hypertension (PAH) (WHO Group 1) to improve exercise capacity, improve WHO functional class and to delay clinical worsening. (1.2)
DOSAGE AND ADMINISTRATION
- Initiate treatment at 1 mg taken three times a day. (2.1)
- For patients who may not tolerate the hypotensive effect of Adempas, consider a starting dose of 0.5 mg, three times a day. (2.1)
- Increase dosage by 0.5 mg at intervals of no sooner than 2-weeks as tolerated to a maximum of 2.5 mg three times a day. (2.1)
- Tablets may be crushed and mixed with water or soft foods for patients who have difficulty swallowing. (2.1)
DOSAGE FORMS AND STRENGTHS
Tablets: 0.5 mg, 1 mg, 1.5 mg, 2 mg and 2.5 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
Adverse reactions occurring more frequently (≥3%) on Adempas compared to placebo are headache, dyspepsia/gastritis, dizziness, nausea, diarrhea, hypotension, vomiting, anemia, gastroesophageal reflux, and constipation. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Bayer HealthCare Pharmaceuticals Inc. at 1-888-842-2937 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
- Nursing mothers: Discontinue drug or breastfeeding. (8.3)
- Renal impairment: Not recommended in patients with creatinine clearance <15 mL/min or on dialysis. (8.7)
- Hepatic impairment: Not recommended in patients with severe (Child Pugh C) hepatic impairment. (8.8)
- Smoking: May require dosages higher than 2.5 mg three times a day if tolerated. Dose decrease may be required in patients who stop smoking. (2.4, 7.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 1/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: EMBRYO-FETAL TOXICITY
1 INDICATIONS AND USAGE
1.1 Chronic-Thromboembolic Pulmonary Hypertension
1.2 Pulmonary Arterial Hypertension
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Adult Patients
2.2 Dosage Interruption
2.3 Pregnancy Testing in Females of Reproductive Potential
2.4 Use in Patients who Smoke
2.5 Strong CYP and P-gp/BCRP Inhibitors
2.6 Transitioning to and from Adempas
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Pregnancy
4.2 Nitrates and Nitric Oxide Donors
4.3 Phosphodiesterase Inhibitors
4.4 Pulmonary Hypertension Associated with Idiopathic Interstitial Pneumonias (PH-IIP)
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
5.2 Adempas REMS Program
5.3 Hypotension
5.4 Bleeding
5.5 Pulmonary Veno-Occlusive Disease
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Pharmacodynamic Interactions with Adempas
7.2 Pharmacokinetic Interactions with Adempas
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology
14 CLINICAL STUDIES
14.1 Chronic-Thromboembolic Pulmonary Hypertension
14.2 Pulmonary Arterial Hypertension
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: EMBRYO-FETAL TOXICITY
Do not administer Adempas to a pregnant female because it may cause fetal harm [see Contraindications (4.1), Warnings and Precautions (5.1) and Use in Specific Populations (8.1)].
Females of reproductive potential: Exclude pregnancy before the start of treatment, monthly during treatment, and 1 month after stopping treatment. To prevent pregnancy, females of reproductive potential must use effective forms of contraception during treatment and for one month after stopping treatment by using effective forms of contraception [see Dosage and Administration (2.3), Warnings and Precautions (5.1, 5.2),and Use in Specific Populations (8.3)].
For all female patients, Adempas is available only through a restricted program called the Adempas Risk Evaluation and Mitigation Strategy (REMS) Program [see Warnings and Precautions (5.1, 5.2)].
-
1 INDICATIONS AND USAGE
1.1 Chronic-Thromboembolic Pulmonary Hypertension
Adempas is indicated for the treatment of adults with persistent/recurrent chronic thromboembolic pulmonary hypertension (CTEPH), (WHO Group 4) after surgical treatment, or inoperable CTEPH, to improve exercise capacity and WHO functional class [see Clinical Studies (14.1)].
1.2 Pulmonary Arterial Hypertension
Adempas is indicated for the treatment of adults with pulmonary arterial hypertension (PAH), (WHO Group 1), to improve exercise capacity, WHO functional class and to delay clinical worsening.
Efficacy was shown in patients on Adempas monotherapy or in combination with endothelin receptor antagonists or prostanoids. Studies establishing effectiveness included predominately patients with WHO functional class II–III and etiologies of idiopathic or heritable PAH (61%) or PAH associated with connective tissue diseases (25%) [see Clinical Studies (14.2)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Adult Patients
The recommended starting dosage is 1 mg taken 3 times a day. For patients who may not tolerate the hypotensive effect of Adempas, consider a starting dose of 0.5 mg taken three times a day. If systolic blood pressure remains greater than 95 mmHg and the patient has no signs or symptoms of hypotension, up-titrate the dose by 0.5 mg taken three times a day. Dose increases should be no sooner than 2 weeks apart. The dose can be increased to the highest tolerated dosage, up to a maximum of 2.5 mg taken three times a day. If at any time, the patient has symptoms of hypotension, decrease the dosage by 0.5 mg taken three times a day.
Crushed Tablets
For patients who are unable to swallow whole tablets, Adempas may be crushed and mixed with water or soft foods (such as applesauce) immediately before administration [see Clinical Pharmacology (12.3)].
2.2 Dosage Interruption
If a dose is missed, advise patients to continue with the next regularly scheduled dose.
In case Adempas is interrupted for 3 days or more, re-titrate Adempas.
2.3 Pregnancy Testing in Females of Reproductive Potential
Obtain pregnancy tests prior to start oftreatment and monthly during treatment [see Use in Specific Populations (8.3)].
2.4 Use in Patients who Smoke
Consider titrating to dosages higher than 2.5 mg three times a day, if tolerated, in patients who smoke. A dose decrease may be required in patients who stop smoking [see Drug Interactions (7.2) and Clinical Pharmacology (12.3)].
2.5 Strong CYP and P-gp/BCRP Inhibitors
Consider a starting dose of 0.5 mg, three times a day when initiating Adempas in patients receiving strong cytochrome P450 (CYP) and P-glycoprotein/breast cancer resistance protein (P-gp/BCRP) inhibitors such as azole antimycotics (for example, ketoconazole, itraconazole) or HIV protease inhibitors (for example, ritonavir). Monitor for signs and symptoms of hypotension on initiation and on treatment with strong CYP and P-gp/BCRP inhibitors [see Warnings and Precautions (5.3), Drug Interactions (7.2) and Clinical Pharmacology (12.3)].
2.6 Transitioning to and from Adempas
- Discontinue sildenafil at least 24 hours prior to administering Adempas [see Contraindications (4.3) and Drug Interactions (7)].
- Discontinue tadalafil at least 48 hours prior to administering Adempas [see Contraindications (4.3) and Drug Interactions (7)]. Consider initiating Adempas at a starting dose of 0.5 mg in patients at risk of hypotension [see Dosage and Administration (2.1)]. Monitor for signs and symptoms of hypotension on initiation.
- Discontinue Adempas at least 24 hours prior to administering a PDE5-inhibitor [see Dosage and Administration (2.1), Contraindications (4.3), and Drug Interactions (7)]. Monitor for signs and symptoms of hypotension on initiation.
-
3 DOSAGE FORMS AND STRENGTHS
Tablets: film-coated, round, bi-convex:
- 0.5 mg, white, with “BAYER” cross on one side and “0.5” and “R” on the other side
- 1 mg, pale-yellow, with “BAYER” cross on one side and “1” and “R” on the other side
- 1.5 mg, yellow-orange, with “BAYER” cross on one side and “1.5” and “R” on the other side
- 2 mg, pale orange, with “BAYER” cross on one side and “2” and “R” on the other side
- 2.5 mg, red-orange, with “BAYER” cross on one side and “2.5” and “R” on the other side
-
4 CONTRAINDICATIONS
4.1 Pregnancy
Based on data from animal reproduction studies, Adempas may cause fetal harm when administered to a pregnant woman and is contraindicated in females who are pregnant. Adempas was consistently shown to have teratogenic effects when administered to animals. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus [see Use in Specific Populations (8.1)].
4.2 Nitrates and Nitric Oxide Donors
Co-administration of Adempas with nitrates or nitric oxide donors (such as amyl nitrite) in any form is contraindicated [see Drug Interactions (7.1) and Clinical Pharmacology (12.2)].
4.3 Phosphodiesterase Inhibitors
Concomitant administration of Adempas with specific PDE-5 inhibitors (such as sildenafil, tadalafil, or vardenafil) or nonspecific PDE 5 inhibitors (such as dipyridamole or theophylline) is contraindicated [see Dosage and Administration (2.6), Drug Interactions (7.1) and Clinical Pharmacology (12.2)]. Do not administer within 24 hours of sildenafil. Do not administer 24 hours before or within 48 hours after tadalafil.
-
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
Based on data from animal reproduction studies, Adempas may cause embryo-fetal toxicity when administered to a pregnant female and is contraindicated in females who are pregnant. Advise females of reproductive potential of the potential risk to a fetus. Obtain a pregnancy test before the start of treatment, monthly during treatment, and for one month after stopping treatment. Advise females of reproductive potential to use effective contraception during treatment with ADEMPAS and for at least one month after the last dose [see Dosage and Administration (2.3) and Use in Specific Populations (8.1, 8.3)].
For females, Adempas is only available through a restricted program under the Adempas REMS Program [see Warnings and Precautions (5.2)].
5.2 Adempas REMS Program
Females can only receive Adempas through the Adempas Risk Evaluation and Mitigation Strategy (REMS) Program, a restricted distribution program [see Warnings and Precautions (5.1)].
Important requirements of the Adempas REMS Program include the following:
- Prescribers must be certified with the program by enrolling and completing training.
- All females, regardless of reproductive potential, must enroll in the Adempas REMS Program prior to initiating Adempas. Male patients are not enrolled in the Adempas REMS Program.
- Female patients of reproductive potential must comply with the pregnancy testing and contraception requirements [see Use in Specific Populations (8.3)].
- Pharmacies must be certified with the program and must only dispense to patients who are authorized to receive Adempas.
Further information, including a list of certified pharmacies, is available at www.AdempasREMS.com or 1-855-4 ADEMPAS.
5.3 Hypotension
Adempas reduces blood pressure. Consider the potential for symptomatic hypotension or ischemia in patients with hypovolemia, severe left ventricular outflow obstruction, resting hypotension, autonomic dysfunction, or concomitant treatment with antihypertensives or strong CYP and P-gp/BCRP inhibitors [see Drug Interactions (7.2) and Clinical Pharmacology (12.3)]. Consider a dose reduction if patient develops signs or symptoms of hypotension.
5.4 Bleeding
In the placebo-controlled clinical trials, serious bleeding occurred in 2.4% of patients taking Adempas compared to 0% of placebo patients. Serious hemoptysis occurred in 5 (1%) patients taking Adempas compared to 0 placebo patients, including one event with fatal outcome. Serious hemorrhagic events also included 2 patients with vaginal hemorrhage, 2 with catheter site hemorrhage, and 1 each with subdural hematoma, hematemesis, and intra-abdominal hemorrhage.
5.5 Pulmonary Veno-Occlusive Disease
Pulmonary vasodilators may significantly worsen the cardiovascular status of patients with pulmonary veno-occlusive disease (PVOD). Therefore, administration of Adempas to such patients is not recommended. Should signs of pulmonary edema occur, the possibility of associated PVOD should be considered and, if confirmed, discontinue treatment with Adempas.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed elsewhere in the labeling:
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.1)]
- Hypotension [see Warnings and Precautions (5.3)]
- Bleeding [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data described below reflect exposure to Adempas in two, randomized, double blind, placebo-controlled trials in patients with inoperable or recurrent/persistent CTEPH (CHEST-1) and treatment naive or pre-treated PAH patients (PATENT-1). The population (Adempas: n = 490; Placebo: n = 214) was between the age of 18 and 80 years [see Clinical Studies (14.1, 14.2)].
The safety profile of Adempas in patients with inoperable or recurrent/persistent CTEPH (CHEST-1) and treatment naive or pre-treated PAH (PATENT-1) were similar. Therefore, adverse drug reactions (ADRs) identified from the 12 and 16 week placebo-controlled trials for PAH and CTEPH respectively were pooled, and those occurring more frequently on Adempas than placebo (≥3%) are displayed in Table 1 below. Most adverse reactions in Table 1 can be ascribed to the vasodilatory mechanism of action of Adempas.
The overall rates of discontinuation due to an adverse event in the pivotal placebo-controlled trials were 2.9% for Adempas and 5.1% for placebo (pooled data).
Table 1: Adverse Reactions Occurring More Frequently (≥3%) on Adempas than Placebo (Pooled from CHEST-1 and PATENT-1) Adverse Reactions
Adempas %
(n=490)
Placebo %
(n=214)
Headache
27
18
Dyspepsia and Gastritis
21
8
Dizziness
20
13
Nausea
14
11
Diarrhea
12
8
Hypotension
10
4
Vomiting
10
7
Anemia
(including laboratory parameters)7
2
Gastroesophageal reflux disease
5
2
Constipation
5
1
Other events that were seen more frequently in Adempas compared to placebo and potentially related to treatment were: palpitations, nasal congestion, epistaxis, dysphagia, abdominal distension and peripheral edema. With longer observation in uncontrolled long-term extension studies the safety profile was similar to that observed in the placebo controlled phase 3 trials.
-
7 DRUG INTERACTIONS
7.1 Pharmacodynamic Interactions with Adempas
Nitrates: Co-administration of Adempas with nitrates or nitric oxide donors (such as amyl nitrite) in any form is contraindicated because of hypotension [see Contraindications (4.2) and Clinical Pharmacology (12.2)].
PDE Inhibitors: Co-administration of Adempas with specific PDE-5 inhibitors (such as sildenafil, tadalafil, or vardenafil) and nonspecific PDE inhibitors (such as dipyridamole or theophylline), is contraindicated because of hypotension. Do not administer within 24 hours of sildenafil. Do not administer 24 hours before or within 48 hours after tadalafil [see Dosage and Administration (2.6)]. Clinical experience with co-administration of Adempas and other phosphodiesterase inhibitors (for example, milrinone, cilostazole, roflumilast) is limited.
7.2 Pharmacokinetic Interactions with Adempas
Smoking: Plasma concentrations in smokers are reduced by 50% to 60% compared to nonsmokers. Based on pharmacokinetic modeling, for patients who are smokers, doses higher than 2.5 mg three times a day may be considered in order to match exposure seen in nonsmoking patients. Safety and effectiveness of Adempas doses higher than 2.5 mg three times a day have not been established. A dose reduction should be considered in patients who stop smoking [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)].
Strong CYP and P-gp/BCRP inhibitors: Concomitant use of riociguat with strong cytochrome CYP inhibitors and P-gp/BCRP inhibitors such as azole antimycotics (for example, ketoconazole, itraconazole) or HIV protease inhibitors (such as ritonavir) increase riociguat exposure and may result in hypotension. Consider a starting dose of 0.5 mg 3 times a day when initiating Adempas in patients receiving strong CYP and P-gp/BCRP inhibitors. Monitor for signs and symptoms of hypotension on initiation and on treatment with strong CYP and P-gp/BCRP inhibitors. A dose reduction should be considered in patients who may not tolerate the hypotensive effect of riociguat [see Dosage and Administration (2.5), Warnings and Precautions (5.3) and Clinical Pharmacology (12.3)].
Strong CYP3A inducers: Strong inducers of CYP3A (for example, rifampin, phenytoin, carbamazepine, phenobarbital or St. John’s Wort) may significantly reduce riociguat exposure. Data are not available to guide dosing of riociguat when strong CYP3A inducers are co-administered. [see Clinical Pharmacology (12.3)].
Antacids: Antacids such as aluminum hydroxide/magnesium hydroxide decrease riociguat absorption and should not be taken within 1 hour of taking Adempas [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on data from animal reproduction studies, Adempas may cause embryo-fetal toxicity and miscarriage when administered to a pregnant woman and is contraindicated during pregnancy [see Contraindications (4.1)]. There are limited available data with ADEMPAS use in pregnant women. In animal reproduction studies, oral administration of riociguat to pregnant rats during organogenesis was teratogenic and embryotoxic at exposures approximately 8 times and 2 times, respectively, the human exposure. In reproduction studies with pregnant rabbits, oral administration of riociguat during organogenesis caused abortions and fetal toxicity at exposures approximately 4 times and 13 times, respectively, the maximum recommended human dose (MRHD). Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Data
Animal Data
In rats administered riociguat orally (1, 5, and 25 mg/kg/day) throughout organogenesis, an increased rate of cardiac ventricular-septal defect was observed at the highest dose tested. The highest dose produced evidence of maternal toxicity (reduced body weight). Post-implantation loss was statistically significantly increased from the mid-dose of 5 mg/kg/day. Plasma exposure at the lowest dose in which no adverse effects were observed is approximately 0.4 times that in humans at the maximally recommended human dose (MRHD) of 2.5 mg three times a day based on area under the time-concentration curve (AUC) for unbound drug in rat and humans. Plasma exposure at the highest dose (25 mg/kg/day) is approximately 8 times that in humans at the MRHD while exposure at the mid-dose (5 mg/kg/day) is approximately 2 times that in humans at the MRHD. In rabbits given doses of 0.5, 1.5 and 5 mg/kg/day, an increase in spontaneous abortions was observed starting at the middle dose of 1.5 mg/kg, and an increase in resorptions was observed at 5 mg/kg/day. Plasma exposures at these doses were 4 times and 13 times, respectively, the human exposure at the MRHD.
8.2 Lactation
Risk Summary
There are no data on the presence of riociguat in human milk, the effects on the breastfed infant, or the effect on milk production. Riociguat is present in rat milk. Because of the potential for serious adverse reactions from ADEMPAS, such as hypotension, in breastfed infants, advise women not to breastfeed during treatment with ADEMPAS.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Female patients of reproductive potential must have a negative pregnancy test prior to starting treatment with Adempas, monthly during treatment, and one month after discontinuation of treatment with Adempas. Advise patients to contact their healthcare provider if they become pregnant or suspect they may be pregnant. Counsel patients on the risk to the fetus [see Boxed Warning, Dosage and Administration (2.3) and Use in Specific Populations (8.1)].
Contraception
Females
Female patients of reproductive potential must use acceptable methods of contraception during treatment with Adempas and for 1 month after treatment with Adempas. Patients may choose one highly effective form of contraception (intrauterine devices [IUD], contraceptive implants or tubal sterilization) or a combination of methods (hormone method with a barrier method or two barrier methods). If a partner’s vasectomy is the chosen method of contraception, a hormone or barrier method must be used along with this method. Counsel patients on pregnancy planning and prevention, including emergency contraception, or designate counseling by another healthcare provider trained in contraceptive counseling [see Boxed Warning].
8.4 Pediatric Use
Safety and effectiveness of Adempas in pediatric patients have not been established [see Nonclinical Toxicology (13.2)].
8.5 Geriatric Use
Of the total number of subjects in clinical studies of Adempas, 23% were 65 and over, and 6% were 75 and over [see Clinical Studies (14)]. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Elderly patients showed a higher exposure to Adempas [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
Safety and efficacy have not been demonstrated in patients with creatinine clearance <15 mL/min or on dialysis [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Safety and efficacy have not been demonstrated in patients with severe hepatic impairment (Child Pugh C) [see Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
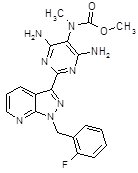
Adempas (riociguat) is a tablet for oral administration. Riociguat is methyl 4,6-diamino-2-[1-(2-fluorobenzyl)-1H-pyrazolo [3,4-b]pyridin-3-yl]-5-pyrimidinyl(methyl)carbamate with the following structural formula:
C20H19FN8O2
Riociguat is a white to yellowish, crystalline, non-hygroscopic substance with a molecular weight of 422.42 g/mol. In solid form it is stable to temperature, light, and humidity.
The solubility at 25°C in water: 4 mg/L, in ethanol: 800 mg/L, in 0.1 HCl (pH 1): 250 mg/L and in buffer (phosphate) pH 7: 3 mg/L. In the pH range of 2 to 4 the solubility showed strong pH-dependency. Solubility increases at lower pH values.
Each round film-coated tablet contains 0.5 mg (1.0, 1.5, 2.0, 2.5 mg) riociguat. The inactive ingredients are cellulose microcrystalline, crospovidone, hypromellose 5cP, lactose monohydrate, magnesium stearate, sodium laurylsulfate, hydroxypropylcellulose, hypromellose 3cP, propylene glycol, and titanium dioxide. Adempas 1, 1.5, 2 and 2.5 mg tablets contain, in addition, ferric oxide yellow. Adempas 2 and 2.5 mg tablets contain, in addition, ferric oxide red..
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Riociguat is a stimulator of soluble guanylate cyclase (sGC), an enzyme in the cardiopulmonary system and the receptor for nitric oxide (NO).
When NO binds to sGC, the enzyme catalyzes synthesis of the signaling molecule cyclic guanosine monophosphate (cGMP). Intracellular cGMP plays an important role in regulating processes that influence vascular tone, proliferation, fibrosis and inflammation.
Pulmonary hypertension is associated with endothelial dysfunction, impaired synthesis of nitric oxide and insufficient stimulation of the NO-sGC-cGMP pathway.
Riociguat has a dual mode of action. It sensitizes sGC to endogenous NO by stabilizing the NO-sGC binding. Riociguat also directly stimulates sGC via a different binding site, independently of NO.
Riociguat stimulates the NO-sGC-cGMP pathway and leads to increased generation of cGMP with subsequent vasodilation.
The active metabolite (M1) of riociguat is 1/3 to 1/10 as potent as riociguat.
12.2 Pharmacodynamics
There is a direct relationship between riociguat plasma concentration and hemodynamic parameters such as systemic vascular resistance, systolic blood pressure, pulmonary vascular resistance (PVR), and cardiac output [see Clinical Studies (14)].
Hemodynamic parameters were assessed in CTEPH patients in CHEST-1 [see Clinical Studies (14.1)]. Right heart catheterization was performed at the beginning and the end of the study period in 233 patients. A statistically significant reduction of PVR (-246 dyn*s*cm-5) was shown in the Adempas group vs. placebo. Improvements in other hemodynamic parameters (not pre-specified as endpoints) are displayed in Table 2 below.
Table 2: CHEST-1, Change In Hemodynamic Parameters from Baseline to Last Visit (Individual Dose Titration to Maximum 2.5 mg Three Times a Day versus placebo) Parameter (unit)
- Mean change
LS mean difference
95% CI
Adempas
Placebo
Pulmonary Capillary Wedge Pressure (mmHg)
0.59
0.18
0.58
–0.36 to 1.53
Right Atrial Pressure (mmHg)
–1.04
–0.55
–0.55
–1.72 to 0.62
Pulmonary Arterial Pressure
Systolic (mmHg)–6.84
0.95
–7.52
–10.88 to –4.16
Pulmonary Arterial Pressure
Diastolic (mmHg)–3.05
0.67
–3.62
–5.30 to –1.95
Pulmonary Arterial Pressure
Mean (mmHg)–4.31
0.76
–4.96
–6.75 to –3.16
Mean Arterial Pressure (mmHg)
–9.27
–0.29
–9.15
–11.83 to –6.46
Mixed Venous Oxygen Saturation (%)
2.95
–0.44
3.85
1.46 to 6.25
Cardiac Output (L/min)
0.81
–0.03
0.86
0.59 to 1.12
Cardiac Index (L/min/m2)
0.45
–0.01
0.47
0.33 to 0.62
Pulmonary Vascular Resistance
(dyn*s*cm-5)–226
23.1
–246
–303 to –190
Pulmonary Vascular Resistance Index (dyn*s*cm-5*m2)
–397
48.3
–449
–554 to –344
Systemic Vascular Resistance
(dyn*s*cm-5)–445
16.6
–478
–602 to –354
Systemic Vascular Resistance Index
(dyn*s*cm-5*m2)–799
53.7
–914
–1141 to –687
Hemodynamic parameters were assessed in PAH patients in PATENT-1 [see Clinical Studies (14.2)]. Right heart catheterization was performed at the beginning and the end of the study period in 339 patients.
A statistically significant reduction of PVR (-226 dyn*sec*cm-5) was shown in the Adempas individual titration group (to maximum dose of 2.5 mg three times a day) vs. placebo. Improvement in other relevant hemodynamic parameters (not pre-specified as endpoints) for the individual dose titration group versus placebo are displayed in Table 3.
Table 3: PATENT-1, Change in Hemodynamic Parameters from Baseline to Last Visit (Individual Dose Titration to Maximum 2.5 mg Three Times a Day versus Placebo) - Parameter (unit)
Mean change
LS mean difference
95% CI
Adempas
Placebo
Pulmonary Capillary Wedge Pressure (mmHg)
1.08
0.46
0.41
–0.36 to 1.18
Right Atrial Pressure (mmHg)
–0.20
0.97
–1.01
–2.15 to 0.13
Pulmonary Arterial Pressure
Systolic (mmHg)–5.39
0.78
–6.73
–9.43 to –4.04
Pulmonary Arterial Pressure Diastolic (mmHg)
–3.19
–1.12
–2.41
–4.15 to –0.68
Pulmonary Arterial Pressure
mean (mmHg)–3.93
–0.5
–3.83
–5.61 to –2.06
Mean Arterial Pressure (mmHg)
–8.54
–1.4
–7.25
–9.6 to –4.90
Mixed Venous Oxygen Saturation (%)
3.15
–2.33
5.02
3.2 to 6.84
Cardiac Output (L/min)
0.93
–0.01
0.93
0.7 to 1.15
Cardiac Index (L/min/m2)
0.54
–0.02
0.56
0.44 to 0.69
Pulmonary Vascular Resistance
(dyn*s*cm-5)–223
–8.9
–226
–281 to –170
Pulmonary Vascular Resistance Index
(dyn*s*cm-5*m2)–374
–22.4
–377
–469 to –285
Systemic Vascular Resistance
(dyn*s*cm-5)–448
–67.5
–395
–473 to –316
Systemic Vascular Resistance Index
(dyn*s*cm-5*m2)–753
–130
–675
–801 to –550
Biomarkers
In the CHEST-1 study, Adempas significantly reduced N-terminal prohormone of brain natriuretic peptide (NT-proBNP), placebo-corrected mean change from baseline -444 ng/L, 95% CI -843 to -45. In the PATENT-1 study Adempas demonstrated a statistically significant reduction of NT-proBNP, placebo‑corrected mean change from baseline: -432 ng/L, 95% CI –782 to –82.
Pharmacodynamic interactions
Nitrates: Riociguat 2.5 mg tablets potentiated the blood pressure lowering effect of sublingual nitroglycerin (0.4 mg) taken 4 and 8 hours after riociguat. Syncope was reported in some patients [see Contraindications (4.2)].
Phosphodiesterase-5 inhibitors: In an exploratory interaction study in 7 patients with PAH on stable sildenafil treatment (20 mg three times a day), single doses of riociguat (0.5 mg and 1 mg sequentially) showed additive hemodynamic effects.
Among patients with PAH on stable sildenafil treatment (20 mg, three times a day) and riociguat (1 to 2.5 mg, three times a day) there was one death, possibly related to the combination of these drugs, and a high rate of discontinuation for hypotension [see Contraindications (4.3)].
Warfarin: Concomitant administration of riociguat and warfarin did not alter prothrombin time.
Acetylsalicylic Acid: Concomitant use of riociguat and aspirin did not affect bleeding time or platelet aggregation.
12.3 Pharmacokinetics
Riociguat pharmacokinetics are dose proportional from 0.5 to 2.5 mg. Inter-individual variability of riociguat exposure (AUC) across all doses is approximately 60%, and within-subject variability is approximately 30%.
Absorption and distribution
The absolute bioavailability of riociguat is about 94%. Peak plasma riociguat concentrations were observed within 1.5 hours after tablet intake. Food does not affect the bioavailability of riociguat.
Bioavailability (AUC and Cmax) of riociguat administered orally as a crushed tablet suspended in applesauce or in water is similar to that of a whole tablet.
The volume of distribution at steady state is approximately 30 L. Plasma protein binding in humans is approximately 95%, with serum albumin and α1–acidic glycoprotein being the main binding components.
Riociguat is a substrate of P-gp and BCRP.
Metabolism and excretion
Riociguat is mainly cleared through metabolism by CYP1A1, CYP3A4, CYP3A5, and CYP2J2. Formation of the major active metabolite, M1, is catalyzed by CYP1A1, which is inducible by polycyclic aromatic hydrocarbons such as those present in cigarette smoke. M1 is further metabolized to the inactive N-glucuronide. Plasma concentrations of M1 in patients with PAH are about half those for riociguat.
Following oral administration of radiolabeled riociguat in healthy individuals, about 40 and 53% of the total radioactivity was recovered in urine and feces, respectively. There appears to be considerable variability in the proportion of metabolites and unchanged riociguat excreted, but metabolites were the major components of the dose excreted in most individuals.
Average systemic clearance of riociguat was about 1.8 L/h in patients with PAH and about 3.4 L/h in healthy subjects. The terminal elimination half-life is about 12 hours in patients and 7 hours in healthy subjects.
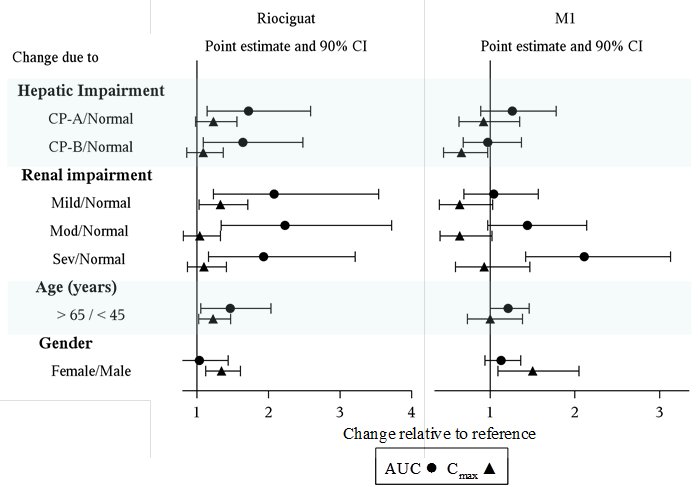
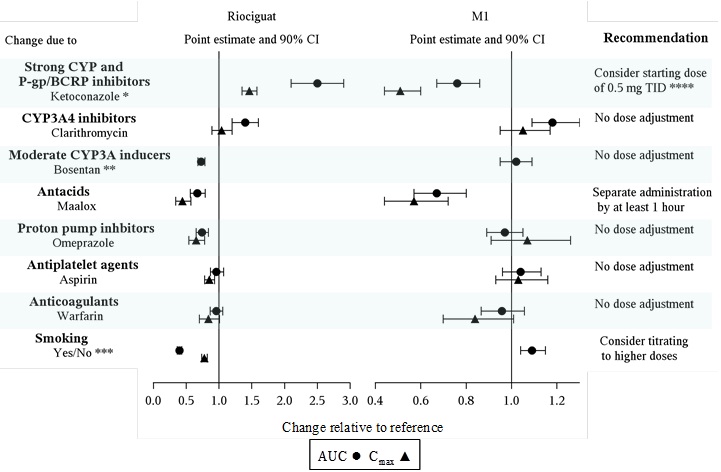
Specific Populations: The effect of intrinsic factors on riociguat and M1 are shown below in Figure 1. There are no clinically relevant effects of age, sex, weight, or race/ethnicity on the pharmacokinetics of riociguat or M1. No dose adjustment is warranted.
Drug interactions: The effect of extrinsic factors on riociguat and M1 were studied in healthy subjects and are shown in Figure 2
*HIV protease inhibitors are strong CYP3A inhibitors and may increase riociguat plasma concentrations to levels similar to those seen with ketoconazole. ** AUC only, estimated using population pharmacokinetics methods *** AUC only for metabolite, estimated using population pharmacokinetics methods. **** Monitor for signs and symptoms of hypotension on initiation and on treatment with strong CYP and P-gp/BCRP inhibitors [see Dosage and Administration (2.4, 2.5), Warnings and Precautions (5.3) and Drug Interactions (7.2)].
Strong CYP3A inducers: Data are not available to inform dosing of riociguat when strong CYP3A inducers are co-administered [see Drug Interactions (7.2)].
Effects of Riociguat on other Drugs: Riociguat did not affect the pharmacokinetics of midazolam, warfarin, or sildenafil [see Contraindications (4.3) and Clinical Pharmacology (12.2)].
Riociguat (2.5 mg three times per day) did not affect the systemic exposure of combined oral contraceptives containing levonorgestrel and ethinyl estradiol when concomitantly administered to healthy female subjects.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis: Carcinogenicity studies of riociguat were conducted in mice and rats. In mice, oral administration of riociguat (up to 25 mg/kg/day in males and 32 mg/kg/day in females) for up to two years did not demonstrate evidence of carcinogenesis. Plasma exposure (AUC) of unbound riociguat at the highest dose was 6 times the human’s exposure.
In rats, oral administration of riociguat (up to 20 mg/kg/day) for up to two years did not demonstrate evidence of carcinogenesis. Plasma exposure (AUC) of unbound riociguat at the highest dose was 7 times the human exposure
Mutagenesis: Riociguat and M1 did not show genotoxic potential in the in vitro bacterial reverse mutation (Ames) assay, the in vitro chromosomal aberration assay in Chinese hamster V79 cells, or the in vivo micronucleus assay in the mouse.
Impairment of fertility: In rats, no effects on male or female fertility were observed.
In male rats, oral administration of riociguat (up to 30 mg/kg/day) prior to and throughout the mating period had no effect on fertility. The no-effect dose for adverse effects is 37 times the human exposure when based on body surface area.
In female rats, oral administration of riociguat (up to 30 mg/kg/day) prior to and during mating and continuing to gestation Day 7 had no effect on fertility. The no-effect dose for adverse effects is 37 times the human exposure when based on body surface area.
13.2 Animal Toxicology
In growing rats, effects on bone formation were observed, including thickening of the growth plates, disorganized trabecular bone, and diffuse hyperostosis [see Use in Specific Populations (8.4)].
-
14 CLINICAL STUDIES
14.1 Chronic-Thromboembolic Pulmonary Hypertension
A double-blind, multi-national, multi-center, study (CHEST-1) was conducted in 261 patients with CTEPH. Patients were included if they:
- were technically inoperable for pulmonary endarterectomy, with PVR >300 dyn*sec*cm-5 and mean pulmonary artery pressure >25 mmHg measured at least 90 days after the start of full anticoagulation, or
- had recurrent or persisting pulmonary hypertension defined as PVR > 300 dyn*sec*cm-5 measured at least 180 days following pulmonary endarterectomy.
Patients were randomized to Adempas titrated up to 2.5 mg three times a day (n=173) or placebo (n=88). All patients were initiated at 1 mg three times a day. Patients with systolic blood pressure < 95 mmHg were excluded from the study. The dose of riociguat was titrated every 2 weeks based on the patient’s systolic blood pressure and signs or symptoms of hypotension. Stable dosages of oral anticoagulants, diuretics, digitalis, calcium channel blockers and oxygen were allowed, but not concomitant therapy with NO donors, endothelin receptor antagonists, prostacyclin analogues (PCA), specific PDE-5 inhibitors (such as, sildenafil, tadalafil, or vardenafil), and nonspecific phosphodiesterase inhibitors (for example, dipyridamole or theophylline).
The primary endpoint of the study was change from baseline in six minute walking distance (6MWD) after 16 weeks. The mean age of the patients enrolled was 59 years (range 18–80 years). In the study, 72% of patients had inoperable CTEPH, 28% had recurrent or persisting pulmonary hypertension following pulmonary endarterectomy. The majority of patients had a World Health Organization (WHO) Functional Class II (31%) or III (64%) at baseline. The mean baseline 6MWD was 347 meters. In the study, 77% of patients were titrated to the maximum dose of 2.5 mg three times a day; 13%, 6%, 4%, and 1% of patients were titrated to riociguat doses of 2, 1.5, 1, and 0.5 mg three times a day, respectively.
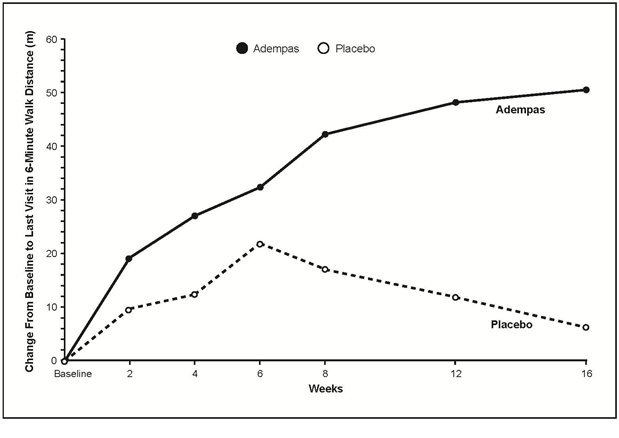
Results of the 6MWD over 16 weeks for the CHEST-1 study are shown in Figure 3.
The pre-specified primary endpoint of the study was the change in 6MWD from baseline to week 16 and was based on imputed values. The imputation for missing values included last observed value, not including follow-up for patients who completed the study or withdrew. For deaths or clinical worsening without a termination visit or a measurement at that visit, the imputed worst value (zero) was used.
Improvements in walking distance were apparent from Week 2 onward. At Week 16, the placebo adjusted mean increase in 6MWD within the Adempas group was 46 m (95% confidence interval [CI]: 25 m to 67 m; p<0.0001). For CHEST-1, the median difference (Hodges-Lehmann non-parametric estimate) in 6MWD was 39 m (95% CI, 25 m to 54 m).
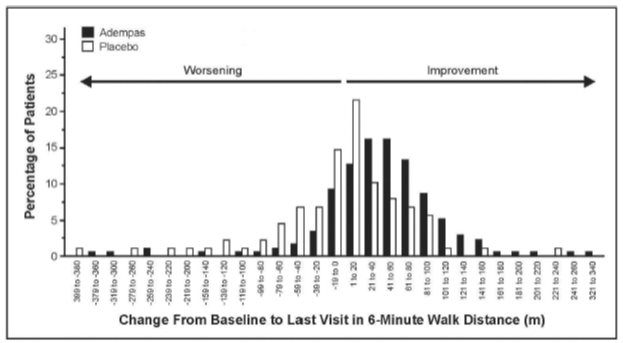
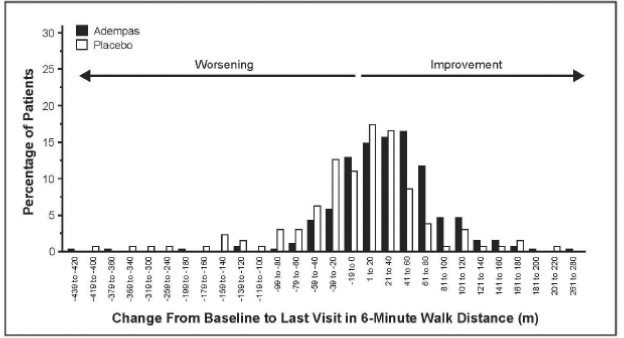
Figure 4 illustrates the results of the Adempas and placebo treatment groups displayed as a histogram summarizing the treatment effect on the 6MWD. The patients are grouped by change in 20 meters from baseline. Overall this figure shows that patients treated with Adempas benefit compared to those treated with placebo. As demonstrated in Figure 4, 143 patients receiving Adempas (83%) experienced an improvement in 6MWD compared to 50 patients (57%) on placebo.
Placebo-adjusted changes in 6MWD at 16 weeks were evaluated in subgroups (see Figure 5).
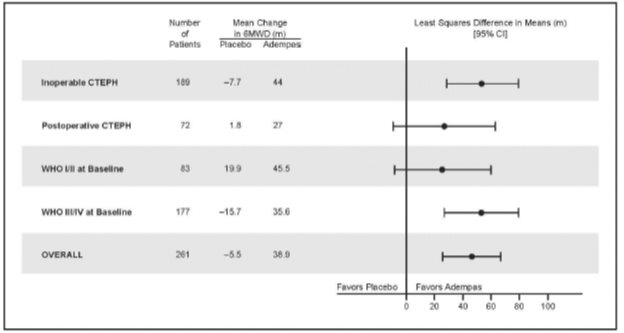
Figure 5: Mean Treatment Difference in Change from Baseline to Last Visit in 6-Minute Walk Distance (meters) by Prespecified Subgroups
WHO Functional Class improvements in the CHEST-1 trial are shown in Table 4.
Table 4: Effects of Adempas on the Change in WHO Functional Class in CHEST-1 from Baseline to Week 16 Change in WHO Functional Class
Adempas (n=173)
Placebo (n=87)
Improved
57 (33%)
13 (15%)
Stable
107 (62%)
68 (78%)
Deteriorated
9 (5%)
6 (7%)
p-value=0.0026
Long Term Treatment of CTEPH
An open-label extension study (CHEST-2) included 237 patients who had completed CHEST-1. At the cut-off date in the CHEST-2 study, the mean treatment duration for the total population was 1077 days (± 433). The probabilities of survival at 1 and 2 years were 97% and 93%, respectively. Without a control group, however, these data must be interpreted cautiously.
14.2 Pulmonary Arterial Hypertension
A double-blind, multi-national, multi-center study (PATENT-1) was conducted in 443 patients with PAH as defined by PVR >300 dyn*sec*cm-5 and a PAP mean >25 mmHg.
Patients were randomized to one of three treatment groups: Adempas titrated up to 1.5 mg (n=63), 2.5 mg (n=254) or placebo (n=126) three times a day. Patients with systolic blood pressure < 95 mmHg were excluded from the study. Patients assigned to Adempas were initiated at 1.0 mg three times a day. The dose of Adempas was up-titrated every 2 weeks based on the patient’s systolic blood pressure and signs or symptoms of hypotension. Oral anticoagulants, diuretics, digitalis, calcium channel blockers, and oxygen were allowed. In this study, 50% of the patients were treatment-naive with respect to PAH therapy, 44% were pre-treated with an endothelin receptor antagonist (ERA) and 6% were pre-treated with a PCA (inhaled, oral or subcutaneous). Pre-treated patients were defined as patients on stable treatment for 3 months with either an ERA or PCA; Adempas was added in combination to these background therapies.
The primary endpoint of the study was change from baseline and placebo in 6MWD after 12 weeks in the 2.5 mg group. The mean age of all patients was 51 years and approximately 80% were female. PAH etiologies were either idiopathic (61%) or familial PAH (2%), PAH associated with connective tissue disease (25%), congenital heart disease (8%), portal hypertension (3%), or anorexigen or amphetamine use (1%). The majority of patients had a WHO Functional Class II (42%) or III (54%) at baseline. The overall mean baseline 6MWD was 363 meters. Approximately 75% of patients were up-titrated to receive the maximum dose of 2.5 mg three times a day by week 12; 15%, 6%, 3%, and 2% were titrated to doses of 2, 1.5, 1, and 0.5 mg 3 times a day, respectively.
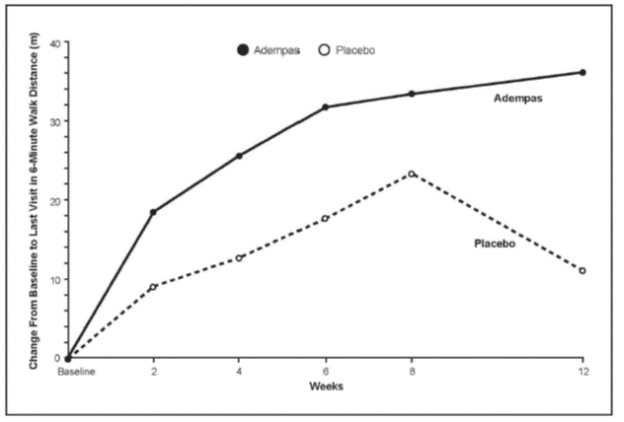
Results of the 6MWD over 12 weeks for the PATENT-1 study are shown in Figure 6.
The pre-specified primary endpoint of the study was the change in 6MWD from baseline to week 12 and was based on imputed values. The imputation for missing values included last observed value, not including follow-up for patients who completed the study or withdrew. In case of death or clinical worsening without a termination visit or a measurement at that termination visit, the imputed worst value (zero) was used.
Figure 7 illustrates the results of the Adempas and placebo treatment groups displayed as a histogram summarizing the treatment effect on the 6MWD. The patients are grouped by change in 20 meters from baseline. Overall this figure shows that patients treated with Adempas benefit compared to those treated with placebo. As demonstrated in Figure 7, 193 patients receiving Adempas (76%) experienced an improvement in 6MWD compared to 74 patients (59%) on placebo.
Improvements 6MWD were apparent from Week 2 onward. At Week 12, the placebo-adjusted mean increase in 6MWD within the Adempas group was 36 m (95% CI: 20 m to 52 m; p<0.0001). For PATENT-1, the median difference (Hodges-Lehmann non-parametric estimate) in 6MWD was 29 m (95% CI, 17 m to 40 m). There was an exploratory 1.5 mg capped titration arm (n = 63). The data did not suggest incremental benefit from escalating dose from 1.5 mg three times a day to 2.5 mg three times a day.
Placebo-adjusted changes in 6MWD at 12 weeks were evaluated in subgroups (see Figure 8).
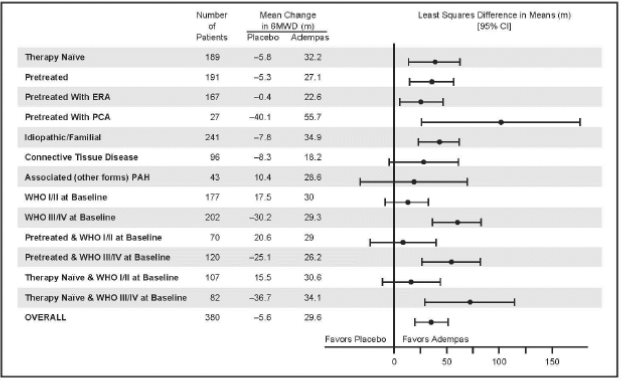
Figure 8: PATENT-1 Mean Treatment Difference in Change from Baseline to Last Visit in 6-Minute Walk Distance (meter) by Prespecified Subgroups
WHO Functional Class improvements in the IDT (individual dose titration) arm of the PATENT-1 trial are shown in Table 5
Table 5: Effects of Adempas on the Change in WHO Functional Class in PATENT-1 from Baseline to Week 12 Change in WHO Functional Class
Adempas (IDT) (n=254)
Placebo (n=125)
Improved
53 (21%)
18 (14%)
Stable
192 (76%)
89 (71%)
Deteriorated
9 (4%)
18 (14%)
p-value = 0.0033
Time to clinical worsening was a combined endpoint defined as death (all-cause mortality), heart/lung transplantation, atrial septostomy, hospitalization due to persistent worsening of pulmonary hypertension, start of new PAH-specific treatment, persistent decrease in 6MWD and persistent worsening of WHO Functional Class.
Effects of Adempas in PATENT-1 on events of clinical worsening are shown in Table 6.
Table 6: Effects of Adempas in PATENT-1 on Events of Clinical Worsening (ITT analysis set) Clinical Worsening Events
Adempas (IDT) (n=254)
Placebo (n=126)
Patients with any clinical worsening*
3 (1.2%)
8 (6.3%)
- Death
2 (0.8%)
3 (2.4%)
- Hospitalizations due to PH
1 (0.4%)
4 (3.2%)
- Decrease in 6MWD due to PH
1 (0.4%)
2 (1.6%)
- Persistent worsening of FC due to PAH
0
1 (0.8%)
- Start of new PAH treatment
1 (0.4%)
5 (4.0%)
* p-value=0.0285 (Mantel-Haenszel estimate)
Note: Patients may have had more than one event of clinical worsening
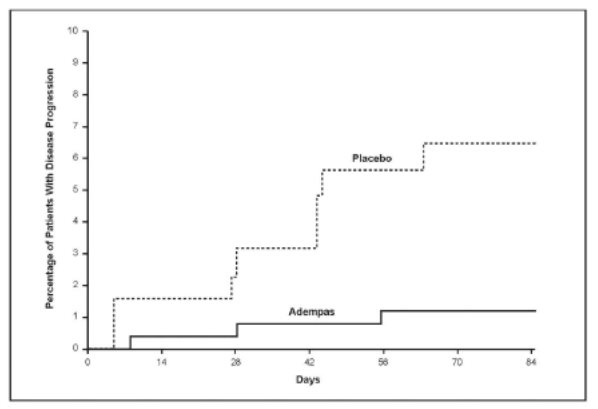
Adempas-treated patients experienced a significant delay in time to clinical worsening versus placebo-treated patients (p=0.0046; Stratified log-rank test). Significantly fewer events of clinical worsening up to week 12 (last visit) were observed in patients treated with Adempas (1.2%) compared to placebo (6.3%) (p=0.0285, Mantel-Haenszel estimate).
The Kaplan-Meier plot of time to clinical worsening is presented in Figure 9.
Long Term Treatment of PAH
An open label extension study (PATENT-2) included 363 patients who had completed PATENT-1. At the cut-off date in the PATENT-2 study, the mean treatment duration for the total population was 663 days (± 319). The probabilities of survival at 1 and 2 years were 97% and 93%, respectively. Without a control group, these data must be interpreted cautiously.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Adempas (riociguat) tablets are film-coated, round, and debossed with the “Bayer cross” on one side.
Color
Debossing
Side 2
NDC 50419-xxx-xx
Bottle of 9
Bottle of 90
Blister of 42
0.5 mg
White
0.5 R
250-91
250-01
250-03
1 mg
Pale yellow
1 R
251-91
251-01
251-03
1.5 mg
Yellow-orange
1.5 R
252-91
252-01
252-03
2 mg
Pale orange
2 R
253-91
253-01
253-03
2.5 mg
Red-orange
2.5 R
254-91
254-01
254-03
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Embryo-Fetal Toxicity
Instruct patients on the risk of fetal harm when Adempas is used during pregnancy [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1)]. Instruct females of reproductive potential to use effective contraception and to contact her healthcare provider immediately if they suspect they may be pregnant. Female patients must enroll in the Adempas REMS Program.
Adempas REMS Program
For female patients, Adempas is available only through a restricted program called the Adempas REMS Program [see Warnings and Precautions (5.2)]. Male patients are not enrolled in the Adempas REMS Program.
Inform female patients (and their guardians, if applicable) of the following important requirements:
- All female patients must sign an enrollment form.
- Advise female patients of reproductive potential that she must comply with the pregnancy testing and contraception requirements [see Use in Specific Populations (8.3)].
- Educate and counsel females of reproductive potential on the use of emergency contraception in the event of unprotected sex or contraceptive failure.
- Advise pre-pubertal females to report any changes in their reproductive status immediately to her prescriber.
Review the Medication Guide and REMS educational materials with female patients.
Lactation
Advise women not to breastfeed during treatment with ADEMPAS [see Use in Specific Populations (8.2)].
Other Risks Associated with Adempas
- Inform patients of the contraindication of Adempas with nitrates or nitric oxide donors or PDE-5 inhibitors.
- Advise patients about the potential risks/signs of hemoptysis and to report any potential signs of hemoptysis to their physicians.
- Instruct patients on the dosing, titration, and maintenance of Adempas.
- Advise patients regarding activities that may impact the pharmacology of Adempas (strong multi pathway CYP inhibitors and P-gp/BCRP inhibitors and smoking). Instruct patients to report all current medications and new medications to their physician.
- Advise patients that antacids should not be taken within 1 hour of taking Adempas.
- Inform patients that Adempas can cause dizziness, which can affect the ability to drive and use machines [see Adverse Reactions (6.1)]. Advise patients to be aware of how they react to Adempas, before driving or operating machinery and if needed, consult their physician. Patients should consult their physicians if dizziness gets worse with Adempas.
-
MEDICATION GUIDE
MEDICATION GUIDE
Adempas (a dem pahs)
(riociguat)tablets
Read this Medication Guide before you start taking Adempas and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking to your doctor about your medical condition or your treatment.
What is the most important information I should know about Adempas?
- Serious birth defects.
- Adempas can cause serious birth defects if taken during pregnancy.
- Females must not be pregnant when they start taking Adempas or become pregnant during treatment with Adempas.
-
Females who are able to get pregnant must have a negative pregnancy test before beginning treatment with Adempas, each month during treatment, and 1 month after you stop treatment with Adempas. Talk to your doctor about your menstrual cycle. Your doctor will decide when to do the tests, and order the tests for you depending on your menstrual cycle.
- Females who are able to get pregnant are females who:
- Have entered puberty, even if they have not started their period, and
-
Have a uterus,and
- Have not gone through menopause (have not had a period for at least 12 months for natural reasons, or who have had their ovaries removed)
- Females who are not able to get pregnant are females who:
- Have not yet entered puberty, or
- Do not have a uterus, or
- Have gone through menopause (have not had a period for at least 12 months for natural reasons, or who have had their ovaries removed)
Females who are able to get pregnant must use 2 acceptable forms of birth control, during treatment with Adempas and for 1 month after stopping Adempas because the medicine may still be in the body.
-
If you have had a tubal sterilization, have a progesterone implant, or have an IUD (intrauterine device), these methods can be used alone and no other form of birth control is needed.
- Talk with your doctor or gynecologist (a doctor who specializes in female reproduction) to find out about options for acceptable birth control that you may use to prevent pregnancy during treatment with Adempas.
- If you decide that you want to change the form of birth control that you use, talk with your doctor or gynecologist to be sure that you choose another acceptable form of birth control.
See the chart below for Acceptable Birth Control Options during treatment with Adempas.
- Do not have unprotected sex. Talk to your doctor or pharmacist right away if you have unprotected sex or if you think your birth control has failed. Your doctor may talk with you about using emergency birth control.
- Tell your doctor right away if you miss a menstrual period or think you may be pregnant for any reason.
If you are the parent or caregiver of a female child who started taking Adempas before reaching puberty, you should check your child regularly to see if she is developing signs of puberty. Tell your doctor right away if you notice that she is developing breast buds or any pubic hair. Your doctor should decide if your child has reached puberty. Your child may reach puberty before having her first menstrual period.
Females can only receive Adempas through a restricted program called the Adempas Risk Evaluation and Mitigation Strategies (REMS) Program. If you are a female who can become pregnant, you must talk to your doctor, understand the benefits and risks of Adempas, and agree to all of the instructions in the Adempas REMS Program.
Males can receive Adempas without taking part in the Adempas REMS Program.
What is Adempas?
Adempas is a prescription medicine used to treat adults with:
-
chronic thromboembolic pulmonary hypertension (CTEPH)
- treated with surgery but who continue to have high pulmonary blood pressure (persistent) or it comes back after surgery (recurrent), or
- that cannot be treated with surgery.
- CTEPH is a type of high blood pressure in the arteries of your lungs caused by blood clots that narrow or block blood flow. Adempas can improve your ability to exercise and can help to improve some of your symptoms.
- pulmonary arterial hypertension (PAH)
- PAH is a type of high blood pressure in the arteries of your lungs. Adempas can improve your ability to exercise, improve some of your symptoms, and help slow down the worsening of your physical condition.
- It is unknown if Adempas is safe and effective in children.
Who should not take Adempas?
Do not take Adempas if:
- you are pregnant, plan to become pregnant, or become pregnant during treatment with Adempas. Adempas can cause serious birth defects. (See the Medication Guide section above called "What is the most important information I should know about Adempas?")
-
you take:
- a nitrate medicine to treat high blood pressure or heart disease, such as nitroglycerin, or a medicine called a nitric oxide donor, such as amyl nitrite
- certain other medicines that contain sildenafil (Revatio or Viagra), tadalafil (Adcirca or Cialis), vardenafil (Levitra or Staxyn), dipyridamole, or theophylline. Revatio and Adcirca are also used to treat PAH
- you have pulmonary hypertension associated with idiopathic interstitial pneumonias (PH-IIP).
Ask your doctor or pharmacist if you are not sure if you take any of the medicines listed above.
What should I tell my doctor before taking Adempas?
Before you take Adempas, tell your doctor if you:
- smoke
- have recently had serious bleeding from your lung, or if you have had a medical procedure called bronchial arterial embolization to stop you from coughing up blood
- have problems with your heart or blood circulation
- have low blood pressure
- have liver problems
- have kidney problems or are on dialysis
- have narrowing of the pulmonary veins, a condition called pulmonary veno-occlusive disease or PVOD
- have any other medical conditions
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. Adempas and other medicines may affect each other causing side effects. Do not start any new medicine until you check with your doctor.
How should I take Adempas?
Adempas will be provided to you by a certified pharmacy. Your doctor will give you complete details.
- Do not take Adempas within 24 hours of sildenafil. Do not take Adempas 24 hours before or within 48 hours after tadalafil.
- Take Adempas exactly as your doctor tells you. Do not stop taking Adempas or change your dose without talking to your doctor.
- When you begin treatment with Adempas, your blood pressure should be monitored about every 2 weeks to help your doctor decide the correct dose of medicine for you.
- Your doctor may change your dose during treatment, especially when you first start taking Adempas. It is important to tell your doctor if you have any symptoms of low blood pressure during this time, such as dizziness, lightheadedness, or fainting.
- Take Adempas 3 times each day, about 6 to 8 hours apart.
- Take Adempas with or without food.
- Do not take more than a total of 7.5 mg of Adempas in 1 day unless your doctor tells you to.
- If you take a heartburn medicine (antacid) that contains aluminum hydroxide or magnesium hydroxide, do not take it within 1 hour of taking Adempas.
- If you take too much Adempas, call your doctor right away or go to the nearest hospital emergency room.
- If you miss a dose, take your next dose of Adempas at the regular time.
- If you miss 3 or more days of treatment with Adempas, call your doctor for instructions before you restart Adempas.
What should I avoid while taking Adempas?
- Do not get pregnant while taking Adempas. (See serious birth defects section of the Medication Guide above called "What is the most important information I should know about Adempas?") If you miss a menstrual period, or think you might be pregnant, call your doctor right away.
- It is not known if Adempas passes into your breast milk. You should not breastfeed if you take Adempas. Talk to your doctor about the best way to feed your baby if you take Adempas.
- Adempas may make you feel dizzy. Do not drive, operate machinery, or do other activities that require mental alertness or coordination until you know how Adempas affects you. Talk with your doctor if you are concerned about when it is safe for you to do these activities.
- Smoking. Adempas may not work as well if you smoke during treatment. Tell your doctor if you stop smoking or start smoking during treatment with Adempas, because your dose of Adempas may need to be changed.
What are the possible side effects of Adempas?
Adempas can cause serious side effects including:
- Serious birth defects. (See "What is the most important information I should know about Adempas?")
- Reduced blood pressure. Adempas reduces blood pressure. This may cause symptoms of low blood pressure, such as lightheadedness, chest pain, and dizziness especially in people who are dehydrated, or have a severe blockage of blood flow out of the heart, or have certain other medical problems. Your doctor will check you for these problems.
- Increased risk of bleeding, including bleeding from the respiratory tract. Tell your doctor right away if you cough up blood during treatment with Adempas.
- Worsening of symptoms in people with Pulmonary Veno-Occlusive Disease (PVOD). If you have PVOD, treatment with Adempas may cause a build-up of fluid in your lungs (pulmonary edema). This may cause you to feel short of breath. Your doctor may tell you to stop taking Adempas and switch you to a different medicine.
- The most common side effects of Adempas are:
- headache
- dizziness
- indigestion
- swelling of your hands, legs, feet, and ankles (peripheral edema)
- nausea, diarrhea, and vomiting
Tell your doctor if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of Adempas.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store Adempas?
- Store Adempas at room temperature between 59° F to 86° F (15° C to 30° C)
Keep Adempas and all medicines out of the reach of children.
General Information about Adempas
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use Adempas for a condition for which it is not prescribed. Do not give Adempas to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about Adempas. If you would like more information about Adempas, talk with your doctor. You can ask your doctor or pharmacist for information about Adempas that is written for health professionals. For more information go to www.Adempas-us.com or call 1-888-842-2937.
What are the ingredients in Adempas?
Active ingredient: riociguat
Inactive ingredients: cellulose microcrystalline, crospovidone, hypromellose 5cP, lactose monohydrate, magnesium stearate, sodium laurylsulfate, hydroxypropylcellulose, hypromellose 3cP, propylene glycol, titanium dioxide. Adempas 1 mg, 1.5 mg, 2 mg and 2.5 mg tablets also contain ferric oxide yellow. Adempas 2 mg and 2.5 mg tablets also contain ferric oxide red.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised 1 2017
Manufactured for:
Bayer HealthCare Pharmaceuticals Inc.
Whippany, NJ 07981Manufactured in Germany
©2013 Bayer HealthCare Pharmaceuticals Inc.
The following are representative examples of Adempas labeling. See the "How Supplied" section for a complete listing of all components.
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC: 50419-250-01
Rx only
Adempas 0.5 mg
(riociguat) tablets
each tablet contains 0.5 mg of riociguatNote to Dispencer: Provide a copy of the Adempas Medication Guide included in this carton to each patient and at each refill.
- 90 tablets
- for oral administration -
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC: 50419-251-01
Rx only
Adempas 1 mg
(riociguat) tablets
each tablet contains 1 mg of riociguatNote to Dispencer: Provide a copy of the Adempas Medication Guide included in this cartonto each patient and at each refill.
- 90 tablets
- for oral administration -
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC: 50419-252-01
Rx only
Adempas 1.5 mg
(riociguat) tablets
each tablet contains 1.5 mg of riociguatNote to Dispencer: Provide a copy of the Adempas Medication Guide included in this cartonto each patient and at each refill.
- 90 tablets
- for oral administration -
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC: 50419-253-01
Rx only
Adempas 2 mg
(riociguat) tablets
each tablet contains 2 mg of riociguatNote to Dispencer: Provide a copy of the Adempas Medication Guide included in this cartonto each patient and at each refill.
- 90 tablets
- for oral administration -
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC: 50419-254-01
Rx only
Adempas 2.5 mg
(riociguat) tablets
each tablet contains 2.5 mg of riociguatNote to Dispencer: Provide a copy of the Adempas Medication Guide included in this cartonto each patient and at each refill.
- 90 tablets
- for oral administration -
INGREDIENTS AND APPEARANCE
ADEMPAS
riociguat tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50419-250 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RIOCIGUAT (UNII: RU3FE2Y4XI) (RIOCIGUAT - UNII:RU3FE2Y4XI) RIOCIGUAT .5 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSPOVIDONE (15 MPA.S AT 5%) (UNII: 68401960MK) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) SODIUM LAURYL SULFATE (UNII: 368GB5141J) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color WHITE Score no score Shape ROUND (biconvex) Size 6mm Flavor Imprint Code 5R;Bayer Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50419-250-01 90 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2013 2 NDC: 50419-250-03 2 in 1 PACKAGE 10/08/2013 2 21 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC: 50419-250-91 9 in 1 BOTTLE; Type 0: Not a Combination Product 09/18/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA204819 10/08/2013 ADEMPAS
riociguat tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50419-251 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RIOCIGUAT (UNII: RU3FE2Y4XI) (RIOCIGUAT - UNII:RU3FE2Y4XI) RIOCIGUAT 1 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSPOVIDONE (15 MPA.S AT 5%) (UNII: 68401960MK) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) SODIUM LAURYL SULFATE (UNII: 368GB5141J) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color YELLOW (pale) Score no score Shape ROUND (biconvex) Size 6mm Flavor Imprint Code 1R;Bayer Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50419-251-01 90 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2013 2 NDC: 50419-251-03 2 in 1 PACKAGE 10/08/2013 2 21 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC: 50419-251-91 9 in 1 BOTTLE; Type 0: Not a Combination Product 09/18/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA204819 10/08/2013 ADEMPAS
riociguat tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50419-252 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RIOCIGUAT (UNII: RU3FE2Y4XI) (RIOCIGUAT - UNII:RU3FE2Y4XI) RIOCIGUAT 1.5 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSPOVIDONE (15 MPA.S AT 5%) (UNII: 68401960MK) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) SODIUM LAURYL SULFATE (UNII: 368GB5141J) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color YELLOW (orange) Score no score Shape ROUND (biconvex) Size 6mm Flavor Imprint Code 15R;Bayer Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50419-252-01 90 in 1 BOTTLE; Type 0: Not a Combination Product 12/08/2013 2 NDC: 50419-252-03 2 in 1 PACKAGE 10/08/2013 2 21 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC: 50419-252-91 9 in 1 BOTTLE; Type 0: Not a Combination Product 09/18/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA204819 10/08/2013 ADEMPAS
riociguat tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50419-253 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RIOCIGUAT (UNII: RU3FE2Y4XI) (RIOCIGUAT - UNII:RU3FE2Y4XI) RIOCIGUAT 2 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSPOVIDONE (15 MPA.S AT 5%) (UNII: 68401960MK) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) SODIUM LAURYL SULFATE (UNII: 368GB5141J) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color ORANGE (pale) Score no score Shape ROUND (biconvex) Size 6mm Flavor Imprint Code 2R;Bayer Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50419-253-01 90 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2013 2 NDC: 50419-253-03 2 in 1 PACKAGE 10/08/2013 2 21 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC: 50419-253-91 9 in 1 BOTTLE; Type 0: Not a Combination Product 09/18/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA204819 10/08/2013 ADEMPAS
riociguat tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50419-254 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RIOCIGUAT (UNII: RU3FE2Y4XI) (RIOCIGUAT - UNII:RU3FE2Y4XI) RIOCIGUAT 2.5 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSPOVIDONE (15 MPA.S AT 5%) (UNII: 68401960MK) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) SODIUM LAURYL SULFATE (UNII: 368GB5141J) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color RED (orange) Score no score Shape ROUND (biconvex) Size 6mm Flavor Imprint Code 25R;Bayer Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50419-254-01 90 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2013 2 NDC: 50419-254-03 2 in 1 PACKAGE 10/08/2013 2 21 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC: 50419-254-91 9 in 1 BOTTLE; Type 0: Not a Combination Product 09/18/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA204819 10/08/2013 Labeler - Bayer HealthCare Pharmaceuticals Inc. (005436809) Establishment Name Address ID/FEI Business Operations Sharp Corporation 143696495 PACK(50419-250, 50419-251, 50419-252, 50419-253, 50419-254) Establishment Name Address ID/FEI Business Operations Bayer AG 314947622 ANALYSIS(50419-250, 50419-251, 50419-252, 50419-253, 50419-254) , API MANUFACTURE(50419-250, 50419-251, 50419-252, 50419-253, 50419-254) , MANUFACTURE(50419-250, 50419-251, 50419-252, 50419-253, 50419-254) , PACK(50419-250, 50419-251, 50419-252, 50419-253, 50419-254) , PARTICLE SIZE REDUCTION(50419-250, 50419-251, 50419-252, 50419-253, 50419-254) , STERILIZE(50419-250, 50419-251, 50419-252, 50419-253, 50419-254)




Trademark Results [Adempas]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ADEMPAS 77723142 3718134 Live/Registered |
Bayer Aktiengesellschaft 2009-04-27 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.