BELVIQ- lorcaserin hydrochloride hemihydrate tablet BELVIQ XR- lorcaserin hydrochloride hemihydrate tablet, film coated, extended release
BELVIQ XR by
Drug Labeling and Warnings
BELVIQ XR by is a Prescription medication manufactured, distributed, or labeled by Eisai Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BELVIQ/BELVIQ XR safely and effectively. See full prescribing information for BELVIQ/BELVIQ XR.
BELVIQ® (lorcaserin hydrochloride) tablets, for oral use, CIV
BELVIQ XR® (lorcaserin hydrochloride) extended-release tablets for oral use, CIV
Initial U.S. Approval: 2012
INDICATIONS AND USAGE
BELVIQ/BELVIQ XR is a serotonin 2C receptor agonist indicated as an adjunct to a reduced-calorie diet and increased physical activity for chronic weight management in adults with an initial body mass index (BMI) of:
- 30 kg/m2 or greater (obese) (1) or
- 27 kg/m2 or greater (overweight) in the presence of at least one weight-related comorbid condition, (e.g., hypertension, dyslipidemia, type 2 diabetes) (1)
Limitations of Use:
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
WARNINGS AND PRECAUTIONS
- Serotonin Syndrome or Neuroleptic Malignant Syndrome (NMS)-like Reactions: The safety of coadministration with other serotonergic or antidopaminergic agents has not been established. Manage with immediate BELVIQ/BELVIQ XR discontinuation and provide supportive treatment. (5.1)
- Valvular heart disease: If signs or symptoms develop consider BELVIQ/BELVIQ XR discontinuation and evaluate the patient for possible valvulopathy. (5.2)
- Cognitive Impairment: May cause disturbances in attention or memory. Caution with use of hazardous machinery when starting BELVIQ/BELVIQ XR treatment. (5.3)
- Psychiatric Disorders, including euphoria and dissociation: Do not exceed recommended dose of BELVIQ 10 mg twice daily or BELVIQ XR 20 mg once daily. (5.4)
- Monitor for depression or suicidal thoughts. Discontinue if symptoms develop. (5.4)
- Use of Antidiabetic Medications: weight loss may cause hypoglycemia. Monitor blood glucose. BELVIQ/BELVIQ XR has not been studied in patients taking insulin. (5.5)
- Priapism: Patients should seek emergency treatment if an erection lasts >4 hours. Use BELVIQ/BELVIQ XR with caution in patients predisposed to priapism. (5.6)
ADVERSE REACTIONS
Most common adverse reactions (greater than 5%) in non-diabetic patients are headache, dizziness, fatigue, nausea, dry mouth, and constipation, and in diabetic patients are hypoglycemia, headache, back pain, cough, and fatigue. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Eisai Inc. at 1-888-274-2378 or FDA at 1-800-FDA-1088 or at www.fda.gov/medwatch.DRUG INTERACTIONS
Serotonergic drugs (selective serotonin reuptake inhibitors (SSRIs), serotonin-norepinephrine reuptake inhibitors (SNRIs), monoamine oxidase inhibitors (MAOIs), triptans, bupropion, dextromethorphan, St. John’s Wort): use with extreme caution due to the risk of serotonin syndrome. (7.1)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2018
- 30 kg/m2 or greater (obese) (1) or
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serotonin Syndrome or Neuroleptic Malignant Syndrome (NMS)-like Reactions
5.2 Valvular Heart Disease
5.3 Cognitive Impairment
5.4 Psychiatric Disorders
5.5 Potential Risk of Hypoglycemia in Patients with Type 2 Diabetes Mellitus on Anti- diabetic Therapy
5.6 Priapism
5.7 Heart Rate Decreases
5.8 Hematological Changes
5.9 Prolactin Elevation
5.10 Pulmonary Hypertension
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-Marketing Experience
7 DRUG INTERACTIONS
7.1 Use with Other Agents that Affect Serotonin Pathways
7.2 Cytochrome P450 (2D6) substrates
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
9.2 Abuse
9.3 Dependence
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1
INDICATIONS AND USAGE
BELVIQ/BELVIQ XR is indicated as an adjunct to a reduced-calorie diet and increased physical activity for chronic weight management in adult patients with an initial body mass index (BMI) of:
- 30 kg/m2 or greater (obese), or
- 27 kg/m2 or greater (overweight) in the presence of at least one weight related comorbid condition (e.g., hypertension, dyslipidemia, type 2 diabetes)
[see Dosage and Administration (2)]
Limitations of Use:
- The safety and efficacy of coadministration of BELVIQ/BELVIQ XR with other products intended for weight loss including prescription drugs (e.g., phentermine), over-the-counter drugs, and herbal preparations have not been established.
- The effect of BELVIQ/BELVIQ XR on cardiovascular morbidity and mortality has not been established.
- 30 kg/m2 or greater (obese), or
-
2
DOSAGE AND ADMINISTRATION
- The recommended dose for BELVIQ is one 10 mg tablet administered orally twice daily.
- The recommended dose for BELVIQ XR is one 20 mg tablet administered orally once daily.
- Do not exceed recommended dose [see Warnings and Precautions (5.4)].
- BELVIQ/BELVIQ XR can be taken with or without food.
- Swallow BELVIQ XR tablets whole and do not chew, crush, or divide.
- Response to therapy should be evaluated by week 12. If a patient has not lost at least 5% of baseline body weight, discontinue BELVIQ/BELVIQ XR, as it is unlikely that the patient will achieve and sustain clinically meaningful weight loss with continued treatment [see Clinical Studies (14)].
- BMI is calculated by dividing weight (in kg) by height (in meters) squared. A BMI chart for height in inches and weight in pounds is provided below:
Table 1. BMI Conversion Chart Weight (lb) 125 130 135 140 145 150 155 160 165 170 175 180 185 190 195 200 205 210 215 220 225 (kg) 56. 8 59. 1 61. 4 63. 6 65. 9 68. 2 70. 5 72. 7 75. 0 77. 3 79. 5 81. 8 84. 1 86. 4 88. 6 90. 9 93. 2 95. 5 97. 7 100. 0 102. 3 Height (in) (cm) 58 147. 3 26 27 28 29 30 31 32 34 35 36 37 38 39 40 41 42 43 44 45 46 47 59 149. 9 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 43 44 45 46 60 152. 4 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 61 154. 9 24 25 26 27 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 62 157. 5 23 24 25 26 27 27 28 29 30 31 32 33 34 35 36 37 38 38 39 40 41 63 160. 0 22 23 24 25 26 27 28 28 29 30 31 32 33 34 35 36 36 37 38 39 40 64 162. 6 22 22 23 24 25 26 27 28 28 29 30 31 32 33 34 34 35 36 37 38 39 65 165. 1 21 22 23 23 24 25 26 27 28 28 29 30 31 32 33 33 34 35 36 37 38 66 167. 6 20 21 22 23 23 24 25 26 27 27 28 29 30 31 32 32 33 34 35 36 36 67 170. 2 20 20 21 22 23 24 24 25 26 27 27 28 29 30 31 31 32 33 34 35 35 68 172. 7 19 20 21 21 22 23 24 24 25 26 27 27 28 29 30 30 31 32 33 34 34 69 175. 3 18 19 20 21 21 22 23 24 24 25 26 27 27 28 29 30 30 31 32 33 33 70 177. 8 18 19 19 20 21 22 22 23 24 24 25 26 27 27 28 29 29 30 31 32 32 71 180. 3 17 18 19 20 20 21 22 22 23 24 24 25 26 27 27 28 29 29 30 31 31 72 182. 9 17 18 18 19 20 20 21 22 22 23 24 24 25 26 27 27 28 29 29 30 31 73 185. 4 17 17 18 19 19 20 20 21 22 22 23 24 24 25 26 26 27 28 28 29 30 74 188. 0 16 17 17 18 19 19 20 21 21 22 23 23 24 24 25 26 26 27 28 28 29 75 190. 5 16 16 17 18 18 19 19 20 21 21 22 23 23 24 24 25 26 26 27 28 28 76 193. 0 15 16 16 17 18 18 19 20 20 21 21 22 23 23 24 24 25 26 26 27 27 - The recommended dose for BELVIQ is one 10 mg tablet administered orally twice daily.
- 3 DOSAGE FORMS AND STRENGTHS
-
4
CONTRAINDICATIONS
● Pregnancy: Weight loss in a pregnant woman offers no benefit and may result in fetal harm [see Use in Specific Populations (8.1)].
● Hypersensitivity: BELVIQ/BELVIQ XR is contraindicated in patients with prior hypersensitivity reactions to lorcaserin or to any of the product components. Hypersensitivity reactions have been reported [see Adverse Reactions (6.2)].
-
5
WARNINGS AND PRECAUTIONS
5.1 Serotonin Syndrome or Neuroleptic Malignant Syndrome (NMS)-like Reactions
BELVIQ/BELVIQ XR is a serotonergic drug. The development of a potentially life-threatening serotonin syndrome or Neuroleptic Malignant Syndrome (NMS)-like reactions have been reported during use of serotonergic drugs, including, but not limited to, selective serotonin-norepinephrine reuptake inhibitors (SNRIs) and selective serotonin reuptake inhibitors (SSRIs), tricyclic antidepressants (TCAs), bupropion, triptans, dietary supplements such as St. John’s Wort and tryptophan, drugs that impair metabolism of serotonin (including monoamine oxidase inhibitors [MAOIs]), dextromethorphan, lithium, tramadol, antipsychotics or other dopamine antagonists, particularly when used in combination [see Drug Interactions (7.1)].
Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g., tachycardia, labile blood pressure, hyperthermia), neuromuscular aberrations (e.g.,hyperreflexia, incoordination) and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). Serotonin syndrome, in its most severe form, can resemble neuroleptic malignant syndrome, which includes hyperthermia, muscle rigidity, autonomic instability with possible rapid fluctuation of vital signs, and mental status changes. Patients should be monitored for the emergence of serotonin syndrome or NMS-like signs and symptoms.
The safety of BELVIQ/BELVIQ XR when coadministered with other serotonergic or antidopaminergic agents, including antipsychotics, or drugs that impair metabolism of serotonin, including MAOIs, has not been systematically evaluated and has not been established.
If concomitant administration of BELVIQ/BELVIQ XR with an agent that affects the serotonergic neurotransmitter system is clinically warranted, extreme caution and careful observation of the patient is advised, particularly during treatment initiation and dose increases. Treatment with BELVIQ/BELVIQ XR and any concomitant serotonergic or antidopaminergic agents, including antipsychotics, should be discontinued immediately if the above events occur and supportive symptomatic treatment should be initiated [see Adverse Reactions (6.1) and Drug Interactions (7.1)].
5.2 Valvular Heart Disease
Regurgitant cardiac valvular disease, primarily affecting the mitral and/or aortic valves, has been reported in patients who took serotonergic drugs with 5-HT2B receptor agonist activity. The etiology of the regurgitant valvular disease is thought to be activation of 5-HT2B receptors on cardiac interstitial cells. At therapeutic concentrations, lorcaserin is selective for 5-HT2C receptors as compared to 5-HT2B receptors. In clinical trials of 1-year duration, 2.4% of patients receiving BELVIQ and 2.0% of patients receiving placebo developed echocardiographic criteria for valvular regurgitation at one year (mild or greater aortic regurgitation and/or moderate or greater mitral regurgitation): none of these patients was symptomatic [see Adverse Reactions (6.1) and Clinical Pharmacology (12.1)].
BELVIQ/BELVIQ XR has not been studied in patients with congestive heart failure or hemodynamically-significant valvular heart disease. Preliminary data suggest that 5HT2B receptors may be overexpressed in congestive heart failure; therefore, BELVIQ/BELVIQ XR should be used with caution in patients with congestive heart failure.
BELVIQ/BELVIQ XR should not be used in combination with serotonergic and dopaminergic drugs that are potent 5-HT2B receptor agonists and are known to increase the risk for cardiac valvulopathy (e.g., cabergoline).
Patients who develop signs or symptoms of valvular heart disease, including dyspnea, dependent edema, congestive heart failure, or a new cardiac murmur while being treated with BELVIQ/BELVIQ XR should be evaluated and discontinuation of BELVIQ/BELVIQ XR should be considered.
5.3 Cognitive Impairment
In clinical trials of at least one year in duration, impairments in attention and memory were reported adverse reactions associated with 1.9% of patients treated with BELVIQ and 0.5% of patients treated with placebo, and led to discontinuation in 0.3% and 0.1% of these patients, respectively. Other reported adverse reactions associated with BELVIQ in clinical trials included confusion, somnolence, and fatigue [see Adverse Reactions (6.1)].
Since BELVIQ/BELVIQ XR have the potential to impair cognitive function, patients should be cautioned about operating hazardous machinery, including automobiles, until they are reasonably certain that BELVIQ/BELVIQ XR therapy does not affect them adversely [see Patient Counseling Information (17)].
5.4 Psychiatric Disorders
Events of euphoria, hallucination, and dissociation were seen with BELVIQ at supratherapeutic doses in short- term studies [see Adverse Reactions (6.1), Drug Abuse and Dependence (9.2), and Overdosage (10)]. In clinical trials of at least 1-year in duration, 6 patients (0.2%) treated with BELVIQ developed euphoria, as compared with 1 patient (<0.1%) treated with placebo. Doses should not exceed BELVIQ 10 mg twice daily or BELVIQ XR 20 mg once daily.
Some drugs that target the central nervous system have been associated with depression or suicidal ideation. Patients treated with BELVIQ/BELVIQ XR should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior. Discontinue BELVIQ/BELVIQ XR in patients who experience suicidal thoughts or behaviors [see Adverse Reactions (6.1)].
5.5 Potential Risk of Hypoglycemia in Patients with Type 2 Diabetes Mellitus on Anti- diabetic Therapy
Weight loss may increase the risk of hypoglycemia in patients with type 2 diabetes mellitus treated with insulin and/or insulin secretagogues (e.g., sulfonylureas); hypoglycemia was observed in clinical trials with BELVIQ. BELVIQ/BELVIQ XR has not been studied in combination with insulin. Measurement of blood glucose levels prior to starting BELVIQ/BELVIQ XR and during BELVIQ/BELVIQ XR treatment is recommended in patients with type 2 diabetes. Decreases in medication doses for anti-diabetic medications which are non-glucose-dependent should be considered to mitigate the risk of hypoglycemia. If a patient develops hypoglycemia after starting BELVIQ/BELVIQ XR, appropriate changes should be made to the anti-diabetic drug regimen [see Adverse Reactions (6.1)].
5.6 Priapism
Priapism (painful erections greater than 6 hours in duration) is a potential effect of 5-HT2C receptor agonism. If not treated promptly, priapism can result in irreversible damage to the erectile tissue. Men who have an erection lasting greater than 4 hours, whether painful or not, should immediately discontinue the drug and seek emergency medical attention.
BELVIQ/BELVIQ XR should be used with caution in men who have conditions that might predispose them to priapism (e.g., sickle cell anemia, multiple myeloma, or leukemia), or in men with anatomical deformation of the penis (e.g., angulation, cavernosal fibrosis, or Peyronie's disease). There is limited experience with the combination of BELVIQ/BELVIQ XR and medication indicated for erectile dysfunction (e.g., phosphodiesterase type 5 inhibitors). Therefore, the combination of BELVIQ/BELVIQ XR and these medications should be used with caution.
5.7 Heart Rate Decreases
In clinical trials of at least 1-year in duration, the mean change in heart rate (HR) was -1.2 beats per minute (bpm) in BELVIQ and -0.4 bpm in placebo-treated patients without diabetes and -2.0 beats per minute (bpm) in BELVIQ and -0.4 bpm in placebo-treated patients with type 2 diabetes. The incidence of HR less than 50 bpm was 5.3% in BELVIQ and 3.2% in placebo-treated patients without diabetes and 3.6% in BELVIQ and 2.0% in placebo-treated patients with type 2 diabetes. In the combined population, adverse reactions of bradycardia occurred in 0.3% of BELVIQ and 0.1% of placebo-treated patients. Use BELVIQ/BELVIQ XR with caution in patients with bradycardia or a history of heart block greater than first degree.
5.8 Hematological Changes
In clinical trials of at least one year in duration, adverse reactions of decreases in white blood cell count (including leukopenia, lymphopenia, neutropenia, and decreased white cell count) were reported in 0.4% of patients treated with BELVIQ as compared to 0.2% of patients treated with placebo. Adverse reactions of decreases in red blood cell count (including anemia and decreases in hemoglobin and hematocrit) were reported by 1.3% of patients treated with BELVIQ as compared to 1.2% treated with placebo [see Adverse Reactions (6.1)]. Consider periodic monitoring of complete blood count during treatment with BELVIQ/BELVIQ XR.
5.9 Prolactin Elevation
Lorcaserin moderately elevates prolactin levels. In a subset of placebo-controlled clinical trials of at least one year in duration, elevations of prolactin greater than the upper limit of normal, two times the upper limit of normal, and five times the upper limit of normal, measured both before and 2 hours after dosing, occurred in 6.7%, 1.7%, and 0.1% of BELVIQ-treated patients and 4.8%, 0.8%, and 0.0% of placebo-treated patients, respectively [see Adverse Reactions (6.1)]. Prolactin should be measured when symptoms and signs of prolactin excess are suspected (e.g., galactorrhea, gynecomastia). There was one patient treated with BELVIQ who developed a prolactinoma during the trial. The relationship of BELVIQ/BELVIQ XR to the prolactinoma in this patient is unknown.
5.10 Pulmonary Hypertension
Certain centrally-acting weight loss agents that act on the serotonin system have been associated with pulmonary hypertension, a rare but lethal disease. Because of the low incidence of this disease, the clinical trial experience with BELVIQ is inadequate to determine if BELVIQ/BELVIQ XR increases the risk for pulmonary hypertension.
-
6
ADVERSE REACTIONS
The following important adverse reactions are described below and elsewhere in labeling:
- Serotonin Syndrome or NMS-like Reactions [see Warnings and Precautions (5.1)]
- Valvular Heart Disease [see Warnings and Precautions (5.2)]
- Cognitive Impairment [see Warnings and Precautions (5.3)]
- Psychiatric Disorders [see Warnings and Precautions (5.4)]
- Hypoglycemia [see Warnings and Precautions (5.5)]
- Heart Rate Decreases [see Warnings and Precautions (5.7)]
- Hematological Changes [see Warnings and Precautions (5.8)]
- Prolactin Elevation [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
In the BELVIQ placebo-controlled clinical database of trials of at least one year in duration, of 6888 patients (3451 BELVIQ vs. 3437 placebo; age range 18-66 years, 79.3% women, 66.6% Caucasians, 19.2% Blacks, 11.8% Hispanics, 2.4% other, 7.4% type 2 diabetics), a total of 1969 patients were exposed to BELVIQ 10 mg twice daily for 1 year and 426 patients were exposed for 2 years.
In clinical trials of at least one year in duration, 8.6% of patients treated with BELVIQ prematurely discontinued treatment due to adverse reactions, compared with 6.7% of placebo-treated patients. The most common adverse reactions leading to discontinuation more often among BELVIQ treated patients than placebo were headache (1.3% vs. 0.8%), depression (0.9% vs. 0.5%), and dizziness (0.7% vs. 0.2%).
Most Common Adverse Reactions
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most common adverse reactions for non-diabetic patients (greater than 5% and more commonly than placebo) treated with BELVIQ compared to placebo were headache, dizziness, fatigue, nausea, dry mouth, and constipation. The most common adverse reactions for diabetic patients were hypoglycemia, headache, back pain, cough, and fatigue. Adverse reactions that were reported by greater than or equal to 2% of patients and were more frequently reported by patients taking BELVIQ compared to placebo are summarized in Table 2 (non-diabetic subjects) and Table 3 (subjects with type 2 diabetes mellitus).
Table 2. Adverse Reactions Reported by Greater Than or Equal to 2% of BELVIQ Patients and More Commonly than with Placebo in Patients without Diabetes Mellitus Number of patients (%) Adverse Reaction BELVIQ
N=3195Placebo
N=3185Gastrointestinal Disorders Nausea 264 (8.3) 170 (5.3) Diarrhea 207 (6.5) 179 (5.6) Constipation 186 (5.8) 125 (3.9) Dry mouth 169 (5.3) 74 (2.3) Vomiting 122 (3.8) 83 (2.6) General Disorders And Administration Site Conditions Fatigue 229 (7.2) 114 (3.6) Infections And Infestations Upper respiratory tract infection 439 (13.7) 391 (12.3) Nasopharyngitis 414 (13.0) 381 (12.0) Urinary tract infection 207 (6.5) 171 (5.4) Musculoskeletal And Connective Tissue Disorders Back pain 201 (6.3) 178 (5.6) Musculoskeletal pain 65 (2.0) 43 (1.4) Nervous System Disorders Headache 537 (16.8) 321 (10.1) Dizziness 270 (8.5) 122 (3.8) Respiratory, Thoracic And Mediastinal Disorders Cough 136 (4.3) 109 (3.4) Oropharyngeal pain 111 (3.5) 80 (2.5) Sinus congestion 93 (2.9) 78 (2.4) Skin And Subcutaneous Tissue Disorders Rash 67 (2.1) 58 (1.8) Table 3. Adverse Reactions Reported by Greater Than or Equal to 2% of BELVIQ Patients and More Commonly than with Placebo in Patients with Type 2 Diabetes Mellitus Number of patients (%) Adverse Reaction BELVIQ
N=256Placebo
N=252Gastrointestinal Disorders Nausea 24 (9.4) 20 (7.9) Toothache 7 (2.7) 0 General Disorders And Administration Site Conditions Fatigue 19 (7.4) 10 (4.0) Peripheral edema 12 (4.7) 6 (2.4) Immune System Disorders Seasonal allergy 8 (3.1) 2 (0.8) Infections And Infestations Nasopharyngitis 29 (11.3) 25 (9.9) Urinary tract infection 23 (9.0) 15 (6.0) Gastroenteritis 8 (3.1) 5 (2.0) Metabolism And Nutrition Disorders Hypoglycemia 75 (29.3) 53 (21.0) Worsening of diabetes mellitus 7 (2.7) 2 (0.8) Decreased appetite 6 (2.3) 1 (0.4) Musculoskeletal And Connective Tissue Disorders Back pain 30 (11.7) 20 (7.9) Muscle spasms 12 (4.7) 9 (3.6) Nervous System Disorders Headache 37 (14.5) 18 (7.1) Dizziness 18 (7.0) 16 (6.3) Psychiatric Disorders Anxiety 9 (3.5) 8 (3.2) Insomnia 9 (3.5) 6 (2.4) Stress 7 (2.7) 3 (1.2) Depression 6 (2.3) 5 (2.0) Respiratory, Thoracic And Mediastinal Disorders Cough 21 (8.2) 11 (4.4) Vascular Disorders Hypertension 13 (5.1) 8 (3.2) Other Adverse Reactions
Serotonin-associated Adverse Reactions
SSRIs, SNRIs, bupropion, tricyclic antidepressants, and MAOIs were excluded from the BELVIQ trials. Triptans and dextromethorphan were permitted: 2% and 15%, respectively, of patients without diabetes and 1% and 12%, respectively, of patients with type 2 diabetes experienced concomitant use at some point during the trials. Two patients treated with BELVIQ in the clinical program experienced a constellation of symptoms and signs consistent with serotonergic excess, including one patient on concomitant dextromethorphan who reported an event of serotonin syndrome. Some symptoms of possible serotonergic etiology that are included in the criteria for serotonin syndrome were reported by patients treated with BELVIQ and placebo during clinical trials of at least 1 year in duration. In both groups, chills were the most frequent of these events (1.0% vs. 0.2%, respectively), followed by tremor (0.3% vs. 0.2%), confusional state (0.2% vs. less than 0.1%), disorientation (0.1% vs. 0.1%) and hyperhidrosis (0.1% vs. 0.2%). Because serotonin syndrome has a very low incidence, an association between BELVIQ/BELVIQ XR and serotonin syndrome cannot be excluded on the basis of clinical trial results [see Warnings and Precautions (5.1)].Hypoglycemia in Patients with Type 2 Diabetes
In a clinical trial of patients with type 2 diabetes mellitus, severe hypoglycemia (requiring the assistance of another person, requiring intravenous glucose, or hospitalization) occurred in 4 (1.6%) of BELVIQ-treated patients and in 1 (0.4%) placebo-treated patient. Of these 4 BELVIQ-treated patients, all were concomitantly using a sulfonylurea (with or without metformin). BELVIQ/BELVIQ XR has not been studied in patients taking insulin. Hypoglycemia defined as blood sugar less than or equal to 65 mg/dL and with symptoms occurred in 19 (7.4%) BELVIQ-treated patients and 16 (6.3%) placebo-treated patients.Cognitive Impairment
In clinical trials of at least 1-year duration, adverse reactions related to cognitive impairment (e.g., difficulty with concentration/attention, difficulty with memory, and confusion) occurred in 2.3% of patients taking BELVIQ and 0.7% of patients taking placebo.Psychiatric Disorders
Psychiatric disorders leading to hospitalization or drug withdrawal occurred more frequently in patients treated with BELVIQ (2.2%) as compared to placebo (1.1%) in non-diabetic patients.Euphoria. In short-term studies with healthy individuals, the incidence of euphoric mood following supratherapeutic doses of BELVIQ (40 and 60 mg) was increased as compared to placebo [see Drug Abuse and Dependence (9.2)]. In clinical trials of at least 1-year duration in obese patients, euphoria was observed in 0.17% of patients taking BELVIQ and 0.03% taking placebo.
Depression and Suicidality. In trials of at least one year in duration, reports of depression/mood problems occurred in 2.6% BELVIQ-treated vs. 2.4% placebo-treated and suicidal ideation occurred in 0.6% BELVIQ- treated vs. 0.4% placebo-treated patients. 1.3% of BELVIQ patients vs. 0.6% of placebo patients discontinued drug due to depression-, mood-, or suicidal ideation-related events.
Laboratory Abnormalities
Lymphocyte and Neutrophil Counts. In clinical trials of at least 1-year duration, lymphocyte counts were below the lower limit of normal in 12.2% of patients taking BELVIQ and 9.0% taking placebo, and neutrophil counts were low in 5.6% and 4.3%, respectively.Hemoglobin. In clinical trials of at least 1-year duration, 10.4% of patients taking BELVIQ and 9.3% taking placebo had hemoglobin below the lower limit of normal at some point during the trials.
Prolactin. In clinical trials, elevations of prolactin greater than the upper limit of normal, two times the upper limit of normal, and five times the upper limit of normal, occurred in 6.7%, 1.7%, and 0.1% of BELVIQ-treated patients and 4.8%, 0.8%, and 0.0% of placebo-treated patients, respectively.
Eye disorders
More patients on BELVIQ reported an eye disorder than patients on placebo in clinical trials of patients without diabetes (4.5% vs. 3.0%) and with type 2 diabetes (5.9% vs. 1.6%). In the population without diabetes, events of blurred vision, dry eye, and visual impairment occurred in BELVIQ-treated patients at an incidence greater than that of placebo. In the population with type 2 diabetes, visual disorders, conjunctival infections, irritations, and inflammations, ocular sensation disorders, and cataract conditions occurred in BELVIQ-treated patients at an incidence greater than placebo.Echocardiographic Safety Assessments
The possible occurrence of regurgitant cardiac valve disease was prospectively evaluated in 7794 patients in three clinical trials of at least one year in duration, 3451 of whom took BELVIQ. The primary echocardiographic safety parameter was the proportion of patients who developed echocardiographic criteria of mild or greater aortic insufficiency and/or moderate or greater mitral insufficiency from baseline to 1 year. At 1 year, 2.4% of patients who received BELVIQ and 2.0% of patients who received placebo developed valvular regurgitation. The relative risk for valvulopathy with BELVIQ is summarized in Table 4. BELVIQ/BELVIQ XR was not studied in patients with congestive heart failure or hemodynamically-significant valvular heart disease [see Warnings and Precautions (5.2)].
Table 4. Incidence of FDA-Defined Valvulopathy at Week 52 by Treatment Group1 Study 1 Study 2 Study 3 BELVIQ
N=1278Placebo
N=1191BELVIQ N=1208 Placebo
N=1153BELVIQ
N=210Placebo
N=209FDA-defined
Valvulopathy, n (%)34 (2.7) 28 (2.4) 24 (2.0) 23 (2.0) 6 (2.9) 1 (0.5) Relative Risk (95% CI) 1.13 (0.69, 1.85) 1.00 (0.57, 1.75) 5.97 (0.73, 49.17) Pooled RR (95% CI) 1.16 (0.81, 1.67) 1 Patients without valvulopathy at baseline who received study medication and had a post-baseline echocardiogram; ITT-intention-to-treat; LOCF-last observation carried forward
6.2 Post-Marketing Experience
The following adverse reactions have been identified during post approval use. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorders: drug hypersensitivity
- Serotonin Syndrome or NMS-like Reactions [see Warnings and Precautions (5.1)]
-
7
DRUG INTERACTIONS
7.1 Use with Other Agents that Affect Serotonin Pathways
Based on the mechanism of action of BELVIQ/BELVIQ XR and the theoretical potential for serotonin syndrome, use with extreme caution in combination with other drugs that may affect the serotonergic neurotransmitter systems, including, but not limited to, triptans, monoamine oxidase inhibitors (MAOIs, including linezolid, an antibiotic which is a reversible non-selective MAOI), selective serotonin reuptake inhibitors (SSRIs), selective serotonin- norepinephrine reuptake inhibitors (SNRIs), dextromethorphan, tricyclic antidepressants (TCAs), bupropion, lithium, tramadol, tryptophan, and St. John’s Wort [see Warnings and Precautions (5.1)].
7.2 Cytochrome P450 (2D6) substrates
Use caution when administering BELVIQ/BELVIQ XR together with drugs that are CYP 2D6 substrates, as BELVIQ/BELVIQ XR can increase exposure of these drugs [see Clinical Pharmacology (12.3)].
-
8
USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
BELVIQ/BELVIQ XR is contraindicated during pregnancy, because weight loss offers no benefit to a pregnant woman and may result in fetal harm [see Clinical Considerations]. Limited data on lorcaserin use in pregnant women are not sufficient to determine a drug-associated risk of major congenital malformations or miscarriage. No adverse developmental effects were observed when lorcaserin was administered to pregnant rats and rabbits during organogenesis at exposures up to 44- and 19-times the clinical dose of 20 mg daily, respectively. In rats, maternal exposure to lorcaserin in late pregnancy resulted in lower body weight in offspring which persisted to adulthood [see Data]. Advise pregnant women of the potential risk to a fetus.The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage of clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryofetal risk
Appropriate weight gain based on pre-pregnancy weight is currently recommended for all pregnant women, including those who are already overweight or obese, due to the obligatory weight gain that occurs in maternal tissues during pregnancy.Data
Animal Data
Reproduction studies were performed in pregnant rats and rabbits that were administered lorcaserin hydrochloride during the period of embryofetal organogenesis. Plasma exposures up to 44 and 19 times the clinical dose of 20 mg daily in pregnant rats and rabbits, respectively, did not reveal evidence of teratogenicity or embryolethality with lorcaserin hydrochloride.In a pre- and postnatal development study, maternal rats were dosed from gestation through post-natal day 21 at 5, 15, and 50 mg/kg lorcaserin hydrochloride; pups were indirectly exposed in utero and throughout lactation. Stillborns and lower pup viability was observed at 50 mg/kg, or 44 times the clinical dose of 20 mg daily, based on AUC. All other doses lowered pup body weight similarly at birth which persisted to adulthood; however, no developmental abnormalities were observed and reproductive performance was not affected.
8.2 Lactation
Risk Summary
There are no data on the presence of lorcaserin in human milk, the effects on the breastfed infant, or the effects on milk production. Because of the potential for serious adverse reactions in a breastfed infant, advise women that use of BELVIQ/BELVIQ XR is not recommended while breastfeeding.8.4 Pediatric Use
The safety and effectiveness of BELVIQ/BELVIQ XR in pediatric patients below the age of 18 have not been established and the use of BELVIQ/BELVIQ XR is not recommended in pediatric patients.
8.5 Geriatric Use
In the BELVIQ clinical trials, a total of 135 (2.5%) of the patients were 65 years of age and older. Clinical studies of BELVIQ did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects, but greater sensitivity of some older individuals cannot be ruled out.
Since elderly patients have a higher incidence of renal impairment, use of BELVIQ/BELVIQ XR in the elderly should be made on the basis of renal function [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)]. Elderly patients with normal renal function should require no dose adjustment.
8.6 Renal Impairment
No dose adjustment of BELVIQ/BELVIQ XR is required in patients with mild renal impairment. Use BELVIQ/BELVIQ XR with caution in patients with moderate renal impairment. Use of BELVIQ/BELVIQ XR in patients with severe renal impairment or end stage renal disease is not recommended [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Dose adjustment is not required for patients with mild hepatic impairment (Child-Pugh score 5-6) to moderate hepatic impairment (Child-Pugh score 7-9). The effect of severe hepatic impairment on lorcaserin was not evaluated. Use BELVIQ/BELVIQ XR with caution in patients with severe hepatic impairment [see Clinical Pharmacology (12.3)].
-
9
DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
BELVIQ/BELVIQ XR is listed in Schedule IV of the Controlled Substances Act.
9.2 Abuse
In a human abuse potential study in recreational drug abusers, supratherapeutic oral doses of BELVIQ (40 and 60 mg) produced up to two- to six-fold increases on measures of “High”, “Good Drug Effects”, “Hallucinations” and “Sedation” compared to placebo. These responses were similar to those produced by oral administration of the positive control drugs, zolpidem (15 and 30 mg) and ketamine (100 mg). In this study, the incidence of the adverse reaction of euphoria following lorcaserin administration (40 and 60 mg; 19%) is similar to the incidence following zolpidem administration (13-16%), but less than the incidence following ketamine administration (50%). The duration of euphoria following lorcaserin administration persisted longer (> 9 hours) than that following zolpidem (1.5 hours) or ketamine (2.5 hours) administration.
Overall, in short-term studies with healthy individuals, the rate of euphoria following oral administration of lorcaserin was 16% following 40 mg (n = 11 of 70) and 19% following 60 mg (n = 6 of 31). However, in clinical studies with obese patients with durations of 4 weeks to 2 years, the incidence of euphoria and hallucinations following oral doses of lorcaserin up to 40 mg was low (< 1.0%).
9.3 Dependence
There are no data from well-conducted animal or human studies that evaluate whether lorcaserin can induce physical dependence, as evidenced by a withdrawal syndrome. However, the ability of lorcaserin to produce hallucinations, euphoria, and positive subjective responses at supratherapeutic doses suggests that lorcaserin may produce psychic dependence.
-
10
OVERDOSAGE
No experience with overdose of BELVIQ/BELVIQ XR is available. In clinical studies that used doses that were higher than the recommended dose, the most frequent adverse reactions associated with BELVIQ were headache, nausea, abdominal discomfort, and dizziness. Single 40- and 60-mg doses of BELVIQ caused euphoria, altered mood, and hallucination in some subjects. Treatment of overdose should consist of BELVIQ/BELVIQ XR discontinuation and general supportive measures in the management of overdosage. Lorcaserin is not eliminated to a therapeutically significant degree by hemodialysis.
-
11
DESCRIPTION
BELVIQ/BELVIQ XR (lorcaserin hydrochloride) is a serotonin 2C receptor agonist for oral administration used for chronic weight management. Lorcaserin hydrochloride is chemically designated as (R)-8-chloro-1-methyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride hemihydrate. The empirical formula is C11H14ClN·HCl·0.5H2O, and the molecular weight of the hemihydrate form is 241.16 g/mol.The structural formula is:
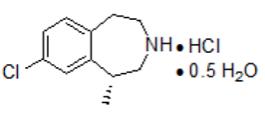
Lorcaserin hydrochloride hemihydrate is a white to off-white powder with solubility in water greater than 400 mg/mL.
Each BELVIQ tablet contains 10.4 mg of crystalline lorcaserin hydrochloride hemihydrate, equivalent to 10.0 mg anhydrous lorcaserin hydrochloride, and the following inactive ingredients: silicified microcrystalline cellulose NF; hydroxypropyl cellulose NF; croscarmellose sodium NF; polyvinyl alcohol USP; polyethylene glycol NF; titanium dioxide USP; talc USP, FD&C blue #2/indigo carmine aluminum lake; and magnesium stearate NF.
Each BELVIQ XR extended-release tablet contains 20.8 mg of crystalline lorcaserin hydrochloride hemihydrate, equivalent to 20.0 mg anhydrous lorcaserin hydrochloride, and the following inactive ingredients: microcrystalline cellulose NF; mannitol USP; hypromellose USP; ethylcellulose dispersion Type B NF; colloidal silicon dioxide NF; polyvinyl alcohol USP; polyethylene glycol NF; titanium dioxide USP; talc USP; FD&C yellow #6/sunset yellow FCF aluminum lake; iron oxide yellow NF; iron oxide red NF; and magnesium stearate NF.
-
12
CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Lorcaserin is believed to decrease food consumption and promote satiety by selectively activating 5-HT2C receptors on anorexigenic pro-opiomelanocortin neurons located in the hypothalamus. The exact mechanism of action is not known.
Lorcaserin at the recommended daily dose selectively interacts with 5-HT2C receptors as compared to 5-HT2A and 5-HT2B receptors (see Table 5), other 5-HT receptor subtypes, the 5-HT receptor transporter, and 5-HT reuptake sites.
Table 5. Lorcaserin Potency (EC50) and Binding Affinity (Ki) to Human 5-HT2A, 5-HT2B, and 5-HT2C Receptor Subtypes Serotonin Receptor Subtype EC50, nM Ki, nM 5HT2C 39 13 5HT2B 2380 147 5HT2A 553 92 12.2 Pharmacodynamics
Cardiac Electrophysiology. The effect of multiple oral doses of lorcaserin 15 mg and 40 mg once daily on QTc interval was evaluated in a randomized, placebo- and active- (moxifloxacin 400 mg) controlled four-treatment arm parallel thorough QT study in 244 healthy subjects. In a study with demonstrated ability to detect small effects, the upper bound of the one-sided 95% confidence interval for the largest placebo adjusted, baseline-corrected QTc based on individual correction method (QTcI) was below 10 ms, the threshold for regulatory concern.
12.3 Pharmacokinetics
Absorption
BELVIQ
Lorcaserin is absorbed from the gastrointestinal tract with peak plasma concentration occurring 1.5 - 2 hours after oral dosing. The absolute bioavailability of lorcaserin has not been determined. Steady state is reached within 3 days after twice daily dosing, and accumulation is estimated to be approximately 70%.Effect of Food. Twelve adult volunteers (6 men and 6 women) were given a single 10 mg oral dose of BELVIQ in a fasted state and after administration of a high fat (approximately 50% of total caloric content of the meal) and high-calorie (approximately 800–1000 calories) meal. The Cmax increased approximately 9% and exposure (AUC) increased approximately 5% under fed conditions. Tmax was delayed approximately 1 hour in the fed state. BELVIQ can be administered with or without food.
BELVIQ XR
In an open label, randomized, crossover clinical trial, single dose and steady state pharmacokinetics of BELVIQ XR 20 mg administered once daily were compared with BELVIQ 10 mg tablet administered twice daily under fasted conditions in 34 healthy subjects. At steady state, the time to reach peak plasma concentrations of lorcaserin (tmax) following BELVIQ XR 20 mg once daily was approximately 10 hours compared with 1.5 hours for BELVIQ 10 mg tablet twice daily. A single dose administration of BELVIQ XR 20 mg resulted in comparable total plasma exposure (AUC0-∞), but approximately 25% lower peak exposures (Cmax) relative to two doses of BELVIQ tablets administered 12 hours apart. At steady state, however, both Cmax,ss and area under the plasma concentration versus time curve (AUC0-24,ss) of BELVIQ XR 20 mg administered once daily were bioequivalent to BELVIQ 10 mg tablets administered twice daily under fasted conditions.Effect of Food. Intake of high fat, high calorie breakfast before a single 20 mg oral dose of BELVIQ XR resulted in approximately 46% increase in Cmax and 17% increase in AUC0-∞ but no change in tmax. At steady state, however, there was no significant food effect on the rate or extent of absorption of BELVIQ XR.
Distribution
Lorcaserin distributes to the cerebrospinal fluid and central nervous system in humans. Lorcaserin hydrochloride is moderately bound (~70%) to human plasma proteins.
Metabolism
Lorcaserin is extensively metabolized in the liver by multiple enzymatic pathways. After oral administration of lorcaserin the major circulating metabolite is lorcaserin sulfamate (M1), with a plasma Cmax that exceeds lorcaserin Cmax by 1- to 5-fold. N-carbamoyl glucuronide lorcaserin (M5) is the major metabolite in urine; M1 is a minor metabolite in urine, representing approximately 3% of dose. Other minor metabolites excreted in urine were identified as glucuronide or sulfate conjugates of oxidative metabolites. The principal metabolites exert no pharmacological activity at serotonin receptors.
Elimination
Lorcaserin is extensively metabolized by the liver and the metabolites are excreted in the urine. In a human mass balance study in which healthy subjects ingested radiolabeled lorcaserin, 94.5% of radiolabeled material was recovered, with 92.3% and 2.2% recovered from urine and feces, respectively. The terminal phase half-life for BELVIQ/ BELVIQ XR is approximately 11 to 12 hours.
Specific Populations
Renal Impairment. The pharmacokinetics of lorcaserin was studied in patients with varying degrees of renal function. Creatinine clearance (CLcr) was calculated by Cockcroft-Gault equation based on ideal body weight (IBW). Impaired renal function decreased Cmax of lorcaserin, with no change in AUC.
Exposure of lorcaserin sulfamate metabolite (M1) was increased in patients with impaired renal function by approximately 1.7-fold in mild (CLcr = 50-80 mL/min), 2.3-fold in moderate (CLcr = 30-50 mL/min) and 10.5-fold in severe renal impairment (CLcr = <30 mL/min) compared to normal subjects (CLcr >80 mL/min).
Exposure of the N-carbamoyl-glucuronide metabolite (M5) was increased in patients with impaired renal function by approximately 1.5-fold in mild (CLcr = 50-80 mL/min), 2.5-fold in moderate (CLcr = 30-50 mL/min) and 5.1-fold in severe renal impairment (CLcr = <30 mL/min) compared to normal subjects (CLcr >80 mL/min).
The terminal half-life of M1 is prolonged by 26%, 96%, and 508% in mild, moderate, and severe renal impairment, respectively. The terminal half-life of M5 is prolonged by 0%, 26%, and 22% in mild, moderate, and severe renal impairment, respectively. The metabolites M1 and M5 accumulate in patients with severely impaired renal function.
Approximately 18% of metabolite M5 in the body was cleared from the body during a standard 4-hour hemodialysis procedure. Lorcaserin and M1 were not cleared by hemodialysis. Lorcaserin is not recommended for patients with severe renal impairment (CLcr <30 mL/min) or patients with end stage renal disease [see Use in Specific Populations (8.6)].
Estimate Ideal Body Weight (IBW) in (kg)
Males: IBW = 50 kg + 2.3 kg for each inch over 5 feet.
Females: IBW = 45.5 kg + 2.3 kg for each inch over 5 feet.
The Cockcroft-Gault calculation using the IBW:
female:
GFR (mL/min) = 0.85 x (140-age) x ideal body weight (kg)
72 x serum creatinine (mg/dL)
male:
GFR (mL/min) = (140-age) x ideal body weight (kg)
72 x serum creatinine (mg/dL)
Hepatic Impairment. The pharmacokinetics of lorcaserin was evaluated in patients with hepatic impairment and subjects with normal hepatic function. Lorcaserin Cmax was 7.8% and 14.3% lower, in subjects with mild (Child-Pugh score 5-6) and moderate (Child-Pugh score 7-9) hepatic impairment, respectively, than that in subjects with normal hepatic function. The half-life of lorcaserin is prolonged by 59% to 19 hours in patients with moderate hepatic impairment. Lorcaserin exposure (AUC) is approximately 22% and 30% higher in patients with mild and moderate hepatic impairment, respectively. Dose adjustment is not required for patients with mild to moderate hepatic impairment. The effect of severe hepatic impairment on lorcaserin was not evaluated [see Use in Specific Populations (8.7)].
Gender. No dosage adjustment based on gender is necessary. Gender did not meaningfully affect the pharmacokinetics of lorcaserin.
Geriatric. No dosage adjustment is required based on age alone. In a clinical trial of 12 healthy elderly (age greater than 65 years) subjects and 12 matched adult patients, lorcaserin exposure (AUC and Cmax) was equivalent in the two groups. Cmax was approximately 18% lower in the elderly group, and Tmax was increased from 2 hours to 2.5 hours in the elderly group as compared to the non-elderly adult group.
Race. No dosage adjustment based on race is necessary. Race did not meaningfully affect the pharmacokinetics of lorcaserin.
Drug-Drug Interactions
Lorcaserin inhibits CYP 2D6-mediated metabolism. In a clinical trial in 21 CYP 2D6 extensive metabolizers, concomitant administration of lorcaserin (10 mg BID for 4 days) increased dextromethorphan peak concentrations (Cmax) by approximately 76% and exposure (AUC) by approximately 2-fold [see Drug Interactions (7.2)].
-
13
NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Mutagenesis
Lorcaserin was not mutagenic in an in vitro bacterial mutation assay (Ames test), was not clastogenic in an in vitro chromosome aberration assay in Chinese hamster ovary cells, and was not genotoxic in an in vivo micronucleus assay in rat bone marrow.
Carcinogenesis
The carcinogenic potential of lorcaserin was assessed in two-year carcinogenicity studies in mice and rats. CD-1 mice received doses of 5, 25 and 50 mg/kg. There were no treatment-related increases in the incidence of any tumor in mice at doses that produced plasma exposure in males and females of 8 and 4-times the daily human clinical dose, respectively.
In the rat carcinogenicity study, male and female Sprague-Dawley rats received 10, 30, and 100 mg/kg lorcaserin hydrochloride. In females, mammary adenocarcinoma increased at 100 mg/kg, which was associated with plasma exposures that were 87-times the daily human clinical dose. The incidence of mammary fibroadenoma was increased in female rats at all doses with no safety margin to the clinical dose. The increases in adenocarcinomas and fibroadenomas may be associated with lorcaserin-induced changes in prolactin homeostasis in rats. The relevance of the increased incidence of mammary adenocarcinomas and fibroadenomas in rats to humans is unknown.
In male rats, treatment-related neoplastic changes were observed in the subcutis (fibroma, Schwannoma), the skin (squamous cell carcinoma), mammary gland (adenocarcinoma and fibroadenoma), and the brain (astrocytoma) at greater than or equal to 30 mg/kg (plasma exposure 17-times human clinical dose). At higher exposure, liver adenoma and thyroid follicular cell adenoma were increased but were considered secondary to liver enzyme induction in rats and are not considered relevant to humans. Human brain exposure (AUC24h,ss) to lorcaserin at the clinical dose is estimated to be 70-fold lower than brain exposure in rats at the dose at which no increased incidence of astrocytoma was observed. Excluding the liver and thyroid tumors, these neoplastic findings in male rats are of unknown relevance to humans.
Impairment of Fertility
Potential effects on fertility were assessed in Sprague-Dawley rats in which males were dosed with lorcaserin hydrochloride for 4 weeks prior to and through the mating period, and females were dosed for 2 weeks prior to mating and through gestation day 7. Lorcaserin had no effects on fertility in rats at exposures up to 29 times the human clinical dose.
-
14
CLINICAL STUDIES
The safety and efficacy of BELVIQ for chronic weight management in conjunction with reduced caloric intake and increased physical activity were evaluated in 3 randomized, double-blind, placebo-controlled trials with durations ranging from 52 to 104 weeks. Two trials in adults without type 2 diabetes mellitus (Study 1 and Study 2) and one study in adults with type 2 diabetes mellitus (Study 3) evaluated the effect of BELVIQ. The primary efficacy parameter in these studies was weight loss at 1 year, which was assessed by percent of patients achieving greater than or equal to 5% weight loss, percent of patients achieving greater than or equal to 10% weight loss, and mean weight change. All patients received one-on-one instruction for a reduced-calorie diet and exercise counseling that began with the first dose of study medication and continued every four weeks throughout the trial.
Study 1 was a 2-year study that enrolled 3182 patients who were obese (BMI 30-45 kg/m2), or who were overweight (BMI 27-29.9 kg/m2) and had at least one weight-related comorbid condition such as hypertension or dyslipidemia. In Year 2, placebo patients were continued on placebo and BELVIQ, patients were re- randomized in a 2:1 ratio to continue BELVIQ or to switch to placebo. The mean age was 44 (range 18-65); 83.5% were women. Sixty-seven percent were Caucasian, 19% were African American and 12% were Hispanic. Mean baseline body weight was 100.0 kg and mean BMI was 36.2 kg/m2.
Study 2 was a 1-year study that enrolled 4008 patients who were obese (BMI 30-45 kg/m2) or were overweight (BMI 27-29.9 kg/m2) with at least one comorbid condition such as hypertension or dyslipidemia. The mean age was 44 (range 18-65); 80% were women. Sixty-seven percent were Caucasian, 20% were African American and 11% were Hispanic. Mean baseline body weight was 100.2 kg and mean BMI was 35.9 kg/m2.
Study 3 was a 1-year study that enrolled 604 adult patients with BMI greater than or equal to 27 kg/m2 and inadequately controlled type 2 diabetes (HbA1c range 7-10%) being treated with metformin and/or a sulfonylurea. Mean age was 53 (range 21-65); 54% were women. Sixty-one percent were Caucasian, 21% African American and 14% were Hispanic. Mean BMI was 36 kg/m2 and mean HbA1C was 8.1%.
A substantial percentage of randomized subjects withdrew from each study prior to week 52: 50% in Study 1, 45% in Study 2 and 36% in Study 3.
One-Year Weight Management in Patients without Diabetes Mellitus
Weight loss at 1 year in Studies 1 and 2 is presented in Table 6. The pooled data are reflective of the individual study results.
Statistically significantly greater weight loss was achieved with BELVIQ compared to placebo at week 52. The Year 1 placebo-adjusted weight loss achieved in patients treated with BELVIQ was 3.3 kg by ITT/LOCF analysis. The time course of weight loss with BELVIQ and placebo through week 52 is depicted in Figure 1.
Patients who did not lose at least 5% of baseline body weight by week 12 were unlikely to achieve at least 5% weight loss at week 52.
Table 6. Weight Loss at 1 Year in Studies 1 and 2 Combined BELVIQ
N=3098Placebo
N=3038Weight (kg) Baseline mean (SD) 100.4 (15.7) 100.2 (15.9) Change from baseline (adjusted mean1) (SE) -5.8 (0.1) -2.5 (0.1) Difference from placebo (adjusted mean1)
(95% CI)-3.3**
(-3.6, -2.9)Percent change from baseline (adjusted mean1) (SE) -5.8 (0.1) -2.5 (0.1) Difference from placebo (adjusted mean1)
(95% CI)-3.3**
( -3.6, -3.0)% of Patients losing greater than or equal to 5% body weight 47.1 22.6 Difference from placebo
(95% CI)24.5**
(22.2, 26.8)% of Patients losing greater than or equal to 10% body weight 22.4 8.7 Difference from placebo
(95% CI)13.8**
(12.0, 15.5)SD=Standard Deviation; SE=Standard Error; CI=Confidence Interval
Intent to Treat Population using last observation carried forward method; All patients who received study medication and had a post-baseline body weight. Forty-four percent (44%) of patients in BELVIQ and 51% in placebo dropped out before the 52-week endpoint.
1Least squares means adjusted for baseline value, treatment, study and treatment by study interaction.
**p<0.001 compared to placebo. Type 1 error was controlled across the three endpoints.
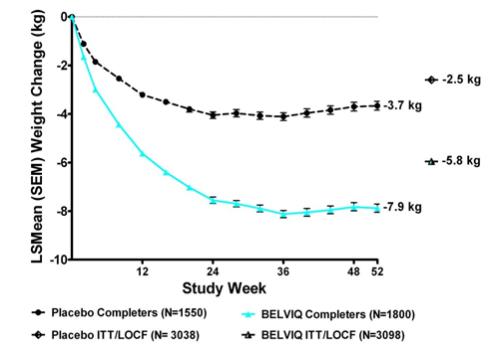
Figure 1. Longitudinal Weight Change (kg) in Completer Population: Studies 1 and 2
Two-Year Weight Management in Patients without Diabetes Mellitus
The safety and efficacy of BELVIQ for weight management during 2 years of treatment were evaluated in Study 1. Of the 3182 patients who were randomized in Year 1, 1553 (48.8%) were randomized in Year 2. Patients in all three Year 2 patient groups (BELVIQ Year 1/ BELVIQ Year 2, BELVIQ Year 1/placebo Year 2, and placebo Year 1/placebo Year 2) regained weight in Year 2 but remained below their Year 1 mean baseline weight (Figure 2).
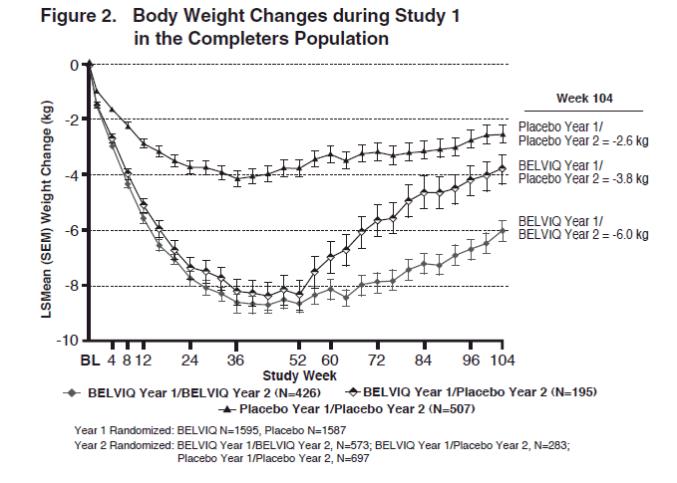
Effect of BELVIQ on Cardiometabolic Parameters and Anthropometry
Changes in lipids, fasting glucose, fasting insulin, waist circumference, heart rate, and blood pressure with BELVIQ are shown in Table 7.
In a substudy of 154 patients conducted as part of Study 2, DEXA analysis showed a 9.9% reduction in fat mass from a baseline of 44.5 kg in patients treated with BELVIQ compared to a 4.6% reduction from a baseline of 45.0 kg in patients treated with placebo. The placebo-adjusted reduction in fat mass achieved on BELVIQ was -5.3%. Reductions in lean body mass were 1.9% and 0.3% from baseline values of 48.0 kg and 51.0 kg, respectively, for BELVIQ- and placebo-treated patients.
Table 7. Mean Changes in Cardiometabolic Parameters and Waist Circumference in Year 1 of Studies 1 and 2 BELVIQ
N=3096Placebo
N=3039BELVIQ minus
Placebo (LSMean)Baseline mg/dL % change from Baseline (LSMean1) Baseline mg/dL % change from Baseline (LSMean) Total Cholesterol 194.4 -0.9 194.8 0.4 -1.2* LDL Cholesterol 114.3 1.6 114.1 2.9 -1.3* HDL Cholesterol 53.2 1.8 53.5 0.6 1.2* Triglycerides 135.4 -5.3 137.0 -0.5 -4.8* Baseline change from Baseline (LSMean) Baseline change from Baseline (LSMean) BELVIQ minus
Placebo (LSMean)Systolic blood pressure
(mmHg)121.4 -1.8 121.5 -1.0 -0.7* Diastolic blood pressure
(mmHg)77.4 -1.6 77.7 -1.0 -0.6* Heart Rate (bpm) 69.5 -1.2 69.5 -0.4 -0.8 Fasting glucose (mg/dL) 92.1 -0.2 92.4 0.6 -0.8 Fasting insulin2 (µIU/mL) 15.9 -3.3 15.8 -1.3 -2.1* Waist Circumference (cm) 109.3 -6.5 109.6 -4.0 -2.5 1 Least squares means adjusted for baseline value, treatment, study and treatment by study interaction
2 Measured in Study 1 only (n=1538)
* Statistically significant versus placebo based on the pre-specified gatekeeping method for controlling Type I error in key secondary endpoints.
One-Year Weight Management in Patients with Type 2 Diabetes Mellitus
Weight loss among patients with type 2 diabetes mellitus who were treated with BELVIQ was statistically significantly greater than that among patients treated with placebo (Table 8).
Table 8. Weight Loss at 1 Year in Study 3 (Type 2 Diabetes Mellitus) BELVIQ
N=251Placebo
N=248Weight loss (kg)
Baseline mean (SD)
Change from baseline (adjusted mean1) (SE)
Difference from placebo (adjusted mean1)
(95% CI)103.5 (17.2)
-4.7 (0.4)
-3.1**
(-4.0, -2.2)102.3 (18.0)
-1.6 (0.4)Percent change from baseline (adjusted mean1) (SE)
Difference from placebo (adjusted mean1)
(95% CI)-4.5 (0.4)
-3.1**
(-3.9, -2.2)-1.5 (0.4) % of Patients losing greater than or equal to 5% body weight
Difference from placebo
(95% CI)37.5
21.3**
(13.8, 28.9)16.1 % of Patients losing greater than or equal to 10% body weight
Difference from placebo
(95% CI)16.3
11.9**
(6.7, 17.1)4.4 SD=Standard Deviation; SE=Standard Error; CI=Confidence Interval
Intent to Treat Population using last observation carried forward method; All patients who received study medication and had a post-baseline body weight. Thirty-four percent (34%) of patients in Belviq and 38% in placebo dropped out before the 52-week endpoint.
1Least squares means adjusted for baseline value, baseline HbA1c stratum and prior antihyperglycemic medication stratum.
**p<0.001 compared to placebo. Type 1 error was controlled across the three endpoints.
Effect of BELVIQ on Cardiometabolic Parameters and Anthropometry in Patients with Type 2
Diabetes Mellitus
Patients in Study 3 were taking either metformin and/or a sulfonylurea at study start, and had inadequate glycemic control (HbA1c range 7-10%). Changes in HbA1c and fasting glucose with BELVIQ use are shown in Table 9.
Table 9. Mean Changes in Cardiometabolic Parameters and Waist Circumference in Patients with Type 2 Diabetes Mellitus BELVIQ
N=256Placebo
N=252BELVIQ minus
Placebo (LSMean)Baseline Change from
Baseline
(LSMean1)Baseline Change from Baseline (LSMean) HbA1C (%) 8.1 -0.9 8.0 -0.4 -0.5* Fasting glucose (mg/dL) 163.3 -27.4 160.0 -11.9 -15.5* Systolic blood pressure (mmHg) 126.6 -0.8 126.5 -0.9 0.1 Diastolic blood pressure (mmHg) 77.9 -1.1 78.7 -0.7 -0.4 Heart Rate (bpm) 72.3 -2.0 72.7 -0.4 -1.6 Waist Circumference (cm) 115.8 -5.5 113.5 -3.3 -2.2 Baseline % Change
from Baseline
(LSMean)Baseline % Change
from Baseline
(LSMean)BELVIQ minus
Placebo (LSMean)Total Cholesterol (mg/dL) 173.5 -0.7 172.0 -0.1 -0.5 LDL Cholesterol (mg/dL) 95.0 4.2 94.6 5.0 -0.8 HDL Cholesterol (mg/dL) 45.3 5.2 45.7 1.6 3.6 Triglycerides (mg/dL) 172.1 -10.7 163.5 -4.8 -5.9 Intent to Treat Population using last observation carried forward method; All patients who received study medication and
had a post-baseline measurement.
* Statistically significant versus placebo based on the pre-specified gatekeeping method for controlling Type I error in key secondary endpoints.
1Least squares means adjusted for baseline value, baseline HbA1c stratum and prior antihyperglycemic medication stratum.
-
16
HOW SUPPLIED/STORAGE AND HANDLING
BELVIQ 10-mg tablets are blue-colored, round, biconvex, film-coated tablets debossed with “A” on one side and “10” on the other side and are available as follows:
NDC: 62856-529-60 Bottle of 60
BELVIQ XR 20-mg extended-release tablets are orange-colored, round, biconvex, film-coated tablets debossed with “A” on one side and “20” on the other side and are available as follows:
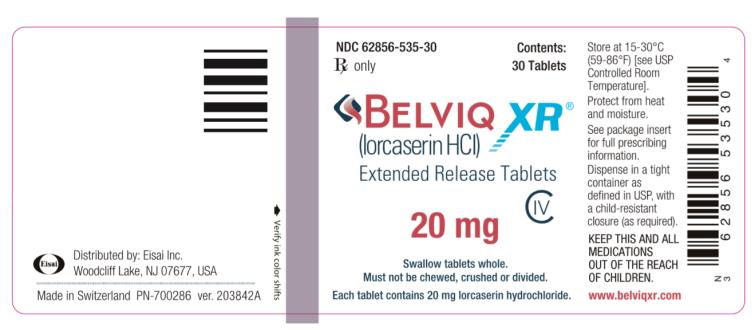
- NDC: 62856-535-30 Bottle of 30
Store at 25°C (77°F): excursions permitted to 15–30°C (59–86°F) [see USP controlled room temperature].
-
17
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
- Inform patients that BELVIQ/BELVIQ XR is indicated for chronic weight management only in conjunction with a reduced-calorie diet and increased physical activity.
- Caution patients not to increase their dose of BELVIQ/BELVIQ XR.
- Instruct patients to discontinue use of BELVIQ/BELVIQ XR if they have not achieved 5% weight loss by 12 weeks of treatment.
- Instruct patients to tell their healthcare provider about all the medications, nutritional supplements and vitamins (including any weight loss products) that they may take while taking BELVIQ/BELVIQ XR.
- Inform patients of the possibility of serotonin syndrome or Neuroleptic Malignant Syndrome (NMS)-like reactions with the combined use of BELVIQ/BELVIQ XR with other serotonergic drugs, including selective serotonin-norepinephrine reuptake inhibitors (SNRIs) and selective serotonin reuptake inhibitors (SSRIs), triptans, drugs that impair metabolism of serotonin (including monoamine oxidase inhibitors [MAOIs]), dietary supplements such as St. John’s Wort and tryptophan, tramadol, or antipsychotics or other dopamine antagonists.
- Inform patients who develop signs or symptoms of valvular heart disease, including dyspnea or dependent edema to seek medical attention.
- Caution patients about operating hazardous machinery, including automobiles, until they are reasonably certain that BELVIQ/BELVIQ XR therapy does not affect them adversely.
- Instruct patients to seek medical attention in the event of emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
- Instruct men who have an erection lasting greater than 4 hours, whether painful or not, to immediately discontinue the drug and seek emergency medical attention.
- Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider with a known or suspected pregnancy [see Contraindications (4), Use in Specific Populations (8.1)].
- Advise women to avoid use of BELVIQ/BELVIQ XR while breastfeeding [see Use in Specific Populations (8.2)].
Distributed by Eisai Inc., Woodcliff Lake, NJ 07677
© 2018
- Inform patients that BELVIQ/BELVIQ XR is indicated for chronic weight management only in conjunction with a reduced-calorie diet and increased physical activity.
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
BELVIQ® (BEL-VEEK)
(lorcaserin hydrochloride)
tablets, CIVBELVIQ XR® (BEL-VEEK Eks-Are)
(lorcaserin hydrochloride)
extended release tablets, CIVWhat is BELVIQ?
BELVIQ is a prescription medicine that may help adults with obesity, or some adults who are overweight and have weight-related medical problems, lose weight and keep the weight off.
BELVIQ should be used with a reduced calorie diet and increased physical activity.
It is not known if BELVIQ is safe and effective when taken with other prescription, over-the-counter, or herbal weight loss products.
It is not known if BELVIQ changes your risk of heart problems or stroke or of death due to heart problems or stroke.
It is not known if BELVIQ is safe when taken with some other medicines that treat depression, migraines, mental problems, or the common cold (serotonergic or antidopaminergic agents).
It is not known if BELVIQ is safe and effective in children under 18 years oldBELVIQ is a federally controlled substance (CIV) because it contains lorcaserin hydrochloride and may be abused or lead to drug dependence. Keep your BELVIQ in a safe place, to protect it from theft. Never give your BELVIQ to anyone else, because it may cause harm to them. Selling or giving away this medicine is against the law. Do not take BELVIQ if you: - are pregnant or planning to become pregnant. BELVIQ may harm your unborn baby.
- are allergic to lorcaserin or any of the ingredients in BELVIQ or BELVIQ XR. See the end of this leaflet for a complete list of ingredients in BELVIQ and BELVIQ XR.
Before you take BELVIQ, tell your healthcare provider about all of your medical conditions, including if you:
- have or have had heart problems including:
○ congestive heart failure
○ heart valve problems
○ slow heart beat or heart block
- have diabetes
- have a condition such as sickle cell anemia, multiple myeloma, or leukemia
- have a deformed penis, Peyronie’s disease, or ever had an erection that lasted more than 4 hours
- have kidney problems
- have liver problems
- are pregnant or plan to become pregnant
- are breastfeeding or plan to breastfeed. It is not known if BELVIQ passes into your breast milk. You and your healthcare provider should decide if you will take BELVIQ or breastfeed. You should not do both.
BELVIQ may affect the way other medicines work, and other medicines may affect how BELVIQ works.
Especially tell your healthcare provider if you take medicines for depression, migraines or other medical conditions such as:
- triptans, used to treat migraine headache
- medicines used to treat mood, anxiety, psychotic or thought disorders, including tricyclics, lithium, selective serotonin uptake inhibitors (SSRIs), selective serotonin-norepinephrine reuptake inhibitors (SNRIs), monoamine oxidase inhibitors (MAOIs), or antipsychotics
- cabergoline
- linezolid, an antibiotic
- tramadol
- dextromethorphan, an over-the-counter medicine used to treat the common cold or cough
- over-the-counter supplements such as tryptophan or St. John’s Wort
- medicines to treat erectile dysfunction
Know all the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.How should I take BELVIQ? - Take BELVIQ exactly as your healthcare provider tells you to take it.
- Your healthcare provider will tell you how much BELVIQ to take and when to take it.
- BELVIQ comes in 2 different dose forms. Your doctor will prescribe the form of BELVIQ that is right for you.
○ BELVIQ: Take one tablet 2 times each day.
○ BELVIQ XR: Take one tablet 1 time each day.
-
Do not increase your dose of BELVIQ.
○ BELVIQ can be taken with or without food.
○ Take the whole BELVIQ XR extended release tablet. Do not chew, crush, or divide the tablet.
- Your healthcare provider should start you on a diet and exercise program when you start taking BELVIQ. Stay on this program while you are taking BELVIQ.
- Your healthcare provider should tell you to stop taking BELVIQ if you do not lose a certain amount of weight within the first 12 weeks of treatment.
- If you take too much BELVIQ or overdose, call your healthcare provider or go to the nearest emergency room right away.
What should I avoid while taking BELVIQ? - Do not drive a car or operate heavy machinery until you know how BELVIQ affects you. BELVIQ can slow your thinking.
What are the possible side effects of BELVIQ?
BELVIQ may cause serious side effects, including:
-
Serotonin Syndrome or Neuroleptic Malignant Syndrome (NMS)-like reactions.
BELVIQ and certain medicines for depression, migraine, the common cold, or other medical problems may affect each other causing serious or life-threatening side effects. Call your healthcare provider right away if you start to have any of the following symptoms while taking BELVIQ:
○ mental changes such as agitation, hallucinations, confusion, or other changes in mental status
○ coordination problems, uncontrolled muscle spasms, or muscle twitching (overactive reflexes)
○ restlessness
○ racing or fast heart beat, high or low blood pressure
○ sweating or fever
○ nausea, vomiting, or diarrhea
○ muscle rigidity (stiff muscles)
-
Valvular heart disease. Some people taking medicines like BELVIQ have had problems with the valves in their heart. Call your healthcare provider right away if you have any of the following symptoms while taking BELVIQ:
○ trouble breathing
○ swelling of the arms, legs, ankles, or feet
○ dizziness, fatigue, or weakness that will not go away
○ fast or irregular heartbeat
-
Changes in your attention or memory.
-
Mental problems. Taking BELVIQ in high doses may cause psychiatric problems such as:
○ hallucinations
○ feeling high or in a very good mood (euphoria)
○ feelings of standing next to yourself or out of your body (disassociation)
-
Depression or thoughts of suicide. You should pay attention to any mental changes, especially sudden changes, in your mood, behaviors, thoughts, or feelings. Call your healthcare provider right away if you have any mental changes that are new, worse, or worry you.
-
Low blood sugar (hypoglycemia) in people with type 2 diabetes mellitus who also take medicines used to treat type 2 diabetes mellitus. Weight loss can cause low blood sugar in people with type 2 diabetes mellitus who also take medicines used to treat type 2 diabetes mellitus (such as insulin or sulfonylureas). You should check your blood sugar before you start taking BELVIQ and while you take BELVIQ.
-
Painful erections (priapism). The medicine in BELVIQ can cause painful erections that last more than 6 hours. If you have an erection lasting more than 4 hours whether it is painful or not, stop using BELVIQ and call your healthcare provider or go to the nearest emergency room right away.
-
Slow heart beat. BELVIQ may cause your heart to beat slower. Tell your healthcare provider if you have a history of your heart beating slow or heart block.
-
Decreases in your blood cell count. BELVIQ may cause your red and white blood cell count to decrease. Your healthcare provider may do tests to check your blood cell count while you are taking BELVIQ.
- Increase in prolactin. The medicine in BELVIQ may increase the amount of a certain hormone your body makes called prolactin. Tell your healthcare provider if your breasts begin to make milk or a milky discharge or if you are a male and your breasts begin to increase in size.
○ headache ○ dizziness ○ fatigue ○ nausea ○ dry mouth ○ constipation ○ cough ○ low blood sugar (hypoglycemia) in patients with diabetes ○ back pain These are not all the possible side effects of BELVIQ.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store BELVIQ?
Store BELVIQ at room temperature between 59°F to 86°F (15°C to 30°C).
Safely throw away medicine that is out of date or no longer needed.
Keep BELVIQ and all medicines out of the reach of children.General information about the safe and effective use of BELVIQ.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use BELVIQ for a condition for which it was not prescribed. Do not give BELVIQ to other people, even if they have the same symptoms you have. It may harm them.
You can ask your healthcare provider or pharmacist for information about BELVIQ that is written for health professionals.What are the ingredients in BELVIQ and BELVIQ XR?
BELVIQ tablets
Active ingredient: lorcaserin hydrochloride hemihydrate
Inactive ingredients: silicified microcrystalline cellulose NF; hydroxypropyl cellulose NF; croscarmellose sodium NF; polyvinyl alcohol USP; polyethylene glycol NF; titanium dioxide USP; talc USP; FD&C blue #2/indigo carmine aluminum lake; and magnesium stearate NF
BELVIQ XR extended-release tablets
Active ingredient: lorcaserin hydrochloride hemihydrate
Inactive ingredients: microcrystalline cellulose NF; mannitol USP; hypromellose USP; ethylcellulose dispersion Type B NF; colloidal silicon dioxide NF; polyvinyl alcohol USP; polyethylene glycol NF; titanium dioxide USP; talc USP; FD&C yellow #6/sunset yellow FCF aluminum lake; iron oxide yellow NF; iron oxide red NF; and magnesium stearate NF
Distributed by Eisai Inc., Woodcliff Lake, NJ 07677. For more information, go to www.BELVIQ.com or call 1-888-274-2378.This Patient Information has been approved by the U.S. Food and Drug Administration Revised: 4/2018
- are pregnant or planning to become pregnant. BELVIQ may harm your unborn baby.
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
BELVIQ
lorcaserin hydrochloride hemihydrate tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 62856-529 Route of Administration ORAL DEA Schedule CIV Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LORCASERIN HYDROCHLORIDE HEMIHYDRATE (UNII: REV26SR2B4) (LORCASERIN - UNII:637E494O0Z) LORCASERIN HYDROCHLORIDE ANHYDROUS 10 mg Inactive Ingredients Ingredient Name Strength CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TALC (UNII: 7SEV7J4R1U) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) MAGNESIUM STEARATE (UNII: 70097M6I30) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) Product Characteristics Color BLUE (blue) Score no score Shape ROUND (ROUND) Size 6mm Flavor Imprint Code A;10 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 62856-529-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 06/27/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022529 06/27/2012 BELVIQ XR
lorcaserin hydrochloride hemihydrate tablet, film coated, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 62856-535 Route of Administration ORAL DEA Schedule CIV Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LORCASERIN HYDROCHLORIDE HEMIHYDRATE (UNII: REV26SR2B4) (LORCASERIN - UNII:637E494O0Z) LORCASERIN HYDROCHLORIDE ANHYDROUS 20 mg Inactive Ingredients Ingredient Name Strength CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) MANNITOL (UNII: 3OWL53L36A) HYPROMELLOSE 2910 (15 MPA.S) (UNII: 36SFW2JZ0W) HYPROMELLOSE 2208 (4000 MPA.S) (UNII: 39J80LT57T) ETHYLCELLULOSES (UNII: 7Z8S9VYZ4B) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) POLYETHYLENE GLYCOL 8000 (UNII: Q662QK8M3B) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TALC (UNII: 7SEV7J4R1U) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) MAGNESIUM STEARATE (UNII: 70097M6I30) Product Characteristics Color ORANGE Score no score Shape ROUND Size 9mm Flavor Imprint Code A;20 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 62856-535-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 07/15/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208524 07/15/2016 Labeler - Eisai Inc. (831600833)
Trademark Results [BELVIQ XR]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 BELVIQ XR 86693918 4908131 Live/Registered |
EISAI INC. 2015-07-15 |
 BELVIQ XR 86693911 4922297 Live/Registered |
Arena Pharmaceuticals GmbH 2015-07-15 |
 BELVIQ XR 86330779 4695303 Live/Registered |
EISAI INC. 2014-07-08 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.