KYGEVVI- doxecitine and doxribtimine powder, for solution
KYGEVVI by
Drug Labeling and Warnings
KYGEVVI by is a Prescription medication manufactured, distributed, or labeled by UCB, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use KYGEVVI safely and effectively. See full prescribing information for KYGEVVI.
KYGEVVI (doxecitine and doxribtimine) powder, for oral solution
Initial U.S. Approval: 2025INDICATIONS AND USAGE
KYGEVVI is a combination of doxecitine and doxribtimine, both pyrimidine nucleosides, indicated for the treatment of thymidine kinase 2 deficiency (TK2d) in adults and pediatric patients with an age of symptom onset on or before 12 years. ( 1)
DOSAGE AND ADMINISTRATION
- Obtain baseline transaminase (alanine aminotransferase [ALT] and aspartate aminotransferase [AST]) levels in all patients prior to treatment initiation. ( 2.1)
- Recommended dosage (
2.2):
KYGEVVI Dosage Level KYGEVVI Dosage (mg/kg/day) Starting 260 mg/kg/day (consisting of 130 mg doxecitine and 130 mg doxribtimine) Intermediate 520 mg/kg/day (consisting of 260 mg doxecitine and 260 mg doxribtimine) Maintenance 800 mg/kg/day (consisting of 400 mg doxecitine and 400 mg doxribtimine) - Titrate to the next dosage level based on tolerability after a minimum of 2 weeks at the current dosage level. ( 2.2)
- Administer KYGEVVI orally in 3 equally divided doses with food. ( 2.2)
- See full prescribing information for dosage and administration modifications, monitoring, and preparation and administration instructions. ( 2.4)
- Use KYGEVVI only with ZX2000 administration kit. ( 2.4)
DOSAGE FORMS AND STRENGTHS
Powder for oral solution: 2 g doxecitine and 2 g doxribtimine. ( 3)
CONTRAINDICATIONS
- None. ( 4)
WARNINGS AND PRECAUTIONS
- Elevated Liver Transaminase Levels: Obtain baseline liver transaminase (ALT, AST) and total bilirubin levels prior to treatment initiation with KYGEVVI. If signs or symptoms consistent with liver injury are observed, interrupt treatment. Consider permanently discontinuing KYGEVVI if signs/symptoms consistent with liver injury persist or worsen. Monitor patients yearly and as clinically indicated. ( 5.1)
- Gastrointestinal Adverse Reactions: Reduce KYGEVVI dosage or interrupt treatment based on severity of diarrhea and/or vomiting. If persistent severe diarrhea and/or vomiting occurs, consider discontinuing KYGEVVI permanently. ( 5.2)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥5%) are diarrhea, abdominal pain (including abdominal pain upper), vomiting, alanine aminotransferase increased (ALT), and aspartate aminotransferase increased (AST). ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact UCB, Inc. at 1-844-599-2273 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 11/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Recommendation Prior to KYGEVVI Treatment Initiation
2.2 Recommended Dosage
2.3 Dosage and Administration Modifications and Monitoring
2.4 Preparation and Administration Instructions
2.5 Storage Instructions for Prepared KYGEVVI Solution
2.6 Missed Dose
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Elevated Liver Transaminase Levels
5.2 Gastrointestinal Adverse Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Recommendation Prior to KYGEVVI Treatment Initiation
Obtain baseline liver transaminase (alanine aminotransferase [ALT] and aspartate aminotransferase [AST]) and total bilirubin levels in patients prior to treatment initiation with KYGEVVI [see Dosage and Administration (2.3)and Warnings and Precautions (5.1)] .
2.2 Recommended Dosage
The recommended dosage of KYGEVVI is based on the patient's weight (Table 1). Titrate to the next dosage level based on tolerability after a minimum of 2 weeks at the current dosage level.
Table 1: Recommended Starting, Intermediate, and Maintenance Dosage of KYGEVVI KYGEVVI Dosage Level KYGEVVI Dosage (mg/kg/day) Starting 260 mg/kg/day (consisting of 130 mg doxecitine and 130 mg doxribtimine) Intermediate 520 mg/kg/day (consisting of 260 mg doxecitine and 260 mg doxribtimine) Maintenance 800 mg/kg/day (consisting of 400 mg doxecitine and 400 mg doxribtimine) Administer KYGEVVI orally in 3 equally divided doses approximately 6 hours apart (plus or minus 2 hours) with food [see Clinical Pharmacology (12.3)] .
After calculating the daily dose, use Table 2 to determine the required number of KYGEVVI packets, volume of water needed to reconstitute the powder from the packet(s), and individual volume that is administered 3 times a day [see Dosage and Administration (2.4)].
2.3 Dosage and Administration Modifications and Monitoring
Liver Test Abnormalities
If signs or symptoms consistent with liver injury are observed, interrupt treatment with KYGEVVI until liver transaminase (ALT, AST) and total bilirubin levels have either returned to baseline or stabilized at a new baseline value. Consider re-starting KYGEVVI at the last tolerated dose and increase the dose based on tolerability [see Dosage and Administration (2.2)] . Consider permanently discontinuing KYGEVVI if signs or symptoms consistent with liver injury persist or worsen. Monitor liver transaminases and total bilirubin levels yearly and as clinically indicated [see Warnings and Precautions (5.1)] .
Gastrointestinal
Based on the severity of the diarrhea and/or vomiting, reduce the dose of KYGEVVI or interrupt treatment until diarrhea and/or vomiting improves or returns to baseline. Consider re-starting KYGEVVI at the last tolerated dose and increase the dose based on tolerability [see Dosage and Administration (2.2)] . For persistent or recurring diarrhea and/or vomiting, consider discontinuing KYGEVVI permanently. Monitor for dehydration and treat promptly with electrolyte replacement [see Warnings and Precautions (5.2)] .
2.4 Preparation and Administration Instructions
Use Table 2 for preparation and administration information.
Table 2: Recommended Dosage - Preparation and Dosing by Daily-Dose Range Total Daily Dose
(mg/day)Volume of Solution
(mL)
(administered 3 times per day)Total mL of Water for Reconstitution Total Number of KYGEVVI Packets for Reconstitution - * The volume of each individual dose, when multiplied by 3, may not match the corresponding water volume used in the preparation of the oral solution as the final volume of the reconstituted oral solution will increase after the powder from the packets is added to the water volume.
750 – 824 2.5 40 1 825 – 974 3 975 – 1,124 3.5 1,125 – 1,299 4 1,300 – 1,449 4.5 1,450 – 1,649 5 1,650 – 1,949 6 1,950 – 2,249 7 2,250 – 2,549 8 2,550 – 2,849 9 2,850 – 3,149 10 3,150 – 3,449 11 3,450 – 3,749 12 3,750 – 4,049 13 4,050 – 4,349 14 80 2 4,350 – 4,649 15 4,650 – 4,949 16 4,950 – 5,249 17 5,250 – 5,549 18 5,550 – 5,849 19 5,850 – 6,149 20 6,150 – 6,449 21 6,450 – 6,749 22 6,750 – 7,049 23 7,050 – 7,349 24 7,350 – 7,649 25 7,650 – 7,949 26 7,950 – 8,249 27 * 8,250 – 8,549 28 120 3 8,550 – 8,849 29 8,850 – 9,749 30 9,750 – 11,249 35 11,250 – 12,749 40 12,750 – 14,249 45 160 4 14,250 – 15,749 50 15,750 – 17,249 55 * 17,250 – 18,749 60 200 5 18,750 – 20,249 65 20,250 – 21,749 70 * 21,750 – 23,249 75 240 6 23,250 – 24,749 80 24,750 – 26,249 85 280 7 26,250 – 27,749 90 27,750 – 29,249 95 * 29,250 – 30,749 100 320 8 30,750 – 32,249 105 32,250 – 33,749 110 * 33,750 – 35,249 115 360 9 35,250 – 36,749 120 36,750 – 38,249 125 * 38,250 – 39,749 130 400 10 39,750 – 41,249 135 * 41,250 – 42,749 140 * 42,750 – 44,249 145 440 11 44,250 – 45,749 150 * 45,750 – 47,249 155 * 47,250 – 48,749 160 480 12 48,750 – 50,249 165 * 50,250 – 51,749 170 * 51,750 – 53,249 175 * 520 13 53,250 – 54,749 180 * 54,750 – 56,249 185 560 14 56,250 – 57,749 190 * 57,750 – 59,249 195 * 59,250 – 60,749 200 600 15 60,750 – 62,249 205 * 62,250 – 63,749 210 * 63,750 – 65,249 215 * 640 16 65,250 – 66,749 220 * 66,750 – 68,249 225 * Use the ZX2000 administration kit provided separately to prepare and administer the prescribed dose [see How Supplied/Storage and Handling (16)] . Refer to the Instructions for Use for full preparation and administration information on use of KYGEVVI with the ZX2000 administration kit. Household devices such as measuring cups or spoons are not adequate measuring devices. KYGEVVI should be prepared and administered by adults only.
Preparation Instructions
Preparation of KYGEVVI with a liquid other than water has not been studied clinically and is not recommended.
- Obtain the required number of KYGEVVI packets to prepare a one-day supply of solution each morning.
- Use 40 mL of water per packet. Pour the prescribed volume of room temperature water (between 20°C - 25°C or 68°F - 77°F) into the mixing bottle.
- Add the powder from the required number of KYGEVVI packets into the mixing bottle.
- Screw the dosing cup tightly onto the mixing bottle and gently invert the mixing bottle back and forth at least 20 times. If powder remains, repeat until the powder dissolves.
- The mixed solution may appear cloudy and have some residual powder (inactive ingredients) remaining at the bottom or top.
Administration Instructions
Oral Administration
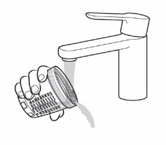
- Before each administration, gently invert the tightly closed mixing bottle slowly back and forth at least 3 times.
- Use 1 of 2 methods (dosing cup or oral syringe) to administer KYGEVVI solution. Choose the method based on the volume of solution to be administered per dose.
- Take KYGEVVI solution in 3 equally divided doses approximately 6 hours apart (plus or minus 2 hours) with food.
- Do not administer another dose if the dose is spit out or if a complete dose is not taken. Take the next dose at the next scheduled time.
- Discard any remaining KYGEVVI solution 16 hours after reconstitution or after taking or giving the 3 doses, whichever comes first.
Feeding Tube Administration
KYGEVVI is compatible with most commonly available feeding tubes. KYGEVVI is compatible with feeding tubes made with polyvinylchloride (PVC) free from DEHP (Phthalates), polyurethane (PUR), and silicone (SIL) material.
- Follow the instructions of the feeding tube manufacturer to administer KYGEVVI.
- Draw up the KYGEVVI solution using a syringe compatible with the feeding tube.
- Administer the solution immediately through the feeding tube.
- Flush any residual solution in the syringe or feeding tube until no solution is left. To flush the tube, a single flushing step with a volume of water equivalent to the tube's priming volume is sufficient.
- Discard any remaining KYGEVVI solution 16 hours after reconstitution or after taking or giving the 3 doses, whichever comes first.
2.5 Storage Instructions for Prepared KYGEVVI Solution
- Store reconstituted KYGEVVI solution at controlled room temperature between 20°C to 25°C (68°F to 77°F) or in the refrigerator between 2°C to 8°C (36°F to 46°F).
- Discard KYGEVVI solution 16 hours after reconstitution or after taking or giving the 3 doses, whichever comes first.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Elevated Liver Transaminase Levels
Elevated liver transaminase [alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST)] levels were reported in patients treated with KYGEVVI [see Adverse Reactions (6.1)] . In Study 1, two patients permanently discontinued treatment with KYGEVVI upon recurrence of elevated liver enzymes after a rechallenge at a reduced dose.
Obtain baseline liver transaminase (ALT, AST) and total bilirubin levels in patients prior to treatment initiation with KYGEVVI. If signs or symptoms consistent with liver injury are observed, interrupt treatment with KYGEVVI until liver transaminase (ALT, AST) and total bilirubin levels have either returned to baseline or stabilized at a new baseline value. Consider permanently discontinuing KYGEVVI if signs or symptoms consistent with liver injury persist or worsen. Monitor liver transaminases and total bilirubin levels yearly and as clinically indicated [see Dosage and Administration (2.3)] .
5.2 Gastrointestinal Adverse Reactions
Diarrhea and vomiting leading to hospitalization, dose reduction, and permanent discontinuation were reported in patients treated with KYGEVVI [see Adverse Reactions (6.1)] .
Based on the severity of the diarrhea and/or vomiting, reduce the dosage of KYGEVVI or interrupt treatment until diarrhea and/or vomiting improves or returns to baseline. Consider restarting KYGEVVI at the last tolerated dose, and increase the dose as tolerated. For persistent or recurring diarrhea and/or vomiting, consider discontinuing KYGEVVI permanently and provide supportive care with electrolyte repletion as clinically indicated.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Elevated Liver Transaminase Levels [see Warnings and Precautions (5.1)]
- Gastrointestinal Adverse Reactions [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of KYGEVVI was evaluated in a prospective, open-label, single-arm study in pediatric and adult patients with genetically confirmed TK2d previously treated with pyrimidine nucleosides (Trial 1). Additional safety information was derived from retrospective chart review studies (Study 1, Study 2) and from an expanded access program [see Clinical Studies (14)] .
Permanent discontinuation of KYGEVVI due to an adverse reaction occurred in 9% of patients (Trial 1, Study 1, and Study 2). The adverse reactions which resulted in permanent discontinuation of KYGEVVI in >2% of patients were diarrhea (3%) and elevated liver enzymes (3%). In the expanded access program, diarrhea resulted in permanent discontinuation in 2 patients.
Dose reductions of KYGEVVI due to an adverse reaction occurred in 22% of patients (Trial 1, Study 1, and Study 2). Adverse reactions which required dose reduction in >2% of patients included diarrhea (21%) and abdominal pain (3%).
Diarrhea resulted in hospitalization in 2 pediatric patients (Study 1 and expanded access program).
Adverse Reactions from Trial 1
A total of 47 patients, between the ages of 0.7 and 74 years of age at enrollment, received KYGEVVI or pyrimidine nucleosides dosages up to 800 mg/kg/day [see Clinical Studies (14)] . KYGEVVI is not approved for use in patients with an age of TK2d symptom onset > 12 years. The mean (SD) KYGEVVI or pyrimidine nucleosides exposure during Trial 1 was 6.6 (2) years.
Table 3 summarizes the adverse reactions reported in ≥ 5% patients treated with KYGEVVI or pyrimidine nucleosides.
Table 3: Adverse Reactions That Occurred in ≥5% Adult and Pediatric Patients with TK2d Treated with KYGEVVI or Pyrimidine Nucleosides (Trial 1) Adverse reactions Treated Patients
(N=47)
n (%)Diarrhea 34 (72) Abdominal pain (including abdominal pain upper) 11 (23) Vomiting 10 (21) Alanine aminotransferase increased (ALT) 10 (21) Aspartate aminotransferase increased (AST) 8 (17) Adverse reactions, vomiting and elevated liver transaminases, were observed in a higher percentage of pediatric patients than in adult patients. In Trial 1, vomiting occurred in 28% (9/32) of pediatric patients compared to 7% (1/15) of adult patients. Elevated liver transaminases occurred in 25% (8/32) for ALT and 22% (7/32) for AST of pediatric patients compared to 13% (2/15) for ALT and 7% (1/15) for AST of adult patients.
Laboratory Adverse Reaction
Elevated liver enzymes have been observed as a clinical manifestation of TK2d. In Trial 1 and Study 1, elevations in alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) occurred in 28% (14/50) and 22% (11/50) of patients respectively. In Trial 1, of all the patients who started treatment with elevated AST/ALT at baseline, 5% had last post-baseline ALT values that were higher severity than the baseline severity while continuing treatment [see Warnings and Precautions (5.1)] .
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on KYGEVVI use during pregnancy to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Endogenous pyrimidine nucleosides are transported across the placenta. There are risks for adverse maternal and fetal outcomes during pregnancy with mitochondrial myopathies, including TK2 deficiency ( see Clinical Considerations). In animal reproduction studies, oral administration of doxecitine and doxribtimine to pregnant rats and rabbits during organogenesis resulted in maternal and fetal toxicities in the rabbit at dose exposures 1233 and 811 times the maximum recommended human dose (MRHD) of 400 mg/kg/day doxecitine and 400 mg/kg/day doxribtimine, respectively, based on plasma exposure, but were not observed in the rat ( see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20% respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Mitochondrial myopathies are associated with increased adverse perinatal outcomes, including preterm birth, pre-eclampsia and gestational diabetes.
Data
Animal Data
In an embryofetal development study in pregnant rats, once daily oral doses of 200, 600, and 2000 mg/kg/day doxecitine and doxribtimine were administered throughout organogenesis between gestation day (GD) 7 to 17. No maternal or embryofetal toxicity was observed up to 2000 mg/kg/day (1223 times and 425 times the MRHD of doxecitine and doxribtimine, respectively, based on plasma exposure).
In an embryofetal development study in pregnant rabbits, once daily oral doses of 200, 600, and 2000 mg/kg/day doxecitine and doxribtimine were administered throughout organogenesis between GD 7 and GD 19. Marked maternal toxicity and fetal malformations (dilated aorta with an associated narrow pulmonary trunk) were observed at the highest dose (1233 times and 811 times the MRHD of doxecitine and doxribtimine, respectively, based on plasma exposure). The maternal and fetal no observed adverse effect level (NOAEL) in rabbits (600 mg/kg/day) was associated with maternal plasma exposures 729 times and 126 times the MRHD of 400 mg/kg/day doxecitine and 400 mg/kg/day doxribtimine, respectively.
8.2 Lactation
Risk Summary
There are no data on the presence of doxecitine and doxribtimine or its metabolites in either human or animal milk, the effects on the breastfed infant, or the effects on milk production. Data from published literature reports the presence of nucleosides and nucleotides in human milk.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for KYGEVVI and any potential adverse effects on the breastfed infant from KYGEVVI or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of KYGEVVI for the treatment of thymidine kinase 2 deficiency (TK2d) have been established in pediatric patients with an age of symptom onset on or before 12 years. Use of KYGEVVI for this indication in this population is supported by evidence from two retrospective studies (Study 1, Study 2), one open-label study (Trial 1), and an expanded access program in which a total of 68 patients 0.7 years of age to less than 17 years of age were treated [see Clinical Studies (14)] .
In Trial 1, compared to adults, a higher percentage of pediatric patients experienced adverse reactions of vomiting and elevated liver transaminases [see Adverse Reactions (6.1)] . Serious adverse reactions in the pediatric population included hospitalization due to diarrhea in two patients [see Warnings and Precautions (5.2)] .
8.5 Geriatric Use
Clinical studies of KYGEVVI did not include sufficient number of patients 65 years of age and older to determine whether they respond differently from younger adult patients.
8.6 Renal Impairment
Plasma concentrations of doxecitine and doxribtimine increased in patients with moderate or severe renal impairment. The pharmacokinetics (PK) of doxecitine and doxribtimine have not been evaluated in patients with mild renal impairment. An appropriate dosage adjustment of KYGEVVI in patients with renal impairment could not be determined because renal impairment had distinct effects on the PK of doxecitine and PK of doxribtimine, and it is not feasible to separately adjust the dosage for doxecitine or doxribtimine contained in KYGEVVI [see Clinical Pharmacology (12.3)] .
-
11 DESCRIPTION
KYGEVVI is a combination of doxecitine and doxribtimine, both of which are pyrimidine nucleosides. KYGEVVI is a powder for oral solution. Both doxecitine and doxribtimine are white to off-white powders and soluble in water.
Doxecitine
The chemical name of doxecitine is 4-Amino-1-((2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)pyrimidin-2(1H)-one. The molecular formula is C 9H 13N 3O 4and the molecular weight is 227.22 g/mol. The chemical structure is:
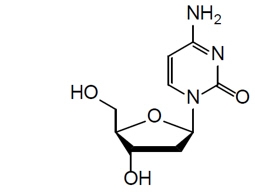
Doxribtimine
The chemical name of doxribtimine is 1-((2R,4S,5R)-4-Hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione. The molecular formula is C 10H 14N 2O 5and the molecular weight is 242.23 g/mol. The chemical structure is:
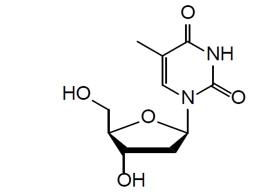
Each packet of KYGEVVI powder contains 2 grams doxecitine and 2 grams doxribtimine. The inactive ingredients are colloidal silicon dioxide and magnesium stearate.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Administration of KYGEVVI is intended to incorporate the pyrimidine nucleosides, deoxycytidine and deoxythymidine, into skeletal muscle mitochondrial deoxyribonucleic acid (DNA). This action restores mitochondrial DNA copy number in TK2d mutant mice.
12.2 Pharmacodynamics
The exposure-response relationship and time course of pharmacodynamic response for the safety and effectiveness of KYGEVVI have not been fully characterized.
12.3 Pharmacokinetics
Following oral administration of doxecitine and doxribtimine in healthy adult subjects, the baseline-adjusted maximum plasma concentration (C max) and area under the plasma concentration-time curve (AUC) increased in a less than dose proportional manner for doxecitine at doses ranging from 43 mg/kg to 133 mg/kg and more than dose proportional manner for doxribtimine at doses ranging from 43 mg/kg to 133 mg/kg. There is minimal or no accumulation of doxecitine and doxribtimine following multiple dose administrations. Following oral administration of doxecitine and doxribtimine at the recommended maintenance dosage of 800 mg/kg/day under fed conditions in 18 TK2d pediatric and adult subjects, the estimated baseline-unadjusted geometric mean C maxat steady state was 12 ng/mL and 19 ng/mL for doxecitine and doxribtimine, respectively, and the geometric mean AUC from time 0 to 24 hours (AUC 0-24hr) was 108 ng∙h/mL and 191 ng∙h/mL for doxecitine and doxribtimine, respectively. Inter-subject variability (geometric CV%) in C maxand AUC 0-24hvalues of doxecitine and doxribtimine were greater than 70%.
Absorption
The absolute bioavailability of doxecitine and doxribtimine following oral administration has not been determined. The median time to peak plasma concentration (T max) was approximately 2 hours for doxecitine and 4 hours for doxribtimine.
Effect of Food
Following an oral administration of 133 mg/kg doxecitine and 133 mg/kg doxribtimine with a high-fat, high-calorie meal in healthy adult subjects, baseline-adjusted plasma C maxand AUC 0-tincreased by 79% and 137%, respectively, for doxecitine; and increased by 27% and 74%, respectively, for doxribtimine, compared to fasted conditions [see Dosage and Administration (2.2)] .
Distribution
In vitro plasma protein binding of doxecitine and doxribtimine was less than 10% over the concentration range between 0.23 mcg/mL and 23 mcg/mL.
Elimination
The mean half-life was approximately 1 hour for doxecitine and 5 hours for doxribtimine following a single oral administration of 133 mg/kg doxecitine and 133 mg/kg doxribtimine under fed conditions in healthy adult subjects.
Metabolism
Doxecitine and doxribtimine are primarily degraded (catabolized) by cytidine deaminase and thymidine phosphorylase, respectively, to their nucleobases and the 2-deoxy-α-D-ribose 1-phosphate moiety. Intermediate products of doxecitine catabolism are deoxyuridine, uracil, and dihydrouracil with the end products β-alanine, ammonia, and carbon dioxide (CO 2). Thymine, the pyrimidine nucleobase of doxribtimine, is subsequently catabolized to dihydrothymine and ultimately to γ-amino-isobutyric acid and CO 2.
Doxecitine and doxribtimine are not known to be metabolized by cytochrome P450 (CYP) isoforms.
Excretion
Urinary excretion of intact doxecitine and doxribtimine was <1% of the dose in healthy subjects following an oral administration of doxecitine and doxribtimine.
Specific Populations
Male and Female Patients
The pharmacokinetics of doxecitine and doxribtimine were not significantly different between male and female subjects.
Patients with Renal Impairment
The pharmacokinetics of doxecitine and doxribtimine in subjects with moderate (estimated glomerular filtration rate [eGFR] ≥ 30 and ≤ 59 mL/min/1.73 m 2) or severe (eGFR ≥ 15 and ≤ 29 mL/min/1.73 m 2) renal impairment were compared with healthy subjects with normal renal function following a single oral administration of 133 mg/kg doxecitine and 133 mg/kg doxribtimine. Baseline-adjusted plasma doxecitine AUC was 122% and 66% higher in subjects with moderate and severe renal impairment, respectively, compared with matched control subjects with normal renal function. Baseline adjusted plasma doxribtimine AUC was 447% and 148% higher in subjects with moderate and severe renal impairment, respectively, compared with matched control subjects with normal renal function [see Use in Specific Populations (8.6)] .
Patients with Hepatic Impairment
No studies have been conducted to evaluate the effect of hepatic impairment on the pharmacokinetics of doxecitine and doxribtimine.
Drug Interaction Studies
In Vitro Studies
CYP enzymes: Doxecitine and doxribtimine are not inducers, inhibitors, or substrates of CYP isozymes at clinically relevant concentrations.
Transporter systems: Doxecitine and doxribtimine do not inhibit P-glycoprotein (P-gp), BCRP, BSEP, OATP1B1, OATP1B3, OAT1, OAT3, OCT1, OCT2, MATE1, or MATE2-K at clinically relevant concentrations. Doxribtimine may be a substrate of BCRP, but its clinical significance is unknown.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Animal studies to evaluate the carcinogenic potential of doxecitine and doxribtimine have not been conducted.
Mutagenesis
Doxecitine and doxribtimine were not mutagenic or clastogenic in an in vitrobacterial reverse mutation (Ames) and an in vivorat micronucleus assay. Doxecitine and doxribtimine induced chromosomal aberrations in the absence of metabolic activation in an in vitrocytogenetic study in human lymphocytes. One compound (alpha-hydroxythymidine) originating from a doxribtimine starting material and present in the final drug product was positive for mutagenesis in the Ames assay and positive for clastogenesis in human peripheral lymphocytes when tested alone.
Impairment of Fertility
Doxecitine and doxribtimine had no effect on male or female fertility or early embryonic development at doses up to 2000 mg/kg/day in rats (1131 times and 1223 times the MRHD in males and females respectively for 400 mg/kg/day doxecitine and 1056 times and 425 times the MRHD in males and females respectively for 400 mg/kg/day doxribtimine, based on plasma exposure).
-
14 CLINICAL STUDIES
The efficacy of KYGEVVI for the treatment of patients with TK2d, with an age of symptom onset on or before 12 years of age, was established based on data from one Phase 2 clinical study (Trial 1), two retrospective chart review studies (Study 1, Study 2), and an expanded access program. The survival in treated patients was compared with survival in an untreated external control group comprised of untreated patients from published literature and Study 2.
Trial 1 (NCT03845712) is a prospective, open-label, single-arm study in 47 patients with genetically confirmed TK2d previously treated with pyrimidine nucleosides. Thirty-eight of these 47 patients have an age of TK2d symptom onset ≤12 years; none of the 38 patients discontinued treatment. The initial oral dose of KYGEVVI was matched to the patient's pyrimidine nucleoside dose of 260-800 mg/kg/day upon entering the study in patients with an age of TK2d symptom onset ≤ 12 years, and dosage was titrated, as needed, over a maximum of 4 weeks to the maintenance dose of 800 mg/kg/day.
Study 1 (NCT03701568) was a retrospective chart review study in 38 patients with genetically confirmed TK2d treated with pyrimidine nucleosides. Twenty-nine of these patients had an age of TK2d symptom onset ≤12 years; none of the 29 patients discontinued treatment. Thirty-five of these 38 patients were later enrolled in Trial 1 to receive treatment with KYGEVVI and one was later enrolled in Study 2. KYGEVVI was not administered in Study 1. Patients enrolled in Study 1 were receiving pyrimidine nucleoside treatment at doses 160-800 mg/kg/day.
Study 2 (NCT05017818) was a retrospective chart review study in 61 patients with genetically confirmed TK2d (43 untreated patients and 18 patients treated with pyrimidine nucleoside therapy). Nine of these 61 patients were also included in the expanded access program and 1 patient was included in Study 1. Twenty-seven of the 43 untreated patients had an age of TK2d symptom onset ≤12 years, and 13 of the 18 treated patients had an age of TK2d symptom onset ≤12 years. Twenty-two untreated patients were included in the untreated external control group used to evaluate survival. Of the 18 treated patients, 6 (33%) discontinued treatment due to an adverse reaction. KYGEVVI was not administered in Study 2. Patients enrolled in Study 2 were receiving pyrimidine nucleoside treatment at doses 200-1200 mg/kg/day.
Expanded Access Program
The expanded access program data included 43 patients receiving KYGEVVI; 9 patients were included in Study 2.
Efficacy Results
A total of 82 patients with genetically confirmed TK2d and the age of symptom onset ≤12 years were treated with KYGEVVI or pyrimidine nucleosides. Efficacy was assessed by comparing overall survival in the treated patients to an external control group of untreated patients matched to treated patients using age of TK2d symptom onset (≤ 2 years or >2 to ≤ 12 years). A total of 78 matched pairs were identified.
Of the 78 treated patients included in the survival analysis, 54% were male and 36% were of Hispanic or Latino ethnicity. Eighty-two percent of patients were White, 4% Black or African American, 5% Asian, 3% Other, and 1% American Indian or Alaska Native. The median age of TK2d symptom onset was 1.5 years (range: 0.01 to 12 years). The median duration of treatment was 4 years (range: 1 day to 12 years) and the median dose received was 762 mg/kg/day (range: 260 to 800 mg/kg/day).
Treatment reduced the overall risk of death from treatment start by approximately 86% (95% CI: 61%, 96%) (Table 4, Figure 1).
Table 4: Overall Survival in Patients with TK2d (Age of Symptom Onset ≤12 Years) Treated with KYGEVVI Versus Matched Untreated Patients (External Control) * Treated Patients
(n= 78)Matched Untreated Patients
(n=78)CI: Confidence Interval - * Treated patients were originally from Trial 1 (n=9), Study 1 (n=27), Study 2 (n=11), and the expanded access program (n=31). Untreated patients were from published literature (n=57) and Study 2 (n=21).
- † An additional censoring step for untreated subjects was performed for each matched pair where the untreated subject died and had a longer follow-up time than the matched treated subject who was censored. The follow-up time of the untreated subject was then censored at the follow-up time of the treated subject.
- ‡ Based on the area under the survival curves up to 4-, 6-, 10-years post treatment start.
- § Estimates based on Cox Proportional Hazard Model with Firth correction that includes matched pair as a strata, age of symptom onset as a continuous covariate, and treatment (treated or untreated) as a time independent variable.
Number of Deaths (%) † 3 (3.8%) 28 (35.9%) Restricted Mean Survival Time in Years
(95% CI) †,‡- At 4 years post treatment start
- At 6 years post treatment start
- At 10 years post treatment start
- 3.8 (3.7, 4)
- 5.8 (5.5, 6)
- 9.6 (9.2, 10)
- 2.6 (2.2, 3)
- 3.7 (3, 4.3)
- 5.7 (4.5, 6.9)
Hazard Ratio § For Risk of death from treatment start 0.14 (0.04, 0.39) (95%CI) Note: Treated patients were matched 1:1 to untreated patients by category of age of TK2d symptom onset (≤2 years or >2 to ≤12 years). Within each category of age of symptom onset, the matching was performed as follows: treated patients were sorted in descending order, according to their age of treatment initiation; the first treated patient in the sorted list was matched with the sorted untreated patient having the highest last known age; this matched untreated patient was then no longer available for matching with any remaining treated patients; the procedure continues in order through the sorted list of treated patients. Time of treatment start in the untreated patient was set to that of the matched treated patient.
- * Treated patients were from Trial 1, Study 1, Study 2, and the expanded access program. Untreated patients were from published literature and Study 2.
- † Kaplan-Meier Curves with 95% confidence intervals using log-log transformation and with treatment group as strata variable; Age of TK2d Symptom Onset ≤ 12 years. An additional censoring step for untreated subjects in the footnote (b) of Table 4 was performed.
Figure 1: Kaplan-Meier Survival Curves for Time to Death from Treatment Start in Patients with TK2d Treated with KYGEVVI and Matched Untreated Patients (External control) *,† 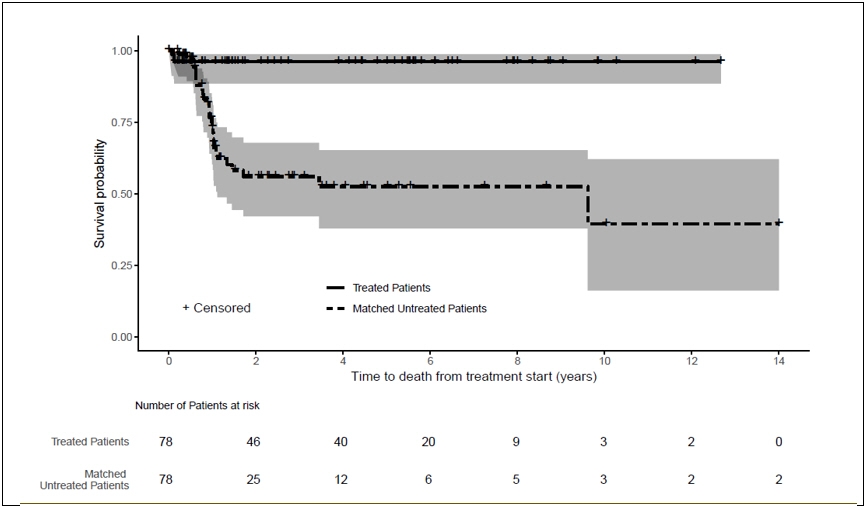
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
KYGEVVI is supplied as follows:
Package 1 of 2
NDC: 50474-350-01: Each single use packet contains 4 g of KYGEVVI as a white to off-white powder (2 g doxecitine and 2 g doxribtimine)
NDC: 50474-350-30: Carton contains 30 packets
Package 2 of 2
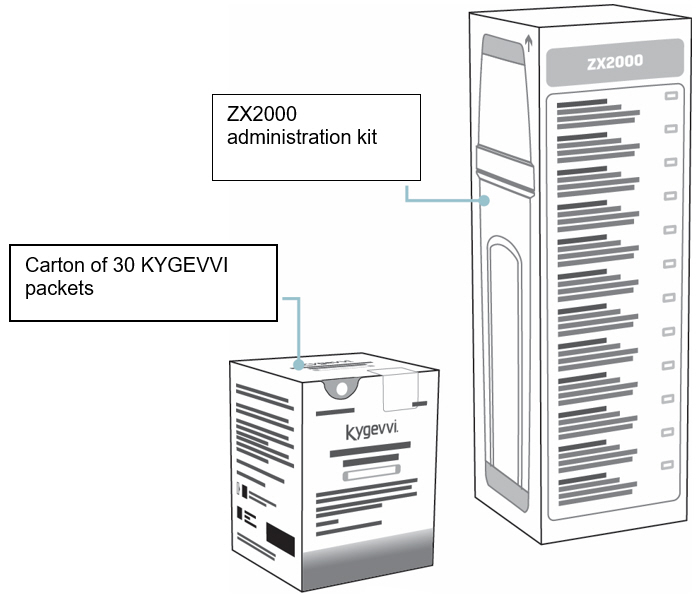
The ZX2000 administration kit for use with KYGEVVI is supplied separately and contains the following:
- One dosing system (includes mixing bottle and dosing cup)
- Two 10 mL oral syringes
- Two spare seals
Storage and Handling
Store KYGEVVI packets at 20°C to 25°C (68°F to 77°F); excursion permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Store reconstituted KYGEVVI solution at controlled room temperature or in the refrigerator. Discard KYGEVVI solution 16 hours after reconstitution or after taking or giving the 3 doses, whichever comes first [see Dosage and Administration (2.5)].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Preparation and Administration Instructions of KYGEVVI Solution
Advise the patient to use the ZX2000 administration kit provided by the pharmacy to prepare and administer KYGEVVI solution.
Inform the patient to prepare a one-day supply of the KYGEVVI solution each morning and take each individual dose with food [see Dosage and Administration (2.4)].
Elevated Liver Transaminase Levels
Inform the patient that KYGEVVI may cause liver enzyme elevations. Instruct the patient to promptly report loss of appetite, abdominal pain, dark urine, or jaundice to his/her healthcare provider [see Warnings and Precautions (5.1)].
Gastrointestinal Adverse Reactions
Inform the patient that KYGEVVI may cause diarrhea and vomiting. Advise the patient to promptly report to his/her healthcare provider diarrhea and vomiting that lasts longer than a few days while taking KYGEVVI [see Warnings and Precautions (5.2)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
KYGEVVI (KY-JEH-vee)
(doxecitine and doxribtimine)
powder, for oral solutionThis Patient Information has been approved by the U.S. Food and Drug Administration. Issued: 11/2025 What is KYGEVVI?
KYGEVVI is a prescription medicine used for the treatment of thymidine kinase 2 deficiency (TK2d) in adults and children with a symptom onset on or before 12 years of age.Before taking or giving KYGEVVI, tell your healthcare provider about all of your medical conditions, including if you: - have or have had liver problems
- are pregnant or plan to become pregnant. It is not known if KYGEVVI will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if KYGEVVI passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby if you take KYGEVVI.
How should I take or give KYGEVVI? - See the detailed Instructions for Usethat comes with KYGEVVI for important information about the correct way to prepare and take or give a dose of KYGEVVI.
- Take or give KYGEVVI exactly as your healthcare provider tells you. Your healthcare provider should tell you how much KYGEVVI to take or give, and when to take or give it.
- Your healthcare provider may change your or your child's dose of KYGEVVI depending on how you or your child respond to treatment and based on your or your child's weight.
- Take or give KYGEVVI 3 times a day about 6 hours apart.
- Take or give KYGEVVI with food.
- KYGEVVI may be given through a feeding tube.
- KYGEVVI comes as a packet containing powder.
- Prepare a one-day supply of KYGEVVI solution each morning.
- KYGEVVI should be prepared and given by adults only.
- Do notopen the KYGEVVI packets until you are ready to use them.
- Mix KYGEVVI powder only with room temperature water. Do notmix with any other liquids or foods.
- Only use the dosing cup, mixing bottle, and oral syringes provided with your ZX2000 administration kit. Do notuse a household measuring cup or measuring spoon.
If you spit out the dose or do not take the full dose of KYGEVVI, do nottake another dose. Take your next dose at your next scheduled time.
What are the possible side effects of KYGEVVI?
KYGEVVI may cause serious side effects, including:- Increased liver enzyme levels.Increased liver enzyme levels in your blood are common with KYGEVVI. Your healthcare provider will do blood tests to check your liver enzyme levels before starting treatment and during treatment with KYGEVVI. Tell your healthcare provider right away if you develop any signs or symptoms of liver problems, including:
- loss of appetite
- pain on the right side of your stomach area
- dark or amber-colored urine
- yellowing of your skin or the white part of your eyes
- nausea and vomiting
- itching
- Stomach and intestinal (gastrointestinal) problems. Diarrhea and vomiting are common with KYGEVVI, but may also be severe and lead to hospitalization. Tell your healthcare provider right away if you have diarrhea or vomiting during treatment with KYGEVVI that lasts longer than a few days.
The most common side effects of KYGEVVI include: - diarrhea
- stomach area (abdominal) pain including pain in the upper stomach area
- vomiting
- increased liver enzyme levels in your blood
These are not all of the possible side effects of KYGEVVI. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store KYGEVVI? - Store KYGEVVI packets at room temperature between 68°F to 77°F (20°C to 25°C).
- Store prepared KYGEVVI solution at room temperature between 68°F to 77°F (20°C to 25°C) orin the refrigerator between 36°F to 46°F (2°C to 8°C).
- Use KYGEVVI solution within 16 hours of preparing. Throw away (discard) any remaining solution 16 hours after preparing orafter taking or giving the 3 doses, whichever comes first.
General information about the safe and effective use of KYGEVVI.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use KYGEVVI for a condition for which it was not prescribed. Do not give KYGEVVI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about KYGEVVI that is written for health professionals.What are the ingredients in KYGEVVI?
Active ingredients:doxecitine and doxribtimine.
Inactive ingredients:colloidal silicon dioxide and magnesium stearate.Manufactured for: UCB, Inc., Smyrna, GA 30080
KYGEVVI® is a trademark of the UCB Group of Companies.
©2025, UCB, Inc., Smyrna, GA 30080
All rights reserved.
For more information about KYGEVVI, go to www.KYGEVVI.com or call 1-844-599-2273. -
INSTRUCTIONS FOR USE
KYGEVVI (KY-JEH-vee)
(doxecitine and doxribtimine)
powder, for oral solutionTable of contents
-
Instructions for use
- Important information
-
Before you start
- Supplies for preparing and taking or giving KYGEVVI
-
Important information
- What you need to know before preparing and taking or giving KYGEVVI
- Storing KYGEVVI packets and oral solution
-
Preparing your one-day supply of KYGEVVI
- Get supplies ready
- Measure water and add powder from packets
- Mix and inspect medicine
-
Dosing methods
- How to measure your individual dose
-
Individual doses 50 mL or more
- Measure and take or give your individual dose
-
Individual doses less than 50 mL
- Measure and take or give your individual dose
-
Between individual doses
- Clean up after first and second individual dose
-
End-of-day clean up
- Pour out and clean up after third individual dose
-
Dosing cup maintenance
- Replacing the seal if misplaced or damaged
- Contact information
Instructions For Use
Important information
This Instructions for Use contains information on how to prepare a one-day supply of KYGEVVI, and take or give an individual dose of KYGEVVI.Read this Instructions for Use before taking or giving KYGEVVI and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your or your child's medical condition or treatment.
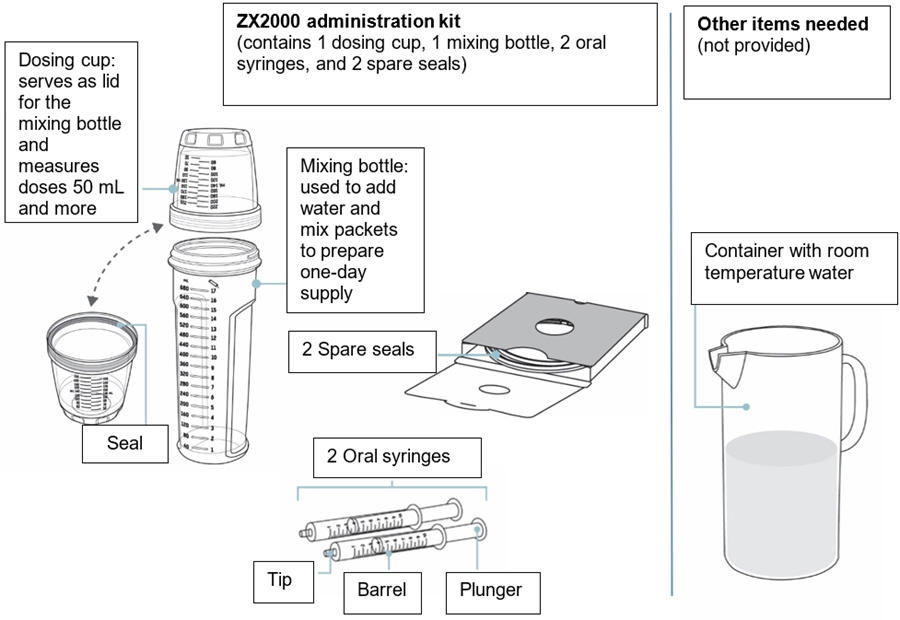
When you are prescribed KYGEVVI for the first time, you will be provided with the carton(s) of 30 KYGEVVI packets and the ZX2000 administration kit (see Figure A). Only use the dosing cup, mixing bottle, and oral syringes provided with your ZX2000 administration kit. Do notuse a household measuring cup or measuring spoon.
Before you start
Supplies for preparing and taking or giving KYGEVVI
Carton of 30 KYGEVVI packets
Before you start
Supplies for preparing and taking or giving KYGEVVI
Important information
What you need to know before preparing and taking or giving KYGEVVI
- You will prepare a one-day supplyof KYGEVVI solution to be taken in 3 equal dosesthroughout the day (about 6 hoursapart).
- KYGEVVI solution should be prepared on the same day as taking or giving your dose.
- KYGEVVI should be prepared and given by adults only.
- Take or give KYGEVVI exactly as prescribed by your healthcare provider.
- Do notchange your dose without talking with your healthcare provider.
- Do nottake or give more than the prescribed amount of KYGEVVI solution.
- Take KYGEVVI solution with food.
- Do notuse KYGEVVI packets after the expiration date on the packet or carton.
- Only use the dosing cup, mixing bottle, and oral syringes provided with your ZX2000 administration kit. Do notuse a household measuring cup or measuring spoon.
- Each ZX2000 administration kit comes with two oral syringes. Keep the second oral syringe as a spare.
- Rinse and dry the mixing bottle and dosing cup before first use. Do notuse the dosing cup, mixing bottle, or oral syringe if it appears dirty or damaged.
- Each ZX2000 administration kit can be used for 6 months. Contact your healthcare provider when you need a replacement.
- Contact your healthcare provider or pharmacist for a replacement if your mixing bottle, dosing cup, or oral syringe is damaged or if the markings are missing or no longer readable.
- Do notuse the packets if the tamper evident seal on the carton is broken.
- Mix KYGEVVI powder only with room temperature water. Do notmix KYGEVVI powder with any other liquids or foods.
- You may have KYGEVVI solution left over after taking or giving the 3 individual doses. Throw away (discard) any remaining KYGEVVI solution 16 hours after preparing or after taking or giving the 3 doses, whichever comes first.
- If powder spills out of a packet before use, do notuse the packet. Throw it awayand use a new KYGEVVI packet.
- If KYGEVVI solution comes in contact with your eyes or skin, rinse under cold water and contact your healthcare provider.
- KYGEVVI solution is compatible with most feeding tubes. Follow the steps in this Instructions for Use booklet to prepare your one-day supply of KYGEVVI. Then follow the feeding tube manufacturer instructions on how to give KYGEVVI using a feeding tube.
- Draw up the prescribed individual dose using a syringe compatible with the feeding tube. Give the KYGEVVI solution right away through the feeding tube.
- Flush any remaining KYGEVVI solution in the syringe or feeding tube until no solution is left. Ask your healthcare provider how much water to use.
- Throw away (discard) any remaining KYGEVVI solution 16 hours after preparing or after taking or giving the 3 doses, whichever comes first.
Important information
Storing KYGEVVI packets and oral solution:
- Store KYGEVVI packets at room temperature between 68°F to 77°F (20°C to 25°C).
- Store the prepared KYGEVVI solution at room temperature between 68°F to 77°F (20°C to 25°C) orin the refrigerator between 36°F to 46°F (2°C to 8°C).
- Make sure the dosing cup is screwed on tightly to the mixing bottle when storing your KYGEVVI solution to avoid spills or leaks.
- When storing or carrying the KYGEVVI solution, keep the mixing bottle upright.
- Use the KYGEVVI solution within 16 hours of preparing. Throw away (discard) any remaining solution 16 hours after preparing or after taking or giving the 3 doses, whichever comes first.
- Keep KYGEVVI and all medicines out of the reach of children.
Preparing your one-day supply of KYGEVVI
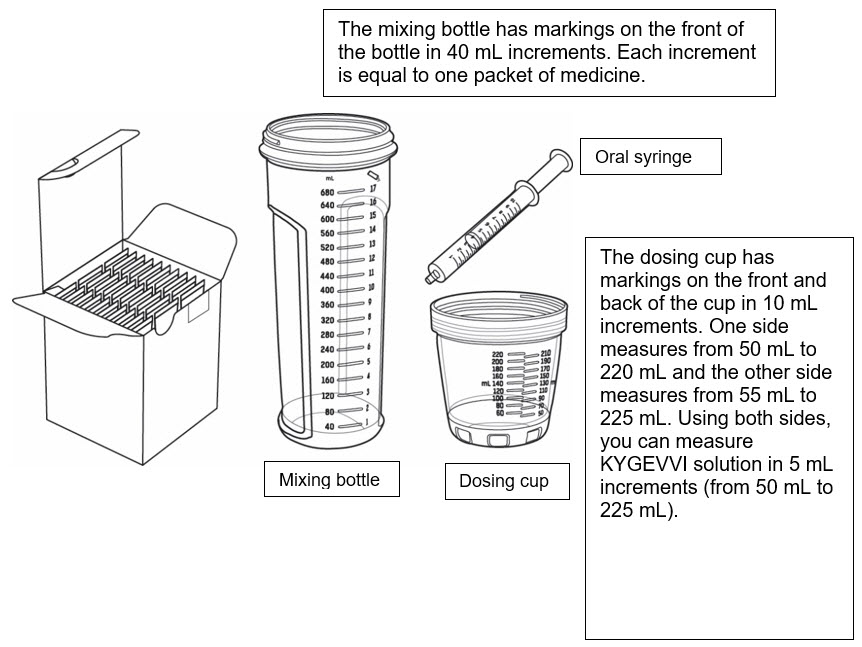
Get supplies ready
Step 1
- Wash your hands well with soap and water.
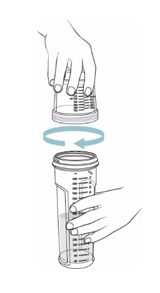
- Place the mixing bottle, dosing cup, and oral syringe (if you need one to measure your individual dose) on a clean, well-lit flat work surface. If the dosing cup is attached to the mixing bottle, unscrew it from the mixing bottle and set it down (see Figure B).
- Make sure "KYGEVVI" appears on the carton.
- Check the expiration date printed on the bottom of the KYGEVVI carton. When opening the KYGEVVI carton for the first time, break the tamper evident seal.
- Remove the prescribed number of KYGEVVI packets needed for your one-day supply of KYGEVVI out of the carton. Your one-day supply of KYGEVVI will be divided into 3 individual doses.
- Do notopen the KYGEVVI packets until Step 2.
Preparing your one-day supply of KYGEVVI
Measure water and add powder from packets
Step 2
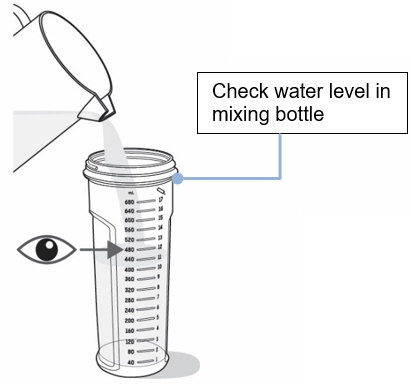
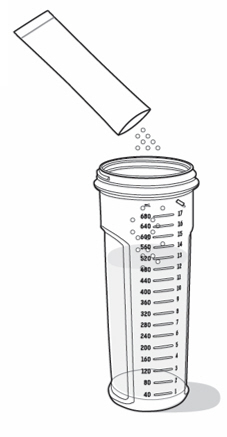
- On a flat surface, pour the prescribed amount of room temperature water into the mixing bottle (see
Figure C).
- Do notpour the water into the dosing cup.
- Important: Do notadd the powder from the packets to the mixing bottle before this step.
- Check to make sure the mixing bottle is filled with water up to the marking that matches the amount prescribed by your healthcare provider. The marking should also match the number of packets needed for your one-day supply (see Figure C).
- Check you have counted out the correct number of KYGEVVI packets for your one-day supply, as shown on your prescription.
- Tap the packet on a hard surface to settle the powder to the bottom of the packet away from the dotted line (see Figure D).
- Carefully fold and tear or cut along the dotted line (see Figure E). If you spill any powder, do notuse it. Throw the packet away and use a new packet.
- Empty the entire packet contents into the mixing bottle containing water. Be careful not to drop the packet into the mixing bottle (see Figure F).
- Pour only 1 packet into the mixing bottle at a time. Repeat Steps 2dto 2f, for each packet until you have poured the prescribed number of packets for your one-day supply.
Preparing your one-day supply of KYGEVVI
Mix and inspect medicine
Step 3
- Screw the dosing cup tightly onto the mixing bottle (see Figure G).
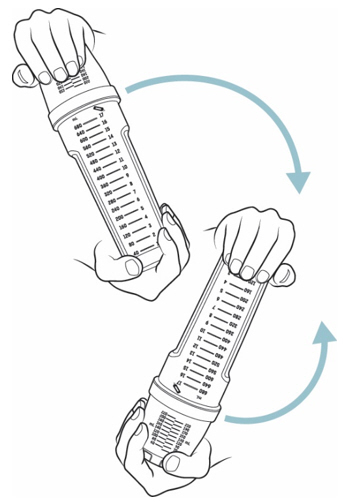
- Place one hand at the end of the mixing bottle and the other hand at the end of the dosing cup. Gently turn the mixing bottle upside down and back at least 20 times(see Figure H).
- Check the solution. If you see any powder, keep turning the mixing bottle upside down and back until the powder dissolves (see
Figure I).
- The mixed solution may be cloudy and have some remaining powder at the bottom or top. This is normal.
You have now prepared your one-day supply of KYGEVVI solution for 3 individual doses. Take KYGEVVI solution with food.
Figure G Figure H Figure I 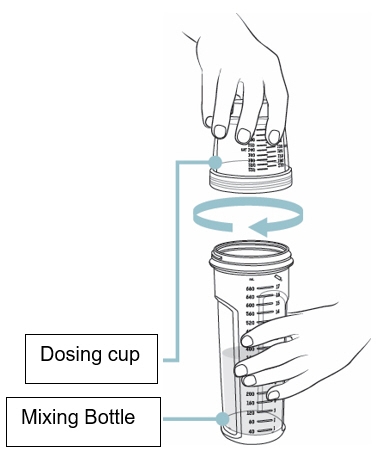
Dosing methods
How to measure your individual dose
There are 2 different methods to take or give KYGEVVI solution depending on your individual dose. Use the table below to identify which steps you should follow:
Individual doses 50 mL or more Individual doses lessthan 50 mL
(dosing cup used for dose preparation only)Example 100 mL Example 14 mL 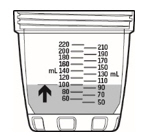
Use dosing cup
Follow Step 4Use oral syringe
Follow Step 5Individual doses 50 mL or more
Measure and take or give your individual dose
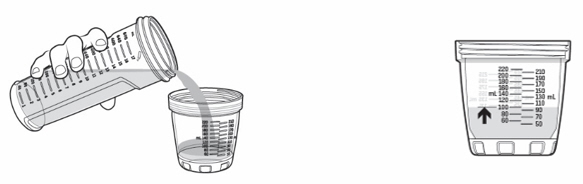
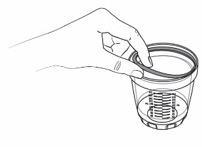
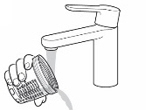
Step 4: Dosing cup
- Check to make sure the dosing cup is screwed on tightly to the mixing bottle.
- Mix the already prepared solution by slowly turning the mixing bottle upside down and back at least 3 times.
- Unscrew the dosing cup from the mixing bottle and place it on a flat surface.
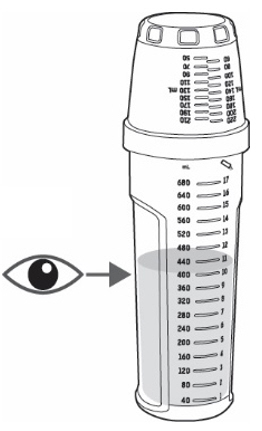
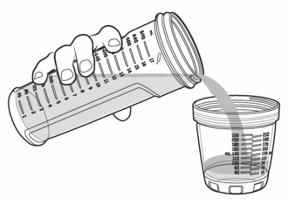
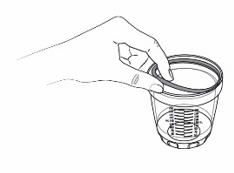
- Pour KYGEVVI solution from the mixing bottle into the dosing cup until it reaches the marking on the dosing cup for your prescribed individual dose (see Figure J). Note:Your dose may be different than the dose shown in Figure J.
- Drink or give the entire solution from the dosing cup (see Figure K).
- When it is time for the second or third individual dose, repeat Steps 4ato 4efor each individual dose.
- After the first or second individual dose, go to Step 6for instructions on how to clean your supplies and store KYGEVVI solution. After the third individual dose, go to Step 7for instructions on how to clean your supplies and dispose of unused KYGEVVI solution.
Individual doses less than 50 mL
Measure and take or give your individual dose
Step 5: Oral syringe
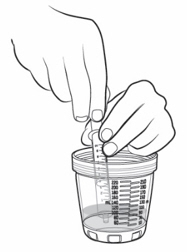
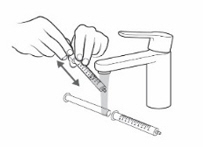
If your individual dose is more than 10 mL, you will need to use the same oral syringe more than 1 time. - Check to make sure the dosing cup is screwed on tightly to the mixing bottle.
- Mix the already prepared solution by slowly turning the mixing bottle upside down and back at least 3 times.
- Unscrew the dosing cup from the mixing bottle and place on a flat surface.
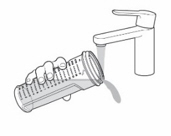
- Pour slightly more than the amount of KYGEVVI solution needed for your prescribed individual dose into the dosing cup (see Figure L).
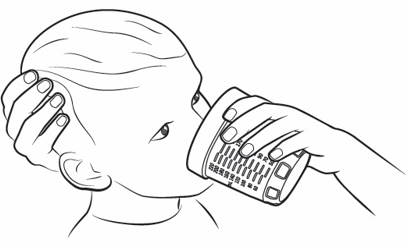
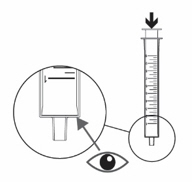
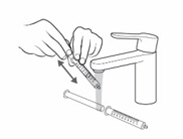
- Push the plunger of the oral syringe all the way down to make sure there is no air in the oral syringe when measuring the dose (see Figure M).
- Place the tip of the oral syringe into the dosing cup with the oral solution. Fill the oral syringe by pulling the plunger back until it reaches the marking on the oral syringe that matches your prescribed individual dose (see
Figure N).
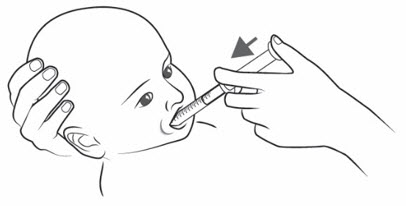
Note:If your individual dose is more than 10 mL, you will need to use the same oral syringe more than 1 time. Follow your healthcare provider's instructions on how to measure your dose. - Place the tip of the oral syringe into the mouth and point the tip towards the inside of either cheek (see
Figure O).
- If you are giving KYGEVVI to a young child, the child must be seated and held in place to prevent the oral solution from going down the wrong way or choking.
- Slowly push the plunger all the way down until the oral syringe is empty (see Figure O).
- If your prescribed individual dose is more than 10 mL, repeat Steps 5dto 5huntil you take or give the full individual dose.
- Pour back any remaining solution from the dosing cup into the mixing bottle.
- When it is time for the second or third individual dose, repeat Steps 5ato 5jfor each individual dose.
- After the first or second individual dose, go to Step 6for instructions on how to clean your supplies and store KYGEVVI solution. After the third individual dose, go to Step 7for instructions on how to clean your supplies and dispose of unused KYGEVVI solution.
Between individual doses
Clean up after first and second individual doseStep 6
After you complete the first or second individual dose:- Rinse the dosing cup with cold water after each use (see Figure P).
- Dry the dosing cup with a clean, dry towel.
- After the dosing cup is dry, screw the dosing cup tightly onto the mixing bottle (see Figure Q) and store it at room temperature or in the refrigerator until it is time for the next individual dose.
- If you used the oral syringe, clean it with cold water:
- Rinse the oral syringe with cold water by filling the oral syringe with water and pushing it back out (see Figure R). Then remove the plunger from the barrel and rinse the plunger and barrel under running tap water until it is clean (see Figure R).
- Let the oral syringe barrel and plunger dry in the open air. After the oral syringe barrel and plunger are dry, put the plunger back into the barrel.
- Do notwash the dosing cup or oral syringe in the dishwasher.
End-of-day clean up
Pour out and clean up after third individual dose
Step 7
After you take or give the third individual dose, throw away any remaining KYGEVVI solution in the sink.
Do not save KYGEVVI solution for another day.- Remove the seal from the dosing cup to thoroughly clean it (see Figure S).
- Clean the mixing bottle, dosing cup and seal by hand with soap and warm water. Use a brush to remove any residue left in the mixing bottle or dosing cup (see Figure T).
- Dry the mixing bottle, dosing cup and seal with a clean towel. Put the dry seal back into the dosing cup, with the thin side of the sealfacing the groove.
- If you used the oral syringe, clean it with cold water:
- Rinse the oral syringe with cold water by filling the oral syringe with water and pushing it back out (see Figure U). Then remove the plunger from the barrel and rinse the plunger and barrel under running tap water until it is clean (see Figure U).
- Let the oral syringe barrel and plunger dry in the open air. After the oral syringe barrel and plunger are dry, put the plunger back into the barrel.
- Do notwash the mixing bottle, dosing cup, seal, or oral syringe in the dishwasher.
- Store all supplies in a clean, dry area out of the reach of children for the next day's use.
Dosing cup maintenance
Replacing the seal if misplaced or damaged
Changing the dosing cup seal
If you misplace the dosing cup seal or you notice leakage when the dosing cup is screwed on tightly to the mixing bottle, change the seal using one of the two spare seals provided in the ZX2000 administration kit. Follow these steps to replace the seal:
- Remove the seal in the dosing cup (see Figure V). Skip this step if you misplaced the seal.
- Wash the dosing cup groove with warm water (see Figure W).
- Get a new seal from the spare seal box (see Figure X).
- Insert the seal into the groove of the dosing cup with the thin side of the sealfacing the groove (see Figure Y).
Contact Information
Contact your healthcare provider or pharmacist if you have any questions about this Instructions for Use. You may also contact UCBCares at 1-844-599-CARE (2273) for assistance.
KYGEVVI manufactured for: UCB, Inc., Smyrna, GA 30080
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Approved: 11/2025 -
Instructions for use
-
PRINCIPAL DISPLAY PANEL - 30 Packet Carton
NDC: 50474-350-30
Rx onlykygevvi ®
(doxecitine and doxribtimine)
Powder for Oral Solution2 g/2 g per packet
Only use the dosing cup, mixing bottle, and oral syringes
provided with your separate ZX2000 administration kit.See enclosed Instructions for Use for important preparation
and administration instructions.30 packets
Each 4 g packet contains 2 g doxecitine and 2 g doxribtimine.
-
INGREDIENTS AND APPEARANCE
KYGEVVI
doxecitine and doxribtimine powder, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50474-350 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DOXECITINE (UNII: 0W860991D6) (DOXECITINE - UNII:0W860991D6) DOXECITINE 2 g DOXRIBTIMINE (UNII: VC2W18DGKR) (DOXRIBTIMINE - UNII:VC2W18DGKR) DOXRIBTIMINE 2 g Product Characteristics Color white (white to off-white) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50474-350-30 30 in 1 CARTON 03/01/2026 1 NDC: 50474-350-01 1 in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219792 03/01/2026 Labeler - UCB, Inc. (028526403)
Trademark Results [KYGEVVI]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 KYGEVVI 98321189 not registered Live/Pending |
ZOGENIX, INC. 2023-12-19 |
 KYGEVVI 97769964 not registered Live/Pending |
ZOGENIX, INC. 2023-01-26 |
 KYGEVVI 97070384 not registered Live/Pending |
ZOGENIX, INC. 2021-10-12 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.