ILARIS- canakinumab injection, powder, lyophilized, for solution ILARIS- canakinumab injection, solution
Ilaris by
Drug Labeling and Warnings
Ilaris by is a Prescription medication manufactured, distributed, or labeled by Novartis Pharmaceuticals Corporation. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ILARIS safely and effectively. See full prescribing information for ILARIS.
ILARIS® (canakinumab) for injection, for subcutaneous use
ILARIS® (canakinumab) injection, for subcutaneous use
Initial U.S. Approval: 2009RECENT MAJOR CHANGES
INDICATIONS AND USAGE
ILARIS is an interleukin-1β blocker indicated for the treatment of:
Periodic Fever Syndromes:
-
Cryopyrin-Associated Periodic Syndromes (CAPS), in adults and children 4 years of age and older including:
-
Tumor Necrosis Factor Receptor Associated Periodic Syndrome (TRAPS) in adult and pediatric patients. (1.1)
-
Hyperimmunoglobulin D Syndrome (HIDS)/Mevalonate Kinase Deficiency (MKD) in adult and pediatric patients. (1.1)
- Familial Mediterranean Fever (FMF) in adult and pediatric patients. (1.1)
Active Systemic Juvenile Idiopathic Arthritis (SJIA) in patients aged 2 years and older (1.2)
DOSAGE AND ADMINISTRATION
- Administer by subcutaneous injection (2.1)
CAPS
150 mg for CAPS patients with body weight greater than 40 kg and 2 mg/kg for CAPS patients with body weight greater than or equal to 15 kg and less than or equal to 40 kg. For children 15 to 40 kg with an inadequate response, the dose can be increased to 3 mg/kg. Administer subcutaneously every 8 weeks. (2.2)
TRAPS, HIDS/ MKD, and FMF
-
Body weight less than or equal to 40 kg
- The recommended starting dose is 2 mg/kg every 4 weeks. The dose can be increased to 4 mg/kg every 4 weeks if the clinical response is not adequate. (2.3)
-
Body weight greater than 40 kg
- The recommended starting dose is 150 mg every 4 weeks. The dose can be increased to 300 mg every 4 weeks if the clinical response is not adequate. (2.3)
SJIA
4 mg/kg (with a maximum of 300 mg) for patients with a body weight greater than or equal to 7.5 kg. Administer subcutaneously every 4 weeks. (2.4)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
Confirmed hypersensitivity to the active substance or to any of the excipients. (4)
WARNINGS AND PRECAUTIONS
- Interleukin-1 blockade may interfere with immune response to infections. Treatment with medications that work through inhibition of IL-1 has been associated with an increased risk of serious infections. ILARIS has been associated with an increased incidence of serious infections. Physicians should exercise caution when administering ILARIS to patients with infections, a history of recurring infections or underlying conditions which may predispose them to infections. Discontinue treatment with ILARIS if a patient develops a serious infection. Do not administer ILARIS to patients during an active infection requiring medical intervention. (5.1)
- Live vaccines should not be given concurrently with ILARIS. Prior to initiation of therapy with ILARIS, patients should receive all recommended vaccinations. (5.4)
ADVERSE REACTIONS
CAPS: The most common adverse reactions greater than 10% reported by patients treated with ILARIS are nasopharyngitis, diarrhea, influenza, rhinitis, nausea, headache, bronchitis, gastroenteritis, pharyngitis, weight increased, musculoskeletal pain, and vertigo. (6)
TRAPS, HIDS/MKD, and FMF: The most common adverse reactions greater than 10% reported by patients treated with ILARIS are injection-site reactions and nasopharyngitis. (6)
SJIA: The most common adverse drug reactions greater than or equal to 10% reported by patients treated with ILARIS are infections (nasopharyngitis and upper respiratory tract infections), abdominal pain and injection-site reactions. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatchDRUG INTERACTIONS
No formal drug interaction studies have been conducted with ILARIS. (7)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2016
-
Cryopyrin-Associated Periodic Syndromes (CAPS), in adults and children 4 years of age and older including:
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Periodic Fever Syndromes
1.2 Systemic Juvenile Idiopathic Arthritis (SJIA)
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
2.2 Cryopyrin-Associated Periodic Syndromes (CAPS)
2.3 Tumor Necrosis Factor Receptor Associated Periodic Syndrome (TRAPS), Hyperimmunoglobulin D Syndrome/Mevalonate Kinase Deficiency (HIDS/MKD), and Familial Mediterranean Fever (FMF)
2.4 Systemic Juvenile Idiopathic Arthritis (SJIA)
2.5 Preparation and Administration of ILARIS Lyophilized Powder
2.6 Administration of ILARIS Solution
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
5.2 Immunosuppression
5.3 Hypersensitivity
5.4 Immunizations
5.5 Macrophage Activation Syndrome
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Hypersensitivity
6.3 Immunogenicity
6.4 Laboratory Findings
7 DRUG INTERACTIONS
7.1 TNF-Blocker and IL-1 Blocking Agent
7.2 Immunization
7.3 Cytochrome P450 Substrates
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Patients with Renal Impairment
8.7 Patients with Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Treatment of CAPS
14.2 Treatment of Periodic Fever Syndromes: TRAPS, HIDS/MKD, and FMF
14.3 Treatment of SJIA
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1
INDICATIONS AND USAGE
1.1 Periodic Fever Syndromes
ILARIS® (canakinumab) is an interleukin-1β (IL-1 β) blocker indicated for the treatment of the following autoinflammatory Periodic Fever Syndromes:
Cryopyrin-Associated Periodic Syndromes (CAPS)
ILARIS is indicated for the treatment of Cryopyrin-Associated Periodic Syndromes (CAPS), in adults and children 4 years of age and older including:
- Familial Cold Autoinflammatory Syndrome (FCAS)
- Muckle-Wells Syndrome (MWS)
Tumor Necrosis Factor Receptor (TNF) Associated Periodic Syndrome (TRAPS)
ILARIS is indicated for the treatment of Tumor Necrosis Factor (TNF) receptor Associated Periodic Syndrome (TRAPS) in adult and pediatric patients.
Hyperimmunoglobulin D Syndrome (HIDS)/Mevalonate Kinase Deficiency (MKD)
ILARIS is indicated for the treatment of Hyperimmunoglobulin D (Hyper-IgD) Syndrome (HIDS)/Mevalonate Kinase Deficiency (MKD) in adult and pediatric patients.
Familial Mediterranean Fever (FMF)
ILARIS is indicated for the treatment of Familial Mediterranean Fever (FMF) in adult and pediatric patients.
-
2
DOSAGE AND ADMINISTRATION
2.2 Cryopyrin-Associated Periodic Syndromes (CAPS)
The recommended dose of ILARIS is 150 mg for CAPS patients with body weight greater than 40 kg. For CAPS patients with body weight greater than or equal to 15 kg and less than or equal to 40 kg, the recommended dose is 2 mg/kg. For children 15 to 40 kg with an inadequate response, the dose can be increased to 3 mg/kg.
ILARIS is administered every eight weeks.
2.3 Tumor Necrosis Factor Receptor Associated Periodic Syndrome (TRAPS), Hyperimmunoglobulin D Syndrome/Mevalonate Kinase Deficiency (HIDS/MKD), and Familial Mediterranean Fever (FMF)
The recommended dose of ILARIS for TRAPS, HIDS/MKD, and FMF patients is based on body weight.
For patients with body weight less than or equal to 40 kg, the recommended dose is 2 mg/kg administered every 4 weeks. The dose can be increased to 4 mg/kg every 4 weeks if the clinical response is not adequate.
For patients with body weight greater than 40 kg, the recommended dose is 150 mg administered every 4 weeks. The dose can be increased to 300 mg every 4 weeks if the clinical response is not adequate.
2.4 Systemic Juvenile Idiopathic Arthritis (SJIA)
The recommended dose of ILARIS for SJIA patients with a body weight greater than or equal to 7.5 kg is 4 mg/kg (with a maximum of 300 mg) administered every 4 weeks.
2.5 Preparation and Administration of ILARIS Lyophilized Powder
STEP 1: Using aseptic technique, reconstitute each vial of ILARIS lyophilized powder by slowly injecting 1 mL of Sterile Water for Injection with a 1-mL syringe and an 18-gauge x 2” needle.
STEP 2: Swirl the vial slowly at an angle of about 45° for approximately 1 minute and allow to stand for 5 minutes. Do not shake. Then gently turn the vial upside down and back again ten times. Avoid touching the rubber stopper with your fingers.
STEP 3: Allow to stand for about 15 minutes at room temperature. The reconstituted solution has a final concentration of 150 mg/mL. Do not shake. Do not use if particulate matter is present in the solution. Tap the side of the vial to remove any residual liquid from the stopper. The reconstituted solution should be clear to opalescent, colorless to a slightly brownish yellow tint, and essentially free from particulates. If the solution has a distinctly brown discoloration, do not use. Slight foaming of the product upon reconstitution is not unusual.
After reconstitution, ILARIS should be kept from light, and can be kept at room temperature if used within 60 minutes of reconstitution. Otherwise, it should be refrigerated at 2°C to 8°C (36°F to 46°F) and used within 4 hours of reconstitution.
STEP 4: Using a sterile 1-mL syringe and needle, carefully withdraw the required volume depending on the dose to be administered and subcutaneously inject using a 27-gauge x 0.5” needle.
Injection into scar tissue should be avoided as this may result in insufficient exposure to ILARIS.
Discard any unused product or waste material in accordance with local requirements.
2.6 Administration of ILARIS Solution
STEP 1: ILARIS solution has a concentration of 150 mg/mL. Do not shake. The solution should be essentially free from particulates, clear to opalescent, colorless to slightly brownish-yellow tint. If the solution has a distinctly brown discoloration, is highly opalescent or contains visible particles, do not use.
STEP 2: Using a sterile 1-mL syringe and 18-gauge x 2” needle, carefully withdraw the required volume depending on the dose to be administered and subcutaneously inject using a 27-gauge x 0.5” needle.
Injection into scar tissue should be avoided as this may result in insufficient exposure to ILARIS.
Discard unused product or waste material in accordance with the local requirements.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5
WARNINGS AND PRECAUTIONS
5.1 Serious Infections
ILARIS has been associated with an increased risk of serious infections. Physicians should exercise caution when administering ILARIS to patients with infections, a history of recurring infections or underlying conditions which may predispose them to infections. ILARIS should not be administered to patients during an active infection requiring medical intervention. Administration of ILARIS should be discontinued if a patient develops a serious infection.
Infections, predominantly of the upper respiratory tract, in some instances serious, have been reported with ILARIS. Generally, the observed infections responded to standard therapy. Isolated cases of unusual or opportunistic infections (e.g., aspergillosis, atypical mycobacterial infections, cytomegalovirus, herpes zoster) were reported during ILARIS treatment. A causal relationship of ILARIS to these events cannot be excluded. In clinical trials, ILARIS has not been administered concomitantly with tumor necrosis factor (TNF) inhibitors. An increased incidence of serious infections has been associated with administration of another IL-1 blocker in combination with TNF inhibitors. Coadministration of ILARIS with TNF inhibitors is not recommended because this may increase the risk of serious infections [see Drug Interactions (7.1)].
Drugs that affect the immune system by blocking TNF have been associated with an increased risk of new tuberculosis and reactivation of latent tuberculosis (TB). It is possible that use of IL-1 inhibitors such as ILARIS increases the risk of reactivation of tuberculosis or of opportunistic infections.
Prior to initiating immunomodulatory therapies, including ILARIS, patients should be evaluated for active and latent tuberculosis infection. Appropriate screening tests should be performed in all patients. ILARIS has not been studied in patients with a positive tuberculosis screen, and the safety of ILARIS in individuals with latent tuberculosis infection is unknown. Patients testing positive in tuberculosis screening should be treated according to standard medical practice prior to therapy with ILARIS. All patients should be instructed to seek medical advice if signs, symptoms, or high risk exposure suggestive of tuberculosis (e.g., persistent cough, weight loss, subfebrile temperature) appear during or after ILARIS therapy.
Healthcare providers should follow current CDC guidelines both to evaluate for and to treat possible latent tuberculosis infections before initiating therapy with ILARIS.
5.2 Immunosuppression
The impact of treatment with anti-interleukin-1 (IL-1) therapy on the development of malignancies is not known. However, treatment with immunosuppressants, including ILARIS, may result in an increase in the risk of malignancies.
5.3 Hypersensitivity
Hypersensitivity reactions have been reported with ILARIS therapy. During clinical trials, no anaphylactic reactions have been reported. It should be recognized that symptoms of the underlying disease being treated may be similar to symptoms of hypersensitivity. ILARIS should not be administered to any patients with known clinical hypersensitivity to ILARIS [see Contraindications (4) and Adverse Reactions (6.2)].
5.4 Immunizations
Live vaccines should not be given concurrently with ILARIS [see Drug Interactions (7.2)]. Since no data are available on either the efficacy or on the risks of secondary transmission of infection by live vaccines in patients receiving ILARIS, live vaccines should not be given concurrently with ILARIS. In addition, because ILARIS may interfere with normal immune response to new antigens, vaccinations may not be effective in patients receiving ILARIS. Limited data are available on the response to vaccinations with inactivated (killed) antigens in patients receiving ILARIS [see Drug Interactions (7.2)].
Because IL-1 blockade may interfere with immune response to infections, it is recommended that prior to initiation of therapy with ILARIS, adult and pediatric patients receive all recommended vaccinations, as appropriate and if feasible, including pneumococcal vaccine and inactivated influenza vaccine. (See current recommended immunization schedules at the website of the Centers for Disease Control, http://www.cdc.gov/vaccines/schedules/index.html).
5.5 Macrophage Activation Syndrome
Macrophage activation syndrome (MAS) is a known, life-threatening disorder that may develop in patients with rheumatic conditions, in particular SJIA, and should be aggressively treated. Physicians should be attentive to symptoms of infection or worsening of SJIA, as these are known triggers for MAS. Eleven cases of MAS were observed in 201 SJIA patients treated with canakinumab in clinical trials. Based on the clinical trial experience, ILARIS does not appear to increase the incidence of MAS in SJIA patients, but no definitive conclusion can be made.
-
6
ADVERSE REACTIONS
Approximately 570 patients have been treated with ILARIS in interventional trials in CAPS, TRAPS, HIDS/MKD, FMF or SJIA. These clinical trials included approximately 350 children up to 17 years of age. The most frequently reported adverse drug reactions were infections predominantly of the upper respiratory tract. The majority of the events were mild to moderate although serious infections were observed.
Opportunistic infections have also been reported in patients treated with ILARIS [see Warnings and Precautions (5.1)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Treatment of Periodic Fever Syndromes: CAPS, TRAPS, HIDS/MKD, and FMF
Treatment of CAPS
The data described herein reflect exposure to ILARIS in 104 adult and pediatric CAPS patients, including 20 FCAS, 72 MWS, 10 MWS/NOMID (Neonatal Onset Multisystem Inflammatory Disorder) overlap, 1 non-FCAS non-MWS, and 1 misdiagnosed in placebo-controlled (35 patients) and uncontrolled trials. Sixty-two patients were exposed to ILARIS for at least 6 months, 56 for at least 1 year, and 4 for at least 3 years. A total of 9 serious adverse reactions were reported for CAPS patients. Among these were vertigo (2 patients), infections (3 patients), including intra-abdominal abscess following appendectomy (1 patient). The most commonly reported adverse reactions associated with ILARIS treatment in greater than 10% of the CAPS patients were nasopharyngitis, diarrhea, influenza, rhinitis, nausea, headache, bronchitis, gastroenteritis, pharyngitis, weight increased, musculoskeletal pain, and vertigo. One patient discontinued treatment due to potential infection.
CAPS Study 1 investigated the safety of ILARIS in an 8-week, open-label period (Part 1), followed by a 24-week, randomized withdrawal period (Part 2), followed by a 16-week, open-label period (Part 3). All patients were treated with ILARIS 150 mg subcutaneously or 2 mg/kg if body weight was greater than or equal to 15 kg and less than or equal to 40 kg (see Table 1).
Since all CAPS patients received ILARIS in Part 1, there are no controlled data on adverse events (AEs). Data in Table 1 are for all AEs for all CAPS patients receiving canakinumab. In CAPS Study 1, no pattern was observed for any type or frequency of adverse events throughout the three study periods.
Table 1: Number (%) of Patients with AEs by Preferred Terms, in Greater Than 10% of Patients in Parts 1 to 3 of the Phase 3 Trial for CAPS Patients
Preferred TermILARIS
N=35
n (%)n % of Patients with Adverse Events 35 (100) Nasopharyngitis 12 (34) Diarrhea 7 (20) Influenza 6 (17) Rhinitis 6 (17) Nausea 5 (14) Headache 5 (14) Bronchitis 4 (11) Gastroenteritis 4 (11) Pharyngitis 4 (11) Weight increased 4 (11) Musculoskeletal pain 4 (11) Vertigo 4 (11) Vertigo
Vertigo has been reported in 9% to 14% of patients in CAPS studies, exclusively in MWS patients, and reported as a serious adverse event in two cases. All events resolved with continued treatment with ILARIS.
Injection-Site Reactions
In CAPS Study 1, subcutaneous injection-site reactions were observed in 9% of patients in Part 1 with mild tolerability reactions; in Part 2, one patient each (7%) had a mild or a moderate tolerability reaction and, in Part 3, one patient had a mild local tolerability reaction. No severe injection-site reactions were reported and none led to discontinuation of treatment.
Treatment of TRAPS, HIDS/MKD, and FMF
A Phase III trial (TRAPS, HIDS/MKD, and FMF Study 1) investigated the safety of ILARIS in 3 cohorts (TRAPS, HIDS/MKD, and FMF) as follows: a 12-week screening period (Part 1), followed by a 16-week, randomized, double-blind, placebo-controlled parallel-arm treatment period (Part 2), followed by a 24-week randomized withdrawal period (Part 3), followed by a 72-week, open-label treatment period (Part 4). All patients randomized to treatment with ILARIS in Part 2 received 150 mg subcutaneously every 4 weeks if body weight was greater than 40 kg (or 2 mg/kg every 4 weeks if body weight was less than or equal to 40 kg).
In Part 2 of the TRAPS, HIDS/MKD, and FMF Study 1, initially 90 patients were randomized to ILARIS treatment and 91 patients were randomized to placebo. Of patients randomized to ILARIS, 55.6% remained on the initial dose through week 16 with 6.7% receiving an additional ILARIS dose between Day 7 and Day 15. Of the patients randomized to placebo, 9.9% remained on placebo through Week 16 with 28.6% switching to active treatment with ILARIS by Day 15.
Overall, there were 43 TRAPS, 68 HIDS/MKD, and 58 FMF patients in the safety set with a cumulative canakinumab exposure of 47.61 patient-years. The cumulative exposure in the placebo group was 8.03 patient-years.
In Part 2 of the TRAPS, HIDS/MKD, and FMF Study 1, a total of 22 TRAPS patients aged 3 to 76 years of age, 37 HIDS/MKD patients aged 2 to 43 years of age, and 31 FMF patients aged 2 to 60 years of age were initially randomized to treatment with ILARIS 150 mg every four weeks in the placebo-controlled period of the clinical trial. In addition, 4 non-randomized patients (2 FMF patients of age 20 and 29 years with non-exon 10 mutations and 2 HIDS/MKD patients both of 1 year of age) received open-label treatment in Part 2.
The most commonly reported adverse reactions (greater than or equal to 10%) associated with ILARIS treatment in TRAPS, HIDS/MKD, and FMF patients were injection-site reactions and nasopharyngitis. The reported adverse reactions (greater than or equal to 3%) associated with ILARIS treatment in TRAPS, HIDS/MKD, and FMF patients were injection-site reactions (10.1%), and infections including nasopharyngitis (10.7%), upper respiratory tract infection (7.1%), rhinitis (5.3%), gastroenteritis (3.0%), and pharyngitis (3.0%). Serious infections (e.g., conjunctivitis, pneumonia, pharyngitis, pharyngotonsillitis) were observed in approximately 2.4% (0.03 per 100 patient-days) of patients receiving ILARIS in Part 2 of the TRAPS, HIDS/MKD, and FMF Study 1.
In the ILARIS treatment group, 1 TRAPS patient discontinued treatment due to adverse events, 2 HIDS/MKD patients discontinued treatment due to adverse events, and no FMF patients discontinued treatment due to an adverse event.
Injection-Site Reactions
In the TRAPS, HIDS/MKD, and FMF Study 1, subcutaneous injection-site reactions were observed in 10.1% of patients in Part 2 who had a mild or a moderate tolerability reaction. No severe injection-site reactions were reported and none led to discontinuation of treatment.
Treatment of SJIA
A total of 201 SJIA patients aged 2 to less than 20 years have received ILARIS in clinical trials. The safety of ILARIS compared to placebo was investigated in two phase 3 studies [see Clinical Studies (14.2)]. Patients in SJIA Study 1 received a single dose of ILARIS 4 mg/kg (n=43) or placebo (n=41) via subcutaneous injection and were assessed at Day 15 for the efficacy endpoints and had a safety analysis up to Day 29. SJIA Study 2 was a two-part study with an open-label, single-arm active treatment period (Part I) followed by a randomized, double-blind, placebo-controlled, event-driven withdrawal design (Part II). Overall, 177 patients were enrolled into the study and received ILARIS 4 mg/kg (up to 300 mg maximum) in Part I, and 100 patients received ILARIS 4 mg/kg (up to 300 mg maximum) every 4 weeks or placebo in Part II. Adverse drug reactions listed in Table 2 showed higher rates than placebo from both trials. The adverse drug reactions associated with ILARIS treatment in greater than 10% of SJIA patients were infections, abdominal pain, and injection-site reactions. Serious infections (e.g., pneumonia, varicella, gastroenteritis, measles, sepsis, otitis media, sinusitis, adenovirus, lymph node abscess, pharyngitis) were observed in approximately 4% to 5% (0.02 to 0.17 per 100 patient-days) of patients receiving ILARIS in both studies.
Adverse reactions are listed according to MedDRA version 15.0 system organ class.
Table 2: Tabulated Summary of Adverse Drug Reactions from Pivotal SJIA Clinical Trials SJIA Study 2 SJIA Study 1 Part I Part II ILARIS
N=177
n (%)
(IR)^ILARIS
N=50
n (%)
(IR)Placebo
N=50
n (%)
(IR)ILARIS
N=43
n (%)
(IR)Placebo
N=41
n (%)
(IR)Infections and infestations All Infections (e.g., nasopharyngitis, (viral) upper respiratory tract infection, pneumonia, rhinitis, pharyngitis, tonsillitis, sinusitis, urinary tract infection, gastroenteritis, viral infection) 97 (54.8%)
(0.91)27 (54%)
(0.59)19 (38%)
(0.63)13 (30.2%)
(1.26)5 (12.2%)
(1.37)Gastrointestinal disorders Abdominal pain (upper) 25 (14.1%)
(0.16)8 (16%)
(0.15)6 (12%)
(0.08)3 (7%)
(0.25)1 (2.4%)
(0.23)Skin and subcutaneous tissue disorders Injection-site reaction* mild 19 (10.7%) 6 (12.0%) 2 (4.0%) 0 3 (7.3%) moderate 2 (1.1%) 1 (2.0%) 0 0 0 n= number of patients
^IR=Exposure adjusted incidence rate per 100 patient-days
*No injection-site reaction led to study discontinuation
6.2 Hypersensitivity
During clinical trials, no anaphylactic reactions have been reported. In CAPS trials one patient discontinued and in TRAPS, HIDS/MKD, FMF, and SJIA trials no patients discontinued due to hypersensitivity reactions. ILARIS should not be administered to any patients with known clinical hypersensitivity to ILARIS [see Contraindications (4) and Warnings and Precautions (5.3)].
6.3 Immunogenicity
A biosensor binding assay or a bridging immunoassay was used to detect antibodies directed against canakinumab in patients who received ILARIS. Antibodies against ILARIS were observed in approximately 1.5% and 3.1% of the patients treated with ILARIS for CAPS and SJIA, respectively. No neutralizing antibodies were detected. No apparent correlation of antibody development to clinical response or adverse events was observed. The CAPS clinical studies employed the biosensor binding assay, and most of the SJIA clinical studies employed the bridging assay. The data obtained in an assay are highly dependent on several factors including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications, underlying disease, and the number of patients tested. For these reasons, comparison of the incidence of antibodies to canakinumab between the CAPS and SJIA clinical studies or with the incidence of antibodies to other products may be misleading.
No TRAPS, HIDS/MKD and FMF patients treated with ILARIS doses of 150 mg and 300 mg over 16 weeks of treatment tested positive for anti-canakinumab antibodies.
6.4 Laboratory Findings
Hematology
TRAPS, HIDS/MKD, and FMF
Overall, in the TRAPS, HIDS/MKD, and FMF Study 1, neutrophil count decreased (greater than or equal to Grade 2) was reported in 6.5% of patients and platelet count decreased (greater than or equal to Grade 2) was reported in 0.6% of patients.
SJIA
During clinical trials with ILARIS, mean values decreased for white blood cells, neutrophils and platelets.
In the randomized, placebo-controlled portion of SJIA Study 2, decreased white blood cell counts (WBC) less than or equal to 0.8 times lower limit of normal (LLN) were reported in 5 patients (10.4%) in the ILARIS group compared to 2 (4.0%) in the placebo group. Transient decreases in absolute neutrophil count (ANC) to less than 1x109/L were reported in 3 patients (6.0%) in the ILARIS group compared to 1 patient (2.0%) in the placebo group. One case of ANC less than 0.5x109/L was observed in the ILARIS group and none in the placebo group.
Mild (less than LLN and greater than 75x109/L) and transient decreases in platelet counts were observed in 3 (6.3%) ILARIS-treated patients versus 1 (2.0%) placebo-treated patient.
Hepatic Transaminases
Elevations of transaminases have been observed in patients treated with ILARIS.
In the randomized, placebo-controlled portion of SJIA Study 2, high ALT and/or AST greater than or equal to 3 times upper limit of normal (ULN) were reported in 2 (4.1%) ILARIS-treated patients and 1 (2.0%) placebo patient. All patients had normal values at the next visit.
Bilirubin
Asymptomatic and mild elevations of serum bilirubin have been observed in patients treated with ILARIS without concomitant elevations of transaminases.
-
7
DRUG INTERACTIONS
Interactions between ILARIS and other medicinal products have not been investigated in formal studies.
7.1 TNF-Blocker and IL-1 Blocking Agent
An increased incidence of serious infections and an increased risk of neutropenia have been associated with administration of another IL-1 blocker in combination with TNF inhibitors in another patient population. Use of ILARIS with TNF inhibitors may also result in similar toxicities and is not recommended because this may increase the risk of serious infections [see Warnings and Precautions (5.1)].
The concomitant administration of ILARIS with other drugs that block IL-1 has not been studied. Based upon the potential for pharmacological interactions between ILARIS and a recombinant IL-1ra, concomitant administration of ILARIS and other agents that block IL-1 or its receptors is not recommended.
7.2 Immunization
No data are available on either the effects of live vaccination or the secondary transmission of infection by live vaccines in patients receiving ILARIS. Therefore, live vaccines should not be given concurrently with ILARIS. It is recommended that, if possible, pediatric and adult patients should complete all immunizations in accordance with current immunization guidelines prior to initiating ILARIS therapy [see Warnings and Precautions (5.4)].
7.3 Cytochrome P450 Substrates
The formation of CYP450 enzymes is suppressed by increased levels of cytokines (e.g., IL-1) during chronic inflammation. Thus it is expected that for a molecule that binds to IL-1, such as canakinumab, the formation of CYP450 enzymes could be normalized. This is clinically relevant for CYP450 substrates with a narrow therapeutic index, where the dose is individually adjusted (e.g., warfarin). Upon initiation of canakinumab, in patients being treated with these types of medicinal products, therapeutic monitoring of the effect or drug concentration should be performed and the individual dose of the medicinal product may need to be adjusted as needed.
-
8
USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The limited human data from postmarketing reports on use of ILARIS in pregnant women are not sufficient to inform a drug-associated risk. Monoclonal antibodies, such as canakinumab, are transported across the placenta in a linear fashion as pregnancy progresses; therefore, potential fetal exposure is likely to be greater during the second and third trimesters of pregnancy. In animal embryo-fetal development studies with marmoset monkeys, there was no evidence of embryotoxicity or fetal malformations with subcutaneous administration of canakinumab during the period of organogenesis and later in gestation at doses that produced exposures approximately 11 times the exposure at the maximum recommended human dose (MRHD) and greater. Delays in fetal skeletal development were observed in marmoset monkeys following prenatal exposure to ILARIS at concentrations approximately 11 times the MRHD and greater. Similar delays in fetal skeletal development were observed in mice administered a murine analog of ILARIS during the period of organogenesis. Delays in skeletal ossification are changes from the expected ossification state in an otherwise normal structure/bone: these findings are generally reversible or transitory and not detrimental to postnatal survival [see Animal Data].
The estimated background risk of major birth defects and miscarriage for the indicated population(s) are unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In embryo-fetal development studies, pregnant marmoset monkeys received canakinumab from gestation days 25 to 140 at doses that produced exposures approximately 11 times that achieved with MRHD and greater (on a plasma area under the curve (AUC) basis with maternal subcutaneous doses of 15, 50, or 150 mg/kg twice weekly). ILARIS did not elicit any evidence of embryotoxicity or fetal malformations. There were increases in the incidence of incomplete ossification of the terminal caudal vertebra and misaligned and/or bipartite vertebra in fetuses at all dose levels when compared to concurrent controls suggestive of delay in skeletal development in the marmoset. Since ILARIS does not cross-react with mouse or rat IL-1β, pregnant mice were subcutaneously administered a murine analog of ILARIS at doses of 15, 50, or 150 mg/kg during the period of organogenesis on gestation days 6, 11, and 17. The incidence of incomplete ossification of the parietal and frontal skull bones of fetuses was increased in a dose-dependent manner at all dose levels tested.
8.2 Lactation
Risk Summary
There is no information regarding the presence of canakinumab in human milk, the effects on the breastfed infant, or the effects on milk production. Human IgG is known to be present in human milk. The effects of canakinumab in breast milk and possible systemic exposure in the breastfed infant are unknown. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ILARIS and any potential adverse effects on the breastfed infant from canakinumab or from the underlying maternal condition.
8.4 Pediatric Use
The CAPS trials with ILARIS included a total of 23 pediatric patients with an age range from 4 years to 17 years (11 adolescents were treated subcutaneously with 150 mg, and 12 children were treated with 2 mg/kg based on body weight greater than or equal to 15 kg and less than or equal to 40 kg). The majority of patients achieved improvement in clinical symptoms and objective markers of inflammation (e.g., Serum Amyloid A and C-Reactive Protein). Overall, the efficacy and safety of ILARIS in pediatric and adult patients were comparable. Infections of the upper respiratory tract were the most frequently reported infection. The safety and effectiveness of ILARIS in CAPS patients under 4 years of age has not been established [see Clinical Pharmacology (12.3)].
The safety and efficacy of ILARIS in SJIA patients under 2 years of age have not been established [see Clinical Pharmacology (12.3)].
The TRAPS, HIDS/MKD, and FMF trial included a total of 102 pediatric patients (TRAPS, HIDS/MKD and FMF patients) with an age range from 2 to 17 years who received ILARIS. Overall, there were no clinically meaningful differences in the efficacy, safety and tolerability profile of ILARIS in pediatric patients compared to the overall TRAPS, HIDS/MKD, and FMF populations (comprised of adult and pediatric patients, N=169). The majority of pediatric patients achieved improvement in clinical symptoms and objective markers of inflammation.
8.5 Geriatric Use
Clinical studies of ILARIS did not include sufficient numbers of subjects aged 65 years and older to determine whether they respond differently from younger subjects.
- 10 OVERDOSAGE
-
11
DESCRIPTION
Canakinumab is a recombinant, human anti-human-IL-1β monoclonal antibody that belongs to the IgG1/κ isotype subclass. It is expressed in a murine Sp2/0-Ag14 cell line and comprised of two 447- (or 448-) residue heavy chains and two 214-residue light chains, with a molecular mass of 145157 Daltons when deglycosylated. Both heavy chains of canakinumab contain oligosaccharide chains linked to the protein backbone at asparagine 298 (Asn 298).
The biological activity of canakinumab is measured by comparing its inhibition of IL-1β-dependent expression of the reporter gene luciferase to that of a canakinumab internal reference standard, using a stably transfected cell line.
ILARIS for Injection
ILARIS (canakinumab) for Injection is supplied as a white, preservative-free, lyophilized powder in a sterile, single-dose, colorless, glass vial with coated stopper and aluminum flip-off cap. Reconstitution with 1 mL of Sterile Water for Injection is required prior to subcutaneous administration of the drug. The reconstituted canakinumab is a 150 mg/mL solution essentially free of particulates, clear to opalescent, and is colorless or may have a slightly brownish-yellow tint. A volume of up to 1 mL can be withdrawn for delivery of 150 mg canakinumab, L-histidine (2.8 mg), L-histidine HCl monohydrate (1.7 mg), polysorbate 80 (0.6 mg), sucrose (92.4 mg), and Sterile Water for Injection.
ILARIS Injection
ILARIS (canakinumab) Injection is supplied as a sterile, preservative-free, clear to opalescent, colorless to slightly brownish-yellow solution for subcutaneous injection in a single-dose, glass vial with coated stopper and aluminum flip-off cap. Each vial delivers 1 mL containing 150 mg canakinumab, L-histidine (2.1 mg), L-histidine HCl monohydrate (1.3 mg), mannitol (49.2 mg), polysorbate 80 (0.4 mg), and Sterile Water for Injection.
-
12
CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Canakinumab is a human monoclonal anti-human IL-1β antibody of the IgG1/κ isotype. Canakinumab binds to human IL-1β and neutralizes its activity by blocking its interaction with IL-1 receptors, but it does not bind IL-1α or IL-1 receptor antagonist (IL-1ra).
CAPS refer to rare genetic syndromes generally caused by mutations in the NLRP-3 [nucleotide-binding domain, leucine rich family (NLR), pyrin domain containing 3] gene (also known as Cold-Induced Autoinflammatory Syndrome-1 [CIAS1]). CAPS disorders are inherited in an autosomal dominant pattern with male and female offspring equally affected. Features common to all disorders include fever, urticaria-like rash, arthralgia, myalgia, fatigue, and conjunctivitis.
The NLRP-3 gene encodes the protein cryopyrin, an important component of the inflammasome. Cryopyrin regulates the protease caspase-1 and controls the activation of IL-1β. Mutations in NLRP-3 result in an overactive inflammasome resulting in excessive release of activated IL-1β that drives inflammation. SJIA is a severe autoinflammatory disease, driven by innate immunity by means of proinflammatory cytokines such as IL-1β.
12.2 Pharmacodynamics
C-reactive protein and Serum Amyloid A (SAA) are indicators of inflammatory disease activity that are elevated in patients with CAPS. Elevated SAA has been associated with the development of systemic amyloidosis in patients with CAPS. Following ILARIS treatment, CRP and SAA levels normalize within 8 days. In SJIA the median percent reduction in CRP from baseline to Day 15 was 91%. Improvement in pharmacodynamic markers may not be representative of clinical response.
12.3 Pharmacokinetics
Absorption
The peak serum canakinumab concentration (Cmax) of 16 ± 3.5 mcg/mL occurred approximately 7 days after subcutaneous administration of a single, 150 mg dose subcutaneously to adult CAPS patients. The mean terminal half-life was 26 days. The absolute bioavailability of subcutaneous canakinumab was estimated to be 66%. Exposure parameters (such as AUC and Cmax) increased in proportion to dose over the dose range of 0.30 to 10 mg/kg given as intravenous infusion or from 150 to 300 mg as subcutaneous injection.
Distribution
Canakinumab binds to serum IL-1β. Canakinumab volume of distribution (Vss) varied according to body weight and was estimated to be 6.01 liters in a typical CAPS patient weighing 70 kg, 3.2 liters in a SJIA patient weighing 33 kg, and 6.34 liters for a Periodic Fever Syndrome (TRAPS, HIDS/MKD, FMF) patient weighing 70 kg. The expected accumulation ratio was 1.3-fold for CAPS patients and 1.6-fold for SJIA patients following 6 months of subcutaneous dosing of 150 mg ILARIS every 8 weeks and 4 mg/kg every 4 weeks, respectively.
Elimination
Clearance (CL) of canakinumab varied according to body weight and was estimated to be 0.174 L/day in a typical CAPS patient weighing 70 kg, 0.11 L/day in a SJIA patient weighing 33 kg, and 0.17 L/day in a Periodic Fever Syndrome (TRAPS, HIDS/MKD, FMF) patient weighing 70 kg. There was no indication of accelerated clearance or time-dependent change in the pharmacokinetic properties of canakinumab following repeated administration. No gender- or age-related pharmacokinetic differences were observed after correction for body weight.
Pediatrics
Pharmacokinetic properties are similar in Periodic Fever Syndromes (CAPS, TRAPS, HIDS/MKD, FMF) and SJIA pediatric populations. In patients less than 2 years of age (n=7), the exposure of canakinumab were comparable to older age groups with the same weight based dose.
In CAPS patients, peak concentrations of canakinumab occurred between 2 to 7 days following single subcutaneous administration of ILARIS 150 mg or 2 mg/kg in pediatric patients. The terminal half-life ranged from 22.9 to 25.7 days, similar to the pharmacokinetic properties observed in adults.
In SJIA, exposure parameters (such as AUC and Cmax) were comparable across age groups from 2 years of age and above following subcutaneous administration of canakinumab 4 mg/kg every 4 weeks.
In TRAPS, HIDS/MKD, and FMF exposure parameter trough concentrations were comparable across age groups from 2 to less than 20 years following subcutaneous administration of canakinumab 2 mg/kg every 4 weeks.
-
13
NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies have not been performed to evaluate the carcinogenic potential of canakinumab.
As canakinumab does not cross-react with rodent IL-1β, male and female fertility was evaluated in a mouse model using a murine analog of canakinumab. Male mice were treated weekly beginning 4 weeks prior to mating and continuing through 3 weeks after mating. Female mice were treated weekly for 2 weeks prior to mating through gestation day 3 or 4. The murine analog of canakinumab did not alter either male or female fertility parameters at subcutaneous doses up to 150 mg/kg.
-
14
CLINICAL STUDIES
14.1 Treatment of CAPS
The efficacy and safety of ILARIS for the treatment of CAPS was demonstrated in CAPS Study 1, a 3-part trial in patients 9 to 74 years of age with the MWS phenotype of CAPS. Throughout the trial, patients weighing more than 40 kg received ILARIS 150 mg and patients weighing 15 to 40 kg received 2 mg/kg. Part 1 was an 8-week open-label, single-dose period where all patients received ILARIS. Patients who achieved a complete clinical response and did not relapse by Week 8 were randomized into Part 2, a 24-week randomized, double-blind, placebo-controlled withdrawal period. Patients who completed Part 2 or experienced a disease flare entered Part 3, a 16-week open-label active treatment phase. A complete response was defined as ratings of minimal or better for physician’s assessment of disease activity (PHY) and assessment of skin disease (SKD) and had serum levels of C-Reactive Protein (CRP) and Serum Amyloid A (SAA) less than 10 mg/L. A disease flare was defined as a CRP and/or SAA values greater than 30 mg/L and either a score of mild or worse for PHY or a score of minimal or worse for PHY and SKD.
In Part 1, a complete clinical response was observed in 71% of patients one week following initiation of treatment and in 97% of patients by Week 8 (see Table 3 and Figure 1). In the randomized withdrawal period, a total of 81% of the patients randomized to placebo flared as compared to none (0%) of the patients randomized to ILARIS. The 95% confidence interval for treatment difference in the proportion of flares was 53% to 96%. At the end of Part 2, all 15 patients treated with ILARIS had absent or minimal disease activity and skin disease (see Table 3).
In a second trial, patients 4 to 74 years of age with both MWS and FCAS phenotypes of CAPS were treated in an open-label manner. Treatment with ILARIS resulted in clinically significant improvement of signs and symptoms and in normalization of high CRP and SAA in a majority of patients within 1 week.
Table 3: Physician’s Global Assessment of Auto Inflammatory Disease Activity and Assessment of Skin Disease: Frequency Table and Treatment Comparison in Part 2 (Using LOCF, ITT Population) ILARIS
N=15Placebo
N=16
BaselineStart of Part 2 (Week 8) End of Part 2 Start of Part 2 (Week 8) End of Part 2 Physician's Global Assessment of Auto Inflammatory Disease Activity – n (%) Absent 0/31 (0) 9/15 (60) 8/15 (53) 8/16 (50) 0/16 (0) Minimal 1/31 (3) 4/15 (27) 7/15 (47) 8/16 (50) 4/16 (25) Mild 7/31 (23) 2/15 (13) 0/15 (0) 0/16 (0) 8/16 (50) Moderate 19/31 (61) 0/15 (0) 0/15 (0) 0/16 (0) 4/16 (25) Severe 4/31 (13) 0/15 (0) 0/15 (0) 0/16 (0) 0/16 (0) Assessment of Skin Disease – n (%) Absent 3/31 (10) 13/15 (87) 14/15 (93) 13/16 (81) 5/16 (31) Minimal 6/31 (19) 2/15 (13) 1/15 (7) 3/16 (19) 3/16 (19) Mild 9/31 (29) 0/15 (0) 0/15 (0) 0/16 (0) 5/16 (31) Moderate 12/31 (39) 0/15 (0) 0/15 (0) 0/16 (0) 3/16 (19) Severe 1/32 (3) 0/15 (0) 0/15 (0) 0/16 (0) 0/16 (0) Markers of inflammation CRP and SAA normalized within 8 days of treatment in the majority of patients. Normal mean CRP (Figure 1) and SAA values were sustained throughout CAPS Study 1 in patients continuously treated with canakinumab. After withdrawal of canakinumab in Part 2 CRP (Figure 1) and SAA values again returned to abnormal values and subsequently normalized after reintroduction of canakinumab in Part 3. The pattern of normalization of CRP and SAA was similar.
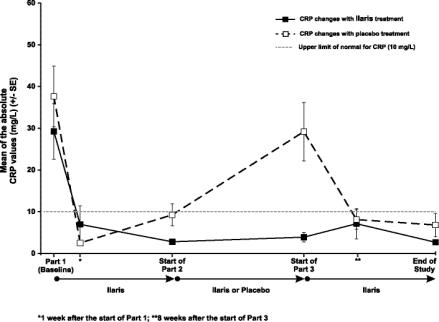
Figure 1. Mean C-Reactive Protein Levels at the End of Parts 1, 2 and 3 of CAPS Study 1
14.2 Treatment of Periodic Fever Syndromes: TRAPS, HIDS/MKD, and FMF
The efficacy and safety of ILARIS for the treatment of TRAPS, HIDS/MKD, and FMF was demonstrated in a 4-Part study (TRAPS, HIDS/MKD, and FMF Study 1) consisting of three separate, disease cohorts (TRAPS, HIDS/MKD and FMF) which enrolled 185 patients aged greater than 28 days. Patients in each cohort entered a 12-week screening period (Part 1) during which they were evaluated for the onset of disease flare. Patients aged 2 to 76 years were then randomized at flare onset into a 16-week double-blind, placebo-controlled treatment period (Part 2) where they received either 150 mg ILARIS (2 mg/kg for patients weighing less than or equal to 40 kg) subcutaneously or placebo every 4 weeks. Part 3 and Part 4 of this study are ongoing.
Randomized patients in Part 2 treated with ILARIS whose disease flare did not resolve, or who had persistent disease activity from Day 8 up to Day 14 (Physician’s Global Assessment [PGA] greater than or equal to 2 or C-reactive Protein [CRP] greater than 10 mg/L and no reduction by at least 40% from baseline) received an additional dose of 150 mg (or 2 mg/kg for patients weighing less than or equal to 40 kg). Patients treated with ILARIS whose disease flare did not resolve, or who had persistent disease activity from Day 15 up to Day 28 (PGA greater than or equal to 2 or CRP greater than 10 mg/L and no reduction by at least 70% from baseline), also received an additional dose of 150 mg (or 2 mg/kg for patients weighing less than or equal to 40 kg). On or after Day 29, patients treated with ILARIS in Part 2 with PGA greater than or equal to 2 and CRP greater than or equal to 30 mg/L were also up-titrated. All up-titrated patients remained at the increased dose of 300 mg (or 4 mg/kg for patients weighing less than or equal to 40 kg) every 4 weeks.
The primary efficacy endpoint of the randomized, 16-week treatment period (Part 2) was the proportion of complete responders within each cohort as defined by patients who had resolution of their index disease flare at Day 15 and did not experience a new disease flare during the remainder of the 16-week treatment period. Resolution of the index disease flare (initial flare at the time of the randomization) was defined at the Day 15 visit as a Physician’s Global Assessment (PGA) Disease Activity score less than 2 (“minimal or no disease”) and C-reactive Protein (CRP) within normal range (less than or equal to 10 mg/L) or reduction greater than or equal to 70% from baseline. The key signs and symptoms assessed in the PGA for each condition were the following: TRAPS: abdominal pain, skin rash, musculoskeletal pain, eye manifestations; HIDS/MKD: abdominal pain; lymphadenopathy, aphthous ulcers; FMF: abdominal pain, skin rash, chest pain, arthralgia/arthritis. A new flare was defined as a PGA score greater than or equal to 2 (“mild, moderate, or severe disease”) and CRP greater to or equal than 30 mg/L. In the 16-week treatment period (Part 2), patients who needed dose escalation, who crossed over from placebo to ILARIS, or who discontinued from the study due to any reason prior to Week 16 were considered as non-responders.
Patients randomized in the TRAPS cohort (N=46) were aged 2 to 76 years (median age at baseline: 15.5 years) and of this population, 57.8% did not have fever at baseline. Randomized TRAPS patients were those with chronic or recurrent disease activity defined as 6 flares per year (median number of flares per year: 9.0) with PGA greater than or equal to 2 and CRP greater than 10 mg/L (median CRP at baseline: 112.5 mg/L). In the TRAPS cohort, 11/22 (50.0%) patients randomized to ILARIS 150 mg every 4 weeks received up-titration to 300 mg every 4 weeks during the 16-week treatment period, while 21/24 (87.5%) patients randomized to placebo crossed over to ILARIS.
Patients randomized in the HIDS/MKD cohort (N=72) were aged 2 to 47 years (median age at baseline: 11.0 years) and of this population, 41.7% did not have fever at baseline. Randomized HIDS/MKD patients were those with a confirmed diagnosis of HIDS according to known genetic MVK/enzymatic (MKD) findings, and documented prior history of greater than or equal to 3 febrile acute flares within a 6 month period (median number of flares per year: 12.0) when not receiving prophylactic treatment and during the study, had active HIDS flares defined as PGA greater than or equal to 2 and CRP greater than 10 mg/L (median CRP at baseline: 113.5 mg/L). In the HIDS/MKD cohort, 19/37 (51.4%) patients randomized to ILARIS 150 mg every 4 weeks received up-titration to 300 mg every 4 weeks during the 16-week treatment period, while 31/35 (88.6%) patients randomized to placebo crossed over to ILARIS.
Patients randomized in the FMF cohort (N=63) were aged 2 to 69 years (median age at baseline: 18.0 years) and of this population, 76.2% did not have fever at baseline. Randomized FMF patients were those with documented active disease despite colchicine therapy or documented intolerance to effective doses of colchicine. Patients had active disease defined as at least one flare per month (median number of flares per year: 18.0) and CRP greater than 10 mg/L (median CRP at baseline: 94.0 mg/L). Patients were allowed to continue their stable dose of colchicine without change. Of the 63 randomized patients, 55 (87.3%) were taking concomitant colchicine therapy on or after randomization. In the FMF cohort, 10/31 (32.3%) patients randomized to ILARIS 150 mg every 4 weeks received up-titration to 300 mg every 4 weeks during the 16-week treatment period, while 27/32 (84.4%) patients randomized to placebo crossed over to ILARIS.
For the primary efficacy endpoint, ILARIS was superior to placebo in the proportion of TRAPS, HIDS/MKD, and FMF patients who resolved their index disease flare at Day 15 and had no new flare over the 16 weeks of treatment from the time of the resolution of the index flare (see Table 4).
Table 4: Proportion of TRAPS, HIDS/MKD, and FMF Patients Who Achieved a Complete Response (Resolution of Index Flare by Day 15 and Maintained Through Week 16) ILARIS 150 mg Placebo Treatment comparison Cohort n/N (%) n/N (%) Odds Ratio
95% CIp-value TRAPS 10/22 (45.5%) 2/24 (8.3%) 9.17
(1.51, 94.61)0.005 HIDS/MKD 13/37 (35.1%) 2/35 (5.7%) 8.94
(1.72, 86.41)0.002 FMF 19/31 (61.3%) 2/32 (6.3%) 23.75
(4.38, 227.53)<0.0001 n=number of patients with the response; N=number of patients evaluated for that response in each cohort; CI: Confidence Interval.
At Day 15, a higher proportion of ILARIS-treated patients compared to placebo-treated patients experienced resolution of their index flare in all disease cohorts (see Table 5).
Table 5: Resolution of Index Flare (Full Analysis Set) Resolution at Day 15* ILARIS 150 mg every 4 weeks Placebo Variable n/N (%) n/N (%) TRAPS 14/22 (63.6%) 5/24 (20.8%) HIDS/MKD 24/37 (64.9%) 13/35 (37.1%) FMF 25/31 (80.7%) 10/32 (31.3%) n=number of patients with the response; N=number of patients evaluated for that response in each cohort
*Resolution of index disease flare (PGA less than 2 and CRP less than or equal to 10 mg/L or reduction greater than or equal to 70% from baseline)There was supportive evidence of efficacy for ILARIS at Day 15, as compared to placebo, for the components of the primary endpoint, CRP and PGA Disease Activity score, as well as for the secondary endpoint Serum Amyloid A (SAA) level (see Table 6).
Table 6: Proportion of TRAPS, HIDS/MKD, and FMF Patients Achieving PGA Less Than 2, CRP Less Than or Equal To 10 mg/L and SAA Less Than or Equal To 10 mg/L at Day 15* TRAPS HIDS/MKD FMF Variable ILARIS
150 mgPlacebo Treatment comparison ILARIS
150 mgPlacebo
Treatment comparison ILARIS
150 mgPlacebo
Treatment comparison n/N (%) n/N (%) Odds Ratio
95% CIn/N (%) n/N (%) Odds Ratio
95% CIn/N (%) n/N (%) Odds Ratio
95% CIPGA Less Than 2 14/22 (63.6%) 8/24 (33.3%) 4.06 (1.12, 14.72) 26/37 (70.3%) 14/35 (40.0%) 3.42 (1.28, 9.16) 27/31 (87.1%) 13/32 (40.6%) 10.07 (2.78, 36.49) CRP Less Than or equal to 10 mg/L 13/22 (59.1%) 8/24 (33.3%) 3.88 (1.05, 14.26) 25/37 (67.6%) 9/35 (25.7%) 6.05 (2.14, 17.12) 28/31 (90.3%) 9/32 (28.1%) 22.51 (5.41, 93.62) SAA Less Than or Equal to 10 mg/L 7/22 (31.8%) 2/24 (8.3%) 5.06 (0.92, 27.91) 10/37 (27.0%) 4/35 (11.4%) 2.94 (0.82, 10.53) 13/31 (41.9%) 5/32 (15.6%) 3.73 (1.11, 12.52) n=number of patients with the response; N=number of patients evaluated for that response in each cohort; CI: Confidence Interval.
*Ilaris-treated patients who up-titrated or discontinued prior to Day 15 and placebo-treated patients who switched over to Ilaris or discontinued prior to Day 15 were classified as non-responders.14.3 Treatment of SJIA
The efficacy of ILARIS for the treatment of active SJIA was assessed in 2 phase 3 studies (SJIA Study 1 and SJIA Study 2). Patients enrolled were aged 2 to less than 20 years (mean age at baseline: 8.5 years) with a confirmed diagnosis of SJIA at least 2 months before enrollment (mean disease duration at baseline: 3.5 years). Patients had active disease defined as greater than or equal to 2 joints with active arthritis (mean number of active joints at baseline: 15.4), documented spiking, intermittent fever (body temperature greater than 38°C) for at least 1 day within 1 week before study drug administration, and CRP greater than 30 mg/L (normal range less than 10 mg/L) (mean CRP at baseline: 200.5 mg/L). Patients were allowed to continue their stable dose of methotrexate, corticosteroids, and/or NSAIDs without change, except for tapering of the corticosteroid dose as per study design in SJIA Study 2 (see below).
SJIA Study 1 was a randomized, double-blind, placebo-controlled, single-dose 4-week study assessing the short-term efficacy of ILARIS in 84 patients randomized to receive a single subcutaneous dose of 4 mg/kg ILARIS or placebo (43 patients received ILARIS and 41 patients received placebo). The primary objective of this study was to demonstrate the superiority of ILARIS versus placebo in the proportion of patients who achieved at least 30% improvement in an adapted pediatric American College of Rheumatology (ACR) response criterion which included both the pediatric ACR core set (ACR30 response) and absence of fever (temperature less than or equal to 38°C in the preceding 7 days) at Day 15.
Pediatric ACR responses are defined by achieving levels of percentage improvement (30%, 50%, and 70%) from baseline in at least 3 of the 6 core outcome variables, with worsening of greater than or equal to 30% in no more than one of the remaining variables. Core outcome variables included a physician global assessment of disease activity, parent or patient global assessment of well-being, number of joints with active arthritis, number of joints with limited range of motion, CRP, and functional ability (Childhood Health Assessment Questionnaire-CHAQ).
Percentages of patients by pediatric ACR response are presented in Table 7.
Table 7: Pediatric ACR Response at Days 15 and 29 Day 15 Day 29 ILARIS Placebo Weighted Difference1 (95% CI)2 ILARIS Placebo Weighted Difference1 (95% CI)2 N=43 N=41 N=43 N=41 ACR30 84% 10% 70% (56%, 84%) 81% 10% 70% (56%, 84%) ACR50 67% 5% 65% (50%, 80%) 79% 5% 76% (63%, 88%) ACR70 60% 2% 64% (49%, 79%) 67% 2% 67% (52%, 81%) 1Weighted difference is the difference between the ILARIS and placebo response rates, adjusted for the stratification factors (number of active joints, previous response to anakinra, and level of oral corticosteroid use)
2CI: confidence interval for the weighted difference
N: Number of patients
Results for the components of the pediatric ACR core set were consistent with the overall ACR response results, for systemic and arthritic components including the reduction in the total number of active joints and joints with limited range of motion. Among the patients who returned for a Day 15 visit, the mean change in patient pain score (0 to 100 mm visual analogue scale) was -50.0 mm on ILARIS (N=43), as compared to +4.5 mm on placebo (N=25). The mean change in pain score among ILARIS-treated patients was consistent through Day 29. All patients treated with ILARIS had no fever at Day 3 compared to 87% of patients treated with placebo.
SJIA Study 2 was a randomized, double-blind, placebo-controlled, withdrawal study of flare prevention by ILARIS in patients with active SJIA. Flare was defined by worsening of greater than or equal to 30% in at least 3 of the 6 core Pediatric ACR response variables combined with improvement of greater than or equal to 30% in no more than 1 of the 6 variables, or reappearance of fever not due to infection for at least 2 consecutive days. The study consisted of 2 major parts. One hundred seventy-seven patients were enrolled in the study and received 4 mg/kg ILARIS subcutaneously every 4 weeks in Part I and 100 of these patients continued into Part II to receive either ILARIS 4 mg/kg or placebo subcutaneously every 4 weeks.
Corticosteroid Dose Tapering
Of the total 128 patients taking corticosteroids who entered the open-label portion of Study 2, 92 attempted corticosteroid tapering. Fifty-seven (62%) of the 92 patients who attempted to taper were able to successfully taper their corticosteroid dose and 42 (46%) discontinued corticosteroids.
Time to Flare
Part II was a randomized withdrawal design to demonstrate that the time to flare was longer with ILARIS than with placebo. Follow-up stopped when 37 events had been observed resulting in patients being followed for different lengths of time. The probability of experiencing a flare over time in Part II was statistically lower for the ILARIS treatment group than for the placebo group (Figure 2). This corresponded to a 64% relative reduction in the risk of flare for patients in the ILARIS group as compared to those in the placebo group (hazard ratio of 0.36; 95% CI: 0.17 to 0.75).
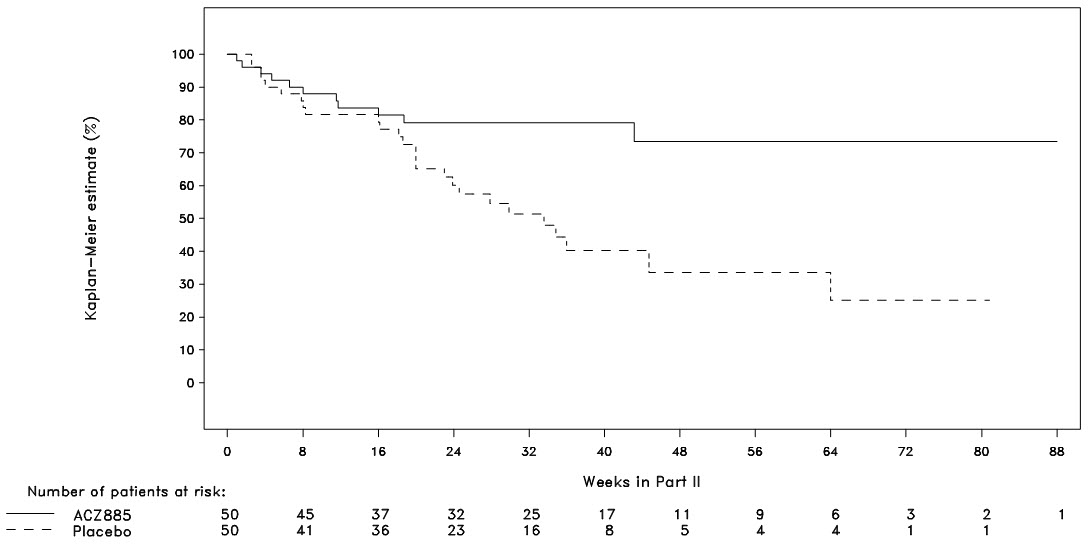
Figure 2. Kaplan-Meier Estimates of the Probability to Stay Flare-Free in Part II of SJIA Study 2 by Treatment
Very few patients were followed for more than 48 weeks
-
16
HOW SUPPLIED/STORAGE AND HANDLING
ILARIS for Injection (Lyophilized Powder)
Carton of 1 vial………………………………………………………………………………………….NDC: 0078-0582-61
Each 150 mg single-dose vial of ILARIS (canakinumab) for Injection contains a sterile, preservative free, white lyophilized powder. After reconstitution with 1 mL of Sterile Water for Injection, the resulting concentration is 150 mg/mL.
ILARIS Injection (Solution)
Carton of 1 vial………………………………………………………………………………………….NDC: 0078-0734-61
Each single-dose vial of ILARIS (canakinumab) Injection delivers 150 mg/mL sterile, preservative-free, clear to slightly opalescent, colorless to a slight brownish to yellow solution.
Special Precautions for Storage
The unopened vial must be stored refrigerated at 2°C to 8°C (36°F to 46° F). Do not freeze. Store in the original carton to protect from light. Do not use beyond the date stamped on the label. ILARIS does not contain preservatives. Discard any unused portions of ILARIS or waste material in accordance with local requirements.
Keep this and all drugs out of the reach of children.
-
17
PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling (Medication Guide)
Patients should be advised of the potential benefits and risks of ILARIS. Physicians should instruct their patients to read the Medication Guide before starting ILARIS therapy.
Drug Administration
Patients should be advised that healthcare providers should perform administration of ILARIS by the subcutaneous injection route.
Infections
Patients should be cautioned that ILARIS use has been associated with serious infections. Patients should be counseled to contact their healthcare professional immediately if they develop an infection after starting ILARIS. Treatment with ILARIS should be discontinued if a patient develops a serious infection. Patients should be counseled not to take any IL-1 blocking drug, including ILARIS, if they are also taking a drug that blocks TNF such as etanercept, infliximab, or adalimumab. Use of ILARIS with other IL-1 blocking agents, such as rilonacept and anakinra is not recommended. Patients should be cautioned not to receive ILARIS if they have a chronic or active infection, including HIV, Hepatitis B or Hepatitis C.
Vaccinations
Prior to initiation of therapy with ILARIS, physicians should review with adult and pediatric patients their vaccination history relative to current medical guidelines for vaccine use, including taking into account the potential of increased risk of infection during treatment with ILARIS.
Injection-site Reactions
Physicians should explain to patients that a very small number of patients in the clinical trials experienced a reaction at the subcutaneous injection-site. Injection-site reactions may include pain, erythema, swelling, pruritus, bruising, mass, inflammation, dermatitis, edema, urticaria, vesicles, warmth, and hemorrhage. Healthcare providers should be cautioned to avoid injecting into an area that is already swollen or red. Any persistent reaction should be brought to the attention of the prescribing physician.
Hypersensitivity
Patients should be counseled to contact their healthcare provider immediately if they develop signs of allergic reaction such as difficulty breathing or swallowing, nausea, dizziness, skin rash, itching, hives, palpitations or low blood pressure.
Pregnancy
Advise female patients of the potential risk to a fetus [see Use in Specific Populations (8.1)].
Manufactured by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936US License Number 1244
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936© Novartis
T2016-102
December 2016
-
MEDICATION GUIDE
Medication Guide
ILARIS® (i-LAHR-us)
(canakinumab)
For injection, for subcutaneous use
Injection, for subcutaneous useWhat is the most important information I should know about ILARIS?
ILARIS can cause serious side effects, including:
-
Increased risk of serious infections. ILARIS can lower the ability of your immune system to fight infections. Your healthcare provider should:
test you for tuberculosis (TB) before you receive ILARIS
monitor you closely for symptoms of TB during treatment with ILARIS
check you for symptoms of any type of infection before, during, and after your treatment with ILARIS
See "What are possible side effects of ILARIS?" for more information about side effects.What is ILARIS?
ILARIS is a prescription medicine injected by your healthcare provider just below the skin (subcutaneous) used to treat:
- The following Periodic Fever Syndromes
- Adults and children 4 years of age and older who have auto-inflammatory diseases called Cryopyrin-Associated Periodic Syndromes (CAPS), including:
- Familial Cold Auto-inflammatory Syndrome (FCAS)
- Muckle-Wells Syndrome (MWS)
- Adults and children who have an auto-inflammatory disease called Tumor Necrosis Factor Receptor Associated Periodic Syndrome (TRAPS)
- Adults and children who have an auto-inflammatory disease called Hyperimmunoglobulin D Syndrome (HIDS) (also known as Mevalonate Kinase Deficiency (MKD).
- Adults and children who have an auto-inflammatory disease called Familial Mediterranean Fever (FMF).
- Adults and children 4 years of age and older who have auto-inflammatory diseases called Cryopyrin-Associated Periodic Syndromes (CAPS), including:
- Systemic Juvenile Idiopathic Arthritis (SJIA) in children 2 years of age and older.
Who should not receive ILARIS?
- Do not receive ILARIS if you are allergic to canakinumab or any of the ingredients in ILARIS. See the end of this Medication Guide for a complete list of ingredients in ILARIS.
Before you receive ILARIS, tell your healthcare provider about all your medical conditions, including if you:
- think you have or are being treated for an active infection
- have symptoms of an infection
- have a history of infections that keep coming back
- have a history of low white blood cells
- have or have had HIV, Hepatitis B, or Hepatitis C
- are scheduled to receive any immunizations (vaccines). You should not get ‘live vaccines’ if you are receiving ILARIS.
- are pregnant or planning to become pregnant. It is not known if ILARIS will harm your unborn baby. Tell your healthcare provider right away if you become pregnant while receiving ILARIS.
- are breastfeeding or planning to breastfeed. It is not known if ILARIS passes into your breast milk. You and your healthcare provider should decide if you will receive ILARIS or breastfeed.
- Medicines that affect your immune system
- Medicines called IL-1 blocking agents such as Kineret® (anakinra), Arcalyst® (rilonacept)
- Medicines called Tumor Necrosis Factor (TNF) inhibitors such as Enbrel® (etanercept), Humira® (adalimumab), Remicade® (infliximab), Simponi® (golimumab), or Cimzia® (certolizumab pegol)
- Medicines that effect enzyme metabolism
How should I receive ILARIS?
- ILARIS is given by your healthcare provider every 8 weeks for CAPS and every 4 weeks for TRAPS, HIDS/MKD, FMF, and SJIA.
What are the possible side effects of ILARIS?
ILARIS can cause serious side effects, including:
- See “What is the most important information I should know about ILARIS?”
-
decreased ability of your body to fight infections (immunosuppression). For people treated with medicines that cause immunosuppression like ILARIS, the chances of getting cancer may increase.
- allergic reactions. Allergic reactions can happen while you are receiving ILARIS. Call your healthcare provider right away if you have any of these symptoms of an allergic reaction:
o rash o itching and hives o difficulty breathing or swallowing o dizziness or feeling faint - risk of infection with live vaccines. You should not get live vaccines if you are receiving ILARIS. Tell your healthcare provider if you are scheduled to receive any vaccines.
When ILARIS is used for the treatment of CAPS:cold symptoms headache feeling like you are spinning (vertigo) diarrhea cough weight gain flu (influenza) body aches injection-site reactions (such as redness, swelling, warmth, or itching) runny nose nausea, vomiting, and diarrhea (gastroenteritis) nausea When ILARIS is used for the treatment of TRAPS, HIDS/MKD, and FMF: cold symptoms runny nose nausea, vomiting, and diarrhea (gastroenteritis) Upper respiratory tract infection sore throat Injection-site reactions (such as redness, swelling, warmth, or itching) When ILARIS is used for the treatment of SJIA: cold symptoms runny nose nausea, vomiting, and diarrhea (gastroenteritis) upper respiratory tract infection sore throat stomach pain pneumonia urinary tract infection injection-site reactions Tell your healthcare provider about any side effect that bothers you or does not go away.
These are not all the possible side effects of ILARIS. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of ILARIS.
Medicines are sometimes prescribed for purposes other than those listed in this Medication Guide. Do not use ILARIS for a condition for which it was not prescribed.
You can ask your healthcare provider or pharmacist for information about ILARIS that was written for health professionals.
What are the ingredients in ILARIS?
Active ingredient: canakinumab
Inactive ingredients:
Powder for Solution for Injection: L-histidine, L-histidine HCl monohydrate, polysorbate 80, sterile water for injection, sucrose
Solution for Injection: L-histidine, L-histidine HCl monohydrate, mannitol, polysorbate 80, sterile water for injectionWhat are Periodic Fever Syndromes?
Periodic Fever Syndromes is the name for several different autoinflammatory diseases, including CAPS, TRAPS, HIDS/MKD, and FMF. People with these diseases cannot keep certain chemicals made by their body (interleukin-1 beta, also called IL-1β) at the correct level. All these diseases have symptoms that often come and go, with irritated body parts (inflammation) and elevated body temperature (fever). These conditions have a dysregulation of IL-1β production and share similar clinical features of recurrent episodes of inflammation and fever such as rash, headache, pain (mostly in the joints, belly, eyes, muscles), fatigue, inflammation of other organs such as heart, lungs, spleen, and brain.
What is SJIA?
SJIA is an autoinflammatory disorder which can be caused by having too much or being too sensitive to certain proteins, including interleukin-1β (IL-1β), and can lead to symptoms such as fever, rash, headache, feeling very tired (fatigue), or painful joints and muscles.
What is Macrophage Activation Syndrome (MAS)?
MAS is a syndrome associated with SJIA and some other autoinflammatory diseases like HIDS/MKD that can lead to death. Tell your healthcare provider right away if your SJIA symptoms get worse or if you have any of these symptoms of an infection:- a fever lasting longer than 3 days
- a cough that does not go away
- redness in one part of your body
- warm feeling or swelling of your skin
Manufactured by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
US License Number 1244
Distributed by: Novartis Pharmaceuticals Corporation, East Hanover, New Jersey 07936 © Novartis
For more information about ILARIS, call 1-877-452-7471 or visit www.ILARIS.com.
Kineret®, Arcalyst®, Enbrel®, Humira®, Remicade®, Simponi®, and Cimzia® are trademarks of Amgen, Regeneron, Immunex Corporation, AbbVie Biotechnology Ltd., Centocor Ortho Biotech Inc., Janssen Biotech Inc., and the UCB Group of companies, respectively.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: December 2016
T2016-103
December 2016 -
Increased risk of serious infections. ILARIS can lower the ability of your immune system to fight infections. Your healthcare provider should:
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
Package Label – 150 mg/vial
Rx Only NDC: 0078-0582-61
ILARIS® (canakinumab)
for Injection
150 mg/vial
For Subcutaneous Use
Reconstitute Prior to Use
Attention: Dispense with enclosed Medication Guide.
Single-Dose Vial. Discard Unused Portion. Sterile, Lyophilized
Reconstitute with 1 mL of Sterile Water for Injection to obtain a concentration of 150 mg/mL canakinumab, L-histidine (2.8 mg/mL), L-histidine hydrochloride monohydrate (1.7 mg/mL), polysorbate 80 (0.6 mg/mL), and sucrose (92.4 mg/mL).

-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
Package Label – 150 mg/mL
Rx Only NDC: 0078-0734-61
ILARIS® (canakinumab)
Injection
150 mg/mL
For Subcutaneous Use
Single-Dose Vial. Discard Unused portion
Attention: Dispense with enclosed Medication Guide

-
INGREDIENTS AND APPEARANCE
ILARIS
canakinumab injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0582 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CANAKINUMAB (UNII: 37CQ2C7X93) (CANAKINUMAB - UNII:37CQ2C7X93) CANAKINUMAB 150 mg in 1 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) 2.8 mg in 1 mL HISTIDINE MONOHYDROCHLORIDE (UNII: 1D5Q932XM6) 1.7 mg in 1 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.6 mg in 1 mL SUCROSE (UNII: C151H8M554) 92.4 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0582-61 1 in 1 CARTON 06/18/2009 02/29/2020 1 1 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125319 06/18/2009 02/29/2020 ILARIS
canakinumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0734 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CANAKINUMAB (UNII: 37CQ2C7X93) (CANAKINUMAB - UNII:37CQ2C7X93) CANAKINUMAB 150 mg in 1 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) 2.1 mg in 1 mL HISTIDINE MONOHYDROCHLORIDE (UNII: 1D5Q932XM6) 1.3 mg in 1 mL MANNITOL (UNII: 3OWL53L36A) 49.2 mg in 1 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.4 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0734-61 1 in 1 CARTON 12/22/2016 1 1 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125319 12/22/2016 Labeler - Novartis Pharmaceuticals Corporation (002147023)
Trademark Results [Ilaris]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ILARIS 79018797 3181206 Live/Registered |
Novartis AG 2005-10-19 |
 ILARIS 78004890 2446597 Dead/Cancelled |
Novartis AG 2000-04-20 |
 ILARIS 77708333 3797205 Live/Registered |
Novartis AG 2009-04-07 |
 ILARIS 77626337 not registered Dead/Abandoned |
Novartis AG 2008-12-04 |
 ILARIS 75638247 2349785 Dead/Cancelled |
Novartis AG 1999-02-16 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.