CASGEVY- exagamglogene autotemcel injection, suspension
CASGEVY by
Drug Labeling and Warnings
CASGEVY by is a Other medication manufactured, distributed, or labeled by Vertex Pharmaceuticals Incorporated. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use CASGEVY™ safely and effectively. See full prescribing information for CASGEVY
CASGEVY (exagamglogene autotemcel), suspension for intravenous infusion
Initial U.S. Approval: 2023RECENT MAJOR CHANGES
Warnings and Precautions, Off-Target Genome Editing Risk (5.4) 08/2025 INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
For autologous use only. For intravenous use only.
- Patients are required to undergo hematopoietic stem cell (HSC) mobilization followed by apheresis to obtain CD34+ cells for CASGEVY manufacturing. (2.2)
- Dosing of CASGEVY is based on body weight. The minimum recommended dose is 3 × 106 CD34+ cells/kg. (2.1, 2.3)
- Full myeloablative conditioning must be administered between 48 hours and 7 days before infusion of CASGEVY. (2.2)
- Prophylaxis for seizures should be considered prior to initiating myeloablative conditioning. (2.2)
- Verify that the patient's identity matches the unique patient identification information on the product labels and Lot Information Sheet prior to thaw and infusion. (2.2)
- Do not sample, alter, or irradiate CASGEVY. (2.2)
- Do not use an in-line blood filter when infusing CASGEVY. (2.3)
- Administer each vial of CASGEVY via intravenous infusion within 20 minutes of thaw. (2.3)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
- None. (4)
WARNINGS AND PRECAUTIONS
- Neutrophil Engraftment Failure: Monitor absolute neutrophil counts (ANC) after CASGEVY infusion. Administer rescue cells in the event of neutrophil engraftment failure. (5.1)
- Delayed Platelet Engraftment: Monitor platelet counts until platelet engraftment and recovery are achieved. Patients should be monitored for bleeding. (5.2)
- Hypersensitivity Reactions: Monitor for hypersensitivity reactions during and after infusion. (5.3)
- Off-Target Genome Editing Risk: The risk of unintended, off-target editing in CD34+cells due to genetic variants cannot be ruled out. (5.4)
ADVERSE REACTIONS
The most common Grade 3 or 4 non-laboratory adverse reactions (incidence ≥ 25%) were mucositis and febrile neutropenia in patients with SCD and TDT, and decreased appetite in patients with SCD. (6)
The most common Grade 3 or 4 laboratory abnormalities (≥ 50%) were neutropenia, thrombocytopenia, leukopenia, anemia, and lymphopenia. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Vertex Pharmaceuticals Incorporated at 1-877-634-8789 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Granulocyte-Colony Stimulating Factor: Granulocyte-Colony Stimulating Factor (G-CSF) must not be used for CD34+ HSC mobilization of patients with SCD. (7.1)
- Hydroxyurea: Discontinue hydroxyurea at least 8 weeks prior to start of mobilization and conditioning. (7.2)
- Voxelotor and Crizanlizumab: Discontinue the use of voxelotor and crizanlizumab at least 8 weeks prior to start of mobilization and conditioning. (7.3)
- Iron Chelators: Discontinue iron chelators at least 7 days prior to initiation of myeloablative conditioning. Avoid the use of non-myelosuppressive iron chelators for at least 3 months and use of myelosuppressive iron chelators for at least 6 months after CASGEVY infusion. (7.4)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1. INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dose
2.2 Preparation Before CASGEVY Infusion
2.3 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Neutrophil Engraftment Failure
5.2 Delayed Platelet Engraftment
5.3 Hypersensitivity Reactions
5.4 Off-Target Genome Editing Risk
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Use of Granulocyte-Colony Stimulating Factor (G-CSF)
7.2 Use of Hydroxyurea
7.3 Use of Voxelotor and Crizanlizumab
7.4 Use of Iron Chelators
7.5 Live Vaccines
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Patients with Renal Impairment
8.7 Patients with Hepatic Impairment
8.8 Patients Seropositive for Human Immunodeficiency Virus (HIV), Hepatitis B Virus (HBV) or Hepatitis C Virus (HCV)
8.9 Patients with Prior HSC Transplant
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Sickle Cell Disease
14.2 Transfusion-dependent β-thalassemia
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1. INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dose
For autologous use only. For one-time, single dose intravenous use only.
The minimum recommended dose of CASGEVY is 3 × 106 CD34+ cells/kg.
CASGEVY is provided as a single dose for infusion containing a suspension of CD34+ cells in one or more vials. See the Lot Information Sheet provided with the product shipment for additional information pertaining to the number of vials required to achieve the patient-specific dose. Administer all vials.
2.2 Preparation Before CASGEVY Infusion
Confirm that hematopoietic stem cell (HSC) transplantation is appropriate for the patient before mobilization, apheresis and myeloablative conditioning are initiated.
Screen patients for HIV-1, HIV-2, HBV, HCV, and any other infectious agents in accordance with local guidelines before collection of cells for manufacturing. CASGEVY should not be used in patients with active HIV-1, HIV-2, HBV or HCV.
Discontinue disease modifying therapies (e.g., hydroxyurea, crizanlizumab, voxelotor) 8 weeks before the planned start of mobilization and conditioning [see Drug Interactions (7.2, 7.3)].
Sickle Cell Disease
Prior to apheresis it is recommended that patients be transfused with a goal to maintain hemoglobin S (HbS) levels < 30% of total hemoglobin (Hb) while keeping total Hb concentration ≤ 11 g/dL.
Transfusion-dependent β-thalassemia
Prior to apheresis procedure it is recommended that patients be transfused with a goal to maintain hemoglobin (Hb) ≥ 11 g/dL.
Mobilization and Apheresis
Patients are required to undergo CD34+ HSC mobilization followed by apheresis to isolate the CD34+ cells needed for CASGEVY manufacturing.
Plerixafor and Granulocyte-Colony Stimulating Factor (G-CSF) were used for mobilization in patients with TDT.
Single agent plerixafor was used for mobilization in patients with SCD. G-CSF should not be administered for mobilization in patients with SCD.
Refer to the prescribing information for the mobilization agent(s) prior to treatment. See Clinical Studies (14) for description of the mobilization regimen used in the clinical trials.
Maximize CD34+ cell collection to obtain as many CD34+ cells as possible for product manufacturing during each mobilization and apheresis cycle. Perform up to three consecutive days of cell collection for product manufacturing per cycle, if clinically tolerated. A total collection target of at least 20 × 106 CD34+ cells/kg is recommended for product manufacture. Collected cells should be sent for product manufacturing even if the total collection target is not achieved. If the minimum dose of CASGEVY (3 × 106 CD34+ cells/kg) is not met after initial product manufacturing, the patient will need to undergo additional cycles of mobilization and apheresis. Each mobilization and apheresis cycle must be separated by a minimum of 14 days.
An additional ≥ 2 × 106 CD34+ cells/kg of unmodified back-up rescue cells must be collected from the patient and be cryopreserved prior to myeloablative conditioning and infusion with CASGEVY. The unmodified back-up cells may be needed for rescue treatment under any one of the following conditions: (1) compromise of CASGEVY after initiation of myeloablative conditioning and before CASGEVY infusion; (2) neutrophil engraftment failure; or (3) loss of engraftment after infusion with CASGEVY [see Warnings and Precautions (5.1)].
Myeloablative Conditioning
In patients with SCD it is recommended that patients be transfused at least 8 weeks prior to the initiation of myeloablative conditioning, with a goal of maintaining hemoglobin S (HbS) levels of < 30% of total Hb while keeping total Hb concentration ≤ 11 g/dL. At initiation of red blood cell exchange or simple transfusions, discontinue disease modifying therapies for sickle cell disease (e.g., hydroxyurea, crizanlizumab, voxelotor).
In patients with TDT it is recommended that patients be transfused to maintain hemoglobin (Hb) ≥ 11 g/dL for 60 days prior to myeloablative conditioning.
Stop iron chelation therapy at least 7 days prior to myeloablative conditioning.
Do not begin myeloablative conditioning until the complete set of vial(s) comprising the total dose of CASGEVY has been received and stored at the treatment center and the availability of the back-up collection of unmodified rescue cells is confirmed. See the Lot Information Sheet provided with the product shipment for confirmation of the total dose of CASGEVY.
Administer full myeloablative conditioning prior to treatment with CASGEVY 1. Refer to the prescribing information for the myeloablative conditioning agent prior to treatment. See Clinical Studies (14) for the conditioning regimen used in the clinical trials.
Consider administration of anti-seizure prophylaxis. Use agents other than phenytoin prior to initiating busulfan conditioning. Consider prophylaxis for hepatic veno-occlusive disease (VOD)/hepatic sinusoidal obstruction syndrome prior to initiating busulfan conditioning.
CASGEVY must be administered between 48 hours and 7 days after the last dose of the myeloablative conditioning.
Receipt and Storage of CASGEVY
- CASGEVY is shipped to the treatment center frozen in a vapor phase of liquid nitrogen shipper.
- Confirm patient identifiers on the product label(s) and Lot Information Sheet.
- If there are any concerns about the product or packaging upon receipt, contact Vertex at +1-877-634-8789.
- Transfer CASGEVY from the vapor phase of nitrogen shipper to the treatment center vapor phase of liquid nitrogen storage at ≤ -135 °C (≤ -211 °F).
Preparing for CASGEVY Administration
CASGEVY contains human cells. Follow universal precautions (wearing gloves, protective clothing, and eye protection) and local biosafety guidelines applicable for handling and disposal of such products to avoid potential transmission of infectious diseases. All material that has been in contact with CASGEVY (solid and liquid waste) should be handled and disposed of as potentially infectious waste in accordance with local biosafety guidelines.
Coordinate the timing of CASGEVY thaw and infusion. Confirm the infusion time in advance and adjust the start time for thaw so that CASGEVY is available for infusion when the patient and healthcare providers are ready. Thaw and infuse one vial at a time.
Premedication: Administer an antipyretic (e.g., acetaminophen) and an antihistamine (e.g., diphenhydramine hydrochloride) prior to administering CASGEVY.
Before thaw, confirm CASGEVY is printed on the vial label and the patient's identity matches the unique patient information located on the CASGEVY vial(s). Do not infuse CASGEVY if the information on the patient-specific label on any of the vials does not match the intended patient, and contact Vertex at +1-877-634-8789.
A dose of CASGEVY may be contained in one or more cryopreserved patient-specific vial(s). Ensure that the correct number of vials are present. Use the accompanying Lot Information Sheet to confirm the total number of vials to be administered and confirm that each vial is within the expiration date prior to preparation of CASGEVY for infusion.
Inspect the vial(s) for any breaks or cracks prior to thawing. If a vial is compromised, do not infuse the contents. Call Vertex at +1-877-634-8789.
When the dose consists of multiple vials, thaw and administer one vial at a time. While thawing and administering a vial, remaining vials must remain in cryostorage at ≤ -135 °C (≤ -211 °F).
Assemble supplies needed to thaw and withdraw the product from the vial(s). With the exception of the water bath, these supplies are single use. Assemble sufficient supplies for each vial to be administered:
- Water bath
- Alcohol swabs
- Vial adapter (to allow for needleless extraction)
- 18-micron stainless steel filter
- 30 mL luer-lock syringe
- 0.9% sodium chloride (saline, 5 to 10 mL needed for each vial)
- 10 mL luer-lock syringe for saline rinse
Thawing the CASGEVY vials
- Thaw each CASGEVY vial at 37 °C (98 °F) using a water bath. Thaw each vial holding the vial neck, gently agitating clockwise and counterclockwise.
- Thawing of each vial can take between 10 to 15 minutes. Thawing is complete when ice crystals are no longer visible in the vial. Ensure water bath temperature does not exceed 40 °C (104 °F) during thawing.
- Do not leave CASGEVY unattended during thaw.
- Remove vial from water bath immediately once thawed.
- The thawed product should appear as a cellular suspension, which may contain proteinaceous particles or cellular aggregates.
- Inspect the vial(s) for any breaks or cracks after thawing.
- Do not wash, spin down and/or resuspend CASGEVY in new media prior to infusion.
- Do not sample, alter, irradiate, or refreeze CASGEVY.
- Infuse within 20 minutes of thaw.
2.3 Administration
CASGEVY is for autologous use only. Before infusion, confirm that the patient's identity matches the unique patient identifiers on the CASGEVY vial(s). Do not infuse CASGEVY if the information on the patient-specific label does not match the intended patient.
A patient's dose may consist of multiple vials. All vials must be administered. Use the Lot Information Sheet to confirm the total number of vials to be administered. The entire volume of each vial provided should be infused. If more than one vial is provided, administer each vial completely before proceeding to thaw and infuse the next vial.
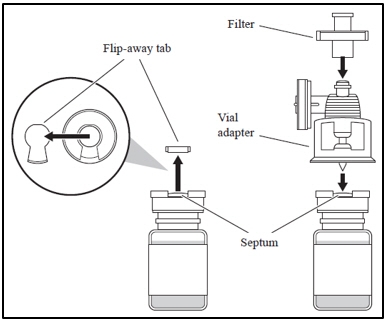
1. Attaching the vial adapter and filter - a. Remove the flip-away tab of the vial cap; clean the septum with an alcohol swab.
- b. Remove the cap on the vial adapter spike.
- c. With the thumb and forefinger of both hands, push the adapter into the vial septum, applying equal pressure until there is a single pop.
- d. Pull up on the adapter until you feel it lock.
- e. Attach the filter to the vial adapter.
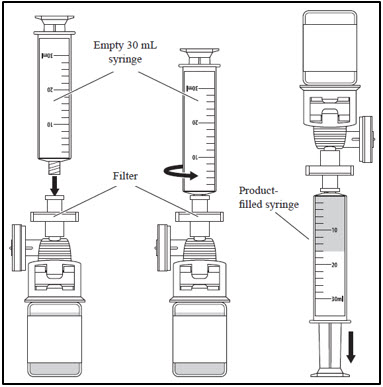
2. Withdrawing CASGEVY from the vial - a. Attach an empty 30 mL syringe to the filter.
- b. Withdraw the entire vial product volume.
- c. Remove the product-filled syringe from the filter and set aside.
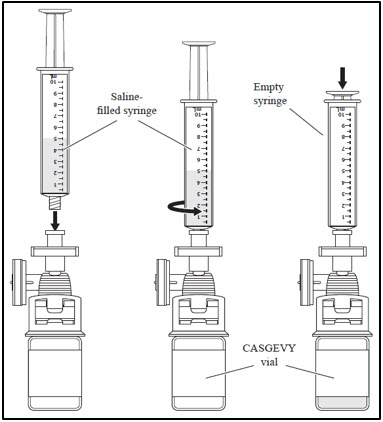
- d. Draw 5 - 10 mL of saline into the empty 10 mL syringe.
- e. Attach the saline-filled syringe to the filter.
- f. Inject the saline into the CASGEVY vial and remove the empty syringe from the filter. Discard the empty syringe.
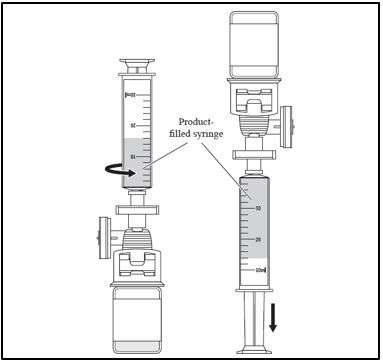
- g. Attach the product-filled syringe to the filter.
- h. Withdraw the contents of the vial into the syringe, then remove the syringe from the filter.
- i. Peel the product/patient identifier label from the Lot Information Sheet and affix to the product-filled syringe.
3. Administer CASGEVY through central venous catheter - a. Perform a two-person confirmation and verification of patient's identification at the bedside prior to the infusion of each vial(s).
- b. Administer CASGEVY as an intravenous bolus (IV push) within 20 minutes of product thaw. Do not use an in-line blood filter or infusion pump when infusing CASGEVY. The total volume of CASGEVY administered within one hour must not exceed 2.6 mL/kg.
- c. After administration of each vial of CASGEVY, flush the primary line with 0.9% sodium chloride solution, using enough volume to flush the tubing and the length of the IV catheter.
Repeat steps 1-3 for each remaining vial. If more than one vial is needed to achieve the patient-specific dose, administer each vial completely before proceeding to thaw and infuse the next vial.
After CASGEVY Administration
Follow standard procedures for patient management after HSC transplantation after CASGEVY infusion.
- Irradiate any blood products required within the first 3 months after CASGEVY infusion.
- Patients treated with CASGEVY should not donate blood, organs, tissues, or cells at any time in the future.
- Restarting iron chelation after CASGEVY infusion may be necessary and should be based on clinical practice. Avoid the use of non-myelosuppressive iron chelators for at least 3 months and use of myelosuppressive iron chelators for at least 6 months after CASGEVY infusion. Phlebotomy can be used in lieu of iron chelation, when appropriate [see Drug Interactions (7.4)].
-
3 DOSAGE FORMS AND STRENGTHS
CASGEVY is a cell suspension for intravenous infusion.
A single dose of CASGEVY is composed of one or more vials. Each vial contains 4 to 13 × 106 CD34+ cells/mL suspended in 1.5 to 20 mL cryopreservation medium [see How Supplied/Storage and Handling (16)]. The minimum recommended dose of CASGEVY is 3 × 106 CD34+ cells per kg of body weight.
See the Lot Information Sheet for actual strength and dose. The Lot Information Sheet is included inside the lid of the liquid nitrogen dry shipper used to transport CASGEVY.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Neutrophil Engraftment Failure
There is potential risk of neutrophil engraftment failure after treatment with CASGEVY. In the clinical trials, all treated patients achieved neutrophil engraftment and no patients received rescue CD34+ cells.
Monitor absolute neutrophil counts (ANC) and manage infections according to standard guidelines and medical judgement. In the event of neutrophil engraftment failure, patients should be infused with rescue CD34+ cells [see Adverse Reactions (6.1)].
5.2 Delayed Platelet Engraftment
Delayed platelet engraftment has been observed with CASGEVY treatment. There is an increased risk of bleeding until platelet engraftment is achieved [see Adverse Reactions (6.1)]. In the clinical trials, there was no association observed between incidence of bleeding events and time to platelet engraftment.
Monitor patients for bleeding according to standard guidelines and medical judgement. Conduct frequent platelet counts until platelet engraftment and platelet recovery are achieved. Perform blood cell count determination and other appropriate testing whenever clinical symptoms suggestive of bleeding arise.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most common Grade 3 or 4 non-laboratory adverse reactions (occurring in ≥ 25%) were mucositis and febrile neutropenia in patients with SCD and patients with TDT, and decreased appetite in patients with SCD.
All (100%) of the patients with TDT and SCD experienced Grade 3 or 4 neutropenia and thrombocytopenia. Other common Grade 3 or 4 laboratory abnormalities (≥ 50%) include leukopenia, anemia and lymphopenia.
Sickle Cell Disease
The safety of CASGEVY in patients with SCD was evaluated in an open-label, single-arm trial (Trial 1) and a long-term follow-up trial (Trial 3), in which 44 adolescent and adult patients with SCD were treated with CASGEVY after undergoing myeloablative conditioning with busulfan. The adverse event profile was generally consistent with that expected from busulfan myeloablative conditioning and HSC transplant.
The median (min, max) duration of follow-up for 44 patients with SCD after being administered CASGEVY was 19.3 (0.8, 48.1) months.
Serious adverse reactions after myeloablative conditioning and CASGEVY infusion were observed in 45% of patients with SCD. The most common serious adverse reactions (≥ 2 patients) were cholelithiasis, pneumonia, abdominal pain, constipation, pyrexia, abdominal pain upper, non-cardiac chest pain, oropharyngeal pain, pain, and sepsis. One (2%) patient died due to a COVID-19 infection and subsequent respiratory failure. The event was not related to CASGEVY.
Table 1 presents the Grade 3 or 4 non-laboratory adverse reactions observed after myeloablative conditioning and CASGEVY infusion in at least 10% of patients in Trial 1. Table 2 presents the Grade 3 or 4 laboratory abnormalities that occurred in at least 10% of patients with SCD.
Table 1: Grade 3 or 4 non-laboratory adverse reactions in ≥ 10% of patients with SCD who underwent busulfan myeloablative conditioning and received CASGEVY in Trial 1: Day 1 to Month 24 after CASGEVY infusion * System organ class, preferred term Patients with SCD (Trial 1)
(N=44)
n (%)- * Table includes adverse events associated with busulfan myeloablative conditioning and treatment with CASGEVY. Adverse event profile generally consistent with that expected from busulfan myeloablative conditioning and HSC transplant.
- † Mucositis includes mucosal inflammation, pharyngeal inflammation, and stomatitis.
- ‡ Encompasses preferred terms that belong to other system organ class.
- § Abdominal pain includes abdominal pain and abdominal pain upper.
- ¶ Musculoskeletal pain includes back pain, musculoskeletal chest pain, neck pain, non-cardiac chest pain, and pain in extremity.
Blood and lymphatic system disorders Febrile neutropenia 21 (48) Gastrointestinal disorders Mucositis †, ‡ 38 (86) Abdominal pain § 5 (11) Hepatobiliary disorders Cholelithiasis 5 (11) Metabolism and nutrition disorders Decreased appetite 18 (41) Musculoskeletal and connective tissue disorders Musculoskeletal pain ¶, ‡ 6 (14) Skin and subcutaneous tissue disorders Pruritus 5 (11) Other clinically important adverse reactions that occurred in less than 10% of patients or were Grade 1 or Grade 2 include the following:
Hepatobiliary disorders: Veno-occlusive liver disease (1 [2%] patient).
Infusion-related reactions: 6 (14%) patients, including preferred terms of abdominal pain in 3 (7%) patients; and infusion-related reaction, nausea, non-cardiac chest pain, pruritus, sinus tachycardia, and vomiting in 1 (2%) patient each.
Table 2: Grade 3 or 4 laboratory abnormalities in ≥ 10% of patients with SCD who underwent busulfan myeloablative conditioning and received CASGEVY in Trial 1: Day 1 to Month 24 after CASGEVY infusion * Laboratory abnormality Patients with SCD (Trial 1)
N=44 †
(%)- * Table includes laboratory abnormalities associated with busulfan myeloablative conditioning and treatment with CASGEVY. Laboratory abnormalities were generally consistent with those expected from busulfan myeloablative conditioning and HSC transplant.
- † The denominator for CD4 lymphocytes decreased is 43 and the denominator for all other laboratory data is 44, based on evaluable data at the time of the interim analysis.
Neutropenia 100 Thrombocytopenia 100 Leukopenia 98 Anemia 84 Lymphopenia 50 CD4 lymphocytes decreased 23 Activated partial thromboplastin time prolonged 16 Hyperbilirubinemia 14 Platelet engraftment
Platelet engraftment in patients with SCD is defined as 3 consecutive measurements of platelet counts ≥ 50 × 109/L, obtained on 3 different days after CASGEVY infusion, without administration of platelet transfusions for 7 days. In Trial 1, the median (min, max) time to platelet engraftment was 35 (23, 126) days (n=43). There was no association observed between bleeding events and time to platelet engraftment.
Neutrophil engraftment
Neutrophil engraftment is defined as 3 consecutive measurements of ANC ≥ 500 cells/µL on 3 different days after CASGEVY infusion, without use of the unmodified rescue CD34+ cells. In Trial 1, the median (min, max) time to neutrophil engraftment was 27 (15, 40) days (n=44). There was no association observed between infections and time to neutrophil engraftment. There was no use of rescue CD34+ cells in any patient.
Transfusion-dependent β-thalassemia
The safety of CASGEVY in patients with TDT was evaluated in an open-label, single-arm trial (Trial 2) and a long-term follow-up trial (Trial 3), in which 52 adolescent and adult patients with TDT were treated with CASGEVY after undergoing myeloablative conditioning with busulfan. The adverse event profile was generally consistent with that expected from busulfan myeloablative conditioning and HSC transplant.
The median (min, max) duration of follow-up for 52 patients with TDT after being administered CASGEVY was 20.4 (2.1, 48.1) months.
Serious adverse reactions after myeloablative conditioning and CASGEVY infusion were observed in 33% of patients with TDT. The most common serious adverse reactions (≥ 2 patients) were veno-occlusive liver disease, pneumonia, hypoxia, thrombocytopenia, viral infection, and upper respiratory tract infection.
Table 3 presents the Grade 3 or 4 non-laboratory adverse reactions observed after myeloablative conditioning and CASGEVY infusion in at least 10% of patients in Trial 2. Table 4 presents the Grade 3 or 4 laboratory abnormalities that occurred in at least 10% of patients with TDT.
Table 3: Grade 3 or 4 non-laboratory adverse reactions in ≥ 10% of patients with TDT who underwent busulfan myeloablative conditioning and received CASGEVY in Trial 2: Day 1 to Month 24 after CASGEVY infusion * System organ class, preferred term Patients with TDT (Trial 2)
(N=52)
n (%)- * Table includes adverse events associated with busulfan myeloablative conditioning and treatment with CASGEVY. Adverse event profile generally consistent with that expected from busulfan myeloablative conditioning and HSC transplant.
- † Mucositis includes anal inflammation, mucosal inflammation, pharyngeal inflammation, and stomatitis.
- ‡ Encompasses preferred terms that belong to other system organ class.
Blood and lymphatic system disorders Febrile neutropenia 28 (54) Gastrointestinal disorders Mucositis †, ‡ 37 (71) Hepatobiliary disorders Veno-occlusive liver disease 5 (10) Metabolism and nutrition disorders Decreased appetite 12 (23) Respiratory, thoracic and mediastinal disorders Epistaxis 7 (13) Other clinically important adverse reactions that occurred in less than 10% of patients or were Grade 1 or Grade 2 include the following:
Immune system disorders: Hemophagocytic lymphohistiocytosis (1 [2%] patient).
Nervous system disorders: Cerebellar hemorrhage (intracranial hemorrhage) (1 [2%] patient).
Infusion-related reactions: 12 (23%) patients, including preferred terms of abdominal pain and nausea in 4 (8%) patients each; pruritus and vomiting in 2 (4%) patients each; and abdominal pain lower, chills, sinus tachycardia, and tachycardia in 1 (2%) patient each.
Table 4: Grade 3 or 4 laboratory abnormalities in ≥ 10% of patients with TDT who underwent busulfan myeloablative conditioning and received CASGEVY in Trial 2: Day 1 to Month 24 after CASGEVY infusion * Laboratory abnormality Patients with TDT (Trial 2)
N=52 †
(%)- * Table includes laboratory abnormalities associated with busulfan myeloablative conditioning and treatment with CASGEVY. Laboratory abnormalities were generally consistent with those expected from busulfan myeloablative conditioning and HSC transplant.
- † The denominator for CD4 lymphocytes decreased is 48 and the denominator for all other laboratory data is 52, based on evaluable data at the time of the interim analysis.
Neutropenia 100 Thrombocytopenia 100 Leukopenia 98 Anemia 92 Lymphopenia 79 CD4 lymphocytes decreased 23 Hyperbilirubinemia 23 Alanine aminotransferase increased 19 Hypokalemia 19 Gamma-glutamyltransferase increased 17 Activated partial thromboplastin time prolonged 13 Hypocalcemia 12 Platelet engraftment
Platelet engraftment in patients with TDT is defined as 3 consecutive measurements of platelet counts ≥ 20 × 109/L, obtained on 3 different days after CASGEVY infusion, without administration of platelet transfusions for 7 days. In Trial 2, the median (min, max) time to platelet engraftment was 44 (20, 200) days (n=52). There is an increased risk of bleeding until platelet engraftment is achieved. In the clinical trials, there was no association observed between incidence of bleeding events and time to platelet engraftment. Patients without a spleen had an earlier median time to platelet engraftment than patients with an intact spleen. Median (min, max) time to platelet engraftment was 34.5 (20, 78) days in patients without a spleen and 47.5 (27, 200) days in patients with an intact spleen. While the use of thrombopoietin (TPO) mimetics was not specified in the trial protocol, five patients (10%) received a TPO mimetic at the time of platelet engraftment. All 5 patients continued TPO mimetic use for thrombocytopenia beyond engraftment. The total duration of TPO mimetic use was 98-457 days.
Neutrophil engraftment
Neutrophil engraftment is defined as 3 consecutive measurements of absolute neutrophil count (ANC) ≥ 500 cells/µL on 3 different days after CASGEVY infusion, without use of the unmodified rescue CD34+ cells. In Trial 2, the median (min, max) time to neutrophil engraftment was 29 (12, 56) days (n=52). There was no association observed between infections and time to neutrophil engraftment. One patient had neutrophil engraftment on Day 56. There was no use of rescue CD34+ cells in any patient.
-
7 DRUG INTERACTIONS
No formal drug interaction studies have been performed. CASGEVY is not expected to interact with the hepatic cytochrome P-450 family of enzymes or drug transporters.
7.1 Use of Granulocyte-Colony Stimulating Factor (G-CSF)
Granulocyte-Colony Stimulating Factor (G-CSF) must not be used for CD34+ HSC mobilization of patients with SCD.
7.2 Use of Hydroxyurea
Discontinue the use of hydroxyurea at least 8 weeks prior to start of each mobilization cycle and conditioning. There is no experience of the use of hydroxyurea after CASGEVY infusion.
7.3 Use of Voxelotor and Crizanlizumab
Discontinue the use of voxelotor and crizanlizumab at least 8 weeks prior to start of mobilization and conditioning, as their interaction potential with mobilization and myeloablative conditioning agents is not known.
7.4 Use of Iron Chelators
Discontinue the use of iron chelators at least 7 days prior to initiation of myeloablative conditioning, due to potential interaction with the conditioning agent. Some iron chelators are myelosuppressive. If iron chelation is required, avoid the use of non-myelosuppressive iron chelators for at least 3 months and use of myelosuppressive iron chelators for at least 6 months after CASGEVY infusion. Phlebotomy can be used instead of iron chelation, when appropriate.
-
8 USE IN SPECIFIC POPULATIONS
Consider the risks of mobilization and myeloablative conditioning agents in patients with reproductive potential and patients that are pregnant or breastfeeding.
8.1 Pregnancy
Risk Summary
There are no clinical data from the use of exagamglogene autotemcel in pregnant women. No animal reproductive and developmental toxicity studies have been conducted with exagamglogene autotemcel to assess whether it can cause fetal harm when administered to a pregnant woman. CASGEVY must not be administered during pregnancy because of the risks associated with myeloablative conditioning. Pregnancy after CASGEVY infusion should be discussed with the treating physician.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.2 Lactation
Risk Summary
There are no data on the presence of exagamglogene autotemcel in human or animal milk, the effects on the breastfed child, or the effects on milk production. Because of the potential risks associated with myeloablative conditioning, breastfeeding should be discontinued during conditioning. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for CASGEVY and any potential adverse effects on the breastfed child from CASGEVY or from the underlying maternal condition. Breastfeeding after CASGEVY infusion should be discussed with the treating physician.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
A negative serum pregnancy test must be confirmed prior to the start of each mobilization cycle and re-confirmed prior to myeloablative conditioning.
Contraception
There are insufficient exposure data to provide a precise recommendation on duration of contraception following treatment with CASGEVY. Women of childbearing potential and men capable of fathering a child should use effective method of contraception from start of mobilization through at least 6 months after administration of CASGEVY. Advise patients of the risks associated with conditioning agents.
Infertility
There are no data on the effects of exagamglogene autotemcel on human fertility. Effects on male and female fertility have not been evaluated in animal studies. Infertility has been observed with myeloablative conditioning therefore, advise patients of fertility preservation options before treatment, if appropriate.
8.4 Pediatric Use
The safety and efficacy of CASGEVY has been established in pediatric patients with SCD and TDT aged 12 years and older. Use of CASGEVY in patients aged 12 to less than 18 years is supported by data in 12 patients with SCD in Trial 1 (6 patients evaluable for the primary efficacy analysis), and 18 patients with TDT in Trial 2 (11 patients evaluable for the primary efficacy analysis). The efficacy and safety profile of CASGEVY in pediatric patients aged 12 years and older were consistent with the efficacy and safety in adult patients [see Adverse Reactions (6.1) and Clinical Studies (14.1, 14.2)].
Patients with SCD
The median (min, max) time to platelet engraftment was 45 (23, 81) days in pediatric patients aged 12 years and older and 32 (23, 126) days in adult patients. The median (min, max) time to neutrophil engraftment was 28 (24, 40) days in pediatric patients aged 12 years and older and 26 (15, 38) days in adult patients.
Patients with TDT
The median (min, max) time to platelet engraftment was 45 (20, 199) days in pediatric patients aged 12 years and older and 41.5 (24, 200) days in adult patients. The median (min, max) time to neutrophil engraftment was 30 (19, 56) days in pediatric patients aged 12 years and older and 28.5 (12, 40) days in adult patients.
The safety and efficacy of CASGEVY in pediatric patients aged less than 12 years has not been established.
8.5 Geriatric Use
CASGEVY has not been studied in patients > 65 years of age. HSC transplantation must be appropriate for a patient to be treated with CASGEVY.
8.6 Patients with Renal Impairment
CASGEVY has not been studied in patients with renal impairment, defined as estimated glomerular filtration rate < 60 mL/min/1.73 m2. Patients should be assessed for renal impairment to ensure HSC transplantation is appropriate.
8.7 Patients with Hepatic Impairment
CASGEVY has not been studied in patients with hepatic impairment. Patients should be assessed for hepatic impairment to ensure HSC transplantation is appropriate.
8.8 Patients Seropositive for Human Immunodeficiency Virus (HIV), Hepatitis B Virus (HBV) or Hepatitis C Virus (HCV)
CASGEVY has not been studied in patients with HIV-1, HIV-2, HBV or HCV. Perform screening for HIV-1, HIV-2, HBV and HCV and any other infectious agents in accordance with local guidelines before collection of cells for manufacturing. CASGEVY should not be used in patients with active HIV-1, HIV-2, HBV or HCV.
-
11 DESCRIPTION
CASGEVY (exagamglogene autotemcel) is a cellular gene therapy consisting of autologous CD34+ HSCs edited by CRISPR/Cas9-technology at the erythroid specific enhancer region of the BCL11A gene to reduce BCL11A expression in erythroid lineage cells, leading to increased fetal hemoglobin (HbF) protein production.
CASGEVY is prepared from the patient's own HSCs, which are obtained via apheresis procedure(s). The autologous cells are enriched for CD34+ cells, and then genome edited ex vivo by introducing the CRISPR/Cas9 ribonucleoprotein (RNP) complex by electroporation. The guide RNA included in the RNP complex enables CRISPR/Cas9 to make a precise DNA double-strand break at a critical transcription factor binding site (GATA1) in the erythroid specific enhancer region of the BCL11A gene. As a result of the editing, GATA1 binding is disrupted and BCL11A expression is reduced. This reduction in BCL11A expression conversely results in an increase in gamma-globin expression and downstream fetal hemoglobin formation.
The edited CD34+ cells are formulated into a suspension in a sterile cryopreservation medium and cryopreserved. CASGEVY is shipped as a frozen suspension in patient-specific vial(s). The product is thawed prior to infusion and administered as a HSC transplant [see Dosage and Administration (2.2) and How Supplied/Storage and Handling (16)]. Due to the presence of cells, the thawed product may be clear to slightly cloudy and may contain small inherent proteinaceous particles or visible cell aggregates.
The formulation contains 5% dimethyl sulfoxide (DMSO) and dextran 40.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
After CASGEVY infusion, the edited CD34+ cells engraft in the bone marrow and differentiate to erythroid lineage cells with reduced BCL11A expression. Reduced BCL11A expression results in an increase in γ-globin expression and HbF protein production in erythroid cells. In patients with severe sickle cell disease, HbF expression reduces intracellular hemoglobin S (HbS) concentration, preventing the red blood cells from sickling and addressing the underlying cause of disease, thereby eliminating VOCs. In patients with transfusion-dependent β-thalassemia, γ-globin production improves the α-globin to non-α-globin imbalance thereby reducing ineffective erythropoiesis and hemolysis and increasing total hemoglobin levels, addressing the underlying cause of disease, and eliminating the dependence on regular red blood cell (RBC) transfusions.
12.2 Pharmacodynamics
Sickle Cell Disease
Fetal Hemoglobin and Total Hemoglobin
HbF and total Hb over time are provided in Table 5 for all patients administered CASGEVY for the treatment of sickle cell disease (full analysis set). HbF and total Hb over time for the subset of patients included in the primary efficacy analysis were consistent with full analysis set.
Table 5: Proportion of hemoglobin comprised by HbF (%) and total Hb (g/dL) over time in patients with SCD in Trial 1 CASGEVY
Full Analysis Set (FAS)
(N=44)Proportion of total Hb comprised by HbF (%) * Total Hb (g/dL) * SD: Standard Deviation. - * %HbF/Hb data not available for all patients at all timepoints.
Month 3 n 43 43 Mean (SD) 36.9 (9.0) 11.9 (1.5) Median (min, max) 36.2 (17.8, 59.6) 11.9 (8.2, 15.4) Month 6 n 38 38 Mean (SD) 43.9 (8.6) 12.5 (1.8) Median (min, max) 44.3 (14.9, 68.4) 12.3 (7.2, 15.9) Month 12 n 32 31 Mean (SD) 43.4 (4.6) 13.0 (1.5) Median (min, max) 42.9 (35.1, 52.1) 12.9 (10.3, 15.7) Month 18 n 27 27 Mean (SD) 42.3 (5.8) 13.3 (1.9) Median (min, max) 43.1 (27.5, 53.3) 12.7 (11.0, 17.3) Month 24 n 17 17 Mean (SD) 42.1 (5.2) 13.1 (1.8) Median (min, max) 42.2 (33.3, 49.1) 13.0 (10.5, 17.3) The mean (SD) proportion of Hb comprised by HbF was 43.9% (8.6%) at Month 6 and was maintained thereafter. Increases in mean (SD) total Hb levels were observed as early as Month 3 after CASGEVY infusion, continued to increase to 12.5 (1.8) g/dL at Month 6, and was maintained thereafter. Of the 44 patients infused with CASGEVY, three male patients reached total Hb levels of at least 16.5 g/dL at one or more time points after Month 9.
Consistent with the increase in HbF levels, the mean (SD) proportion of circulating erythrocytes expressing HbF (F-cells) at Month 3 was 70.1% (13.8%) and continued to increase over time to 94.0% (12.4%) at Month 6, with levels remaining stable thereafter, indicating sustained pan-cellular expression of HbF.
Proportion of Alleles with Intended Genetic Modification
The mean (SD) proportion of alleles with intended genetic modification in the bone marrow and in peripheral blood is shown in Table 6 for all patients administered CASGEVY for the treatment of sickle cell disease in Trial 1.
Table 6: Proportion of alleles with intended genetic modification over time in patients with SCD in Trial 1 CASGEVY
Full Analysis Set (FAS)
(N=44)Proportion of Alleles with Intended Genetic Modification in CD34+ Cells in Bone Marrow * Proportion of Alleles with Intended Genetic Modification in Peripheral Blood - * Allelic editing data not available for all patients at all timepoints. Allelic editing in bone marrow was first assessed at Month 6.
Month 1 n -- 42 Mean (SD) 53.5 (18.2) Month 3 n -- 42 Mean (SD) 70.8 (10.6) Month 6 n 37 38 Mean (SD) 86.1 (7.5) 73.4 (8.1) Month 12 n 31 31 Mean (SD) 86.1 (8.6) 74.2 (8.7) Month 24 n 16 17 Mean (SD) 88.5 (4.6) 79.2 (5.6) Subgroup analyses evaluating the effects of age (adolescent versus adult) and sex (male versus female) showed consistent results on total hemoglobin, fetal hemoglobin and allelic editing in patients with SCD.
Transfusion-dependent β-thalassemia
Fetal Hemoglobin and Total Hemoglobin
Total Hb and HbF levels over time are provided in Table 7 for all patients administered CASGEVY for the treatment of transfusion-dependent β-thalassemia (full analysis set). HbF and total Hb over time for the subset of patients included in the primary efficacy analysis were consistent with full analysis set.
Table 7: Total Hb (g/dL) and HbF levels over time in patients with TDT in Trial 2 CASGEVY
Full Analysis Set (FAS)
(N=52)Total Hb (g/dL) * Total HbF (g/dL) * - * Hb/HbF data not available for all patients at all timepoints.
Month 3 n 47 46 Mean (SD) 11.4 (2.2) 7.7 (2.9) Median (min, max) 11.5 (7.1, 17.6) 8.4 (0.3, 13.0) Month 6 n 45 45 Mean (SD) 12.2 (2.0) 10.9 (2.8) Median (min, max) 12.5 (6.5, 16.4) 11.6 (1.1, 14.5) Month 12 n 43 42 Mean (SD) 12.8 (2.1) 11.5 (2.5) Median (min, max) 12.9 (6.2, 17.2) 12.3 (4.4, 15.3) Month 18 n 30 27 Mean (SD) 12.9 (2.1) 11.5 (2.4) Median (min, max) 13.1 (6.5, 17.7) 12.0 (4.3, 15.0) Month 24 n 15 15 Mean (SD) 13.2 (2.1) 11.9 (2.5) Median (min, max) 13.5 (10.1, 16.9) 12.1 (6.7, 15.4) Increases in mean (SD) total Hb and HbF levels were observed as early as Month 3 after CASGEVY infusion and continued to increase to 12.2 (2.0) g/dL and 10.9 (2.8) g/dL respectively at Month 6. After Month 6, levels of total Hb and HbF were maintained thereafter, with HbF comprising ≥ 88% of total Hb.
Consistent with the increase in HbF levels, the mean (SD) proportion of circulating erythrocytes expressing HbF (F-cells) at Month 3 was 73.8% (19.7%) and continued to increase over time to 95.9% (15.2%) at Month 6, with levels remaining stable from thereafter, indicating sustained pan-cellular expression of HbF.
Proportion of Alleles with Intended Genetic Modification
The mean (SD) proportion of alleles with intended genetic modification in the bone marrow and in peripheral blood is shown in Table 8 for all patients administered CASGEVY for the treatment of transfusion-dependent β-thalassemia in Trial 2.
Table 8: Proportion of alleles with intended genetic modification over time in patients with TDT in Trial 2 CASGEVY
Full Analysis Set (FAS)
(N=52)Proportion of Alleles with Intended Genetic Modification in CD34+ Cells in Bone Marrow * Proportion of Alleles with Intended Genetic Modification in Peripheral Blood - * Allelic editing data not available for all patients at all timepoints. Allelic editing in bone marrow was first assessed at Month 6.
Month 1 n -- 46 Mean (SD) 50.2 (20.6) Month 3 n -- 46 Mean (SD) 66.2 (11.4) Month 6 n 41 44 Mean (SD) 78.0 (82.3) 66.7 (11.3) Month 12 n 41 43 Mean (SD) 78.7 (12.6) 67.7 (10.2) Month 24 n 13 15 Mean (SD) 75.4 (16.4) 65.3 (12.6) Subgroup analyses evaluating the effects of age (adolescent versus adult), sex (male versus female), race, and genotype showed consistent results on total hemoglobin, fetal hemoglobin, and allelic editing in patients with TDT.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
14.1 Sickle Cell Disease
Trial 1 (NCT03745287) is an ongoing single-arm, multi-center trial evaluating the safety and efficacy of a single dose of CASGEVY in adult and adolescent patients with sickle cell disease. Eligible patients underwent mobilization and apheresis to collect CD34+ stem cells for CASGEVY manufacture, followed by myeloablative conditioning and infusion of CASGEVY. Patients were then followed in Trial 1 for 24 months after CASGEVY infusion. Patients who complete or discontinue from Trial 1 are encouraged to enroll in Trial 3 (NCT04208529), an ongoing long-term follow-up trial for additional follow-up for a total of 15 years after CASGEVY infusion.
Patients were eligible for the trial if they had a history of at least 2 protocol-defined severe vaso-occlusive crisis (VOC) events during each of the 2 years prior to screening. In this trial severe VOC is defined as an occurrence of at least one of the following events:
- Acute pain event requiring a visit to a medical facility and administration of pain medications (opioids or intravenous [IV] non-steroidal anti-inflammatory drugs [NSAIDs]) or RBC transfusions
- Acute chest syndrome
- Priapism lasting > 2 hours and requiring a visit to a medical facility
- Splenic sequestration.
Patients were excluded if they had advanced liver disease, history of untreated Moyamoya disease, or presence of Moyamoya disease that in the opinion of the investigator put the patient at risk of bleeding. Patients aged 12 to 16 years were required to have normal transcranial doppler (TCD), and patients aged 12 to 18 years were excluded if they had any history of abnormal TCD in the middle cerebral artery and the internal carotid artery. Patients with an available 10/10 human leukocyte antigen matched related hematopoietic stem cell donor were excluded. Patients with more than 10 unplanned hospitalizations or emergency department visits related to chronic pain rather than SCD-related acute pain crises in the year before screening were excluded.
At the time of the interim analysis, a total of 63 patients enrolled in the trial, of which 58 (92%) patients started mobilization. A total of 44 (76%) patients received CASGEVY infusion and formed the full analysis set (FAS). Thirty-one patients from the FAS (70%) had adequate follow-up to allow evaluation of the primary efficacy endpoint and formed the primary efficacy set (PES). The key demographics and baseline characteristics for all patients administered CASGEVY in Trial 1 are shown in Table 9 below. The baseline characteristics and demographics are consistent between the PES and the FAS.
Table 9: Demographics and baseline characteristics of patients treated with CASGEVY at the interim analysis in Trial 1 Demographics and disease characteristics Full Analysis Set (FAS) *
(N=44)Primary Efficacy Set (PES) *, †
(N=31)- * Interim analysis conducted based on June 2023 data cut-off date.
- † The primary efficacy set (PES), is a subset of the full analysis set (FAS). The PES was defined as all patients who had been followed for at least 16 months after CASGEVY infusion. Patients who had less than 16 months follow-up due to death or discontinuation due to CASGEVY-related adverse events, or continuously received RBC transfusions for more than 10 months after CASGEVY were also included in this set. An additional patient who had less than 16 months of follow-up but was otherwise determined to be a non-responder for the primary efficacy endpoint, was also included in PES.
Age, n (%) Adults (≥ 18 and ≤ 35 years) 32 (73) 24 (77) Adolescents (≥ 12 and < 18 years) 12 (27) 7 (23) All ages (≥ 12 and ≤ 35 years) Median (min, max) 20 (12, 34) 21 (12, 34) Sex, n (%) Female 20 (45) 14 (45) Male 24 (55) 17 (55) Race, n (%) Black or African American 38 (86) 27 (87) White 3 (7) 1 (3) Other 3 (7) 3 (10) Genotype, n (%) βS/βS 40 (91) 30 (97) βS/β0 3 (7) 1 (3) βS/β+ 1 (2) 0 Annualized rate of severe VOCs in the 2 years prior to enrollment (events/year) Median (min, max) 3.5 (2.0, 18.5) 3.5 (2.0, 18.5) Annualized rate of hospitalizations due to severe VOCs in the 2 years prior to enrollment (events/year) Median (min, max) 2.5 (0.5, 9.5) 2.0 (0.5, 8.5) Mobilization and Apheresis
Patients underwent RBC exchange or simple transfusions for a minimum of 8 weeks before the planned start of mobilization and continued receiving transfusions or RBC exchanges until the initiation of myeloablative conditioning. Hemoglobin S (HbS) levels were maintained at < 30% of total Hb while keeping total Hb concentration ≤ 11 g/dL.
To mobilize stem cells for apheresis, patients in Trial 1 were administered plerixafor at a planned dose of 0.24 mg/kg via subcutaneous injection approximately 2 to 3 hours prior to each planned apheresis. Apheresis was carried out for up to 3 consecutive days to achieve the target collection of cells for manufacture and for the unmodified rescue CD34+ cells.
The mean (SD) and median (min, max) number of mobilization and apheresis cycles required for the manufacture of CASGEVY and for the back-up collection of rescue CD34+ cells were 2.3 (1.41) and 2 (1, 6), respectively. Six (10%) patients were unable to receive CASGEVY therapy due to not achieving the minimum dose.
Pre-treatment Conditioning
All patients received full myeloablative conditioning with busulfan prior to treatment with CASGEVY. Busulfan was administered for 4 consecutive days intravenously (IV) via a central venous catheter at a planned starting dose of 3.2 mg/kg/day once daily (qd) or 0.8 mg/kg every 6 hours (q6h). Busulfan plasma levels were measured by serial blood sampling and the dose adjusted to maintain exposure in the target range.
For the once daily dosing, four-day target cumulative busulfan exposure was 82 mg*h/L (range: 74 to 90 mg*h/L), corresponding to AUC0-24h of 5000 µM*min (range: 4500 to 5500 µM*min). For the every 6 hours dosing, the four-day target cumulative busulfan exposure was 74 mg*h/L (range: 59 to 89 mg*h/L), corresponding to AUC0-6h of 1125 µM*min (range: 900 to 1350 µM*min).
All patients received anti-seizure prophylaxis with agents other than phenytoin prior to initiating busulfan conditioning. Phenytoin was not used for anti-seizure prophylaxis because of its induction of cytochrome P-450 and resultant increased clearance of busulfan.
Prophylaxis for hepatic veno-occlusive disease (VOD)/hepatic sinusoidal obstruction syndrome was administered, per regional and institutional guidelines.
CASGEVY Administration
Patients were administered CASGEVY with a median (min, max) dose of 4.0 (2.9, 14.4) × 106 CD34+ cells/kg as an IV infusion.
All patients were administered an antihistamine and an antipyretic prior to CASGEVY infusion.
After CASGEVY Administration
G-CSF was not recommended within the first 21 days after CASGEVY infusion.
As CASGEVY is an autologous therapy, immunosuppressive agents were not required after initial myeloablative conditioning.
Efficacy Results
An interim analysis (IA) was conducted with 31 patients eligible for the primary efficacy analysis, i.e., the primary efficacy set (PES). The median (min, max) total duration of follow-up was 26.0 (17.8, 48.1) months from the time of CASGEVY infusion in PES. There were no cases of graft failure or graft rejection.
The primary efficacy outcome was the proportion of VF12 responders, defined as patients who did not experience any protocol-defined severe VOCs for at least 12 consecutive months within the first 24 months after CASGEVY infusion in Trial 1. The proportion of patients who did not require hospitalization due to severe VOCs for at least 12 consecutive months within the 24-month evaluation period (HF12) was also assessed. The evaluation of VF12 and HF12 began 60 days after the last RBC transfusion for post-transplant support or SCD management. The median (min, max) time to the last RBC transfusion was 19 (11, 52) days following CASGEVY infusion for patients in the primary efficacy set.
The interim analysis occurred at the time when the alpha spending was approximately 0.02 for a one-sided test, when 31 patients were evaluable for VF12 responder status. The VF12 response rate was 29/31 (93.5%, 98% one-sided CI: 77.9%, 100.0%). The 29 VF12 responders did not experience protocol-defined severe VOCs during the evaluation period with a median duration of 22.2 months at the time of the interim analysis. One VF12 responder, after initially achieving a VF12 response, experienced an acute pain episode meeting the definition of a severe VOC at Month 22.8 requiring a 5-day hospitalization; this patient was reported to have a parvovirus B19 infection at the time. Of the 31 patients evaluable for VF12 response, one patient was not evaluable for HF12 response; the remaining 30 patients (100%, 98% one-sided CI: 87.8%, 100.0%) achieved the endpoint of HF12.
14.2 Transfusion-dependent β-thalassemia
Trial 2 (NCT03655678) is an ongoing open-label, multi-center, single-arm trial to evaluate the safety and efficacy of CASGEVY in adult and adolescent patients with transfusion-dependent β-thalassemia. Eligible patients underwent mobilization and apheresis to collect CD34+ stem cells for CASGEVY manufacture, followed by myeloablative conditioning and infusion of CASGEVY. Patients were then followed in Trial 2 for 24 months after CASGEVY infusion. Patients who complete or discontinue from Trial 2 are encouraged to enroll in Trial 3 (NCT04208529), an ongoing long-term follow-up trial for additional follow-up for a total of 15 years after CASGEVY infusion.
Patients were eligible for the trial if they had a history of requiring at least 100 mL/kg/year or 10 units/year of RBC transfusions in the 2 years prior to enrollment.
Patients were excluded if they had severely elevated iron in the heart (i.e., patients with cardiac T2* less than 10 msec by magnetic resonance imaging [MRI] or left ventricular ejection fraction [LVEF] < 45% by echocardiogram) or advanced liver disease (aspartate transaminase [AST] or alanine transaminase [ALT] > 3 × the upper limit of normal [ULN], or direct bilirubin value > 2.5 × ULN, or if a liver biopsy demonstrated bridging fibrosis or cirrhosis [liver biopsy was performed if liver iron content was ≥ 15 mg/g by MRI]). Patients were also excluded if they had an available 10/10 human leukocyte antigen matched related hematopoietic stem cell donor.
At the time of the interim analysis, a total of 59 patients enrolled in the trial, of which 59 (100%) patients started mobilization. A total of 52 (88%) patients received CASGEVY infusion and formed the full analysis set (FAS). Thirty-five patients from the FAS (67%) had adequate follow-up to allow evaluation of the primary efficacy endpoint and formed the primary efficacy set (PES). The key demographics and baseline characteristics for all patients administered CASGEVY in Trial 2 are shown in Table 10, below. The baseline characteristics and demographics are consistent between the PES and the FAS.
Table 10: Demographics and baseline characteristics of patients treated with CASGEVY at the interim analysis in Trial 2 Demographics and disease characteristics Full Analysis Set (FAS) *
(N=52)Primary Efficacy Set (PES)
(N=35) * †- * Interim analysis conducted based on January 2023 data cut-off date.
- † The primary efficacy set (PES), is a subset of the full analysis set (FAS). The PES was defined as all patients who had been followed for at least 16 months after CASGEVY infusion. Patients who continuously received RBC transfusions for more than 10 months after CASGEVY infusion were also included in this set.
- ‡ Race was not collected per regional regulatory requirements in 7 (13.5%) patients in the FAS and 4 (11.4%) patients in the PES.
- § Low to no endogenous β-globin production (β0/β0, β0/IVS-I-110 and IVS-I-110/IVS-I-110).
Age, n (%) Adults (≥ 18 and ≤ 35 years) 34 (65.4) 24 (68.6) Adolescents (≥ 12 and < 18 years) 18 (34.6) 11 (31.4) All ages (≥ 12 and ≤ 35 years) Median (min, max) 20 (12, 35) 20 (12, 33) Sex, n (%) Female 25 (48.1) 17 (48.6) Male 27 (51.9) 18 (51.4) Race, n (%) ‡ Asian 22 (42.3) 13 (37.1) White 18 (34.6) 15 (42.9) Multiracial 3 (5.8) 3 (8.6) Other 2 (3.8) 0 Genotype, n (%) β0/β0-like § 31 (59.6) 20 (57.1) Non-β0/β0-like 21 (40.4) 15 (42.9) Baseline annualized RBC transfusion volume (mL/kg) Median (min, max) 201 (48, 331) 205 (115, 331) Baseline annualized RBC transfusion episodes Median (min, max) 17 (5, 35) 17 (11, 35) Spleen intact, n (%) 36 (69.2) 26 (74.3) Baseline liver iron concentration (mg/g) Median (min, max) 3.5 (1.2, 14.0) 4.0 (1.4, 14.0) Baseline cardiac iron T2* (msec) Median (min, max) 34.0 (12.4, 61.1) 34.8 (19.6, 61.1) Baseline serum ferritin (pmol/L) Median (min, max) 2892 (584, 10837) 2654 (674, 10741) Mobilization and Apheresis
To maintain a total Hb concentration ≥ 11 g/dL, patients underwent RBC transfusions prior to mobilization and apheresis and continued receiving transfusions until the initiation of myeloablative conditioning.
To mobilize stem cells for apheresis, patients in Trial 2 were administered G-CSF and plerixafor. Patients with a spleen were administered a planned dose of 5 µg/kg G-CSF approximately every 12 hours via intravenous or subcutaneous injection for 5 to 6 days. Splenectomized patients were administered a planned dose of 5 µg/kg G-CSF once daily for 5 to 6 days. The dose was increased to every 12 hours in splenectomized patients if there was no increase in white blood cell (WBC) or peripheral blood CD34+ counts. After 4 days of G-CSF administration, all patients received plerixafor at a planned dose of 0.24 mg/kg administered via subcutaneous injection approximately 4 to 6 hours prior to each planned apheresis.
Apheresis was carried out for up to 3 consecutive days to achieve the target collection of cells for manufacture and for the unmodified rescue CD34+ cells.
The mean (SD) and median (min, max) number of mobilization and apheresis cycles required for manufacture CASGEVY and for the back-up collection of rescue CD34+ cells were 1.3 (0.7) and 1 (1, 4), respectively.
Pre-treatment Conditioning
All patients received full myeloablative conditioning with busulfan prior to treatment with CASGEVY. Busulfan was administered for 4 consecutive days intravenously (IV) via a central venous catheter at a planned starting dose of 3.2 mg/kg/day once daily (qd) or 0.8 mg/kg every 6 hours (q6h). Busulfan plasma levels were measured by serial blood sampling and the dose adjusted to maintain exposure in the target range.
For the once-daily dosing, four-day target cumulative busulfan exposure was 82 mg*h/L (range: 74 to 90 mg*h/L), corresponding to AUC0-24h of 5000 µM*min (range: 4500 to 5500 µM*min). For the every 6 hours dosing, the four-day target cumulative busulfan exposure was 74 mg*h/L (range: 59 to 89 mg*h/L), corresponding to AUC0-6h of 1125 µM*min (range: 900 to 1350 µM*min).
All patients received anti-seizure prophylaxis with agents other than phenytoin prior to initiating busulfan conditioning. Phenytoin was not used for anti-seizure prophylaxis because of its induction of cytochrome P-450 and resultant increased clearance of busulfan.
Prophylaxis for hepatic veno-occlusive disease (VOD)/hepatic sinusoidal obstruction syndrome was administered, per regional and institutional guidelines.
CASGEVY Administration
Patients were administered CASGEVY with a median (min, max) dose of 7.5 (3.0, 19.7) × 106 CD34+ cells/kg as an IV infusion.
All patients were administered an antihistamine and an antipyretic prior to CASGEVY infusion.
After CASGEVY Administration
G-CSF was not recommended within the first 21 days after CASGEVY infusion.
As CASGEVY is an autologous therapy, immunosuppressive agents were not required after initial myeloablative conditioning.
Efficacy Results
An interim analysis (IA) was conducted with 35 patients eligible for the primary efficacy analysis, i.e., the primary efficacy set (PES). The median (min, max) total duration of follow-up was 23.8 (16.1, 48.1) months from the time of CASGEVY infusion in the PES. There were no cases of graft failure or graft rejection.
The primary outcome was the proportion of patients achieving transfusion independence for 12 consecutive months (TI12), defined as maintaining weighted average Hb ≥ 9 g/dL without RBC transfusions for at least 12 consecutive months any time within the first 24 months after CASGEVY infusion in Trial 2, evaluated starting 60 days after the last RBC transfusion for post-transplant support or TDT disease management.
The interim analysis occurred at the time when the alpha spending was approximately 0.017 for a one-sided test, when 35 patients were evaluable for TI12 responder status. The TI12 responder rate was 32/35 (91.4%, 98.3% one-sided CI: 75.7%, 100%). All patients who achieved TI12 remained transfusion-independent, with a median (min, max) duration of transfusion-independence of 20.8 (13.3, 45.1) months and normal mean weighted average total Hb levels (mean [SD] 13.1 [1.4] g/dL). The median (min, max) time to last RBC transfusion for patients who achieved TI12 was 30 (11, 91) days following CASGEVY infusion. Three patients did not achieve TI12. These patients had reductions in annualized RBC transfusion volume requirements of 79.8%, 83.9% and 97.9%, and reductions in annualized transfusion frequency of 78.6%, 67.4% and 94.6%, respectively, compared to baseline requirements.
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
CASGEVY is supplied in one or more vials containing a frozen suspension of genome edited autologous CD34+ cells in a cryopreservation medium containing 5% DMSO and dextran 40.
CASGEVY is stored in the vapor phase of liquid nitrogen and is shipped from the manufacturing facility to the treatment center storage facility in a cryoshipper. CASGEVY is supplied in vial(s) packaged in carton(s). One carton contains a single lot of CASGEVY consisting of 1 to 9 vials. A single dose of CASGEVY may consist of multiple CASGEVY lots, and therefore may consist of multiple cartons. A Lot Information Sheet listing the total dose of CASGEVY is affixed inside the shipper.
NDC: 51167-290-09
- Match the identity of the patient with the patient identifiers on each carton, vial, and Lot Information Sheet upon receipt.
- Store the vial(s) in the vapor phase of liquid nitrogen at ≤ -135 °C (≤ -211 °F) until ready for thaw and administration.
- Thaw CASGEVY prior to administration. Thaw and infuse one vial of CASGEVY at a time [see Dosage and Administration (2.2, 2.3)].
- Once thawed, CASGEVY must be administered within 20 minutes [see Dosage and Administration (2.2, 2.3)].
- Do not re-freeze CASGEVY after thawing.
- Do not irradiate CASGEVY.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Prior to treatment, advise patients of the following:
- Risks of mobilization and conditioning agents - Advise patients of the risks associated with mobilization and myeloablative conditioning agents and to read the FDA-approved patient labeling (Patient Information) for these agents.
- Potential need for additional cell collection - Ensure patients understand that if the minimum dose of CASGEVY is not met after initial product manufacturing, additional cycles of mobilization and apheresis will be needed to collect additional cells for product manufacture [see Dosage and Administration (2.2)].
-
Concomitant medications - Advise patients of the need to avoid the following medications:
- Disease modifying therapies (e.g., hydroxyurea, crizanlizumab, voxelotor) should be discontinued 8 weeks before the planned start of mobilization and conditioning [see Drug Interactions (7.2, 7.3)].
- Iron chelation should be stopped at least 7 days prior to myeloablative conditioning. If iron chelation is required, the use of non-myelosuppressive iron chelators should be avoided for at least 3 months after CASGEVY administration and use of myelosuppressive iron chelators should be avoided for at least 6 months after CASGEVY administration. Phlebotomy can be used in lieu of iron chelation, when appropriate [see Drug Interactions (7.4)].
After treatment, advise patients of the following:
- Risk of neutrophil engraftment failure - Advise patients they will need to receive rescue treatment with their collection of unmodified CD34+ cells if they do not achieve neutrophil engraftment after CASGEVY administration [see Dosage and Administration (2.3) and Warnings and Precautions (5.1)].
- Risk of bleeding - There is an increased risk of bleeding from initiation of myeloablative conditioning until platelet engraftment is achieved. Advise patients to monitor for signs and symptoms of new or worsening bleeding or bruising and have frequent blood draws for platelet counts, until platelet recovery has been achieved [see Warnings and Precautions (5.2)].
- Donation of blood products - Advise patients that they should not donate blood, organs, tissues, or cells at any time in the future [see Dosage and Administration (2.3)].
-
SPL UNCLASSIFIED SECTION
Manufactured for:
Vertex Pharmaceuticals Incorporated
Boston, MA 02210
US License No 2279CASGEVY word mark and design are trademarks of Vertex Pharmaceuticals Incorporated.
VERTEX and the VERTEX triangle logo are registered trademarks of Vertex Pharmaceuticals Incorporated.
All other trademarks referenced herein are the property of their respective owners.
©2026 Vertex Pharmaceuticals Incorporated
ALL RIGHTS RESERVED -
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised 01/2024 Patient Information
CASGEVY™ (cass-JEH-vee)
(exagamglogene autotemcel)
suspension for intravenous infusionWhat is the most important information I should know about CASGEVY?
After treatment with CASGEVY you will have fewer blood cells for a while, until CASGEVY takes hold (engrafts) into your bone marrow. This includes low levels of platelets (cells that usually help the blood to clot) and white blood cells (cells that usually fight infections). Your doctor will monitor this and give you treatment as required. The doctor will tell you when blood cell levels return to safe levels.-
Tell your healthcare provider right away if you experience any of the following, which could be signs of low levels of platelet cells:
- severe headache
- abnormal bruising
- prolonged bleeding
- bleeding without injury such as nosebleeds, bleeding from gums, blood in your urine, stool, or vomit, or coughing up blood.
-
Tell your healthcare provider right away if you experience any of the following, which could be signs of low levels of white blood cells:
- fever
- chills
- infections
What is CASGEVY?
CASGEVY is a one-time therapy used to treat people aged 12 years and older with:- sickle cell disease (SCD) who have frequent vaso-occlusive crises or VOCs
- beta-thalassemia (β-thal) who need regular blood transfusions.
How will I receive CASGEVY?
Your healthcare provider will give you other medicines, including a conditioning medicine, as part of your treatment with CASGEVY. It's important to talk to your healthcare provider about the risks and benefits of all medicines involved in your treatment.
After receiving the conditioning medicine, it may not be possible for you to become pregnant or father a child. You should discuss options for fertility preservation with your healthcare provider before treatment.
STEP 1: Before CASGEVY treatment, a doctor will give you mobilization medicine(s). This medicine moves blood stem cells from your bone marrow into the blood stream. The blood stem cells are then collected in a machine that separates the different blood cells (this is called apheresis). This entire process may happen more than once. Each time, it can take up to one week.
During this step 'rescue cells' are also collected and stored at the hospital. These are your existing blood stem cells and are kept untreated just in case there is a problem in the treatment process. If CASGEVY cannot be given after the conditioning medicine, or if the modified blood stem cells do not take hold (engraft) in the body, these rescue cells will be given back to you. If you are given rescue cells, you will not have any treatment benefit from CASGEVY.
STEP 2: After they are collected, your blood stem cells will be sent to the manufacturing site where they are used to make CASGEVY. It may take up to 6 months from the time your cells are collected to manufacture and test CASGEVY before it is sent back to your healthcare provider.
STEP 3: Shortly before your stem cell transplant, your healthcare provider will give you a conditioning medicine for a few days in hospital. This will prepare you for treatment by clearing cells from the bone marrow, so they can be replaced with the modified cells in CASGEVY. After you are given this medicine, your blood cell levels will fall to very low levels. You will stay in the hospital for this step and remain in the hospital until after the CASGEVY infusion.
STEP 4: One or more vials of CASGEVY will be given into a vein (intravenous infusion) over a short period of time.
After the CASGEVY infusion, you will stay in hospital so that your healthcare provider can closely monitor your recovery. This can take 4-6 weeks, but times can vary. Your healthcare provider will decide when you can go home.What should I avoid after receiving CASGEVY? - Do not donate blood, organs, tissues, or cells at any time in the future.
What are the possible or reasonably likely side effects of CASGEVY?
The most common side effects of CASGEVY include:- Low levels of platelet cells, which may reduce the ability of blood to clot and may cause bleeding.
- Low levels of white blood cells, which may make you more susceptible to infection.
- fever
- chills
- infections
- severe headache
- abnormal bruising
- prolonged bleeding
- bleeding without injury such as nosebleeds, bleeding from gums, blood in your urine, stool, or vomit, or coughing up blood.
General information about the safe and effective use of CASGEVY
Talk to your healthcare provider about any health concerns. You can ask your healthcare provider for information about CASGEVY that is written for healthcare professionals.
For more information, go to Casgevy.com or call 1-833-837-8395.
Manufactured for: Vertex Pharmaceuticals Incorporated; 50 Northern Avenue, Boston, MA 02210
CASGEVY word mark and design are trademarks of Vertex Pharmaceuticals Incorporated.
VERTEX and the VERTEX triangle logo are registered trademarks of Vertex Pharmaceuticals Incorporated.
All other trademarks referenced herein are the property of their respective owners.
©2024 Vertex Pharmaceuticals Incorporated -
Tell your healthcare provider right away if you experience any of the following, which could be signs of low levels of platelet cells:
-
PRINCIPAL DISPLAY PANEL - 20 mL Vial Carton
NDC: 51167-290-09
Rx Onlyexagamglogene autotemcel
casgevy™4-13 × 106 CD34+ cells/mL
suspension for IV infusion
For autologous and intravenous use only
Each vial contains autologous genome edited hematopoietic stem cells
at a concentration of 4-13 × 106 CD34+ cells/mL suspended in 1.5 to 20 mL
of cryopreservation medium containing 5% DMSO and dextran 40.The full dose is provided in one or more vial(s).
Patient ID:
V0000000First Name:
XXXXXXXXXXXXXXXXXXXX
Last Name:
XXXXXXXXXXXXXXXXXXXXPatient DOB:
YYYY-MMM-DDCOI ID:
000000000000000LOT:
0000000000000
EXP:
YYYY-MMM-DDDIN 1:
0000000000000
DIN 2:
0000000000000
DIN 3:
0000000000000Number of vials: X
DIN 1:
DIN 2:
DIN 3:
1643-1537-1538-01

-
INGREDIENTS AND APPEARANCE
CASGEVY
exagamglogene autotemcel injection, suspensionProduct Information Product Type CELLULAR THERAPY Item Code (Source) NDC: 51167-290 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength exagamglogene autotemcel (UNII: S53L777GM8) (exagamglogene autotemcel - UNII:S53L777GM8) exagamglogene autotemcel 13000000 in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 51167-290-09 9 in 1 CARTON 1 NDC: 51167-290-01 20 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125787 12/08/2023 Labeler - Vertex Pharmaceuticals Incorporated (602478257)
Trademark Results [CASGEVY]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 CASGEVY 97096682 not registered Live/Pending |
Vertex Pharmaceuticals Incorporated 2021-10-28 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.