SPIRIVA RESPIMAT- tiotropium bromide inhalation spray spray, metered
Spiriva Respimat by
Drug Labeling and Warnings
Spiriva Respimat by is a Prescription medication manufactured, distributed, or labeled by Boehringer Ingelheim Pharmaceuticals, Inc., Boehringer Ingelheim Pharma GmbH and Co. KG. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SPIRIVA RESPIMAT safely and effectively. See full prescribing information for SPIRIVA RESPIMAT.
SPIRIVA® RESPIMAT® (tiotropium bromide) inhalation spray, for oral inhalation
Initial U.S. Approval: 2004
INDICATIONS AND USAGE
SPIRIVA RESPIMAT is an anticholinergic indicated for:
- The long-term, once-daily, maintenance treatment of bronchospasm associated with chronic obstructive pulmonary disease (COPD), and for reducing COPD exacerbations (1.1)
- The long-term, once-daily, maintenance treatment of asthma in patients 6 years of age and older (1.2)
Limitation of Use:
DOSAGE AND ADMINISTRATION
For oral inhalation only
To receive the full dose of medication, SPIRIVA RESPIMAT must be administered as two inhalations once-daily.DOSAGE FORMS AND STRENGTHS
- Inhalation spray: 1.25 mcg or 2.5 mcg tiotropium per actuation with the SPIRIVA RESPIMAT inhaler. Two actuations equal one dose (2.5 mcg or 5 mcg). (3)
CONTRAINDICATIONS
Hypersensitivity to tiotropium, ipratropium, or any component of this product (4)
WARNINGS AND PRECAUTIONS
- Not for acute use, i.e., not a rescue medication (5.1)
- Immediate hypersensitivity reactions: Discontinue SPIRIVA RESPIMAT at once and consider alternatives if immediate hypersensitivity reactions, including angioedema, urticaria, rash, bronchospasm, or anaphylaxis, occur. (5.2)
- Paradoxical bronchospasm: Discontinue SPIRIVA RESPIMAT and consider other treatments if paradoxical bronchospasm occurs. (5.3)
- Worsening of narrow-angle glaucoma may occur. Use with caution in patients with narrow-angle glaucoma and instruct patients to consult a physician immediately if this occurs. (5.4)
- Worsening of urinary retention may occur. Use with caution in patients with prostatic hyperplasia or bladder-neck obstruction and instruct patient to consult a physician immediately if this occurs. (5.5)
ADVERSE REACTIONS
The most common adverse reactions in:
- COPD: (>3% incidence in the placebo-controlled trials with treatment durations of between 4 and 48 weeks) were pharyngitis, cough, dry mouth, and sinusitis (6.1).
- Asthma: (>2% incidence in the placebo-controlled trials with treatment durations of between 12 and 52 weeks) were pharyngitis, headache, bronchitis, and sinusitis in adults (6.2).
To report SUSPECTED ADVERSE REACTIONS, contact Boehringer Ingelheim Pharmaceuticals, Inc. at (800) 542-6257 or (800) 459-9906 TTY or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Anticholinergics: May interact additively with concomitantly used anticholinergic medications. Avoid administration of SPIRIVA RESPIMAT with other anticholinergic-containing drugs. (7.2)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Maintenance Treatment of Chronic Obstructive Pulmonary Disease
1.2 Maintenance Treatment of Asthma
2 DOSAGE AND ADMINISTRATION
2.1 Chronic Obstructive Pulmonary Disease
2.2 Asthma
2.3 Special Populations
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Not for Acute Use
5.2 Immediate Hypersensitivity Reactions
5.3 Paradoxical Bronchospasm
5.4 Worsening of Narrow-Angle Glaucoma
5.5 Worsening of Urinary Retention
5.6 Renal Impairment
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience in Chronic Obstructive Pulmonary Disease
6.2 Clinical Trials Experience in Asthma
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Concomitant Respiratory Medications
7.2 Anticholinergics
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Chronic Obstructive Pulmonary Disease
14.2 Asthma
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Maintenance Treatment of Chronic Obstructive Pulmonary Disease
SPIRIVA RESPIMAT (tiotropium bromide) is indicated for the long-term, once-daily, maintenance treatment of bronchospasm associated with chronic obstructive pulmonary disease (COPD), including chronic bronchitis and emphysema. SPIRIVA RESPIMAT is indicated to reduce exacerbations in COPD patients.
Important Limitation of Use:
SPIRIVA RESPIMAT is NOT indicated for the relief of acute bronchospasm. -
2 DOSAGE AND ADMINISTRATION
To receive the full dose of medication, SPIRIVA RESPIMAT must be administered as two inhalations once-daily. Do not take more than one dose (2 inhalations) in 24 hours.
Prior to first use, the SPIRIVA RESPIMAT cartridge is inserted into the SPIRIVA RESPIMAT inhaler and the unit is primed. When using the unit for the first time, patients are to actuate the inhaler toward the ground until an aerosol cloud is visible and then repeat the process three more times. The unit is then considered primed and ready for use. If not used for more than 3 days, patients are to actuate the inhaler once to prepare the inhaler for use. If not used for more than 21 days, patients are to actuate the inhaler until an aerosol cloud is visible and then repeat the process three more times to prepare the inhaler for use [see Patient Counseling Information (17)].
2.1 Chronic Obstructive Pulmonary Disease
The recommended dosage for patients with COPD is 2 inhalations of SPIRIVA RESPIMAT 2.5 mcg per actuation once-daily; total dose equals 5 mcg of SPIRIVA RESPIMAT.
2.2 Asthma
The recommended dosage for patients with asthma is 2 inhalations of SPIRIVA RESPIMAT 1.25 mcg per actuation once-daily; total dose equals 2.5 mcg of SPIRIVA RESPIMAT. In the treatment of asthma, the maximum benefits in lung function may take up to 4 to 8 weeks of dosing [see Patient Counseling Information (17)].
2.3 Special Populations
No dosage adjustment is required for geriatric, hepatically-impaired, or renally-impaired patients. However, patients with moderate to severe renal impairment given SPIRIVA RESPIMAT should be monitored closely for anticholinergic effects [see Warnings and Precautions (5.6), Use in Specific Populations (8.5, 8.6, 8.7), and Clinical Pharmacology (12.3)].
-
3 DOSAGE FORMS AND STRENGTHS
SPIRIVA RESPIMAT consists of a SPIRIVA RESPIMAT inhaler and an aluminum cylinder (SPIRIVA RESPIMAT cartridge) containing tiotropium bromide (as the monohydrate). The SPIRIVA RESPIMAT cartridge is only intended for use with the SPIRIVA RESPIMAT inhaler.
SPIRIVA RESPIMAT is available in two dosage strengths. Each actuation from the SPIRIVA RESPIMAT inhaler delivers 1.25 mcg or 2.5 mcg of tiotropium (equivalent to 1.562 mcg or 3.124 mcg, respectively, of tiotropium bromide monohydrate) from the mouthpiece. Two actuations equal one dose (2.5 mcg or 5 mcg).
-
4 CONTRAINDICATIONS
SPIRIVA RESPIMAT is contraindicated in patients with a hypersensitivity to tiotropium, ipratropium, or any component of this product [see Warnings and Precautions (5.2)]. In clinical trials with SPIRIVA RESPIMAT, immediate hypersensitivity reactions, including angioedema (including swelling of the lips, tongue, or throat), itching, or rash have been reported [see Warnings and Precautions (5.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Not for Acute Use
SPIRIVA RESPIMAT is intended as a once-daily maintenance treatment for COPD and asthma and should not be used for the relief of acute symptoms, i.e., as rescue therapy for the treatment of acute episodes of bronchospasm. In the event of an acute attack, a rapid-acting beta2-agonist should be used.
5.2 Immediate Hypersensitivity Reactions
Immediate hypersensitivity reactions, including urticaria, angioedema (including swelling of the lips, tongue or throat), rash, bronchospasm, anaphylaxis, or itching may occur after administration of SPIRIVA RESPIMAT. If such a reaction occurs, therapy with SPIRIVA RESPIMAT should be stopped at once and alternative treatments should be considered. Given the similar structural formula of atropine to tiotropium, patients with a history of hypersensitivity reactions to atropine or its derivatives should be closely monitored for similar hypersensitivity reactions to SPIRIVA RESPIMAT.
5.3 Paradoxical Bronchospasm
Inhaled medicines, including SPIRIVA RESPIMAT, may cause paradoxical bronchospasm. If this occurs, it should be treated immediately with an inhaled short-acting beta2-agonist such as albuterol. Treatment with SPIRIVA RESPIMAT should be stopped and other treatments considered.
5.4 Worsening of Narrow-Angle Glaucoma
SPIRIVA RESPIMAT should be used with caution in patients with narrow-angle glaucoma. Prescribers and patients should be alert for signs and symptoms of acute narrow-angle glaucoma (e.g., eye pain or discomfort, blurred vision, visual halos or colored images in association with red eyes from conjunctival congestion and corneal edema). Instruct patients to consult a physician immediately should any of these signs or symptoms develop.
5.5 Worsening of Urinary Retention
SPIRIVA RESPIMAT should be used with caution in patients with urinary retention. Prescribers and patients should be alert for signs and symptoms of urinary retention (e.g., difficulty passing urine, painful urination), especially in patients with prostatic hyperplasia or bladder-neck obstruction. Instruct patients to consult a physician immediately should any of these signs or symptoms develop.
5.6 Renal Impairment
As a predominantly renally excreted drug, patients with moderate to severe renal impairment (creatinine clearance of <60 mL/min) treated with SPIRIVA RESPIMAT should be monitored closely for anticholinergic side effects [see Clinical Pharmacology (12.3)].
-
6 ADVERSE REACTIONS
The following adverse reactions are described, or described in greater detail, in other sections:
- Immediate hypersensitivity reactions [see Warnings and Precautions (5.2)]
- Paradoxical bronchospasm [see Warnings and Precautions (5.3)]
- Worsening of narrow-angle glaucoma [see Warnings and Precautions (5.4)]
- Worsening of urinary retention [see Warnings and Precautions (5.5)]
Because clinical trials are conducted under widely varying conditions, the incidence of adverse reactions observed in the clinical trials of a drug cannot be directly compared to the incidences in the clinical trials of another drug and may not reflect the incidences observed in practice.
Since the same active ingredient (tiotropium bromide) is administered to COPD and asthma patients, prescribers and patients should take into account that the observed adverse reactions could be relevant for both patient populations independent of dosage strength.
6.1 Clinical Trials Experience in Chronic Obstructive Pulmonary Disease
The SPIRIVA RESPIMAT clinical development program included ten placebo controlled clinical trials in COPD. Two trials were four-week cross-over trials and eight were parallel group trials. The parallel group trials included a three week dose-ranging trial, two 12-week trials, three 48-week trials, and two trials of 4-week and 24-week duration conducted for a different program that contained tiotropium bromide 5 mcg treatment arms. The primary safety database consists of pooled data from the 7 randomized, parallel-group, double-blind, placebo-controlled studies of 4-48 weeks in treatment duration. These trials included 6565 adult COPD patients (75% males and 25% females) 40 years of age and older. Of these patients, 3282 patients were treated with SPIRIVA RESPIMAT 5 mcg and 3283 received placebo. The SPIRIVA RESPIMAT 5 mcg group was composed mostly of Caucasians (78%) with a mean age of 65 years and a mean baseline percent predicted post-bronchodilator FEV1 of 46%.
In these 7 clinical trials, 68.3% of patients exposed to SPIRIVA RESPIMAT 5 mcg reported an adverse event compared to 68.7% of patients in the placebo group. There were 68 deaths in the SPIRIVA RESPIMAT 5 mcg treatment group (2.1%) and 52 deaths (1.6%) in patients who received placebo [see Clinical Studies (14) Long-term Active-Controlled Mortality Trial: Survival]. The percentage of SPIRIVA RESPIMAT patients who discontinued due to an adverse event were 7.3% compared to 10% with placebo patients. The percentage of SPIRIVA RESPIMAT 5 mcg patients who experienced a serious adverse event were 15.0% compared to 15.1% with placebo patients. In both groups, the adverse event most commonly leading to discontinuation was COPD exacerbation (SPIRIVA RESPIMAT 2.0%, placebo 4.0%) which was also the most frequent serious adverse event. The most commonly reported adverse reactions were pharyngitis, cough, dry mouth, and sinusitis (Table 1). Other adverse reactions reported in individual patients and consistent with possible anticholinergic effects included constipation, dysuria, and urinary retention.
Table 1 shows all adverse reactions that occurred with an incidence of >3% in the SPIRIVA RESPIMAT 5 mcg treatment group, and a higher incidence rate on SPIRIVA RESPIMAT 5 mcg than on placebo.
Table 1 Number (Percentage) of COPD Patients Exposed to SPIRIVA RESPIMAT 5 mcg with Adverse Reactions >3% (and Higher than Placebo): Pooled Data from 7 Clinical Trials with Treatment Periods Ranging between 4 and 48 Weeks in COPD Patients *Adverse reactions include a grouping of similar terms Body System (Reaction)* SPIRIVA RESPIMAT 5 mcg
[n=3282]Placebo
[n=3283]Gastrointestinal Disorders Dry mouth 134 (4.1) 52 (1.6) Infections and Infestations Pharyngitis 378 (11.5) 333 (10.1) Respiratory, Thoracic, and Mediastinal Disorders Cough 190 (5.8) 182 (5.5) Sinusitis 103 (3.1) 88 (2.7) Other reactions that occurred in the SPIRIVA RESPIMAT 5 mcg group at an incidence of 1% to 3% and at a higher incidence rate on SPIRIVA RESPIMAT 5 mcg than on placebo included: Cardiac disorders: palpitations; Gastrointestinal disorders: constipation, gastroesophageal reflux disease, oropharyngeal candidiasis; Nervous system disorders: dizziness; Respiratory, thoracic, and mediastinal disorders: dysphonia; Skin and subcutaneous tissue disorders: pruritus, rash; Renal and urinary disorders: urinary tract infection.
Less Common Adverse Reactions
Among the adverse reactions observed in the clinical trials with an incidence of <1% and at a higher incidence rate on SPIRIVA RESPIMAT 5 mcg than on placebo were: dysphagia, gingivitis, intestinal obstruction including ileus paralytic, joint swelling, dysuria, urinary retention, epistaxis, laryngitis, angioedema, dry skin, skin infection, and skin ulcer.6.2 Clinical Trials Experience in Asthma
Adult Patients
SPIRIVA RESPIMAT 2.5 mcg has been compared to placebo in four placebo-controlled parallel-group trials ranging from 12 to 52 weeks of treatment duration in adult patients (aged 18 to 75 years) with asthma. The safety data described below are based on one 1-year, two 6-month and one 12-week randomized, double-blind, placebo-controlled trials in a total of 2849 asthma patients on background treatment of at least ICS or ICS and long-acting beta agonist (ICS/LABA). Of these patients, 787 were treated with SPIRIVA RESPIMAT at the recommended dose of 2.5 mcg once-daily; 59.7% were female and 47.5% were Caucasian with a mean age of 43.7 years and a mean post-bronchodilator percent predicted forced expiratory volume in 1 second (FEV1) of 90.0% at baseline.Table 2 shows all adverse reactions that occurred with an incidence of >2% in the SPIRIVA RESPIMAT 2.5 mcg treatment group, and a higher incidence rate on SPIRIVA RESPIMAT 2.5 mcg than on placebo.
Table 2 Number (Percentage) of Asthma Patients Exposed to SPIRIVA RESPIMAT 2.5 mcg with Adverse Reactions >2% (and Higher than Placebo): Pooled Data from 4 Adult Clinical Trials with Treatment Periods Ranging between 12 and 52 Weeks in Asthma Patients *Adverse reactions include a grouping of similar terms Body System (Reaction)* SPIRIVA RESPIMAT 2.5 mcg
[n=787]Placebo
[n=735]Respiratory, Thoracic, and Mediastinal Disorders Pharyngitis 125 (15.9) 91 (12.4) Sinusitis 21 (2.7) 10 (1.4) Bronchitis 26 (3.3) 10 (1.4) Nervous System Disorders Headache 30 (3.8) 20 (2.7) Other reactions that occurred in the SPIRIVA RESPIMAT 2.5 mcg group at an incidence of 1% to 2% and at a higher incidence rate on SPIRIVA RESPIMAT 2.5 mcg than on placebo included: Nervous system disorders: dizziness; Gastrointestinal disorders: oropharyngeal candidiasis, diarrhea; Respiratory, thoracic, and mediastinal disorders: cough, rhinitis allergic; Renal and urinary disorders: urinary tract infection; General disorders and administration site conditions: pyrexia; and Vascular disorders: hypertension.
Less Common Adverse Reactions
Among the adverse reactions observed in the clinical trials with an incidence of 0.5% to <1% and at a higher incidence rate on SPIRIVA RESPIMAT 2.5 mcg than on placebo were: palpitations, dysphonia, acute tonsillitis, tonsillitis, rhinitis, herpes zoster, gastroesophageal reflux disease, oropharyngeal discomfort, abdominal pain upper, insomnia, hypersensitivity (including immediate reactions), angioedema, dehydration, arthralgia, muscle spasms, pain in extremity, chest pain, hepatic function abnormal, liver function test abnormal.Adolescent Patients Aged 12 to 17 years
SPIRIVA RESPIMAT 2.5 mcg has been compared to placebo in two placebo-controlled parallel-group trials ranging from 12 to 48 weeks of treatment duration in adolescent patients with asthma. The safety data described below are based on one 48-week and one 12-week double-blind, placebo-controlled trials in a total of 789 adolescent asthma patients on background treatment of at least ICS or ICS plus one or more controller. Of these patients, 252 were treated with SPIRIVA RESPIMAT at the recommended dose of 2.5 mcg once-daily; 63.9% were male and 95.6% were Caucasian with a mean age of 14.3 years and a mean post-bronchodilator percent predicted FEV1 of 98.3% at baseline. The adverse reaction profile for adolescent patients with asthma was comparable to that observed in adult patients with asthma.Pediatric Patients Aged 6 to 11 years
SPIRIVA RESPIMAT 2.5 mcg has been compared to placebo in two placebo-controlled parallel-group trials ranging from 12 to 48 weeks of treatment duration in pediatric patients aged 6 to 11 years with asthma. The safety data are based on one 48-week and one 12-week double-blind, placebo-controlled trials in a total of 801 pediatric asthma patients aged 6 to 11 years on background treatment of at least ICS or ICS plus one or more controller. Of these patients, 271 were treated with SPIRIVA RESPIMAT at the recommended dose of 2.5 mcg once-daily; 71.2% were male and 86.7% were Caucasian with a mean age of 8.9 years and a mean post-bronchodilator percent predicted FEV1 of 97.9% at baseline. The adverse reaction profile for pediatric patients aged 6 to 11 years with asthma was comparable to that observed in adult patients with asthma.SPIRIVA RESPIMAT 5 mcg also has been compared to placebo in seven placebo-controlled parallel-group trials ranging from 12 to 52 weeks of treatment duration in 4149 adult patients (aged 18 to 75 years) with asthma and in two placebo-controlled parallel-group trials ranging from 12 to 48 weeks of treatment duration in 789 adolescent patients (1370 adults and 264 adolescents receiving SPIRIVA RESPIMAT 5 mcg once-daily). The adverse reaction profile for SPIRIVA RESPIMAT 5 mcg in patients with asthma was comparable to that observed with SPIRIVA RESPIMAT 2.5 mcg in patients with asthma.
6.3 Postmarketing Experience
In addition to the adverse reactions observed during the SPIRIVA RESPIMAT clinical trials in COPD, the following adverse reactions have been observed during post-approval use of SPIRIVA RESPIMAT 5 mcg and another tiotropium formulation, SPIRIVA® HandiHaler® (tiotropium bromide inhalation powder). Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Glaucoma, intraocular pressure increased, vision blurred,
- Atrial fibrillation, tachycardia, supraventricular tachycardia,
- Bronchospasm,
- Glossitis, stomatitis,
- Dehydration,
- Insomnia,
- Hypersensitivity (including immediate reactions), and urticaria.
-
7 DRUG INTERACTIONS
7.1 Concomitant Respiratory Medications
SPIRIVA RESPIMAT has been used concomitantly with short-acting and long-acting sympathomimetic (beta-agonists) bronchodilators, methylxanthines, oral and inhaled steroids, antihistamines, mucolytics, leukotriene modifiers, cromones, and anti-IgE treatment without increases in adverse reactions.
7.2 Anticholinergics
There is potential for an additive interaction with concomitantly used anticholinergic medications. Therefore, avoid coadministration of SPIRIVA RESPIMAT with other anticholinergic-containing drugs as this may lead to an increase in anticholinergic adverse effects [see Warnings and Precautions (5.4, 5.5) and Adverse Reactions (6)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
The limited human data with SPIRIVA RESPIMAT use during pregnancy are insufficient to inform a drug-associated risk of adverse pregnancy-related outcomes. There are risks to the mother and the fetus associated with poorly controlled asthma in pregnancy [see Clinical Considerations]. Based on animal reproduction studies, no structural abnormalities were observed when tiotropium was administered by inhalation to pregnant rats and rabbits during the period of organogenesis at doses 790 and 8 times, respectively, the maximum recommended human daily inhalation dose (MRHDID). Increased post-implantation loss was observed in rats and rabbits administered tiotropium at maternally toxic doses 430 times and 40 times the MRHDID, respectively [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo-Fetal Risk
Poorly or moderately controlled asthma in pregnancy increases the maternal risk of preeclampsia and infant prematurity, low birth weight, and small for gestational age. The level of asthma control should be closely monitored in pregnant women and treatment adjusted as necessary to maintain optimal control.Data
Animal Data
In 2 separate embryo-fetal development studies, pregnant rats and rabbits received tiotropium during the period of organogenesis at doses up to approximately 790 and 8 times the maximum recommended human daily inhalation dose (MRHDID), respectively (on a mcg/m2 basis at inhalation doses of 1471 and 7 mcg/kg/day in rats and rabbits, respectively). No evidence of structural abnormalities was observed in rats or rabbits. However, in rats, tiotropium caused fetal resorption, litter loss, decreases in the number of live pups at birth and the mean pup weights, and a delay in pup sexual maturation at tiotropium doses of approximately 40 times the MRHDID (on a mcg/m2 basis at a maternal inhalation dose of 78 mcg/kg/day). In rabbits, tiotropium caused an increase in post-implantation loss at a tiotropium dose of approximately 430 times the MRHDID (on a mcg/m2 basis at a maternal inhalation dose of 400 mcg/kg/day). Such effects were not observed at approximately 5 and 95 times the MRHDID, respectively (on a mcg/m2 basis at inhalation doses of 9 and 88 mcg/kg/day in rats and rabbits, respectively).8.2 Lactation
There are no data on the presence of tiotropium in human milk, the effects on the breastfed infant, or the effects on milk production. Tiotropium is present in milk of lactating rats; however, due to species-specific differences in lactation physiology, the clinical relevance of these data are not clear [see Data]. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for SPIRIVA RESPIMAT and any potential adverse effects on the breastfed child from SPIRIVA RESPIMAT or from the underlying maternal condition.
Data
The distribution of tiotropium bromide into milk was investigated after a single intravenous administration of 10 mg/kg to lactating rats. Tiotropium and/or its metabolites are present in the milk of lactating rats at concentrations above those in plasma.8.4 Pediatric Use
The safety and efficacy of SPIRIVA RESPIMAT 2.5 mcg have been established in pediatric patients aged 6 to 17 years with asthma in 6 clinical trials up to 1 year in duration. In three clinical trials, 327 patients aged 12 to 17 years with asthma were treated with SPIRIVA RESPIMAT 2.5 mcg; in three additional clinical trials, 345 patients aged 6 to 11 years with asthma were treated with SPIRIVA RESPIMAT 2.5 mcg. Patients in these age groups demonstrated efficacy results similar to those observed in patients aged 18 years and older with asthma [see Clinical Studies (14.2)].
The safety and efficacy of SPIRIVA RESPIMAT have not been established in pediatric patients less than 6 years of age. The safety of SPIRIVA RESPIMAT 2.5 mcg has been studied in pediatric patients with asthma aged 1 to 5 years who were on background treatment of at least ICS in one placebo-controlled clinical trial of 12 weeks duration (36 treated with SPIRIVA RESPIMAT 2.5 mcg and 34 with placebo RESPIMAT). In this study, SPIRIVA RESPIMAT or placebo RESPIMAT was delivered with the AeroChamber Plus Flow-Vu® valved holding chamber with facemask once daily. The majority of the patients in the trial were male (60.4%) and Caucasian (76.2%) with a mean age of 3.1 years. The adverse reaction profile was similar to that observed in adults and older pediatric patients [see Adverse Reactions (6.2)].
In Vitro Characterization Studies with Valved Holding Chamber
Dose delivery and fine particle fraction of SPIRIVA RESPIMAT when administered via a valved holding chamber (AeroChamber Plus Flow-Vu® with or without face mask) was assessed by in vitro studies.Inspiratory flow rates of 4.9, 8.0, and 12.0 L/min in combination with holding times of 0, 2, 5, and 10 seconds were tested. The flow rates were selected to be representative of inspiratory flow rates of children aged 6 to 12 months, 2 to 5 years, and over 5 years, respectively.
Table 3 summarizes the results for delivered dose under the respective test conditions and configurations.
Table 3 In Vitro Medication Delivery through AeroChamber Plus Flow-Vu® Valved Holding Chamber with Face Mask at Different Flow Rates and Holding Times Using the Dose 2.5 mcg (as two actuations) a Centers for Disease Control growth charts, developed by the National Center for Health Statistics in collaboration with the National Center for Chronic Disease Prevention and Health Promotion (2009).
Body weight values correspond to the average of the 50 percentile weight for boys and girls at the ages indicated.
b Inhalation of SPIRIVA RESPIMAT 2.5 mcg dose (as two actuations) in a 70-kg adult without use of a valved holding chamber and mask delivers approximately 2.5 mcg, or 36 ng/kg.Flow Rate
(L/min)
and corresponding ageMask Holding Time
(seconds)Mean Medication Delivery through AeroChamber Plus Flow-Vu® per Dose (mcg) Body Weight 50th Percentile (kg)a 4.9
(6 to 12 Months)small 0
2
5
100.85
0.86
0.55
0.627.5-9.9 86-113
87-115
56-73
63-838.0
(2 to 5 Years)medium 0
2
5
100.74
0.93
0.72
0.5712.3-18.0 41-60
52-76
40-59
32-4612.0
(> 5 Years)medium 0
2
5
101.16
0.96
0.78
0.6118.0 64
53
43
34The in vitro study data show a reduction of the absolute delivered dose through the valved holding chamber. However, in terms of dose per kilogram of body weight the data suggest that under all tested conditions the dose of SPIRIVA RESPIMAT delivered by the AeroChamber Plus Flow-Vu® valved holding chamber with mask will at least lead to a dosing comparable to that of adults without use of a holding chamber and mask (Table 3). The fine particle fraction (< 5 μm) across the flow rates used in these studies was 69-89% of the delivered dose through the valved holding chamber, consistent with the removal of the coarser fraction by the holding chamber. In contrast, the fine particle fraction for SPIRIVA RESPIMAT delivered without a holding chamber typically represents approximately 60% of the delivered dose.
8.5 Geriatric Use
Based on available data, no adjustment of SPIRIVA RESPIMAT dosage in geriatric patients is warranted [see Clinical Pharmacology (12.3)].
Thirty nine percent of SPIRIVA RESPIMAT clinical trial patients with COPD were between 65 and 75 years of age and 14% were greater than or equal to 75 years of age. Approximately seven percent of SPIRIVA RESPIMAT clinical trial patients with asthma were greater than or equal to 65 years of age. The adverse drug reaction profiles were similar in the older population compared to the patient population overall.
8.6 Renal Impairment
Patients with moderate to severe renal impairment (creatinine clearance of <60 mL/min) treated with SPIRIVA RESPIMAT should be monitored closely for anticholinergic side effects [see Dosage and Administration (2), Warnings and Precautions (5.6), and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
High doses of tiotropium may lead to anticholinergic signs and symptoms. However, there were no systemic anticholinergic adverse effects following a single inhaled dose of up to 282 mcg tiotropium dry powder in 6 healthy volunteers. Dry mouth/throat and dry nasal mucosa occurred in a dose-dependent [10-40 mcg daily] manner, following 14-day dosing of up to 40 mcg tiotropium bromide inhalation solution in healthy subjects.
Treatment of overdosage consists of discontinuation of SPIRIVA RESPIMAT together with institution of appropriate symptomatic and/or supportive therapy.
-
11 DESCRIPTION
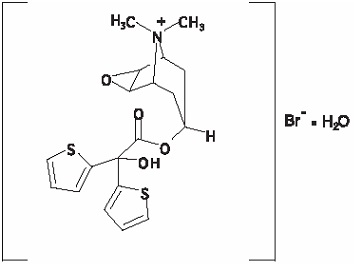
The active component of SPIRIVA RESPIMAT is tiotropium. The drug substance, tiotropium bromide monohydrate, is an anticholinergic with specificity for muscarinic receptors. It is chemically described as (1α, 2ß, 4ß, 5α, 7ß)-7-[(Hydroxydi-2-thienylacetyl)oxy]-9,9-dimethyl-3-oxa-9-azoniatricyclo[3.3.1.02,4] nonane bromide monohydrate. It is a synthetic, non-chiral, quaternary ammonium compound. Tiotropium bromide is a white or yellowish white powder. It is sparingly soluble in water and soluble in methanol.
Tiotropium bromide (monohydrate) has a molecular mass of 490.4 and a molecular formula of C19H22NO4S2Br H2O.
The drug product, SPIRIVA RESPIMAT, is composed of a sterile, aqueous solution of tiotropium bromide filled into a 4.5 mL plastic container crimped into an aluminum cylinder (SPIRIVA RESPIMAT cartridge) for use with the SPIRIVA RESPIMAT inhaler. Excipients include water for injection, edetate disodium, benzalkonium chloride and hydrochloric acid. The SPIRIVA RESPIMAT cartridge is only intended for use with the SPIRIVA RESPIMAT inhaler. The RESPIMAT inhaler is a hand held, pocket sized oral inhalation device that uses mechanical energy to generate a slow moving aerosol cloud of medication from a metered volume of the drug solution.
When used with the SPIRIVA RESPIMAT inhaler, each cartridge containing 4 grams of sterile aqueous solution delivers the labeled number of metered actuations after preparation for use. Each dose (one dose equals two actuations) from the SPIRIVA RESPIMAT inhaler delivers 2.5 mcg or 5 mcg of tiotropium in 22.1 mcL from the mouthpiece. As with all inhaled drugs, the actual amount of drug delivered to the lung may depend on patient factors, such as the coordination between the actuation of the inhaler and inspiration through the delivery system. The duration of inspiration should be at least as long as the spray duration (1.5 seconds).
Prior to first use, the SPIRIVA RESPIMAT cartridge is inserted into the SPIRIVA RESPIMAT inhaler and the unit is primed. When using the unit for the first time, patients are to actuate the inhaler toward the ground until an aerosol cloud is visible and then repeat the process three more times. The unit is then considered primed and ready for use. If not used for more than 3 days, patients are to actuate the inhaler once to prepare the inhaler for use. If not used for more than 21 days, patients are to actuate the inhaler until an aerosol cloud is visible and then repeat the process three more times to prepare the inhaler for use [see Patient Counseling Information (17)].
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tiotropium is a long-acting, antimuscarinic agent, which is often referred to as an anticholinergic. It has similar affinity to the subtypes of muscarinic receptors, M1 to M5. In the airways, it exhibits pharmacological effects through inhibition of M3-receptors at the smooth muscle leading to bronchodilation. The competitive and reversible nature of antagonism was shown with human and animal origin receptors and isolated organ preparations. In preclinical in vitro as well as in vivo studies, prevention of methacholine-induced bronchoconstriction effects was dose-dependent and lasted longer than 24 hours. The bronchodilation following inhalation of tiotropium is predominantly a site-specific effect.
12.2 Pharmacodynamics
Cardiac Electrophysiology
In a multicenter, randomized, double-blind trial using tiotropium dry powder for inhalation that enrolled 198 patients with COPD, the number of subjects with changes from baseline-corrected QT interval of 30 to 60 msec was higher in the SPIRIVA group as compared with placebo. This difference was apparent using both the Bazett (QTcB) [20 (20%) patients vs. 12 (12%) patients] and Fredericia (QTcF) [16 (16%) patients vs. 1 (1%) patient] corrections of QT for heart rate. No patients in either group had either QTcB or QTcF of >500 msec. Other clinical trials with SPIRIVA did not detect an effect of the drug on QTc intervals.The effect of tiotropium dry powder for inhalation on QT interval was also evaluated in a randomized, placebo- and positive-controlled crossover study in 53 healthy volunteers. Subjects received tiotropium inhalation powder 18 mcg, 54 mcg (3 times the recommended dose), or placebo for 12 days. ECG assessments were performed at baseline and throughout the dosing interval following the first and last dose of study medication. Relative to placebo, the maximum mean change from baseline in study-specific QTc interval was 3.2 msec and 0.8 msec for tiotropium inhalation powder 18 mcg and 54 mcg, respectively. No subject showed a new onset of QTc >500 msec or QTc changes from baseline of ≥60 msec.
12.3 Pharmacokinetics
Tiotropium is administered as an inhalation spray. Some of the pharmacokinetic data described below were obtained with higher doses than recommended for therapy. A dedicated pharmacokinetic study in patients with COPD evaluating once-daily tiotropium delivered from the RESPIMAT inhaler (5 mcg) and as inhalation powder (18 mcg) from the HandiHaler resulted in a similar systemic exposure between the two products.
Absorption
Following inhalation of the solution by young healthy volunteers, urinary excretion data suggests that approximately 33% of the inhaled dose reaches the systemic circulation. Oral solutions of tiotropium have an absolute bioavailability of 2% to 3%. Food is not expected to influence the absorption of tiotropium for the same reason. Following 4-week SPIRIVA RESPIMAT once daily dosing, maximum tiotropium plasma concentrations were observed 5-7 minutes after inhalation in COPD and asthma patients.Distribution
The drug has a plasma protein binding of 72% and shows a volume of distribution of 32 L/kg after an intravenous dose to young healthy volunteers. Local concentrations in the lung are not known, but the mode of administration suggests substantially higher concentrations in the lung. Studies in rats have shown that tiotropium does not penetrate the blood-brain barrier.Elimination
Metabolism
The extent of metabolism is small. This is evident from a urinary excretion of 74% of unchanged substance after an intravenous dose to young healthy volunteers. Tiotropium, an ester, is nonenzymatically cleaved to the alcohol N-methylscopine and dithienylglycolic acid, neither of which binds to muscarinic receptors.In vitro experiments with human liver microsomes and human hepatocytes suggest that a fraction of the administered dose (74% of an intravenous dose is excreted unchanged in the urine, leaving 25% for metabolism) is metabolized by cytochrome P450-dependent oxidation and subsequent glutathione conjugation to a variety of Phase II metabolites. This enzymatic pathway can be inhibited by CYP450 2D6 and 3A4 inhibitors, such as quinidine, ketoconazole, and gestodene. Thus, CYP450 2D6 and 3A4 are involved in the metabolic pathway that is responsible for the elimination of a small part of the administered dose. In vitro studies using human liver microsomes showed that tiotropium in supra-therapeutic concentrations does not inhibit CYP450 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1, or 3A4.
Excretion
The terminal half-life of tiotropium in COPD and asthma patients following once daily inhalation is 25 and 44 hours, respectively. Total clearance was 880 mL/min after an intravenous dose in young healthy volunteers. Intravenously administered tiotropium bromide is mainly excreted unchanged in urine (74%). Following 21-day once daily inhalation of 5 mcg of the solution by patients with COPD, 24-hour urinary excretion is 18.6% (0.93 mcg) of the dose. The renal clearance of tiotropium exceeds the creatinine clearance, indicating secretion into the urine. In comparison, 12.8% (0.32 mcg) of the dose was excreted unchanged in the urine over 24 hours at steady state after inhalation of 2.5 mcg in patients with asthma. After chronic once-daily inhalation by COPD and asthma patients, pharmacokinetic steady-state was reached by day 7 with no accumulation thereafter.Specific Populations
Geriatric Patients
As expected for all predominantly renally excreted drugs, advancing age was associated with a decrease of tiotropium renal clearance (347 mL/min in COPD patients <65 years to 275 mL/min in COPD patients ≥65 years). This did not result in a corresponding increase in AUC0-6,ss and Cmax,ss values following inhalation of the solution. Exposure to tiotropium was not found to differ with age in patients with asthma.Pediatric Patients
The peak and total exposure to tiotropium was not found to differ between pediatric patients (aged 6 to 17 years) and adults with asthma.Renal Impairment
Following 4-week SPIRIVA RESPIMAT 5 mcg once daily dosing in patients with COPD, mild renal impairment (creatinine clearance 60 - <90 mL/min) resulted in 23% higher AUC0‑6,ss and 17% higher Cmax,ss values; moderate renal impairment (creatinine clearance 30 - <60 mL/min) resulted in 57% higher AUC0‑6,ss and 31% higher Cmax,ss values compared to COPD patients with normal renal function (creatinine clearance >90 mL/min). The influence of mild or moderate renal impairment on the systemic exposure to SPIRIVA RESPIMAT 2.5 mcg in patients with asthma was similar to what has been described for COPD above. There lacks sufficient data of tiotropium exposure in patients with severe renal impairment (creatinine clearance <30 mL/min) following inhalation of SPIRIVA RESPIMAT. However AUC0-4 and Cmax were 94% and 52% higher, respectively, in patients with severe renal impairment following intravenous infusion of tiotropium bromide.Hepatic Impairment
The effects of hepatic impairment on the pharmacokinetics of tiotropium were not studied.Drug Interactions
An interaction study with tiotropium (14.4 mcg intravenous infusion over 15 minutes) and cimetidine 400 mg three times daily or ranitidine 300 mg once-daily was conducted. Concomitant administration of cimetidine with tiotropium resulted in a 20% increase in the AUC0-4h, a 28% decrease in the renal clearance of tiotropium and no significant change in the Cmax and amount excreted in urine over 96 hours. Co-administration of tiotropium with ranitidine did not affect the pharmacokinetics of tiotropium.Common concomitant medications (LABA, ICS) used by patients with COPD were not found to alter the exposure to tiotropium. Similarly, common concomitant medications (LABA, ICS+LABA combinations, oral corticosteroids and leukotriene modifiers) used by patients with asthma were not found to alter the exposure to tiotropium.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No evidence of tumorigenicity was observed in a 104-week inhalation study in rats at tiotropium doses up to 59 mcg/kg/day, in an 83-week inhalation study in female mice at doses up to 145 mcg/kg/day, and in a 101‑week inhalation study in male mice at doses up to 2 mcg/kg/day. These doses correspond to approximately 30, 40, and 0.5, times the maximum recommended human daily inhalation dose (MRHDID) on a mcg/m2 basis, respectively.
Tiotropium bromide demonstrated no evidence of mutagenicity or clastogenicity in the following assays: the bacterial gene mutation assay, the V79 Chinese hamster cell mutagenesis assay, the chromosomal aberration assays in human lymphocytes in vitro and mouse micronucleus formation in vivo, and the unscheduled DNA synthesis in primary rat hepatocytes in vitro assay.
In rats, decreases in the number of corpora lutea and the percentage of implants were noted at inhalation tiotropium doses of 78 mcg/kg/day or greater (approximately 40 times the MRHDID on a mcg/m2 basis). No such effects were observed at 9 mcg/kg/day (approximately 5 times the MRHDID on a mcg/m2 basis). The fertility index, however, was not affected at inhalation doses up to 1689 mcg/kg/day (approximately 910 times the MRHDID on a mcg/m2 basis).
-
14 CLINICAL STUDIES
14.1 Chronic Obstructive Pulmonary Disease
The efficacy of SPIRIVA RESPIMAT compared to placebo was evaluated in 6 clinical trials: one dose-ranging trial and 5 confirmatory trials (Trials 1-5). In addition, SPIRIVA RESPIMAT was compared to SPIRIVA HandiHaler in a long-term active-controlled trial in COPD (Trial 6).
Dose-Ranging Trial
Dose selection for the Phase III clinical program was supported by a 3-week randomized, double-blind, placebo and active-controlled, parallel group trial in 202 COPD patients. A total of five doses of tiotropium RESPIMAT (1.25 to 20 mcg) were evaluated compared to placebo. Results demonstrated numerical improvements in FEV1 at all doses compared to placebo. The difference in trough FEV1 from placebo for the 1.25, 2.5, 5, 10 and 20 mcg once daily doses were 0.08 L (95% CI -0.03, 0.20), 0.03 L (-0.08, 0.15), 0.13 L (0.02, 0.25), 0.11 L (-0.004, 0.224), and 0.13 L (0.01, 0.24), respectively. Based on these results, the 5 and 10 mcg doses were further evaluated in the confirmatory COPD trials.Confirmatory Trials
A total of 6614 COPD patients (2801 receiving SPIRIVA RESPIMAT 5 mcg and 2798 receiving placebo) were studied in the five confirmatory trials of SPIRIVA RESPIMAT. Trials 1 and 2 were 12-week, randomized, double-blind, placebo- and active- (ipratropium) controlled trials that evaluated bronchodilation. Trials 3-5 were 48-week, randomized, double-blind, placebo-controlled, trials that evaluated bronchodilation and effects on COPD exacerbations. Trials 1-4 included both the tiotropium RESPIMAT 5 mcg and 10 mcg doses, whereas Trial 5 included only the 5 mcg dose. These trials enrolled patients who had a clinical diagnosis of COPD, were 40 years of age or older, had a history of smoking greater than 10 pack-years, had an FEV1 less than or equal to 60% of predicted and a ratio of FEV1/FVC of less than or equal to 0.7. All treatments were administered once-daily in the morning. Change from baseline in trough FEV1 was a primary endpoint in all trials. Trials 3-5 included COPD exacerbations as primary endpoints.Baseline patient characteristics were similar across the five individual confirmatory trials, except for race in Trial 5 in which there were more Asian patients (30%) compared to other trials (<1%). The mean age ranged from 62 to 66 years. Most patients were male (64-78%), ex-smokers (57-65%) and Caucasian (69-99%). Mean pre-bronchodilator FEV1 was between 1.03 and 1.26 L with a mean FEV1/FVC ratio of 42-50%. Except for LABAs and other inhaled anticholinergic agents, other pulmonary medications were allowed as concomitant therapy in Trials 1-4. LABA use was permitted in Trial 5.
Effect on Lung Function
SPIRIVA RESPIMAT 5 mcg demonstrated significant improvement in trough FEV1 compared to placebo in all 5 confirmatory trials (Table 4). The change from baseline in trough FEV1 over time from Trial 4 is depicted in Figure 1 and is representative of the other two 48-week trials. In Trials 3 and 4 patients treated with SPIRIVA RESPIMAT 5 mcg also used less rescue medication compared to patients on placebo.Table 4 Mean Change from Baseline in Trough FEV1 (L) at End of Treatment † at week 12 ‡ at week 48 Trial SPIRIVA
RESPIMAT
5 mcg
NPlacebo
NTrough FEV1 (L) at End of Treatment
Difference from placebo (95% CI)Trial 1† 85 87 0.11 (0.04, 0.18) Trial 2† 90 84 0.13 (0.07, 0.18) Trial 3‡ 326 296 0.14 (0.10, 0.18) Trial 4‡ 324 307 0.11 (0.08, 0.15) Trial 5‡ 1889 1870 0.10 (0.09, 0.12) Figure 1 Trough FEV1 Change from Baseline over 48 weeks (Trial 4), SPIRIVA RESPIMAT 5 mcg
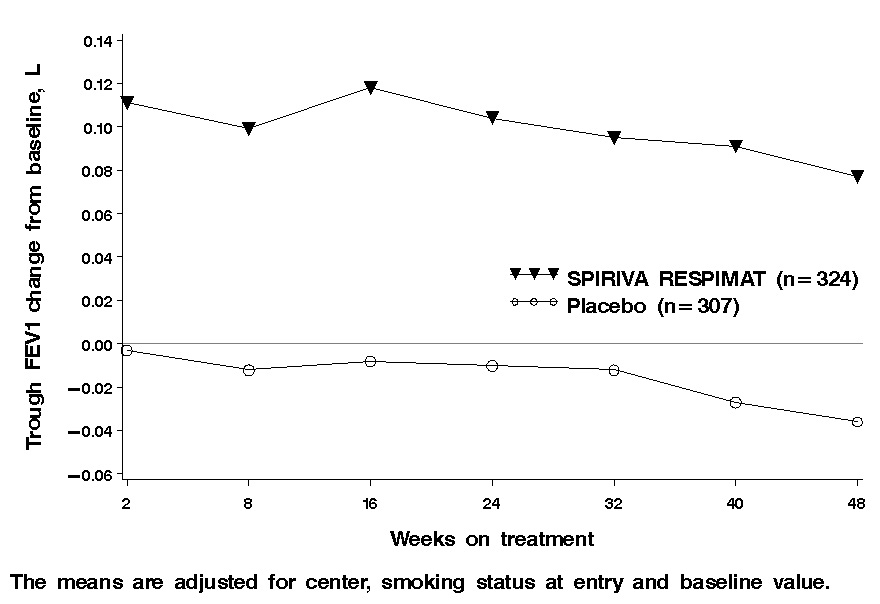
Exacerbations
Trials 3, 4, and 5 also evaluated the effect of SPIRIVA RESPIMAT 5 mcg on COPD exacerbations. For Trials 3 and 4, a pooled analysis of exacerbation rate per patient year was pre-specified as a primary endpoint, while the primary endpoint for Trial 5 was time to first exacerbation. Trial 5 also included exacerbation rate per patient year as a secondary endpoint. Exacerbations were defined as a complex of respiratory events/symptoms with a duration of ≥3 days with ≥2 of the following (increase of symptoms or new onset): shortness of breath/dyspnea/shallow, rapid breathing; sputum production (volume); occurrence of purulent sputum; cough; wheezing; chest tightness.In the pooled analysis of Trials 3 and 4, SPIRIVA RESPIMAT 5 mcg significantly reduced the number of COPD exacerbations compared to placebo with 0.78 exacerbations/patient year versus 1.0 exacerbations/patient year, respectively, with a rate ratio of 0.78 (95% CI 0.67, 0.92). Time to first exacerbation was also delayed in SPIRIVA RESPIMAT 5 mcg patients. For Trial 5, in addition to the definition above, an exacerbation also had to result in a change in or requirement of treatment. In Trial 5, treatment with SPIRIVA RESPIMAT 5 mcg delayed the time to first COPD exacerbation compared to treatment with placebo [hazard ratio of 0.69 (95% CI 0.63, 0.77)]. Consistent with the pooled analysis of Trials 3 and 4, for Trial 5, exacerbation rate was also lower in SPIRIVA RESPIMAT 5 mcg compared to placebo. In Trial 5, SPIRIVA RESPIMAT 5 mcg also reduced the risk of COPD exacerbation-related hospitalization (HR = 0.73; 95% CI = 0.59, 0.90) compared to placebo.
Long-term Active-Controlled Mortality Trial
Survival
In a pooled analysis of SPIRIVA RESPIMAT placebo-controlled clinical trials with complete vital status (mortality) follow-up, including the three 48-week trials (Trial 3, 4, and 5) and one 24-week placebo-controlled trial, 68 deaths (Incidence Rate 2.64 deaths per 100 patient years) were observed in the SPIRIVA RESPIMAT 5 mcg treatment group compared to 51 deaths (Incidence Rate 1.98 deaths per 100 patient years) in those treated with placebo. In a 4-year, randomized, double-blind, placebo-controlled, multicenter clinical trial of tiotropium bromide inhalation powder (SPIRIVA HandiHaler) in 5992 COPD patients a similar incidence rate of death had been observed between SPIRIVA HandiHaler and placebo treated groups.For clarification of the observed difference in fatal events, a long-term, randomized, double-blind, double dummy, active-controlled trial with an observation period up to 3 years was conducted to evaluate the risk of all-cause mortality associated with the use of SPIRIVA RESPIMAT compared to SPIRIVA HandiHaler (Trial 6). The objective of this trial was to rule out a relative excess mortality risk of 25% for SPIRIVA RESPIMAT versus SPIRIVA HandiHaler. The primary endpoints were all-cause mortality and time to first COPD exacerbation. Trial 6 also included a lung function sub-study which measured trough FEV1 measured every 24 weeks for 120 weeks (461 patients receiving SPIRIVA RESPIMAT 5 mcg, 445 patients receiving SPIRIVA HandiHaler).
In Trial 6, 5711 patients received SPIRIVA RESPIMAT 5 mcg and 5694 patients received SPIRIVA HandiHaler. All patients were followed for vital status (mortality) at the end of the trial. At baseline, patient characteristics were balanced between the two treatment arms. The mean age was 65 years and approximately 70% of subjects were male. Approximately, 82% of patients were Caucasian, 14% were Asian, and 2% were Black. Mean post-bronchodilator FEV1 was 1.34 L with a mean FEV1/FVC ratio of 50%. The majority of patients were GOLD II or III (48% and 40%, respectively).
The vital status was confirmed in 99.7% of patients. The median exposure to treatment was 835 days for both treatment groups. All-cause mortality was similar between SPIRIVA RESPIMAT 5 mcg and SPIRIVA HandiHaler with an estimated hazard ratio of 0.96 [(95% CI of (0.84 to 1.09), Table 5].
Table 5 All-cause Mortality of SPIRIVA RESPIMAT vs SPIRIVA HandiHaler (Trial 6) a Hazard ratios were estimated from a Cox proportional hazard model. SPIRIVA RESPIMAT 5 mcg
(N = 5711)SPIRIVA HandiHaler
(N = 5694)Number (%) of Deaths 423 (7.4) 439 (7.7) Incidence Rate per 100 patient years 3.22 3.36 HR (95% CI)a 0.96 (0.84, 1.09) Cause of death was adjudicated by a blinded, independent committee. Cardiovascular deaths included cardiac death, sudden cardiac death, and sudden death; as well as fatal events caused by a cardiac disorder, vascular disorder, or stroke. There were 113 patients (2%) treated with SPIRIVA RESPIMAT 5 mcg who had cardiovascular deaths compared to 101 (2%) patients treated with SPIRIVA HandiHaler. Of the cardiovascular deaths, 11 (0.2%) and 3 (0.1%) deaths were due to myocardial infarction in SPIRIVA RESPIMAT 5 mcg patients and SPIRIVA HandiHaler patients, respectively. For cardiac deaths, sudden cardiac death, and sudden death, there were a total of 69 (1.2%) and 68 (1.2%) deaths in SPIRIVA RESPIMAT 5 mcg patients and SPIRIVA HandiHaler patients, respectively.
Effect on Lung Function and Exacerbations
In the lung function sub-study the effect of SPIRIVA RESPIMAT 5 mcg on trough FEV1 over 120 weeks was similar to SPIRIVA HandiHaler with a mean difference of -0.010 L (95% CI -0.038 to 0.018 L).Trial 6 also included time to first exacerbation as a co-primary endpoint (exacerbations defined as in Trials 3-5). SPIRIVA RESPIMAT 5 mcg failed to demonstrate superiority to SPIRIVA HandiHaler with a similar time to first COPD exacerbation between treatment groups [hazard ratio of 0.98 (95% CI 0.93 to 1.03)].
14.2 Asthma
The SPIRIVA RESPIMAT clinical development program included six 4-week to 8-week cross-over design trials and ten 12-week to 48-week parallel-arm design trials in adult, adolescent (aged 12 to 17 years) and pediatric (aged 1 to 11 years) patients with asthma symptomatic on at least ICS. In all trials, SPIRIVA RESPIMAT was administered on a background of ICS therapy.
Dose Selection
Dose selection for the confirmatory trials was based on three randomized, double-blind, placebo-controlled, 4-week to 8-week, cross-over trials in 256 adult patients, 105 adolescent (age 12 to 17 years) patients, and 101 pediatric (age 6 to 11 years) patients that assessed doses ranging from 1.25 mcg to 10 mcg once daily. Results demonstrated numerical improvements in FEV1 at all doses compared to placebo; however, across the trials, the response was not dose-ordered. For adult patients, in the 4-week trial the difference in peak FEV1 within 3 h post-dosing (peak FEV1, 0-3hr) from placebo for the tiotropium RESPIMAT 1.25, 2.5, and 5 mcg doses were 0.138 L (95% CI 0.090, 0.186), 0.128 L (0.080, 0.176), and 0.188 L (0.140, 0.236), respectively. For adolescent patients, the difference in peak FEV1, 0-3hr from placebo for the tiotropium RESPIMAT 1.25, 2.5, and 5 mcg doses were 0.067 L (95% CI −0.005, 0.138), 0.057 L (−0.021, 0.135), and 0.113 L (0.036, 0.190), respectively. For pediatric patients, the difference in peak FEV1, 0-3h from placebo for the tiotropium RESPIMAT 1.25, 2.5, and 5 mcg doses were 0.075 L (95% CI, 0.030, 0.120), 0.104 L (0.059, 0.149), and 0.087 L (0.042, 0.132), respectively. The 10 mcg dose offered no substantial benefit over lower doses and resulted in more systemic anticholinergic side effects (e.g., dry mouth).The two dose regimen trials in adults with asthma were randomized, double-blind, 4-week, cross-over trials comparing tiotropium RESPIMAT 2.5 mcg twice-daily with 5 mcg once-daily. 24-hour FEV1 results demonstrated comparable treatment effects for twice-daily and once-daily dosing.
12-week to 48-week Parallel-Arm Design Trials in Adults
The program for persistent asthma in adult patients included one 12-week (Trial 1), two replicate 24-week (Trials 2 and 3), and two replicate 48-week (Trials 4 and 5) randomized, double-blind, placebo-controlled trials in a total of 3476 asthma patients (673 receiving SPIRIVA RESPIMAT 2.5 mcg once-daily, 1128 receiving SPIRIVA RESPIMAT 5 mcg once-daily, 541 receiving salmeterol 50 mcg twice daily, and 1134 receiving placebo) on background treatment of at least ICS. Trial 1 evaluated three treatments: SPIRIVA RESPIMAT 2.5 mcg once-daily, SPIRIVA RESPIMAT 5 mcg once-daily, and placebo. Trials 2 and 3 evaluated four treatments: SPIRIVA RESPIMAT 2.5 mcg once-daily, SPIRIVA RESPIMAT 5 mcg once-daily, salmeterol 50 mcg twice daily, and placebo. Trials 4 and 5 evaluated two treatments: SPIRIVA RESPIMAT 5 mcg once-daily and placebo. All trials enrolled patients who had a diagnosis of asthma, were 18 to 75 years of age, and were not current smokers. Patients enrolled in Trials 4 and 5 were required to have airway obstruction that was not fully reversible (post-bronchodilator FEV1/FVC, 0.70). The majority of the 3476 patients in the adult asthma trials were female (60%), Caucasian (61%) or Asian (31%), and had never smoked (81%) with a mean age of 46 years. The patient characteristics for the 12 week to 48 week trials in adult patients with asthma are summarized in Table 6.Table 6 Summary of Baseline Patient Characteristics, Adult Confirmatory Studies Adults, 18 yrs and older Trial 1 Trial 2 Trial 3 Trial 4 Trial 5 Demographics Mean age in years (range) 42.9 (18 – 74) 43.3 (18 – 75) 42.9 (18 – 75) 53.4 (18 – 75) 52.5 (19 – 75) Mean duration of asthma (years) 16.2 21.7 21.8 31.5 29.1 Smoking status, ex-smoker (%) 18 14 19 22 26 Laboratory (median) Absolute eosinophils (109/L) 0.33 0.36 0.35 0.35 0.38 Total IgE (microgram/L) 536 638 641 601 449 Pulmonary function test (mean) Pre-bronchodilator FEV1 (L) 2.30 2.18 2.21 1.55 1.59 Reversibility (%) 24.8 22.8 22.0 15.4 15.0 Absolute reversibility (mL) 556 488 477 215 218 Post-bronchodilator FEV1/FVC (%) 74 72 72 60 59 The primary efficacy endpoint in Trial 1 was change from pre-treatment baseline in peak FEV1, 0-3h at week 12. The co-primary efficacy endpoints in Trials 2 and 3 were change from pre-treatment baseline in peak FEV1, 0-3 hr and change from pre-treatment baseline in trough FEV1 at week 24. Additional efficacy measures included asthma exacerbation, Asthma Control Questionnaire (ACQ), and Asthma Quality of Life Questionnaire (AQLQ).
For Trials 1, 2, and 3, SPIRIVA RESPIMAT 2.5 mcg showed statistically significant improvements in lung function over placebo when used in addition to background treatment of ICS (Table 7).
Table 7 Differences from Placebo in Peak FEV1, 0-3 and Trough FEV1, Adult Confirmatory Studies at Primary Endpoint Time Evaluation a Means adjusted for treatment, center/country, visit, visit*treatment, baseline, baseline*visit.
b Additional asthma medications allowed in stable doses prior to and throughout the trials.
c Low dose ICS = 200–400 mcg budesonide-equivalent. Medium dose ICS = 400–800 mcg budesonide-equivalent.Treatment
(Duration)
ICS Background
Treatment b,cTreatment in
mcg/dayn Peak FEV1, 0- 3hr, in L a Trough FEV1 , in L a Δ from
baselineDifference from placebo Δ from
baselineDifference from placebo Mean 95% CI Mean 95% CI Adult patients, age 18 years and older Trial 1
(12 weeks)
Low dose ICSSPIRIVA RESPIMAT 2.5 mcg
Placebo154
1550.29
0.130.16 0.09, 0.23 0.13
0.020.11 0.04, 0.18 Trial 2
(24 weeks)
Medium dose ICSSPIRIVA RESPIMAT 2.5 mcg
Salmeterol 100 mcg
Placebo259
271
2650.29
0.27
0.050.24
0.210.18, 0.29
0.16, 0.270.15
0.09
–0.030.19
0.120.13, 0.24
0.06, 0.18Trial 3
(24 weeks)
Medium dose ICSSPIRIVA RESPIMAT 2.5 mcg
Salmeterol 100 mcg
Placebo256
264
2530.29
0.25
0.080.21
0.180.16, 0.26
0.12, 0.230.16
0.09
–0.010.18
0.110.12, 0.23
0.05, 0.16Trials 1, 2, and 3 also included a SPIRIVA RESPIMAT 5 mcg once daily treatment arm. In these asthma trials, the FEV1 response (change from baseline for tiotropium compared to placebo) was generally lower for the 5 mcg dose compared to the 2.5 mcg dose. The peak FEV1, 0-3hr response was 16% to 20% lower for the 5 mcg dose compared to the 2.5 mcg dose in all three trials, and, the trough FEV1 response was 11% higher for the 5 mcg dose compared to the 2.5 mcg dose for one trial (Trial 1) and 18% and 24% lower for the 5 mcg dose compared to the 2.5 mcg dose for the other two trials (Trials 2 and 3).
Improvements in morning and evening peak expiratory flow (PEF) were consistent with the observed FEV1 treatment response. Examination of age, gender, smoking history, and serum IgE level subgroups did not identify differences in response among these subgroups.
The improvement of lung function compared to placebo was maintained for 24 hours (Figure 2). The bronchodilator effects of SPIRIVA RESPIMAT 2.5 mcg were apparent after first dose; however, maximum bronchodilator effect took up to 4 to 8 weeks to be achieved.
Figure 2 FEV1 Response over 24-Hours following 24-Weeks of Treatment, Trial 3
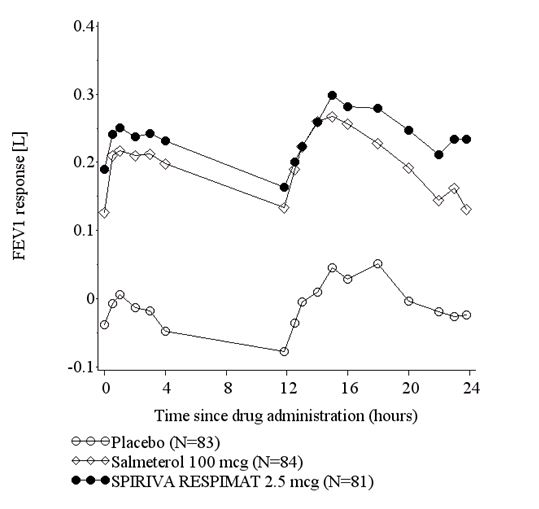
Asthma exacerbation was assessed in Trials 2 and 3 over the 24-week treatment periods. An asthma exacerbation was defined as an episode of progressive increase in ≥1 asthma symptom(s), such as shortness of breath, cough, wheezing, chest tightness or some combination of these symptoms or a decrease of a patient's best morning PEF of 30% from a patient's mean morning PEF for ≥2 consecutive days that required the initiation or increase in treatment with systemic steroids for ≥3 days. Results of asthma exacerbation are shown in Table 8.
Table 8 Exacerbations in Patients on ICS over 24-Weeks Trial 2 Trial 3 SPIRIVA
RESPIMAT 2.5 mcg
(N=259)Placebo
(N=265)SPIRIVA
RESPIMAT 2.5 mcg
(N=256)Placebo
(N=253)Number of patients with at least 1 event, n (%) 9 (3.5) 24 (9.1) 13 (5.1) 19 (7.5) Rate of exacerbations per patient year Mean rate of events 0.08 0.24 0.13 0.18 0.32 (0.20, 0.51) 0.70 (0.46, 1.08) Time to first asthma exacerbation 0.37 (0.17, 0.80) 0.66 (0.33, 1.34) Trials 2 and 3 also evaluated the rate of exacerbations and time to first asthma exacerbation for the SPIRIVA RESPIMAT 5 mcg dose. The rate of asthma exacerbations compared to placebo for SPIRIVA RESPIMAT 5 mcg was 0.78 (95% CI 0.55, 1.10) in Trial 2 and 0.76 (0.50, 1.16) in Trial 3. The hazard ratio for time to first asthma exacerbation for SPIRIVA RESPIMAT 5 mcg compared to placebo was 0.72 (95% CI 0.39, 1.35), in Trial 2 and 0.72 (0.36, 1.43) in Trial 3.
ACQ and AQLQ were assessed in Trials 2 and 3 at week 24. In Trial 2, the ACQ-7 (7 items) responder rate (defined as a change in score >0.5) for the SPIRIVA RESPIMAT 2.5 mcg treatment arm was 63% compared to 53% for placebo with an odds ratio of 1.47 (95% CI 1.02, 2.11). The ACQ-5 (derived from ACQ 7 by removing the FEV1 component and rescue bronchodilator component) results also had a similar trend. In Trial 2, the AQLQ responder rate (defined as a change in score >0.5) for the SPIRIVA RESPIMAT 2.5 mcg treatment arm was 58% compared to 50% for placebo with an odds ratio of 1.34 (95% CI 0.94, 1.93).
12-week and 48-week Parallel-Arm Design Trials in Adolescents 12-17 Years of Age
Efficacy in adolescents was based on partial extrapolation of efficacy in adults and two randomized, double-blind, placebo-controlled trials of 12 and 48 weeks duration in a total of 789 asthma patients 12 to 17 years of age (252 receiving SPIRIVA RESPIMAT 2.5 mcg once-daily, 264 receiving 5 mcg once-daily, and 273 receiving placebo). The 12-week trial enrolled patients with severe asthma who were on background treatment of ICS plus one or more controller medications (e.g. LABA). The 48-week trial enrolled patients with moderate asthma on background treatment of at least ICS. The majority of the patients in the trials were male (63.4%), Caucasian (93.7%) and had never smoked (99.9%) with a mean age of 14.3 years.The primary efficacy endpoint in both trials was change from pre-treatment baseline in peak FEV1, 0-3hr. The primary endpoint evaluation for FEV1 was defined at week 24 for the 48-week trial and at end of the treatment period (week 12) for the 12-week trial. Given the demonstration of efficacy in the adult population, the results of the 2 trials support the efficacy of SPIRIVA RESPIMAT 2.5 mcg once daily in adolescent patients 12-17 years of age with asthma (mean difference in peak FEV1, 0-3hr from placebo for SPIRIVA RESPIMAT 2.5 mcg were 0.13 L (95% CI 0.03, 0.23) and 0.11 L (0.002, 0.22) for the 48-week and 12-week trials, respectively).
12-week and 48-week Parallel-Arm Design Trials in Pediatric Patients 6-11 Years of Age
Efficacy in pediatric patients 6-11 years of age was based on partial extrapolation of efficacy in adults and two randomized, double-blind, placebo-controlled trials of 12 and 48 weeks duration in a total of 801 asthma patients 6 to 11 years of age (271 receiving SPIRIVA RESPIMAT 2.5 mcg once-daily, 265 receiving 5 mcg once-daily, and 265 receiving placebo). The 12-week trial enrolled patients with severe asthma who were on background treatment of ICS plus one or more controller medications (e.g. LABA). The 48-week trial enrolled patients with moderate asthma on background treatment of at least ICS. The primary efficacy endpoint in both trials was change from pre-treatment baseline in peak FEV1, 0-3hr with the evaluation defined at week 24 for the 48-week trial and at end of the treatment period (week 12) for the 12-week trial. The majority of the patients in the trials were male (67.8%) and Caucasian (87.0%) with a mean age of 9.0 years.Compared to placebo, SPIRIVA RESPIMAT 2.5 mcg once daily had a significant effect on the primary endpoint in the 48 week, but not the 12 week trial, with mean differences in peak FEV1, 0-3hr from placebo of 0.17 L (95% CI 0.11, 0.23) and 0.04 L (95% CI -0.03, 0.10) for the 48-week and 12-week trials, respectively. Given the demonstration of efficacy in the adult and adolescent population, the results support the efficacy of SPIRIVA RESPIMAT 2.5 mcg once daily in pediatric patients 6-11 years of age with asthma.
-
16 HOW SUPPLIED/STORAGE
AND HANDLING
SPIRIVA RESPIMAT Inhalation Spray is supplied in a carton containing one SPIRIVA RESPIMAT cartridge and one SPIRIVA RESPIMAT inhaler.
The SPIRIVA RESPIMAT cartridge is provided as an aluminum cylinder with a tamper protection seal on the cap. The SPIRIVA RESPIMAT cartridge is only intended for use with the SPIRIVA RESPIMAT inhaler and should not be interchanged with any other RESPIMAT device delivered product.
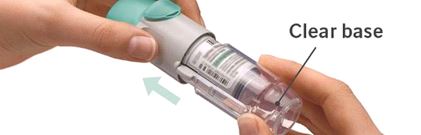
The SPIRIVA RESPIMAT inhaler is a cylindrical shaped plastic inhalation device with a gray colored body and a clear base. The clear base is removed to insert the cartridge. The inhaler contains a dose indicator. The written information on the label of the gray inhaler body indicates that it is labeled for use with the SPIRIVA RESPIMAT cartridge.
SPIRIVA RESPIMAT Inhalation Spray is available in two dosage strengths, identified by dose delivered per actuation and by the color of the cap and associated container label: aqua represents 2.5 mcg per actuation; blue represents 1.25 mcg per actuation.
To deliver the recommended dosage for COPD:
- SPIRIVA RESPIMAT Inhalation Spray 2.5 mcg/actuation 60 metered actuations (NDC: 0597-0100-61)
- SPIRIVA RESPIMAT Inhalation Spray 2.5 mcg/actuation 28 metered actuations (NDC: 0597-0100-31) (institutional pack)
- SPIRIVA RESPIMAT Inhalation Spray 2.5 mcg/actuation 10 metered actuations (NDC: 0597-0100-51) (institutional pack)
To deliver the recommended dosage for asthma:
- SPIRIVA RESPIMAT Inhalation Spray 1.25 mcg/actuation 60 metered actuations (NDC: 0597-0160-61)
The SPIRIVA RESPIMAT cartridge for each strength has a net fill weight of 4 grams and when used with the SPIRIVA RESPIMAT inhaler, is designed to deliver the labeled number of metered actuations after preparation for use. Each actuation from the SPIRIVA RESPIMAT inhaler delivers 1.25 or 2.5 mcg of tiotropium (equivalent to 1.562 or 3.124 mcg, respectively, of tiotropium bromide monohydrate) from the mouthpiece.
When the labeled number of actuations has been dispensed from the inhaler, the RESPIMAT locking mechanism will be engaged and no more actuations can be dispensed.
After assembly, the SPIRIVA RESPIMAT inhaler should be discarded at the latest 3 months after first use or when the locking mechanism is engaged, whichever comes first.
Keep out of reach of children. Do not spray into eyes.
Storage
Store at 25°C (77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Avoid freezing. -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Instructions for Use).
Not for Acute Use
Instruct patients that SPIRIVA RESPIMAT is a once-daily maintenance bronchodilator and should not be used for immediate relief of breathing problems, (i.e., as a rescue medication).Immediate Hypersensitivity Reactions
Inform patients that anaphylaxis, angioedema (including swelling of the lips, tongue, or throat), urticaria, rash, bronchospasm, or itching, may occur after administration of SPIRIVA RESPIMAT. Advise patient to immediately discontinue treatment and consult a physician should any of these signs or symptoms develop.Paradoxical Bronchospasm
Inform patients that SPIRIVA RESPIMAT can produce paradoxical bronchospasm. Advise patients that if paradoxical bronchospasm occurs, patients should discontinue SPIRIVA RESPIMAT.Worsening of Narrow-Angle Glaucoma
Instruct patients to be alert for signs and symptoms of narrow-angle glaucoma (e.g., eye pain or discomfort, blurred vision, visual halos or colored images in association with red eyes from conjunctival congestion and corneal edema). Instruct patients to consult a physician immediately should any of these signs and symptoms develop.Inform patients that care must be taken not to allow the aerosol cloud to enter into the eyes as this may cause blurring of vision and pupil dilation.
Since dizziness and blurred vision may occur with the use of SPIRIVA RESPIMAT, caution patients about engaging in activities such as driving a vehicle or operating appliances or machinery.
Worsening of Urinary Retention
Instruct patients to be alert for signs and symptoms of urinary retention (e.g., difficulty passing urine, painful urination). Instruct patients to consult a physician immediately should any of these signs or symptoms develop.Treatment of Asthma
Instruct asthma patients that the maximum benefits may only be apparent after 4 to 8 weeks of SPIRIVA RESPIMAT treatment.Instructions for Administering SPIRIVA RESPIMAT
It is important for patients to understand how to correctly administer SPIRIVA inhalation spray using the SPIRIVA RESPIMAT inhaler. Instruct patients that SPIRIVA inhalation spray should only be administered via the SPIRIVA RESPIMAT inhaler and the SPIRIVA RESPIMAT inhaler should not be used for administering other medications.Instruct patients that priming SPIRIVA RESPIMAT is essential to ensure appropriate content of the medication in each actuation.
When using the unit for the first time, the SPIRIVA RESPIMAT cartridge is inserted into the SPIRIVA RESPIMAT inhaler and the unit is primed. SPIRIVA RESPIMAT patients are to actuate the inhaler toward the ground until an aerosol cloud is visible and then to repeat the process three more times. The unit is then considered primed and ready for use. If not used for more than 3 days, patients are to actuate the inhaler once to prepare the inhaler for use. If not used for more than 21 days, patients are to actuate the inhaler until an aerosol cloud is visible and then repeat the process three more times to prepare the inhaler for use.
-
SPL UNCLASSIFIED SECTION
Instruct caregivers of children that SPIRIVA RESPIMAT should be used with an adult’s assistance.
Distributed by:
Boehringer Ingelheim Pharmaceuticals, Inc.
Ridgefield, CT 06877 USAAddress medical inquiries to: (800) 542-6257 or (800) 459-9906 TTY.
SPIRIVA®, HANDIHALER®, and RESPIMAT® are registered trademarks and are used under license from Boehringer Ingelheim International GmbH
Copyright © 2019 Boehringer Ingelheim International GmbH
ALL RIGHTS RESERVED -
PATIENT PACKAGE INSERT
Turn - Keep the cap closed.
- Turn the clear base in the direction of the arrows on the label until it clicks (half a turn).

Open - Open the cap until it snaps fully open.

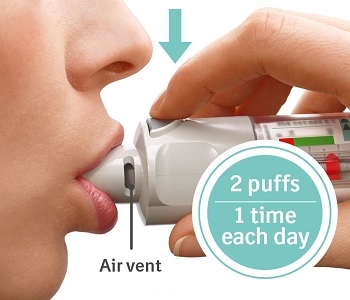
Press - Breathe out slowly and fully.
- Close your lips around the mouthpiece without covering the air vents.
- Point the inhaler to the back of your throat.
- While taking a slow, deep breath through your mouth, Press the dose-release button and continue to breathe in.
- Hold your breath for 10 seconds or for as long as comfortable.
- Repeat Turn, Open, Press (TOP) for a total of 2 puffs.
- Close the cap until you use your inhaler again.
It is difficult to insert the cartridge deep enough:
Did you accidentally turn the clear base before inserting the cartridge? Open the cap, press the dose-release button, then insert the cartridge.
Did you insert the cartridge with the wide end first? Insert the cartridge with the narrow end first.
I cannot press the dose-release button:
Did you turn the clear base? If not, turn the clear base in a continuous movement until it clicks (half a turn).
Is the dose indicator on the SPIRIVA RESPIMAT pointing to 0 (zero)? The SPIRIVA RESPIMAT inhaler is locked after the labeled number of puffs have been used. Prepare and use your new SPIRIVA RESPIMAT inhaler.
Did you turn the clear base already? If the clear base has already been turned, follow steps “Open” and “Press” under “Daily use” to get your medicine.
Is the dose indicator on the SPIRIVA RESPIMAT pointing to 0 (zero)? The SPIRIVA RESPIMAT inhaler is locked after the labeled number of puffs have been used. Prepare and use your new SPIRIVA RESPIMAT inhaler.
The dose indicator on the SPIRIVA RESPIMAT reaches 0 (zero) too soon:
Did you use SPIRIVA RESPIMAT as indicated (2 puffs 1 time each day)?
Did you turn the clear base before you inserted the cartridge? The dose indicator counts each turn of the clear base regardless whether a cartridge has been inserted or not.
Did you spray in the air often to check whether the SPIRIVA RESPIMAT is working? After you have prepared SPIRIVA RESPIMAT, no test-spraying is required if used daily.
Did you insert the cartridge into a used SPIRIVA RESPIMAT? Always insert a new cartridge into a new SPIRIVA RESPIMAT.
My SPIRIVA RESPIMAT sprays automatically:
Was the cap open when you turned the clear base? Close the cap, then turn the clear base.
Did you press the dose-release button when turning the clear base? Close the cap, so the dose-release button is covered, then turn the clear base.
Did you stop when turning the clear base before it clicked? Turn the clear base in a continuous movement until it clicks (half a turn).
My SPIRIVA RESPIMAT does not spray:
Did you insert a cartridge? If not, insert a cartridge.
Did you repeat Turn, Open, Press (TOP) less than 3 times after inserting the cartridge? Repeat Turn, Open, Press (TOP) 3 times after inserting the cartridge as shown in steps 4 to 6 under “Prepare for first use”.
Is the dose indicator on the SPIRIVA RESPIMAT pointing to 0 (zero)? You have used up all your medicine and the inhaler is locked.
For more information about SPIRIVA RESPIMAT, including current prescribing information, or a video demonstration on how to use SPIRIVA RESPIMAT, go to www.spiriva.com, or scan the code below. You may also call 1-800-542-6257 or (TTY) 1-800-459-9906 for further information about SPIRIVA RESPIMAT.

This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Distributed by: Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT 06877 USA
SPIRIVA® and RESPIMAT® are registered trademarks and are used under license from Boehringer Ingelheim International GmbH
Copyright © 2019 Boehringer Ingelheim International GmbH
ALL RIGHTS RESERVED - PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
SPIRIVA RESPIMAT
tiotropium bromide inhalation spray spray, meteredProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0597-0160 Route of Administration RESPIRATORY (INHALATION) Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TIOTROPIUM BROMIDE (UNII: XX112XZP0J) (TIOTROPIUM - UNII:0EB439235F) TIOTROPIUM BROMIDE 1.562 ug Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0597-0160-61 1 in 1 CARTON 09/15/2015 1 60 in 1 CARTRIDGE; Type 0: Not a Combination Product 2 NDC: 0597-0160-28 1 in 1 CARTON 09/15/2015 2 28 in 1 CARTRIDGE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA207070 09/15/2015 SPIRIVA RESPIMAT
tiotropium bromide inhalation spray spray, meteredProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0597-0100 Route of Administration RESPIRATORY (INHALATION) Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TIOTROPIUM BROMIDE (UNII: XX112XZP0J) (TIOTROPIUM - UNII:0EB439235F) TIOTROPIUM BROMIDE 3.124 ug Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0597-0100-61 1 in 1 CARTON 09/30/2014 1 60 in 1 CARTRIDGE; Type 0: Not a Combination Product 2 NDC: 0597-0100-28 1 in 1 CARTON 09/30/2014 2 28 in 1 CARTRIDGE; Type 0: Not a Combination Product 3 NDC: 0597-0100-31 1 in 1 CARTON 07/02/2018 3 28 in 1 CARTRIDGE; Type 0: Not a Combination Product 4 NDC: 0597-0100-51 1 in 1 CARTON 03/15/2019 4 10 in 1 CARTRIDGE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021936 09/30/2014 Labeler - Boehringer Ingelheim Pharmaceuticals, Inc. (603175944) Registrant - Boehringer Ingelheim Pharmaceuticals, Inc. (603175944) Establishment Name Address ID/FEI Business Operations Boehringer Ingelheim Pharma GmbH and Co. KG 551147440 API MANUFACTURE(0597-0160, 0597-0100) , MANUFACTURE(0597-0100, 0597-0160) , ANALYSIS(0597-0160, 0597-0100)












© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.