ALYFTREK- vanzacaftor, tezacaftor, and deutivacaftor tablet, film coated
ALYFTREK by
Drug Labeling and Warnings
ALYFTREK by is a Prescription medication manufactured, distributed, or labeled by Vertex Pharmaceuticals Incorporated. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ALYFTREK safely and effectively. See full prescribing information for ALYFTREK.
ALYFTREK (vanzacaftor, tezacaftor, and deutivacaftor tablets), for oral use
Initial U.S. Approval: 2024WARNING: DRUG-INDUCED LIVER INJURY AND LIVER FAILURE
See full prescribing information for complete boxed warning.
- Elevated transaminases have been observed in patients treated with ALYFTREK (5.1, 6).
- Cases of serious and potentially fatal drug-induced liver injury and liver failure leading to transplantation and death were reported in patients who were taking ELX/TEZ/IVA, a drug containing the same or similar active ingredients as ALYFTREK (5.1).
- Assess liver function tests (ALT, AST, alkaline phosphatase, bilirubin) in all patients prior to initiating ALYFTREK, every month for first 6 months, every 3 months for next 12 months, then at least annually (2.1, 5.1).
- Interrupt ALYFTREK for significant elevations in LFTs or signs or symptoms of liver injury. Follow patients closely with clinical and laboratory monitoring until abnormalities resolve (5.1).
- Resume ALYFTREK if abnormalities resolve and only if the benefit is expected to outweigh the risk (5.1).
- ALYFTREK should not be used in patients with severe hepatic impairment (Child-Pugh Class C). ALYFTREK is not recommended in patients with moderate hepatic impairment (Child-Pugh Class B) (2.4, 5.1, 8.7, 12.3).
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
ALYFTREK is a combination of deutivacaftor, a CFTR potentiator, tezacaftor, and vanzacaftor indicated for the treatment of cystic fibrosis (CF) in adult and pediatric patients aged 6 years and older who have a clinical diagnosis of CF and who have at least one variant in the CFTR gene that is either responsive based on clinical and/or in vitro data or results in production of CFTR protein. (1, 12.1)
If the patient's genotype is unknown, an FDA-cleared CF genetic test should be used to confirm the presence of at least one variant in the CFTR gene that is either responsive based on clinical and/or in vitro data or results in production of CFTR protein. (1)
DOSAGE AND ADMINISTRATION
Prior to initiating ALYFTREK obtain liver function tests (ALT, AST, alkaline phosphatase, and bilirubin) in all patients. Monitor liver function tests every month during the first 6 months of treatment, then every 3 months during the next 12 months, then at least annually thereafter. (2.1, 5.1)
Recommended Dosage for Adult and Pediatric Patients Aged 6 Years and Older (with fat-containing food) (2.2) Age Weight Once Daily Oral Dosage 6 to less than 12 years old Less than 40 kg Three tablets of vanzacaftor 4 mg/tezacaftor 20 mg/deutivacaftor 50 mg Greater than or equal to 40 kg Two tablets of vanzacaftor 10 mg/tezacaftor 50 mg/deutivacaftor 125 mg 12 years and older Any Weight Two tablets of vanzacaftor 10 mg/tezacaftor 50 mg/deutivacaftor 125 mg - Should not be used in patients with severe hepatic impairment. Use not recommended in patients with moderate hepatic impairment unless the benefit outweighs the risk. If used, no dose adjustment is recommended. Liver function tests should be closely monitored. (2.4, 5.1, 6.1, 8.7)
- See full prescribing information for dosage modifications for concomitant use of ALYFTREK with strong or moderate CYP3A inhibitors. (2.3, 5.7, 7.1)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Drug-Induced Liver Injury and Liver Failure: Elevated transaminases have been observed in patients treated with ALYFTREK. Cases of serious and potentially fatal drug-induced liver injury and liver failure have been reported with a drug that contains the same or similar active ingredients as ALYFTREK. Assess liver function tests (ALT, AST, alkaline phosphatase, and bilirubin) in all patients prior to initiating and throughout treatment with ALYFTREK. Interrupt ALYFTREK in the event of significant elevations in liver function tests or signs or symptoms of liver injury. ALYFTREK should not be used in patients with severe hepatic impairment (Child-Pugh Class C). ALYFTREK is not recommended in patients with moderate hepatic impairment (Child-Pugh Class B). (2.4, 5.1, 8.7)
- Hypersensitivity Reactions: Hypersensitivity reactions, including anaphylaxis, have been reported in the postmarketing setting for drugs containing elexacaftor, tezacaftor, and/or ivacaftor. If signs or symptoms of serious hypersensitivity reactions develop during ALYFTREK treatment, discontinue ALYFTREK and initiate appropriate therapy. (5.2)
- Patients Who Discontinued or Interrupted Elexacaftor-, Tezacaftor-, or Ivacaftor-Containing Drugs Due to Adverse Reactions: Consider benefits and risks before using ALYFTREK in patients who discontinued or interrupted elexacaftor-, tezacaftor-, or ivacaftor-containing drugs due to adverse reactions. If ALYFTREK is used, closely monitor for adverse reactions as clinically appropriate. (5.3)
- Intracranial Hypertension: Intracranial hypertension (IH) has been reported in the postmarketing setting with use of drugs containing the same or similar active ingredients as ALYFTREK. If an unusual headache or visual disturbances occur during treatment, and IH is suspected, interrupt ALYFTREK and refer for prompt medical evaluation. (5.4)
- Neuropsychiatric Events, Including Suicidal Thoughts and Behaviors: Serious neuropsychiatric events, including symptoms of anxiety, depression, suicidal ideation and behavior, and sleep disturbances, have been reported in the postmarketing setting for ALYFTREK or drugs containing the same or similar active ingredients. Monitor patients closely for new or worsening symptoms. Consider the risks and benefits for the individual patient to determine if therapy with ALYFTREK should be interrupted at the occurrence of neuropsychiatric symptoms. (5.5)
- Reduced Effectiveness in Patients with Concomitant Use with CYP3A Inducers: Concomitant use with strong and moderate CYP3A inducers decreased vanzacaftor, tezacaftor, and deutivacaftor exposure, which may reduce ALYFTREK efficacy. Therefore, concomitant use is not recommended. (5.6, 7.1)
- Adverse Reactions with Concomitant Use with CYP3A Inhibitors: Concomitant use with strong or moderate CYP3A inhibitors increased vanzacaftor, tezacaftor, and deutivacaftor exposure, which may increase the risk of ALYFTREK associated adverse reactions. Reduce the ALYFTREK dosage with concomitant use. (2.3, 5.7, 7.1)
- Cataracts: Non-congenital lens opacities/cataracts have been reported in patients with CF aged 18 years or less treated with drugs containing ivacaftor. Baseline and follow up ophthalmological examinations are recommended in pediatric patients treated with ALYFTREK. (5.8, 8.4)
ADVERSE REACTIONS
Most common adverse reactions to ALYFTREK (≥5% of patients and at a frequency higher than ELX/TEZ/IVA by ≥1%) were cough, nasopharyngitis, upper respiratory tract infection, headache, oropharyngeal pain, influenza, fatigue, increased ALT, rash, increased AST, and sinus congestion. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Vertex Pharmaceuticals Incorporated at 1-877-634-8789 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: DRUG-INDUCED LIVER INJURY AND LIVER FAILURE
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Laboratory Testing Prior to ALYFTREK Initiation and During Treatment
2.2 Recommended Dosage
2.3 Dosage Modification for Strong or Moderate CYP3A Inhibitors
2.4 Recommended Dosage for Patients with Hepatic Impairment
2.5 Recommendations Regarding Missed Dose(s)
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Drug-Induced Liver Injury and Liver Failure
5.2 Hypersensitivity Reactions, Including Anaphylaxis
5.3 Patients Who Discontinued or Interrupted Elexacaftor-, Tezacaftor-, or Ivacaftor-Containing Drugs Due to Adverse Reactions
5.4 Intracranial Hypertension
5.5 Neuropsychiatric Events, Including Suicidal Thoughts and Behaviors
5.6 Reduced Effectiveness with Concomitant Use with CYP3A Inducers
5.7 Adverse Reactions with Concomitant Use with CYP3A Inhibitors
5.8 Cataracts
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs and Grapefruit on ALYFTREK
7.2 Effect of ALYFTREK on Other Drugs
7.3 Drugs with No Clinically Significant Interactions with ALYFTREK
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: DRUG-INDUCED LIVER INJURY AND LIVER FAILURE
Elevated transaminases have been observed in patients treated with ALYFTREK. Cases of serious and potentially fatal drug-induced liver injury and liver failure were reported in patients who were taking a fixed-dose combination drug containing elexacaftor, tezacaftor, and ivacaftor, which contains the same or similar active ingredients as ALYFTREK. Liver injury has been reported within the first month of therapy and up to 15 months following initiation of elexacaftor/tezacaftor/ivacaftor [see Warnings and Precautions (5.1) and Adverse Reactions (6)].
Assess liver function tests (ALT, AST, alkaline phosphatase, and bilirubin) in all patients prior to initiating ALYFTREK, every month during the first 6 months of treatment, then every 3 months for the next 12 months, then at least annually thereafter. Consider more frequent monitoring for patients with a history of liver disease or elevated liver function tests at baseline [see Dosage and Administration (2.1), Warnings and Precautions (5.1), Adverse Reactions (6), and Use in Specific Populations (8.7)].
Interrupt ALYFTREK for significant elevations in liver function tests or in the event of signs or symptoms of liver injury. Consider referral to a hepatologist. Follow patients closely with clinical and laboratory monitoring until abnormalities resolve. If abnormalities resolve, resume treatment only if the benefit is expected to outweigh the risk. Closer monitoring is advised after resuming ALYFTREK [see Warnings and Precautions (5.1)].
ALYFTREK should not be used in patients with severe hepatic impairment (Child-Pugh Class C). ALYFTREK is not recommended in patients with moderate hepatic impairment (Child-Pugh Class B) and should only be considered when there is a clear medical need, and the benefit outweighs the risk. If used, monitor patients closely [see Dosage and Administration (2.4), Warnings and Precautions (5.1), Adverse Reactions (6), Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
-
1 INDICATIONS AND USAGE
ALYFTREK is indicated for the treatment of cystic fibrosis (CF) in adult and pediatric patients 6 years of age and older who have a clinical diagnosis of CF and who have at least one variant in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that is either responsive based on clinical and/or in vitro data (see Table 5) or results in production of CFTR protein [see Clinical Pharmacology (12.1)].
If the patient's genotype is unknown, an FDA-cleared CF genetic test should be used to confirm the presence of at least one variant in the CFTR gene that is either responsive based on clinical and/or in vitro data or results in production of CFTR protein.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Laboratory Testing Prior to ALYFTREK Initiation and During Treatment
Prior to initiating ALYFTREK, obtain liver function tests (ALT, AST, alkaline phosphatase, and bilirubin) for all patients. Monitor liver function tests every month during the first 6 months of treatment, then every 3 months for the next 12 months, then at least annually thereafter. Consider more frequent monitoring for patients with a history of liver disease, elevated liver function tests at baseline, or a history of elevated liver function tests with drugs containing elexacaftor, tezacaftor, and/or ivacaftor [see Warnings and Precautions (5.1) and Use in Specific Populations (8.7)].
2.2 Recommended Dosage
The recommended ALYFTREK dosage in adult and pediatric patients aged 6 years and older is provided in Table 1. Administer ALYFTREK orally (swallow the tablets whole) with fat-containing food, once daily, at approximately the same time each day [see Clinical Pharmacology (12.3)]. Examples of meals or snacks that contain fat are those prepared with butter or oils or those containing eggs, peanut butter, cheeses, nuts, whole milk, or meats.
Table 1: Recommended Dosage of ALYFTREK in Adult and Pediatric Patients Aged 6 Years and Older Age Weight Once Daily Oral Dosage 6 to less than 12 years old Less than 40 kg Three tablets of vanzacaftor 4 mg/tezacaftor 20 mg/deutivacaftor 50 mg (total dose of vanzacaftor 12 mg/tezacaftor 60 mg/ deutivacaftor 150 mg) Greater than or equal to 40 kg Two tablets of vanzacaftor 10 mg/tezacaftor 50 mg/deutivacaftor 125 mg (total dose of vanzacaftor 20 mg/tezacaftor 100 mg/ deutivacaftor 250 mg) 12 years and older Any weight Two tablets of vanzacaftor 10 mg/tezacaftor 50 mg/deutivacaftor 125 mg (total dose of vanzacaftor 20 mg/tezacaftor 100 mg/ deutivacaftor 250 mg) 2.3 Dosage Modification for Strong or Moderate CYP3A Inhibitors
Table 2 describes the recommended dosage modification for ALYFTREK when used concomitantly with strong or moderate CYP3A inhibitors [see Warnings and Precautions (5.7)]. Administer ALYFTREK orally (swallow the tablets whole) with fat-containing food, once daily, at approximately the same time each day [see Clinical Pharmacology (12.3)].
Table 2: Dosage Modification for Concomitant Use of ALYFTREK with Strong or Moderate CYP3A Inhibitors in Adult and Pediatric Patients Aged 6 Years and Older Age Weight Moderate CYP3A Inhibitors Strong CYP3A Inhibitors 6 to less than 12 years old Less than 40 kg Two tablets of vanzacaftor 4 mg/tezacaftor 20 mg/deutivacaftor 50 mg every other day (total dose of vanzacaftor 8 mg/tezacaftor 40 mg/deutivacaftor 100 mg) Two tablets of vanzacaftor 4 mg/tezacaftor 20 mg/deutivacaftor 50 mg once a week (total dose of vanzacaftor 8 mg/tezacaftor 40 mg/deutivacaftor 100 mg) Greater than or equal to 40 kg One tablet of vanzacaftor 10 mg/tezacaftor 50 mg/deutivacaftor 125 mg every other day One tablet of vanzacaftor 10 mg/tezacaftor 50 mg/deutivacaftor 125 mg once a week 12 years and older Any weight One tablet of vanzacaftor 10 mg/tezacaftor 50 mg/deutivacaftor 125 mg every other day One tablet of vanzacaftor 10 mg/tezacaftor 50 mg/deutivacaftor 125 mg once a week 2.4 Recommended Dosage for Patients with Hepatic Impairment
- Severe Hepatic Impairment (Child-Pugh Class C): ALYFTREK should not be used in patients with severe hepatic impairment (HI) (Child-Pugh Class C).
- Moderate Hepatic Impairment (Child-Pugh Class B): The use of ALYFTREK in patients with moderate HI (Child-Pugh Class B) is not recommended. Use of ALYFTREK should only be considered in patients with moderate HI when there is a clear medical need, and the benefit outweighs the risk. If used, the recommended dosage in patients with moderate HI is the same as for patients with normal hepatic function. Liver function tests should be closely monitored [see Dosage and Administration (2.1, 2.2)].
- Mild Hepatic Impairment (Child-Pugh Class A): The recommended dosage of ALYFTREK in patients with mild HI (Child-Pugh Class A) is the same as in patients with normal hepatic function. Liver function tests should be closely monitored [see Dosage and Administration (2.1, 2.2)].
2.5 Recommendations Regarding Missed Dose(s)
If 6 hours or less have passed since the missed dose, take the missed dose as soon as possible and continue on the original schedule.
If more than 6 hours have passed since the missed dose, skip the missed dose, and continue on the original schedule the next day.
-
3 DOSAGE FORMS AND STRENGTHS
Tablets:
- Fixed-dose combination containing vanzacaftor 4 mg (equivalent to 4.24 mg of vanzacaftor calcium dihydrate), tezacaftor 20 mg, and deutivacaftor 50 mg. Each tablet is purple, round-shaped, film-coated, debossed with "V4" on one side and plain on the other.
- Fixed-dose combination containing vanzacaftor 10 mg (equivalent to 10.6 mg of vanzacaftor calcium dihydrate), tezacaftor 50 mg, and deutivacaftor 125 mg. Each tablet is purple, oblong-shaped, film-coated, debossed with "V10" on one side and plain on the other.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Drug-Induced Liver Injury and Liver Failure
Elevated transaminases have been observed in patients treated with ALYFTREK [see Adverse Reactions (6.1)]. Cases of serious and potentially fatal drug-induced liver injury and liver failure have been reported in patients with and without a history of liver disease who were taking a fixed-dose combination drug containing elexacaftor, tezacaftor, and ivacaftor (ELX/TEZ/IVA), which contains the same or similar active ingredients as ALYFTREK. Liver injury has been reported within the first month of therapy and up to 15 months following initiation of ELX/TEZ/IVA.
Assess liver function tests (ALT, AST, alkaline phosphatase, and bilirubin) in all patients prior to initiating ALYFTREK. Assess liver function tests every month during the first 6 months of treatment, then every 3 months for the next 12 months, then at least annually thereafter. Consider more frequent monitoring in patients with a history of liver disease, elevated liver function tests at baseline, or a history of elevated liver function tests with drugs containing ELX, TEZ and/or IVA. [see Dosage and Administration (2.4) and Use in Specific Populations (8.7)].
Interrupt ALYFTREK in the event of signs or symptoms of liver injury. These may include:
- Significant elevations in liver function tests (e.g., ALT or AST >5× the upper limit of normal (ULN) or ALT or AST >3× ULN with bilirubin >2× ULN)
- Clinical signs or symptoms suggestive of liver injury (e.g., jaundice, right upper quadrant pain, nausea, vomiting, altered mental status, ascites)
Consider referral to a hepatologist and follow patients closely with clinical and laboratory monitoring until abnormalities resolve. If abnormalities resolve and if the benefit is expected to outweigh the risk, resume ALYFTREK treatment with close monitoring.
ALYFTREK should not be used in patients with severe hepatic impairment (Child-Pugh Class C). ALYFTREK is not recommended in patients with moderate hepatic impairment (Child-Pugh Class B) and should only be considered when there is a clear medical need, and the benefit outweighs the risk. If used, monitor patients closely [see Dosage and Administration (2.4), Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
5.2 Hypersensitivity Reactions, Including Anaphylaxis
Hypersensitivity reactions, including cases of anaphylaxis, have been reported in the postmarketing setting of drugs containing ELX, TEZ, and/or IVA (the same or similar active ingredients in ALYFTREK). If signs or symptoms of serious hypersensitivity reactions develop during ALYFTREK treatment, discontinue ALYFTREK and institute appropriate therapy. Consider the benefits and risks for the individual patient to determine whether to resume treatment with ALYFTREK.
5.3 Patients Who Discontinued or Interrupted Elexacaftor-, Tezacaftor-, or Ivacaftor-Containing Drugs Due to Adverse Reactions
There are no available safety data for ALYFTREK in patients who previously discontinued or interrupted treatment with drugs containing elexacaftor, tezacaftor, or ivacaftor due to adverse reactions. Consider the benefits and risks before using ALYFTREK in these patients. If ALYFTREK is used in these patients, closely monitor for adverse reactions as clinically appropriate.
5.4 Intracranial Hypertension
Cases of intracranial hypertension (IH) have been reported in the postmarketing setting with the use of drugs containing the same or similar active ingredients as ALYFTREK [see Adverse Reactions (6.2)]. Clinical manifestations of IH include headache, blurred vision, diplopia, and potential vision loss; papilledema can be found on fundoscopy. If an unusual headache or visual disturbances occur during treatment, and IH is suspected, interrupt ALYFTREK and refer for prompt medical evaluation. Consider the benefits and risks for the individual patient to determine whether to resume treatment with ALYFTREK. Patients should be monitored until IH resolution and for recurrence. Patients with elevated vitamin A levels may be at increased risk.
5.5 Neuropsychiatric Events, Including Suicidal Thoughts and Behaviors
Serious neuropsychiatric events, including symptoms of anxiety, depression, suicidal ideation and behavior, and sleep disturbances, have been reported in the postmarketing setting in patients taking ALYFTREK or drugs containing the same or similar active ingredients [see Adverse Reactions (6.2)]. The events were reported in adult and pediatric patients with and without a previous history of neuropsychiatric symptoms. Symptoms may occur within the first three months of treatment initiation.
Assess patients for baseline neuropsychiatric symptoms and monitor for new or worsening symptoms of anxiety, depression, suicidal ideation or behavior, or sleep disturbances. Consider the benefits and risks for the individual patient to determine if therapy with ALYFTREK should be interrupted at the occurrence of neuropsychiatric symptoms and whether to resume therapy with symptom improvement.
5.6 Reduced Effectiveness with Concomitant Use with CYP3A Inducers
Following concomitant use of strong or moderate CYP3A inducers with ALYFTREK, exposures of vanzacaftor, tezacaftor, and deutivacaftor were decreased, which may reduce ALYFTREK effectiveness. Concomitant use with strong or moderate CYP3A inducers is not recommended [see Drug Interactions (7.1)].
5.7 Adverse Reactions with Concomitant Use with CYP3A Inhibitors
Following concomitant use of strong or moderate CYP3A inhibitors with ALYFTREK, exposures of vanzacaftor, tezacaftor, and deutivacaftor were increased, which may increase the risk of ALYFTREK-associated adverse reactions. Reduce the ALYFTREK dosage with concomitant use of strong or moderate CYP3A inhibitors [see Dosage and Administration (2.3) and Drug Interactions (7.1)].
5.8 Cataracts
Cases of non-congenital lens opacities have been reported in pediatric patients treated with drugs containing ivacaftor (which is similar to an active ingredient in ALYFTREK). Although other risk factors were present (such as corticosteroid use, exposure to radiation) in some cases, a possible risk attributable to ivacaftor treatment cannot be excluded. Baseline and follow-up ophthalmological examinations are recommended in pediatric patients treated with ALYFTREK [see Use in Specific Populations (8.4)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Drug-Induced Liver Injury and Liver Failure [see Warnings and Precautions (5.1)]
- Hypersensitivity Reactions, Including Anaphylaxis [see Warnings and Precautions (5.2)]
- Patients Who Discontinued or Interrupted Elexacaftor-, Tezacaftor-, or Ivacaftor-Containing Drugs Due to Adverse Reactions [see Warnings and Precautions (5.3)]
- Intracranial Hypertension [see Warnings and Precautions (5.4)]
- Neuropsychiatric Events, Including Suicidal Thoughts and Behaviors [see Warnings and Precautions (5.5)]
- Cataracts [see Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The adverse reactions data below are from clinical trials of ALYFTREK in patients 6 years of age and older with CF with at least one responsive CFTR variant who were able to tolerate ELX/TEZ/IVA. Adverse reactions data in patients who previously discontinued or interrupted ELX/TEZ/IVA due to adverse reactions are not available.
Adverse Reactions in Patients Aged 12 Years and Older with CF
The safety of ALYFTREK is based on 480 patients with CF aged 12 years and older who have at least one F508del variant or another responsive variant in the CFTR gene in two, 52-week, active-controlled trials (Trials 1 and 2) [see Clinical Studies (14)]. In both trials, patients received a fixed-dose combination drug containing elexacaftor, tezacaftor, and ivacaftor (ELX/TEZ/IVA) in a 4-week run-in period and then were subsequently randomized to continue ELX/TEZ/IVA (elexacaftor 200 mg/tezacaftor 100 mg/ivacaftor 150 mg in the morning and ivacaftor 150 mg in the evening) or receive ALYFTREK (vanzacaftor 20 mg/tezacaftor 100 mg/deutivacaftor 250 mg) once daily. Patients with a history of prior intolerance to ELX/TEZ/IVA (i.e., patients who discontinued or interrupted treatment due to adverse reactions) were excluded. Trials 1 and 2 were not designed to evaluate meaningful comparisons of the incidence of adverse reactions between the ALYFTREK and ELX/TEZ/IVA treatment groups. For additional information regarding ELX/TEZ/IVA adverse reactions, refer to ELX/TEZ/IVA Prescribing Information.
In Trial 1 and Trial 2 combined, the proportion of patients who discontinued treatment prematurely due to adverse reactions were 3.8% and 3.7% in ALYFTREK and ELX/TEZ/IVA treatment groups, respectively.
Serious adverse reactions that occurred more frequently with ALYFTREK treatment than with ELX/TEZ/IVA treatment that occurred in 2 or more patients (≥0.4%) were influenza (1.5%), increased AST (0.4%), increased GGT (0.4%), depression (0.4%), and syncope (0.4%).
Table 3: Adverse Reactions Occurring in ≥5% of ALYFTREK-Treated Patients and ≥1% Higher than ELX/TEZ/IVA-Treated Patients Aged 12 Years and Older with CF Who Had at Least One F508del Variant or Responsive Variant in the CFTR Gene (Trials 1 and 2) Adverse Reactions ALYFTREK
N=480ELX/TEZ/IVA
N=491Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; ELX, elexacaftor; IVA, ivacaftor; TEZ, tezacaftor - * Cough is composed of several similar terms including productive cough.
- † Upper respiratory tract infection is composed of several similar terms including viral upper respiratory tract infection.
Cough* 120 (25%) 116 (24%) Nasopharyngitis 102 (21%) 95 (19%) Upper respiratory tract infection† 101 (21%) 97 (20%) Headache 76 (16%) 63 (13%) Oropharyngeal pain 69 (14%) 60 (12%) Influenza 52 (11%) 26 (5%) Fatigue 51 (11%) 46 (9%) ALT increased 38 (8%) 29 (6%) Rash 37 (8%) 22 (4%) AST increased 33 (7%) 27 (5%) Sinus congestion 32 (7%) 15 (3%) Adverse events that occurred in ≥5% of ELX/TEZ/IVA-treated patients at a similar or higher incidence than the ALYFTREK-treated patients included: infective pulmonary exacerbation of CF, COVID-19, diarrhea, abdominal pain, pyrexia, nasal congestion, increased sputum, increased blood creatinine phosphokinase, rhinorrhea, hemoptysis, nausea, back pain, arthralgia, constipation, sinusitis, dyspnea, and vomiting.
Liver Function Test Elevations
The incidence of adverse reactions of transaminase elevations was 9% in ALYFTREK-treated patients and 7.1% in ELX/TEZ/IVA-treated patients in Trials 1 and 2. In these trials, 1.5% of ALYFTREK-treated patients and 0.6% of ELX/TEZ/IVA-treated patients discontinued treatment for elevated transaminases. Table 4 shows the incidence of maximum transaminase (ALT or AST) elevations in Trials 1 and 2.
Table 4: Number and Incidence of Maximum Transaminase Elevation in Patients Aged 12 Years and Older with CF Who Had at Least One F508del Variant or Responsive Variant in the CFTR Gene (Trials 1 and 2) Maximum ALT or AST Elevation ALYFTREK
N=480ELX/TEZ/IVA*
N=491Abbreviations: ALT: alanine aminotransferase; AST, aspartate aminotransferase; ELX, elexacaftor; IVA, ivacaftor; TEZ, tezacaftor. - * Trials 1 and 2 were not designed to evaluate meaningful comparisons of safety between the ALYFTREK and ELX/TEZ/IVA treatment groups. For additional information regarding ELX/TEZ/IVA transaminase elevations, refer to ELX/TEZ/IVA Prescribing Information.
>3× ULN 29 (6%) 15 (3.1%) >5× ULN 12 (2.5%) 6 (1.2%) >8× ULN 6 (1.3%) 1 (0.2%) Rash
In Trials 1 and 2, the incidence of rash (e.g., rash, rash pruritic) was 11% in ALYFTREK-treated patients and 7.7% in ELX/TEZ/IVA-treated patients. The rashes were generally mild to moderate in severity. The incidence of rash was 9.4% in males and 13% in females with ALYFTREK treatment and 7.6% in males and 7.9% in females with ELX/TEZ/IVA treatment. A role of hormonal contraceptives in the occurrence of rash cannot be excluded [see Drug Interactions (7.3)].
Increased Creatine Phosphokinase
In Trials 1 and 2, the incidence of maximum creatine phosphokinase >5× the ULN was 7.9% with ALYFTREK treatment and 6.5% with ELX/TEZ/IVA treatment. Discontinuation due to increased creatinine phosphokinase was 0.2% for ALYFTREK-treated patients and 0.2% for ELX/TEZ/IVA-treated patients. Cases of rhabdomyolysis without renal involvement have been reported in patients who had recently exercised taking a fixed-dose combination drug containing ELX/TEZ/IVA (the same or similar active ingredients as ALYFTREK).
Increased Blood Pressure
Elevations in mean systolic and diastolic blood pressure have been reported in patients taking a fixed-dose combination drug containing ELX/TEZ/IVA (the same or similar active ingredients as ALYFTREK). The proportion of patients who had systolic blood pressure >140 mmHg and >10 mmHg increase from baseline on at least two occasions was 3.5% in ALYFTREK-treated patients and 3.3% in ELX/TEZ/IVA-treated patients. The proportion of patients who had diastolic blood pressure >90 mmHg and >5 mmHg increase from baseline on at least two occasions was 1.7% in ALYFTREK-treated patients and 1.8% in ELX/TEZ/IVA-treated patients. The mean systolic and diastolic blood pressures remained in the normal range from both ALYFTREK and ELX/TEZ/IVA treatment arms.
Adverse Reactions in Pediatric Patients Aged 6 to Less Than 12 Years with CF
A 24-week, open-label trial of ALYFTREK was conducted in 78 patients with CF aged 6 to less than 12 years with at least one variant responsive to ELX/TEZ/IVA (Trial 3). In Trial 3, patients who weighed less than 40 kg received ALYFTREK (vanzacaftor 12 mg/tezacaftor 60 mg/deutivacaftor 150 mg once daily) and patients who weighed 40 kg or more received ALYFTREK (vanzacaftor 20 mg/tezacaftor 100 mg/deutivacaftor 250 mg once daily). Adverse reactions for these patients were generally similar to those reported in Trial 1 and Trial 2. In Trial 3, the incidence of maximum transaminase (ALT or AST) >3×, >5×, and >8× ULN were 3.8%, 1.3%, and 0%, respectively.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of ALYFTREK or drugs containing the same or similar active ingredients as ALYFTREK. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Nervous System Disorders: intracranial hypertension
Psychiatric Disorders: anxiety, depression, suicidal ideation and behavior, insomnia
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs and Grapefruit on ALYFTREK
Strong or Moderate CYP3A Inducers
Concomitant use of ALYFTREK with strong or moderate CYP3A inducers is not recommended.
Vanzacaftor, tezacaftor, and deutivacaftor are substrates of CYP3A. Concomitant use of ALYFTREK with a strong or moderate CYP3A inducer decreases vanzacaftor, tezacaftor, and deutivacaftor exposure [see Clinical Pharmacology (12.3)] which may reduce ALYFTREK effectiveness [see Warnings and Precautions (5.6)].
Strong or Moderate CYP3A Inhibitors
Reduce the ALYFTREK dosage when used concomitantly with a strong or moderate CYP3A inhibitor [see Dosage and Administration (2.3)].
Vanzacaftor, tezacaftor, and deutivacaftor are CYP3A substrates. Concomitant use with a strong CYP3A inhibitor increases vanzacaftor, tezacaftor, and deutivacaftor exposure [see Clinical Pharmacology (12.3)], which may increase the risk of ALYFTREK adverse reactions [see Warnings and Precautions (5.7)]. Concomitant use with a moderate CYP3A inhibitor is predicted to increase vanzacaftor, tezacaftor, and deutivacaftor exposure [see Clinical Pharmacology (12.3)], which may increase the risk of ALYFTREK adverse reactions [see Warnings and Precautions (5.7)].
7.2 Effect of ALYFTREK on Other Drugs
P-glycoprotein (P-gp) Substrates
Unless otherwise recommended in the P-gp substrate Prescribing Information, monitor more frequently for adverse reactions with concomitant use of ALYFTREK with P-gp substrates where minimal concentration changes may lead to serious adverse reactions related to P-gp substrates.
Tezacaftor and deutivacaftor (components of ALYFTREK) are P-gp inhibitors. Administration of tezacaftor/ivacaftor increases exposure of P-gp substrates [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions related to these substrates.
Breast Cancer Resistance Protein (BCRP) Substrates
Unless otherwise recommended in the BCRP substrate Prescribing Information, monitor more frequently for adverse reactions with concomitant use of ALYFTREK with BCRP substrates where minimal concentration changes may lead to serious adverse reactions related to BCRP substrates.
Vanzacaftor (VNZ) and deutivacaftor (D-IVA) (components of ALYFTREK) are inhibitors of BCRP in vitro. Concomitant use of ALYFTREK with BCRP substrates may increase exposure of these substrates; however, this has not been studied clinically [see Clinical Pharmacology (12.3)].
CYP2C9 Substrates
Use caution when ALYFTREK is used concomitantly with CYP2C9 substrates. Monitor the international normalized ratio (INR) more frequently with concomitant use of ALYFTREK with warfarin.
This recommendation is based upon a mechanistic understanding of deutivacaftor pharmacokinetics (it is an inhibitor of CYP2C9 in vitro) [see Clinical Pharmacology (12.3)]. Concomitant use of ALYFTREK with CYP2C9 substrates may increase exposure of these substrates; however, this has not been studied clinically.
7.3 Drugs with No Clinically Significant Interactions with ALYFTREK
Ciprofloxacin
No clinically relevant effect on the exposure of tezacaftor was observed when tezacaftor/ivacaftor was used concomitantly with ciprofloxacin [see Clinical Pharmacology (12.3)].
Hormonal Contraceptives
No clinically significant differences in the pharmacokinetics of ethinyl estradiol/norethindrone containing hormonal contraceptives were observed when used concomitantly with tezacaftor in combination with ivacaftor and ivacaftor alone [see Clinical Pharmacology (12.3)]. No clinically significant differences in the pharmacokinetics of ethinyl estradiol/norethindrone containing hormonal contraceptives are expected when used in combination with ALYFTREK based upon a mechanistic understanding of vanzacaftor, tezacaftor, and deutivacaftor pharmacokinetics [see Clinical Pharmacology (12.3)]; however, this has not been studied clinically.
A role for hormonal contraceptives contributing to rash cannot be excluded [see Adverse Reactions (6.1)]. For patients with CF taking hormonal contraceptives who develop rash, consider interrupting ALYFTREK and hormonal contraceptives. Following the resolution of rash, consider resuming ALYFTREK without the hormonal contraceptives. If rash does not recur, resumption of hormonal contraceptives can be considered.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on ALYFTREK use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Although there are no animal reproduction studies with the concomitant administration of vanzacaftor, tezacaftor, and deutivacaftor, separate reproductive and developmental studies were conducted with vanzacaftor and tezacaftor in pregnant rats and rabbits. Deutivacaftor is a deuterated isotopologue of ivacaftor with a toxicity profile similar to ivacaftor. Reproductive and development studies were conducted with ivacaftor in pregnant rats and rabbits.
In animal embryo fetal development (EFD) studies, oral administration of vanzacaftor to pregnant rats and rabbits during organogenesis demonstrated no adverse developmental effects at doses that produced maternal exposures up to approximately 30 times the exposure at the maximum recommended human dose (MRHD) in rats and 22 times the MRHD in rabbits. Oral administration of tezacaftor to pregnant rats and rabbits during organogenesis demonstrated no adverse developmental effects at doses that produced maternal exposures up to approximately 3 times the exposure at the MRHD in rats and 0.2 times the MRHD in rabbits (based on summed AUCs of tezacaftor and the metabolite M1-TEZ). Oral administration of ivacaftor to pregnant rats and rabbits during organogenesis demonstrated no adverse developmental effects at doses that produced maternal exposures up to approximately 8 and 9 times the exposure at the MRHD, respectively (based on AUC of ivacaftor for rats and rabbits). No adverse developmental effects were observed after oral administration of vanzacaftor, tezacaftor, or ivacaftor to pregnant rats from the period of organogenesis through lactation at doses that produced maternal exposures approximately 18 times, 1 time, and 8 times the exposures at the MRHD, respectively (based on AUCs of vanzacaftor, tezacaftor and M1-TEZ, and ivacaftor) (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Vanzacaftor: In an EFD study, pregnant rats were administered vanzacaftor at oral doses of 2.5, 5, and 10 mg/kg/day during the period of organogenesis from gestation Days 6-17. Vanzacaftor did not cause adverse effects to the fetus at exposures up to 30 times the MRHD (based on AUC for vanzacaftor at maternal doses up to 10 mg/kg/day). In an EFD study, pregnant rabbits were administered vanzacaftor at oral doses of 10, 40, and 70 mg/kg/day during the period of organogenesis from gestation Days 7-20. Vanzacaftor did not cause adverse effects to the fetus at exposures up to 22 times the MRHD (based on AUC of vanzacaftor at maternal doses up to 40 mg/kg/day). The high dose of 70 mg/kg/day (71 times the exposure at the MRHD) produced maternal toxicity (i.e., mortality, abortion, decreased mean body weight or body weight gains) and was associated with findings of increased post-implantation loss, decreased live fetuses, decreased fetal body weight, and increased kidney malformations. In a pre- and postnatal development (PPND) study in pregnant rats administered vanzacaftor at oral doses of 2.5, 5, and 10 mg/kg/day from gestation Day 6 through lactation Day 18, vanzacaftor did not cause adverse developmental effects in pups at maternal doses up to 10 mg/kg/day (approximately 18 times the exposure at the MRHD). Placental transfer of vanzacaftor was observed in pregnant rats.
Tezacaftor: In an EFD study, pregnant rats were administered tezacaftor at oral doses of 25, 50, and 100 mg/kg/day during the period of organogenesis from gestation Days 6-17. Tezacaftor did not cause adverse effects to the fetus at exposures up to 3 times the MRHD (based on summed AUCs of tezacaftor and M1-TEZ). Maternal toxicity in rats was observed at ≥50 mg/kg/day (approximately ≥1 time the MRHD). In an EFD study, pregnant rabbits were administered tezacaftor at oral doses of 10, 25, and 50 mg/kg/day during the period of organogenesis from gestation Days 7-20. Tezacaftor did not cause adverse effects to the fetus at exposures up to 0.2 times the MRHD (based on summed AUCs of tezacaftor and M1-TEZ). Lower fetal body weights were observed in rabbits at a maternally toxic dose that produced exposures approximately 1 time the MRHD (based on summed AUCs of tezacaftor and M1-TEZ at a maternal dose of 50 mg/kg/day). In a PPND study, pregnant rats were administered tezacaftor at oral doses of 25, 50, and 100 mg/kg/day from gestation Day 6 through lactation Day 18. Tezacaftor had no adverse developmental effects on pups at an exposure of approximately 1 time the MRHD (based on summed AUCs for tezacaftor and M1-TEZ at a maternal dose of 25 mg/kg/day). Decreased fetal body weights and early developmental delays in pinna detachment, eye opening, and righting reflex occurred at a maternally toxic dose (based on maternal weight loss) that produced exposures approximately 2 times the exposure at the MRHD (based on summed AUCs for tezacaftor and M1-TEZ). Placental transfer of tezacaftor was observed in pregnant rats.
Deutivacaftor: Animal reproduction studies have not been conducted with deutivacaftor. However, as a deuterated isotopologue of ivacaftor with a toxicity profile similar to ivacaftor based on a 13-week single-agent repeat dose toxicity study, the reproductive and developmental toxicity data from ivacaftor can inform the developmental and reproductive risks associated with deutivacaftor.
In an EFD study, pregnant rats were administered ivacaftor at oral doses of 50, 100, and 200 mg/kg/day during the period of organogenesis from gestation Days 7-17. Ivacaftor did not cause adverse effects to the fetus at exposures up to 8 times the MRHD for deutivacaftor (based on AUC of ivacaftor in animal studies up to 200 mg/kg/day). In an EFD study, pregnant rabbits were administered ivacaftor at oral doses of 25, 50, and 100 mg/kg/day during the period of organogenesis from gestation Days 7-19. Ivacaftor did not cause adverse effects to the fetus at exposures up to 9 times the MRHD for deutivacaftor (based on AUC of ivacaftor in animal studies). Maternal toxicity (i.e., death, decreased food consumption, decreased mean body weight and body weight gain, decreased clinical condition, abortions) was observed at doses greater than or equal to 50 mg/kg/day (approximately 3 times the MRHD). In a PPND study, pregnant rats were administered ivacaftor at oral doses of 50, 100, and 200 mg/kg/day from gestation Day 7 through lactation Day 20. Ivacaftor had no effects on delivery or growth and development of offspring at exposures up to 8 times the MRHD (based on AUC for ivacaftor at maternal oral doses up to 100 mg/kg/day). Decreased fetal body weights were observed at a maternally toxic dose (200 mg/kg/day, 13 times the exposure at MHRD). Placental transfer of ivacaftor was observed in pregnant rats and rabbits.
8.2 Lactation
Risk Summary
There are no data on the presence of vanzacaftor, tezacaftor, or deutivacaftor or their metabolites in human milk, the effects on the breastfed infant, or the effects on milk production.
Vanzacaftor and tezacaftor are excreted into the milk of lactating female rats. Deutivacaftor has not been evaluated; however, ivacaftor is excreted into the milk of lactating female rats (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ALYFTREK and any potential adverse effects on the breastfed child from ALYFTREK or from the underlying maternal condition.
Data
The concentration of vanzacaftor, tezacaftor, or deutivacaftor in animal milk does not necessarily predict the concentration of drug in human milk.
Vanzacaftor: Lacteal excretion of vanzacaftor in rats was demonstrated following a single oral dose (10 mg/kg) of 14C-vanzacaftor administered 6 to 10 days postpartum to lactating dams. Exposure of 14C-vanzacaftor in milk was approximately 0.2 times the value observed in plasma (based on AUC0-72h).
Tezacaftor: Lacteal excretion of tezacaftor in rats was demonstrated following a single oral dose (30 mg/kg) of 14C-tezacaftor administered 6 to 10 days postpartum to lactating dams. Exposure of 14C-tezacaftor in milk was approximately 3.0 times higher than in plasma (based on AUC0-72h).
Deutivacaftor: Deutivacaftor has not been evaluated; however, ivacaftor is excreted into the milk of lactating female rats. Lacteal excretion of ivacaftor in rats was demonstrated following a single oral dose (100 mg/kg) of 14C-ivacaftor administered 9 to 10 days postpartum to lactating dams. Exposure of 14C-ivacaftor in milk was approximately 1.5 times higher than in plasma (based on AUC0-24h).
8.4 Pediatric Use
The safety and effectiveness of ALYFTREK for the treatment of CF have been established in pediatric patients aged 6 years and older who have a clinical diagnosis of CF and who have at least one variant in the CFTR gene that is either responsive based on clinical and/or in vitro data or results in production of CFTR protein. Use of ALYFTREK for this indication was supported by evidence from two adequate and well-controlled trials (Trials 1 and 2) in patients with CF aged 12 years and older who had at least one F508del variant or another responsive variant in the CFTR gene and additional pharmacokinetic and safety data in pediatric patients with CF aged 6 to less than 12 years who had at least one F508del variant or another responsive variant in the CFTR gene (Trial 3). In these trials, a total of 145 patients with CF aged 6 to less than 18 years received ALYFTREK including:
- In Trial 1, 26 adolescents aged 12 to less than 18 years who were heterozygous for F508del and a CFTR variant that is not responsive to ivacaftor or tezacaftor/ivacaftor (minimal function variant) [see Adverse Reactions (6.1) and Clinical Studies (14)].
- In Trial 2, 41 adolescents aged 12 to less than 18 years who were homozygous for F508del variant, heterozygous for F508del variant and either a gating or a residual function variant, or with at least one variant responsive to ELX/TEZ/IVA with no F508del variant [see Adverse Reactions (6.1) and Clinical Studies (14)].
- In Trial 3, 78 pediatric patients with CF aged 6 to less than 12 years (mean age 9.1 years) with at least one variant that is responsive to ELX/TEZ/IVA [see Adverse Reactions (6.1)]. In Trial 3, patients who weighed less than 40 kg patients received ALYFTREK (vanzacaftor 12 mg/tezacaftor 60 mg/deutivacaftor 150 mg once daily) and patients who weighed 40 kg or more received ALYFTREK (vanzacaftor 20 mg/tezacaftor 100 mg/deutivacaftor 250 mg once daily).
The efficacy of ALYFTREK in patients aged 6 to less than 12 years for this indication was extrapolated from patients aged 12 years and older with support from population pharmacokinetic analyses showing vanzacaftor, tezacaftor, and deutivacaftor exposure levels in patients aged 6 to less than 12 years to be within the range of exposures observed in patients aged 12 years and older [see Clinical Pharmacology (12.3)].
Safety of ALYFTREK in patients aged 6 to less than 12 years for this indication was based on Trial 3. The overall safety profile of patients in Trial 3 was generally similar to the safety data in adult and pediatric patients 12 years of age and older observed in Trials 1 and 2 [see Adverse Reactions (6.1)].
There is a risk of cataracts in pediatric patients treated with ALYFTREK. Perform baseline and follow-up ophthalmological examination in pediatric patients prior to and during treatment with ALYFTREK [see Warnings and Precautions (5.8)].
The safety and effectiveness of ALYFTREK in patients younger than 6 years of age have not been established.
Juvenile Animal Toxicity Data
Findings of cataracts were observed in juvenile rats dosed from postnatal Day 7 through 35 with ivacaftor dose levels of 10 mg/kg/day and higher (0.21 times the MRHD based on systemic exposure of ivacaftor and its metabolites). This finding has not been observed in older animals [see Warnings and Precautions (5.8)].
Studies were conducted with tezacaftor in juvenile rats starting at postnatal day (PND) 21 and ranging up to PNDs 35 to 49. Findings of convulsions and death were observed in juvenile rats that received a tezacaftor dose level of 100 mg/kg/day (approximately equivalent to 1.9 times the MRHD based on summed AUCs of tezacaftor and its metabolite, M1-TEZ). A no effect dose level was identified at 30 mg/kg/day (approximately equivalent to 0.8 times the MRHD based on summed AUCs of tezacaftor and its metabolite, M1-TEZ). Findings were dose related and generally more severe when dosing with tezacaftor was initiated earlier in the postnatal period (PND 7, which would be approximately equivalent to a human neonate). Tezacaftor and its metabolite, M1-TEZ, are substrates for P-glycoprotein. Lower brain levels of P-glycoprotein activity in younger rats resulted in higher brain levels of tezacaftor and M1-TEZ. These findings are not relevant for the indicated pediatric population 6 to 11 years of age, for whom levels of P-glycoprotein activity are equivalent to levels observed in adults.
8.5 Geriatric Use
Clinical studies of ALYFTREK did not include a sufficient number of patients with CF aged 65 years and older (n=2, 0.4% of patients treated with ALYFTREK in Trials 1 and 2) to determine whether they respond differently from younger adult patients with CF.
8.6 Renal Impairment
The recommended ALYFTREK dosage in patients with CF with mild to moderate renal impairment (RI) (eGFR 30 to < 90 mL/min/1.73 m2) is the same in patients with CF with normal kidney function. Use of ALYFTREK in patients with CF with severe RI (eGFR <30 mL/min/1.73 m2) or end-stage renal disease is recommended only if the benefits are expected to outweigh the risks.
No clinically significant differences in the pharmacokinetics of vanzacaftor, tezacaftor, or deutivacaftor were observed in patients with mild to moderate RI (eGFR 30 to <90 mL/min/1.73 m2) [see Clinical Pharmacology (12.3)]. The effect of severe RI (eGFR <30 mL/min/1.73 m2) on vanzacaftor, tezacaftor, or deutivacaftor pharmacokinetics is unknown [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Severe Hepatic Impairment
ALYFTREK should not be used in patients with severe hepatic impairment (HI) (Child-Pugh Class C). ALYFTREK has not been studied in patients with CF with severe HI [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
Moderate Hepatic Impairment
The use of ALYFTREK is not recommended in patients with moderate HI (Child-Pugh Class B). Use of ALYFTREK should only be considered in patients with HI when there is a clear medical need, and the benefit outweighs the risk. If used, the recommended dosage in patients with moderate HI is the same as for patients with normal hepatic function. Liver function tests should be closely monitored [see Dosage and Administration (2.2), Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
Mild Hepatic Impairment
The recommended dosage of ALYFTREK in patients with mild HI (Child-Pugh Class A) is the same as in patients with normal hepatic function. Liver function tests should be closely monitored [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
ALYFTREK (vanzacaftor, tezacaftor, and deutivacaftor tablets) are fixed-dose combination tablets for oral use available as:
- 10 mg of vanzacaftor (equivalent to 10.6 mg of vanzacaftor calcium dihydrate), 50 mg of tezacaftor, 125 mg of deutivacaftor or
- 4 mg of vanzacaftor (equivalent to 4.24 mg of vanzacaftor calcium dihydrate), 20 mg of tezacaftor, 50 mg of deutivacaftor.
The tablets contain the following inactive ingredients: croscarmellose sodium, hypromellose, hypromellose acetate succinate, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. The tablet film coating contains Brilliant Blue FCF aluminum lake/FD&C Blue #1, carmine, hydroxypropyl cellulose, hypromellose, iron oxide red, talc, and titanium dioxide.
The active ingredients of ALYFTREK are described below:
Vanzacaftor
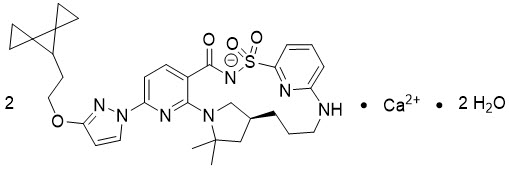
Vanzacaftor is provided as a calcium salt. Vanzacaftor calcium dihydrate is a white solid that is practically insoluble in water (< 0.1 mg/mL). Its chemical name is calcium bis((14S)-8-[3-(2-{dispiro[2.0.24.13]heptan-7-yl}ethoxy)pyrazol-1-yl]-12,12-dimethyl-2,2,4-trioxo-2λ6-thia-3,9,11,18,23-pentaazatetracyclo[17.3.1.111,14.05,10]tetracosa-1(23),5,7,9,19,21-hexaen-3-ide) dihydrate. Its molecular formula is C32H38N7O4S∙Ca0.5∙H2O and its molecular weight is 654.82. Vanzacaftor calcium dihydrate has the following structural formula:
Tezacaftor
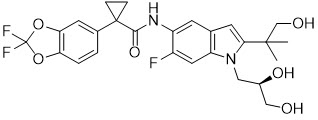
Tezacaftor is a white to off-white solid that is practically insoluble in water (< 5 microgram/mL). Its chemical name is 1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)-N-{1-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl}cyclopropane-1-carboxamide. Its molecular formula is C26H27N2F3O6 and its molecular weight is 520.50. Tezacaftor has the following structural formula:
Deutivacaftor
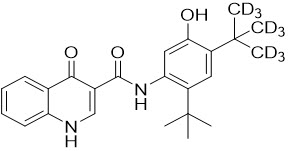
Deutivacaftor is a white to off-white solid that is practically insoluble in water (< 0.1 mg/mL). Pharmacologically, it is a CFTR potentiator. Its chemical name is N-(2-(tert-butyl)-5-hydroxy-4-(2-(methyl-d3)propan-2-yl-1,1,1,3,3,3-d6)phenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide. Its molecular formula is C24H19D9N2O3 and its molecular weight is 401.55. Deutivacaftor has the following structural formula:
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Vanzacaftor and tezacaftor bind to different sites on the CFTR protein and have an additive effect in facilitating the cellular processing and trafficking of select mutant forms of CFTR (including F508del-CFTR) to increase the amount of CFTR protein delivered to the cell surface compared to either molecule alone. Deutivacaftor potentiates the channel open probability (or gating) of the CFTR protein at the cell surface.
The combined effect of vanzacaftor, tezacaftor and deutivacaftor is increased quantity and function of CFTR at the cell surface, resulting in increased CFTR activity as measured both by CFTR-mediated chloride transport in vitro and by sweat chloride in patients with CF.
CFTR Chloride Transport Assay in Fischer Rat Thyroid Cells Expressing Mutant CFTR Protein
Effects of vanzacaftor/tezacaftor/deutivacaftor on chloride transport for mutant CFTR protein was determined in Ussing chamber electrophysiology studies using a panel of Fischer Rat Thyroid (FRT) cell lines stably expressing CFTR protein from individual variants. Vanzacaftor/tezacaftor/deutivacaftor increased chloride transport in FRT cells expressing select CFTR variants, as identified in Table 5.
The threshold that the treatment-induced increase in chloride transport must exceed for the mutant CFTR protein to be considered responsive is ≥10% of normal over baseline. This threshold was used because it is expected to predict clinical benefit. For individual variants, the magnitude of the net change over baseline in CFTR-mediated chloride transport in vitro is not correlated with the magnitude of clinical response.
CFTR Chloride Transport Assay in Human Bronchial Epithelial Cells Expressing Mutant CFTR Protein
Homozygous and heterozygous N1303K- Human Bronchial Epithelial (HBE) cells showed greater chloride transport in the presence of vanzacaftor/tezacaftor/deutivacaftor than F508del/F508del-HBE cells treated with tezacaftor/ivacaftor which has shown clinical benefit in people homozygous for F508del.
Patient Selection
Select adult and pediatric patients 6 years of age and older for the treatment of CF with ALYFTREK based on a clinical diagnosis of CF and the presence of at least one variant in the CFTR gene that is either responsive based on clinical and/or in vitro data or results in production of CFTR protein [see Indications and Usage (1)].
ALYFTREK should only be used in patients with a clinical diagnosis of CF. The presence of eligible CFTR variant(s) should not be the sole determinant for using ALYFTREK.
Table 5 lists CFTR variants responsive to ALYFTREK based on clinical and/or in vitro data in FRT or HBE cells [see Clinical Studies (14)].
Table 5: List of CFTR Gene Variants Responsive to ALYFTREK The list of responsive CFTR variants is non-exhaustive. There may be protein-producing CFTR variants not listed that respond to treatment with ALYFTREK. - * Clinical data is obtained from Trials 1 and 2.
- † This variant is also predicted to be responsive by FRT assay with ALYFTREK.
- ‡ The N1303K variant is predicted to be responsive only by HBE assay. All other variants predicted to be responsive with in vitro data are supported by FRT assay.
- § Complex/compound variants where a single allele of the CFTR gene has multiple variants; these exist independent of the presence of variants on the other allele.
Based on Clinical Data* A455E G551D† L1077P† R352Q S1251N S945L W1098C† D1152H G85E† L206W R75Q S549N V562I W1282R F508del† H1054D M1101K† S1159F S549R V754M Y563N† G1244E I336K R1066H Based on in vitro Data‡ 1140-1151dup D443Y G1265V I1398S M150R R1283M S977F 1461insGAT D443Y;G576A;R668C§ G126D I148L M152L R1283S S977F;R1438W§ 1507_1515del9 D513G G1298V I148N M152V R1438W T1036N 2055del9 D529G G1349D I148T M265R R170H T1053I 2183A→G D565G G149R I148T;H609R§ M348K R248K T1057R 2851A/G D567N G149R;G576A;R668C§ I175V M394L R258G T1086A 293A→G D572N G178E I331N M469V R297Q T1086I 3007del6 D579G G178R I336L M498I R31C T1246I 3131del15 D58H G194R I444S M952I R31L T1299I 3132T→G D58V G194V I497S M952T R334L T1299K 3141del9 D614G G213E I502T M961L R334Q T164P 3143del9 D651H G213E;R668C§ I506L N1088D R347H T338I 314del9 D651N G213V I506T N1195T R347L T351I 3195del6 D806G G226R I506V N1303I R347P T351S 3199del6 D836Y G239R I506V;D1168G§ N1303K‡ R352W T351S;R851L§ 3331del6 D924N G253R I521S N186K R516G T388M 3410T→C D979A G27E I530N N187K R516S T465I 3523A→G D979V G27R I556V N396Y R553Q T465N 3601A→C D985H G314E I586V N418S R555G T501A 3761T→G D985Y G314R I601F N900K R560S T582S 3791C/T D993A G424S I601T P1013H R560T T604I 3850G→A D993G G437D I618N P1013L R600S T908N 3978G→C D993Y G451V I618T P1021L R668C T990I 4193T→G E1104K G461R I807M P1021T R709Q V1008D 546insCTA E1104V G461V I86M P111L R74Q V1010D 548insTAC E1126K G463V I980K P1372T R74Q;R297Q§ V1153E A1006E E116K G480C K1060T P140S R74Q;V201M;D1270N§ V11I A1025D E116Q G480D K162E P205S R74W V1240G A1067P E1221V G480S K464E P439S R74W;D1270N§ V1293G A1067T E1228K G500D K464N P499A R74W;R1070W;D1270N§ V1293I A1067V E1409K G545R K522E P574H R74W;S945L§ V1415F A107G E1433K G551A K522Q P5L R74W;V201M§ V201M A1081V E193K G551R K951E P67L R74W;V201M;D1270N§ V232A A1087P E217G G551S L1011S P750L R74W;V201M;L997F§ V232D A120T E264V G576A L102R P798S R751L V317A A1319E E282D G576A;R668C§ L102R;F1016S§ P988R R75L V322M A1374D E292K G576A;S1359Y§ L1065P P99L R75Q;L1065P§ V392G A141D E384K G622D L1065R Q1012P R75Q;N1088D§ V456A A1466S E403D G622V L1227S Q1100P R75Q;S549N§ V456F A155P E474K G628A L1324P Q1209P R792G V520F A234D E527G G628R L1335P Q1291H R792Q V520I A234V E56K G85V L137P Q1291R R810G V562I;A1006E§ A238V E588V G91R L137R Q1313K R851L V562L A309D E60K G930E L1388P Q1352H R933G V591A A349V E822K G970D L1480P Q151K S1045Y V603F A357T E92K G970S L159S Q179K S108F V920L A455V F1016S G970V L15P Q237E S1118F V920M A457T F1052V H1079P L15P;L1253F§ Q237H S1159P V93D A462P F1074L H1085P L165S Q237P S1188L W1282G A46D F1078S H1085R L167R Q30P S1235R W202C A534E F1099L H1375N L210P Q359K/T360K§ S1255P W361R A554E F1107L H1375P L293P Q359R S13F W496R A559T F191V H139L L320V Q372H S13P Y1014C A559V F200I H139R L327P Q452P S158N Y1032C A561E F311del H146R L32P Q493L S182R Y1032N A566D F311L H147del L333F Q493R S18I Y1073C A613T F312del H147P L333H Q552P S18N Y1092H A62P F433L H199Q L346P Q98P S308P Y109C A72D F508C H199R L441P Q98R S341P Y109H A872E F508C;S1251N§ H199Y L453S R1048G S364P Y109N c.1367_1369dupTTG F508del;R1438W§ H609L L467F R1066C S434P Y122C C225R F575Y H609R L558F R1066G S492F Y1381H C491R F587I H620P L594P R1066L S50P Y161C C590Y F587L H620Q L610S R1066M S519G Y161D C866Y F693L(TTG) H939R L619S R1070P S531P Y161S D110E F87L H939R;H949L§ L633P R1070Q S549I Y301C D110H F932S H954P L636P R1070W S557F Y517C D110N G1047D I1023R L88S R1162L S589I Y569C D1152A G1047R I1027T L927P R1162Q S589N Y89C D1270N G1061R I105N L967F;L1096R§ R117C S624R Y913C D1270Y G1069R I1139V L967S R117C;G576A;R668C§ S686Y Y913S D1312G G1123R I1203V L973F R117G S737F Y919C D1377H G1173S I1234L L997F R117H S821G D1445N G1237V I1234Vdel6aa M1101R R117L S898R D192G G1244R I125T M1137R R117L;L997F§ S912L D192N G1247R I1269N M1137V R117P S912L;G1244V§ D373N G1249E I1366N M1210K R1239S S912T D426N G1249R I1366T M150K R1283G S955P 12.2 Pharmacodynamics
Effects on Sweat Chloride
- In patients with CF heterozygous for F508del and a CFTR variant that results in a protein that is not responsive to ivacaftor or tezacaftor/ivacaftor [minimal function variant] (Trial 1) the treatment difference of ALYFTREK compared to ELX/TEZ/IVA for mean absolute change in sweat chloride from baseline through Week 24 was -8.4 mmol/L (95% CI: -10.5, -6.3; P <0.0001).
- In patients with CF homozygous for the F508del variant, heterozygous for the F508del variant and either a gating or a residual function variant, or at least one variant responsive to ELX/TEZ/IVA with no F508del variant (Trial 2), the treatment difference of ALYFTREK compared to ELX/TEZ/IVA for mean absolute change in sweat chloride from baseline through Week 24 was -2.8 mmol/L (95% CI: -4.7, -0.9; P = 0.0034).
- In an open-label trial in patients with CF aged 6 to less than 12 years with at least one variant that is responsive to ELX/TEZ/IVA (Trial 3) [see Adverse Reactions (6.1)], the mean absolute change in sweat chloride from baseline through Week 24 was -8.6 mmol/L (95% CI: -11.0, -6.3).
The clinical relevance of these differences in sweat chloride has not been established in interventional clinical trials.
Cardiac Electrophysiology
At approximately 6 times the maximum recommended dose of vanzacaftor, clinically significant QTc interval prolongation was not observed. Similarly, in separate studies of tezacaftor and ivacaftor evaluating up to 3 times the respective maximum recommended doses, clinically significant QTc interval prolongation was not observed.
12.3 Pharmacokinetics
The pharmacokinetic parameters for vanzacaftor, tezacaftor, and deutivacaftor in patients with CF aged 12 years and older are provided in Table 6 as mean (SD) unless otherwise specified. No clinically significant differences in the pharmacokinetics of vanzacaftor, tezacaftor, and deutivacaftor were observed between healthy adult subjects and patients with CF.
Table 6: Pharmacokinetics Parameters of ALYFTREK Components Vanzacaftor Tezacaftor Deutivacaftor Abbreviations: AUC: area under the concentration versus time curve; SD: Standard Deviation; Cmax: maximum observed concentration; Tmax: time of maximum concentration; ss: steady state - * Median (range)
- † When administered with fat-containing meals relative to fasted conditions. Note: The high-fat meal was approximately 800-1000 calories with 50% fat. The low-fat meal was approximately 400-500 calories with 25% fat.
- ‡ Vanzacaftor, tezacaftor, deutivacaftor do not partition preferentially into human red blood cells.
- § Vanzacaftor and deutivacaftor bind primarily to albumin and alpha 1-acid glycoprotein. Tezacaftor binds primarily to albumin.
- ¶ The mean (SD) terminal half-lives of vanzacaftor, tezacaftor, and deutivacaftor are 54.0 (10.1) hours, 92.4 (23.1) hours and 17.3 (2.67) hours, respectively based on a single dose of vanzacaftor/tezacaftor/deutivacaftor tablets in healthy subjects in the fed state.
- # Following radiolabeled doses.
Exposure Cmax,ss (mcg/mL) 0.812 (0.344) 6.77 (1.24) 2.33 (0.637) AUC0-24h,ss (mcg∙h/mL) 18.6 (8.08) 89.5 (28.0) 39.0 (15.3) Time to steady state within 20 days within 8 days within 8 days AUC Accumulation Ratio 6.09 (1.81) 1.92 (0.337) 1.74 (0.497) Absorption Tmax* (hours) 7.80 (3.70, 11.9) 1.60 (1.40, 1.70) 3.7 (2.7, 11.4) Effect of food AUCinf† Increase 4- (low-fat meal) to 6- (high-fat meal) fold No clinically significant change Increase 3- (low-fat meal) to 4- (high-fat meal) fold Distribution‡ Apparent (oral) volume of distribution (L) 121 (28.6) 73.1 (13.3) 159 (26.1) Protein Binding§ > 99% Approximately 99% > 99% Elimination Effective Half-life (hours)¶ 92.8 (30.2) 22.5 (5.85) 19.2 (8.71) Apparent (oral) Clearance (L/hours) 1.34 (0.819) 1.22 (0.390) 7.29 (2.68) Metabolism Primary Pathway CYP3A4/5 CYP3A4/5 CYP3A4/5 Active metabolites None M1-TEZ M1-D-IVA Metabolite potency (relative to parent) Not applicable Similar Approximately 20% Excretion # Feces 91.6%
(primarily metabolites)72% (unchanged or M2-TEZ)
[0.79% as unchanged drug]Not available Urine 0.50% 13.7% Not available Specific Populations
No clinically significant differences in the pharmacokinetics of vanzacaftor, tezacaftor, or deutivacaftor were observed based on age, sex, race, CFTR genotype, or mild to moderate renal impairment (eGFR 30 to <90 mL/min/1.73m2 as estimated by modification of diet in renal disease (MDRD) equation). The effect of severe renal impairment (eGFR less than 30 mL/min/1.73m2) on vanzacaftor, tezacaftor, or deutivacaftor pharmacokinetics is unknown.
Weight was identified as the key covariate having a clinically meaningful impact on pharmacokinetics of vanzacaftor, tezacaftor, and deutivacaftor.
Pediatric Patients Aged 6 to Less Than 18 Years
Vanzacaftor, tezacaftor and deutivacaftor exposures observed in clinical trials are presented by age group and dosage administered in Table 7. No clinically significant differences in vanzacaftor, tezacaftor, and deutivacaftor exposures were observed in patients with CF aged 6 to less than 18 years compared to adults following the recommended dosages.
Table 7: Mean (SD) Vanzacaftor, Tezacaftor and Deutivacaftor Exposures by Age Group Age Group Weight Dosage (once daily) AUC0-24h (mcg∙h/mL) Vanzacaftor Tezacaftor Deutivacaftor Abbreviations: SD: Standard Deviation; AUC0-24h: Area Under the Concentration versus time curve at steady state 6 to <12 years <40 kg
(N = 70)vanzacaftor 12 mg
tezacaftor 60 mg
deutivacaftor 150 mg13.0 (4.90) 69.1 (20.7) 30.2 (11.6) ≥40 kg
(N = 8)vanzacaftor 20 mg
tezacaftor 100 mg
deutivacaftor 250 mg18.6 (7.49) 101 (33.7) 48.5 (18.7) 12 to <18 years -
(N = 66)vanzacaftor 20 mg
tezacaftor 100 mg
deutivacaftor 250 mg15.8 (6.52) 93.0 (32.5) 37.1 (15.3) ≥18 years -
(N = 414)19.0 (8.22) 89.0 (27.2) 39.3 (15.3) Patients with Hepatic Impairment
Vanzacaftor AUC was approximately 30% lower, tezacaftor AUC was comparable, and deutivacaftor AUC was approximately 20% lower in subjects with moderate hepatic impairment (Child-Pugh Class B) compared to subjects with normal liver function matched for demographics [see Use in Specific Populations (8.7)].
The effect of mild hepatic impairment (Child-Pugh Class A) or severe hepatic impairment (Child-Pugh Class C) on vanzacaftor, tezacaftor, or deutivacaftor pharmacokinetics is unknown.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Exposure changes associated with concomitant use of vanzacaftor, tezacaftor, ivacaftor and/or deutivacaftor with other drugs are shown in Table 8.
Table 8: Observed or Predicted Exposure Changes Associated with Concomitant Use of Vanzacaftor, Tezacaftor, Ivacaftor and/or Deutivacaftor with Other Drugs Dosage Effected Drug Geometric Mean Ratio (90% CI)
No Effect = 1.0AUC Cmax Abbreviations: CI = Confidence Interval; ELX = elexacaftor; VNZ = vanzacaftor; TEZ = tezacaftor; IVA = ivacaftor; D-IVA = deutivacaftor; PK = Pharmacokinetics; qd = once daily - * The itraconazole dosing (200 mg qd for 14 days) did not fully cover the elimination of vanzacaftor. A 10.5-fold increase in vanzacaftor AUC is predicted by physiologically based pharmacokinetic modeling and simulations when itraconazole fully covers the elimination.
- † Effect is not clinically significant [see Drug Interactions (7.3)].
- ‡ Predicted by physiologically based pharmacokinetic modeling and simulations. Data presented as geometric mean ratio and 5th to 95th percentiles of individuals in the simulated population [see Drug Interactions (7.1)].
Itraconazole
200 mg q12h on Day 1, followed by 200 mg dailyTEZ 25 mg daily + IVA 50 mg daily Tezacaftor 4.02
(3.71, 4.63)2.83
(2.62, 3.07)Itraconazole
200 mg dailyELX 20 mg + TEZ 50 mg + D-IVA 50 mg single dose Tezacaftor 4.51
(3.85, 5.29)1.48
(1.33, 1.65)Deutivacaftor 11.1
(8.72, 14.1)1.96
(1.70, 2.26)Itraconazole
200 mg daily*VNZ 5 mg single dose Vanzacaftor 6.37
(5.53, 7.35)1.55
(1.41, 1.70)Ciprofloxacin†
750 mg twice dailyTEZ 50 mg q12h + IVA 150 mg q12h Tezacaftor 1.08
(1.03, 1.13)1.05
(0.99, 1.11)Digoxin 0.5 mg
single doseTEZ 25 mg daily + IVA 50 mg daily Digoxin 1.3
(1.17, 1.45)1.32
(1.07, 1.64)Fluconazole
200 mg dailyVNZ 20 mg qd + TEZ 100 mg + D-IVA 250 mg qd Vanzacaftor 2.55
(2.12, 3.12)‡2.48
(2.04, 3.01)‡Deutivacaftor 3.13
(2.44, 3.95)‡2.27
(1.82, 2.93)‡Erythromycin
500 mg four times dailyVNZ 20 mg qd + TEZ 100 mg + D-IVA 250 mg qd Vanzacaftor 3.29
(1.62, 7.55)‡3.19
(1.60, 7.29)‡Deutivacaftor 4.13
(1.80, 9.73)‡2.89
(1.52, 6.97)‡Verapamil
80 mg three times dailyVNZ 20 mg qd + TEZ 100 mg + D-IVA 250 mg qd Vanzacaftor 3.93
(1.84, 8.75)‡3.80
(1.78, 8.33)‡Deutivacaftor 5.11
(2.06, 12.5)‡3.43
(1.64, 7.65)‡Rifampin
600 mg dailyVNZ 20 mg qd + TEZ 100 mg + D-IVA 250 mg qd Vanzacaftor 0.18
(0.10, 0.34)‡0.22
(0.12, 0.38)‡Deutivacaftor 0.10
(0.04, 0.26)‡0.20
(0.08, 0.44)‡Carbamazepine
400 mg twice dailyVNZ 20 mg qd + TEZ 100 mg + D-IVA 250 mg qd Vanzacaftor 0.44
(0.28, 0.61)‡0.46
(0.31, 0.64)‡Deutivacaftor 0.24
(0.11, 0.47)‡0.32
(0.17, 0.57)‡Efavirenz
600 mg dailyVNZ 20 mg qd + TEZ 100 mg + D-IVA 250 mg qd Vanzacaftor 0.31
(0.16, 0.57)‡0.35
(0.19, 0.59)‡Deutivacaftor 0.27
(0.11, 0.50)‡0.44
(0.23, 0.68)‡Other Drugs: No clinically significant differences in tezacaftor pharmacokinetics were observed when tezacaftor/ivacaftor was used concomitantly with ciprofloxacin. No clinically significant differences in the pharmacokinetics of the following drugs were observed when used concomitantly with tezacaftor/ivacaftor: midazolam (CYP3A4 substrate) or ethinyl estradiol/norethindrone containing hormonal contraceptives.
In Vitro Studies
CYP450 Enzymes: Vanzacaftor, tezacaftor, and deutivacaftor are CYP3A substrates. Deutivacaftor inhibits CYP2C8, CYP2C9, and CYP3A4. Vanzacaftor and tezacaftor do not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Vanzacaftor, tezacaftor, and deutivacaftor do not induce CYP3A4.
Transporter Systems: Tezacaftor and deutivacaftor are substrates of P-gp, but vanzacaftor is not a substrate of P-gp. Tezacaftor is a substrate of BCRP, OATP1B1, but not OATP1B3. Vanzacaftor and deutivacaftor are not substrates for OATP1B1 or OATP1B3. Vanzacaftor and deutivacaftor are BCRP inhibitors. Vanzacaftor, tezacaftor and deutivacaftor are P-gp inhibitors. Vanzacaftor, tezacaftor, and deutivacaftor do not inhibit OATP1B1 nor OATP1B3.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies of carcinogenicity, mutagenicity, or impairment of fertility were conducted with the combination of vanzacaftor, tezacaftor, and deutivacaftor; however, separate studies of vanzacaftor, tezacaftor, deutivacaftor, and ivacaftor are described below.
Vanzacaftor
A 6-month study in Tg.rasH2 mice showed no evidence of tumorigenicity at 30 mg/kg/day dose, the highest dose tested.
Vanzacaftor was negative for genotoxicity in the following assays: Ames test for bacterial gene variant, in vitro micronucleus assay in TK6 cells, and in vivo rat micronucleus assay.
Administration of oral vanzacaftor had no effects on fertility and early embryonic development in male and female rats at up to 12.5 and 10 mg/kg/day, respectively (approximately 19 times for males and 30 times for females the exposure at the MRHD based on AUCs of vanzacaftor).
Tezacaftor
A two-year study in Sprague-Dawley rats and a 6-month study in Tg.rasH2 transgenic mice were conducted to assess the carcinogenic potential of tezacaftor. No evidence of tumorigenicity from tezacaftor was observed in male and female rats at oral doses up to 50 and 75 mg/kg/day (approximately 2 and 4 times the MRHD based on summed AUCs of tezacaftor and M1-TEZ in males and females, respectively). No evidence of tumorigenicity was observed in male and female Tg.rasH2 transgenic mice at tezacaftor doses up to 500 mg/kg/day.
Tezacaftor was negative for genotoxicity in the following assays: Ames test for bacterial gene variant, in vitro chromosomal aberration assay in Chinese hamster ovary cells and in vivo mouse micronucleus test.
There were no effects on male or female fertility and early embryonic development in rats at oral tezacaftor doses up to 100 mg/kg/day (approximately 3 times the MRHD based on summed AUC of tezacaftor and M1-TEZ).
Deutivacaftor
Deutivacaftor is a deuterated isotopologue of ivacaftor with an established toxicity profile similar to ivacaftor based on a 13-week single-agent repeat dose toxicity study; therefore, reproductive and developmental toxicity data and carcinogenicity data from ivacaftor are expected to be equivalent to deutivacaftor.
Ivacaftor
Two-year studies were conducted in CD-1 mice and Sprague-Dawley rats to assess the carcinogenic potential of ivacaftor. No evidence of tumorigenicity from ivacaftor was observed in mice or rats at oral doses up to 200 mg/kg/day and 50 mg/kg/day, respectively (approximately equivalent to 3 and 11 times the MRHD, respectively, based on summed AUCs of ivacaftor).
Ivacaftor was negative for genotoxicity in the following assays: Ames test for bacterial gene variant, in vitro chromosomal aberration assay in Chinese hamster ovary cells and in vivo mouse micronucleus test.
Ivacaftor impaired fertility and reproductive performance indices in male and female rats at 200 mg/kg/day (approximately 15 and 13 times, respectively, the MRHD based on AUCs of ivacaftor). Increases in prolonged diestrus were observed in females at 200 mg/kg/day. Ivacaftor also increased the number of females with all nonviable embryos and decreased corpora lutea, implantations and viable embryos in rats at 200 mg/kg/day when dams were dosed prior to and during early pregnancy. Slight decreases of the seminal vesicle weights were observed in males at 200 mg/kg/day dose (approximately 15 times the MRHD based on summed AUCs of ivacaftor). These impairments of fertility and reproductive performance in male and female rats at 200 mg/kg/day were attributed to severe toxicity.
-
14 CLINICAL STUDIES
The efficacy of ALYFTREK in patients aged 12 years and older with cystic fibrosis (CF) who have at least one F508del variant or a responsive variant in the CFTR gene was evaluated in two 52-week randomized, double-blind, active-controlled trials comparing ALYFTREK and a fixed-dose combination drug containing elexacaftor, tezacaftor, and ivacaftor (ELX/TEZ/IVA) (Trial 1, NCT05033080 and Trial 2, NCT05076149). The two trials enrolled a total of 971 patients aged 12 years and older with CF who have at least one F508del variant or other ELX/TEZ/IVA-responsive variants in the CFTR gene. Because patients in Trial 1 and Trial 2 would receive ELX/TEZ/IVA, patients with a history of intolerance to ELX/TEZ/IVA were excluded from these trials.
- Trial 1 enrolled patients with CF heterozygous for F508del and a CFTR variant that results in a protein that was not responsive to ivacaftor or tezacaftor/ivacaftor (minimal function variant). A total of 398 patients with CF aged 12 years and older received a daily oral dosage of ELX/TEZ/IVA (elexacaftor 200 mg/tezacaftor 100 mg/ivacaftor 150 mg in the morning and ivacaftor 150 mg in the evening) during a 4-week run-in period and were then randomized to receive ALYFTREK (total once daily oral dosage of vanzacaftor 20 mg/tezacaftor 100 mg/deutivacaftor 250 mg) or ELX/TEZ/IVA (same dosage as in the run-in period) during the 52-week treatment period. Patients had a mean age of 30.8 years (range: 12.2 to 71.6 years), were 59% male, 97.5% White, 1.3% Black/African American, 0.3% Asian, 0.3% Other race, and 6% Hispanic or Latino ethnicity. After the 4₋week run-in, the mean ppFEV1 at baseline was 67.1 percentage points (range: 28.0, 108.6) and the mean sweat chloride at baseline was 53.9 mmol/L (range: 10.0 mmol/L, 113.5 mmol/L).
- Trial 2 enrolled patients with CF who had one of the following genotypes: homozygous for the F508del variant, heterozygous for the F508del variant and either a gating or a residual function variant, or at least one variant responsive to ELX/TEZ/IVA with no F508del variant. A total of 573 patients with CF aged 12 years and older received a daily oral dosage of ELX/TEZ/IVA (elexacaftor 200 mg/tezacaftor 100 mg/ivacaftor 150 mg in the morning and ivacaftor 150 mg in the evening) during a 4-week run-in period and were then randomized to receive ALYFTREK (total once daily oral dosage of vanzacaftor 20 mg/tezacaftor 100 mg/ deutivacaftor 250 mg) or ELX/TEZ/IVA (same dosage as during the run-in period) during the 52-week treatment period. Patients had a mean age of 33.7 years (range: 12.2 to 71.2 years), were 51.1% male, 92.8% White, 0% Black/African American, 0.3% Asian, 0.2% American Indian or Alaska Native, 0.3% Other race, and 1.6% Hispanic or Latino ethnicity. After the 4-week run-in, the mean ppFEV1 at baseline was 66.8 percentage points (range: 36.4, 112.5) and the mean sweat chloride at baseline was 42.8 mmol/L (range: 10.0 mmol/L, 113.3 mmol/L).
Efficacy Endpoints
In both trials, the primary endpoint evaluated non-inferiority in mean absolute change in ppFEV1 from baseline through Week 24 and a key secondary endpoint evaluated the mean absolute change from baseline in sweat chloride through Week 24 in the ALYFTREK and ELX/TEZ/IVA treatment groups.
Trials 1 and 2 also assessed other secondary endpoints including pulmonary exacerbation rate and change in Cystic Fibrosis Questionnaire-Revised respiratory domain (CFQ-R RD) score from baseline.
Efficacy Results
- In Trial 1, treatment with ALYFTREK resulted in an LS mean difference of 0.2 percentage points (95% CI: -0.7, 1.1) in absolute change in ppFEV1 from baseline through Week 24 compared to ELX/TEZ/IVA.
- In Trial 2, treatment with ALYFTREK resulted in an LS mean difference of 0.2 percentage points (95% CI: -0.5, 0.9) in absolute change in ppFEV1 from baseline through Week 24 compared to ELX/TEZ/IVA.
As the lower bounds of the 95% CI of the LS mean difference in absolute change from baseline in ppFEV1 through Week 24 were greater than -3.0 percentage points (the pre-specified non-inferiority margin) in Trial 1 and Trial 2, these results demonstrate non-inferiority of ALYFTREK to ELX/TEZ/IVA.
Table 9 provides the primary and key secondary efficacy endpoints results for Trials 1 and 2.
Table 9: Efficacy Results in Patients Aged 12 Years and Older with CF Who Had at Least One F508del Variant or Responsive Variant in the CFTR Gene (Trials 1 and 2) Analysis* Statistic Trial 1 Trial 2 ALYFTREK
N = 196ELX/TEZ/IVA
N = 202ALYFTREK
N = 284ELX/TEZ/IVA
N = 289ppFEV1: percent predicted Forced Expiratory Volume in 1 second; CI: Confidence Interval; SE: Standard Error; SwCl: sweat chloride Note: Analyses were based on the full analysis set (FAS). FAS was defined as all randomized subjects who carry the intended CFTR allele variant and received at least 1 dose of study drug. - * A 4-week ELX/TEZ/IVA run-in-period was performed to establish an on-treatment baseline.
- † The pre-specified non-inferiority margin was -3.0 percentage points.
Primary Endpoint Absolute change from baseline in ppFEV1 through Week 24 (percentage points) n 187 193 268 276 LS mean (SE) 0.5 (0.3) 0.3 (0.3) 0.2 (0.3) 0.0 (0.2) LS mean difference, 95% CI† 0.2 (-0.7, 1.1) 0.2 (-0.5, 0.9) Key Secondary Endpoint Absolute change from baseline in SwCl through Week 24 (mmol/L) n 185 194 270 276 LS mean (SE) -7.5 (0.8) 0.9 (0.8) -5.1 (0.7) -2.3 (0.7) LS mean difference, 95% CI -8.4 (-10.5, -6.3) -2.8 (-4.7, -0.9) P-value (2-sided) < 0.0001 0.0034 The trials were not designed to demonstrate a difference between the treatment groups or to support non-inferiority of the other secondary endpoints. In Trial 1 and Trial 2, mean absolute change from baseline ppFEV1 through Week 52, the rate in pulmonary exacerbations through Week 52, and the absolute change from baseline in the CFQ-R RD through Week 24 were similar between the ALYFTREK-treated and the ELX/TEZ/IVA-treated patients. The results were not tested for statistical significance as they were not in the pre-specified multiple testing procedure.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
ALYFTREK (vanzacaftor, tezacaftor, and deutivacaftor) tablets are supplied as follows:
Table 10: ALYFTREK Tablets and Package Configuration Strengths Tablet Description Package Configuration NDC 4 mg of vanzacaftor/ 20 mg of tezacaftor / 50 mg of deutivacaftor purple, round-shaped, film-coated, debossed with "V4" on one side and plain on the other 84-count carton containing 4 wallets, each wallet containing 21 tablets in blister packs NDC: 51167-135-01 10 mg of vanzacaftor/ 50 mg of tezacaftor / 125 mg of deutivacaftor purple, oblong-shaped, film-coated, debossed with "V10" on one side and plain on the other 56-count carton containing 4 wallets, each wallet containing 14 tablets in blister packs NDC: 51167-121-01 -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Drug-Induced Liver Injury and Liver Failure
Inform patients that elevations of transaminases have occurred in patients with CF treated with ALYFTREK and that cases of drug-induced liver injury and failure have been observed with fixed-dose combination drug containing elexacaftor, tezacaftor, and ivacaftor, which contains the same or similar active ingredients as ALYFTREK.
Advise all patients that liver function tests should be assessed prior to initiating ALYFTREK, and then assessed every month during the first 6 months of treatment, then every 3 months for the next 12 months, then at least annually thereafter. Inform patients with a history of liver disease or liver function test elevations at baseline that more frequent monitoring may be necessary. Instruct patients to interrupt treatment with ALYFTREK if symptoms of liver injury occur (e.g., jaundice, right upper quadrant pain, nausea, vomiting, altered mental status, ascites) and notify their healthcare provider immediately [see Dosage and Administration (2.1), Warnings and Precautions (5.1), Adverse Reactions (6.1), Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
Hypersensitivity Reactions, Including Anaphylaxis
Inform patients that hypersensitivity reactions including anaphylaxis have been reported in patients who received drugs containing elexacaftor, tezacaftor, and/or ivacaftor (the same or similar active ingredients as ALYFTREK). Instruct patients to discontinue ALYFTREK and notify their healthcare provider if they experience signs and symptoms of a hypersensitivity reaction, including rash, hives, itching, facial swelling, tightness of the chest and wheezing [see Warnings and Precautions (5.2)].
Patients Who Discontinued or Interrupted Elexacaftor-, Tezacaftor-, or Ivacaftor-Containing Drugs Due to Adverse Reactions
Inform patients that there is no available safety data for ALYFTREK in patients who previously discontinued or interrupted treatment with elexacaftor-, tezacaftor-, or ivacaftor-containing drugs due to adverse reactions. These patients who start treatment with ALYFTREK may require closer and more frequent monitoring [see Warnings and Precautions (5.3)].
Intracranial Hypertension
Inform patients that intracranial hypertension has occurred in patients who received drugs containing the same or similar active ingredients as ALYFTREK. Instruct patients to notify their healthcare provider right away if they experience signs and symptoms of intracranial hypertension, including headache, blurred vision, diplopia, and vision loss [see Warnings and Precautions (5.4)].
Neuropsychiatric Events, Including Suicidal Thoughts and Behaviors
Inform patients that neuropsychiatric symptoms, including anxiety, depression, suicidal thoughts and behaviors, and sleep disturbances (e.g., insomnia), have been reported with the use of ALYFTREK or drugs containing the same or similar active ingredients as ALYFTREK. The symptoms have been observed in patients with and without a history of similar symptoms and may occur within three months of ALYFTREK initiation. Instruct patients to contact their healthcare provider immediately if changes in behavior or thinking that are not typical for the patient occur, or if the patient develops suicidal ideation or behavior [see Warnings and Precautions (5.5)].
Drug Interactions with CYP3A Inducers and Inhibitors
Inform patients that certain medications, herbal supplements, or vitamins, when used concomitantly with ALYFTREK, may reduce the effectiveness of ALYFTREK or increase the risk of adverse reactions associated with ALYFTREK. Instruct patients to report all concomitant medications, herbal supplements, or vitamins, to their healthcare providers while taking ALYFTREK [see Dosage and Administration (2.3), Warnings and Precautions (5.6, 5.7) and Drug Interactions (7.1)].
Instruct patients to avoid food or drink containing grapefruit when using ALYFTREK [see Drug Interactions (7.1)].
Cataracts
Inform patients that abnormality of the eye lens (cataract) has been noted in some pediatric patients receiving drugs containing ivacaftor (which is similar to an active ingredient in ALYFTREK) and baseline and follow-up ophthalmological examinations are needed in pediatric patients receiving ALYFTREK [see Warnings and Precautions (5.8) and Nonclinical Toxicology (13.1)].
Administration
Inform patients that ALYFTREK is best absorbed by the body when taken with food that contains fat. Examples include eggs, butter, peanut butter, whole-milk dairy products (such as whole milk, cheese and yogurt), etc. [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
Inform patients of the following if they miss an ALYFTREK dose [see Dosage and Administration (2.5)]:
- If 6 hours or less have passed since the missed dose is usually taken, patients with CF should be instructed to take the prescribed dose with fat-containing food as soon as possible.
- If more than 6 hours have passed since the missed dose, patients with CF should be instructed to skip the missed dose and continue on the original schedule the next day.
-
SPL UNCLASSIFIED SECTION
Manufactured for
Vertex Pharmaceuticals Incorporated
50 Northern Avenue
Boston, MA 02210ALYFTREK, VERTEX and associated logos are trademarks of Vertex Pharmaceuticals Incorporated.
All other trademarks referenced herein are the property of their respective owners.
©2026 Vertex Pharmaceuticals Incorporated
All Rights Reserved -
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 03/2026 MEDICATION GUIDE
ALYFTREK™ (ah-LIF-trek)
(vanzacaftor, tezacaftor, and deutivacaftor tablets), tablets for oral useWhat is the most important information I should know about ALYFTREK? Elevated liver enzymes have been observed in patients taking ALYFTREK. Cases of serious liver damage and liver failure leading to transplantation and death have been seen in some people with or without a history of liver problems taking elexacaftor/tezacaftor/ivacaftor (TRIKAFTA), a medicine which has the same or similar active ingredients as ALYFTREK. Your healthcare provider will do blood tests to check your liver: - before you start ALYFTREK
- every month during your first 6 months of taking ALYFTREK
- then every 3 months during the next 12 months of taking ALYFTREK
- then at least every year while you are taking ALYFTREK
Stop taking ALYFTREK and call your healthcare provider right away if you have any of the following symptoms of liver problems:- pain, swelling, or discomfort in the upper right stomach (abdominal) area
- yellowing of your skin or the white part of your eyes
- mental changes
- nausea or vomiting
- dark, amber-colored urine
- loss of appetite
- have fluid in your stomach area (ascites)
What is ALYFTREK? - ALYFTREK is a prescription medicine for people aged 6 years and older who have a diagnosis of cystic fibrosis (CF) and who have at least one genetic change (variant) in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that is either responsive to ALYFTREK or results in the production of protein.
- Talk to your healthcare provider to learn if you have an indicated CF gene variant.
It is not known if ALYFTREK is safe and effective in children under 6 years of age. What should I tell my healthcare provider before taking ALYFTREK? Before taking ALYFTREK, tell your healthcare provider about all of your medical conditions, including if you: - have or have had liver problems.
- are allergic to ALYFTREK or any ingredients in ALYFTREK. See the end of this medication guide for a complete list of ingredients in ALYFTREK.
- have taken another medicine with elexacaftor, tezacaftor, or ivacaftor before and temporarily or permanently stopped because of side effects. Your healthcare provider may want to see you more often.
- have kidney problems.
- have or have had mental health problems.
- are pregnant or plan to become pregnant. It is not known if ALYFTREK will harm your unborn baby. You and your healthcare provider should decide if you will take ALYFTREK while you are pregnant.
- are breastfeeding or planning to breastfeed. It is not known if ALYFTREK passes into your breast milk. You and your healthcare provider should decide if you will take ALYFTREK while you are breastfeeding.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. ALYFTREK may affect the way other medicines work and other medicines may affect how ALYFTREK works. The dose of ALYFTREK may need to be adjusted when taken with certain medicines. Ask your healthcare provider or pharmacist for a list of these medicines if you are not sure. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. How should I take ALYFTREK? - Take ALYFTREK exactly as your healthcare provider tells you to take it.
- Take ALYFTREK by mouth only.
- Always take ALYFTREK tablets with food that contains fat. Examples of fat-containing foods include butter, oil, eggs, peanut butter, nuts, meat, and whole-milk dairy products such as whole milk, cheese, and yogurt.
- ALYFTREK tablets (age 6 to less than 12 years weighing less than 88 pounds (40 kg)):
- The purple round-shaped tablet is marked with 'V4' and each tablet contains the medicines vanzacaftor, tezacaftor, and deutivacaftor. Take 3 tablets at the same time each day.
- ALYFTREK tablets (age 6 to less than 12 years weighing 88 pounds (40 kg) or more, or 12 years and older):
- The purple oblong-shaped tablet is marked with 'V10' and each tablet contains the medicines vanzacaftor, tezacaftor, and deutivacaftor. Take 2 tablets at the same time each day.
- Take ALYFTREK tablets whole.
- Take the doses about the same time every day.
- If you miss a dose of ALYFTREK and:
- it is 6 hours or less from the time you usually take the dose, take the missed dose with food that contains fat as soon as you can. Then take your next dose at your usual time.
- it is more than 6 hours from the time you usually take the dose, do not take the missed dose. Take your dose the next day at your usual time.
- Do not take more than your usual dose of ALYFTREK to make up for a missed dose.
If you are not sure about your dosing, call your healthcare provider. What should I avoid while taking ALYFTREK? Avoid food or drink that contains grapefruit while you are taking ALYFTREK. What are the possible or reasonably likely side effects of ALYFTREK? ALYFTREK can cause serious side effects, including: - See "What is the most important information I should know about ALYFTREK?"
-
Serious Allergic Reactions can happen to people who are treated with ALYFTREK. Call your healthcare provider or go to the emergency room right away if you have any symptoms of an allergic reaction. Symptoms of an allergic reaction may include:
- rash or hives
- tightness of the chest or throat or difficulty breathing
- light-headedness or dizziness
- Increased pressure around the brain (intracranial hypertension) has happened in people treated with medicines containing the same or similar ingredients as ALYFTREK. If you experience an unusual headache, blurred vision, double vision, or vision loss, call your healthcare provider right away.
- Serious mental health problems such as anxiety, depression, suicidal thoughts and behaviors, and trouble sleeping have happened in people treated with ALYFTREK or medicines containing the same or similar ingredients as ALYFTREK. If you experience new or worsening mental health problems, call your healthcare provider right away.
- Abnormality of the eye lens (cataract) has happened in some children and adolescents treated with ALYFTREK. If you are a child or adolescent, your healthcare provider should perform eye examinations before and during treatment with ALYFTREK to look for cataracts.
The most common side effects of ALYFTREK include: - cough
- pain or swelling of your nose or throat (nasopharyngitis)
- upper respiratory tract infection (common cold) including stuffy and runny nose
- headache
- mouth or throat pain
- flu (influenza)
- tiredness
- increase in liver enzymes
- rash
- sinus congestion
Your healthcare provider should monitor you during treatment with ALYFTREK. You may require additional monitoring if your treatment with a medicine that works like ALYFTREK has been previously stopped or interrupted because of side effects. Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all of the possible side effects of ALYFTREK. For more information, ask your healthcare provider or pharmacist.Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store ALYFTREK? - Store ALYFTREK at room temperature between 68°F to 77°F (20°C to 25°C).
Keep ALYFTREK and all medicines out of the reach of children. General information about the safe and effective use of ALYFTREK. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use ALYFTREK for a condition for which it was not prescribed. Do not give ALYFTREK to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about ALYFTREK that is written for health professionals. What are the ingredients in ALYFTREK? Active ingredients: vanzacaftor, tezacaftor, and deutivacaftor. Inactive ingredients: croscarmellose sodium, hypromellose, hypromellose acetate succinate, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. The tablet film coat contains Brilliant Blue FCF aluminum lake/FD&C Blue #1, carmine, hydroxypropyl cellulose, hypromellose, iron oxide red, talc, and titanium dioxide. Manufactured for: Vertex Pharmaceuticals Incorporated; 50 Northern Avenue, Boston, MA 02210
ALYFTREK, VERTEX and associated logos are trademarks of Vertex Pharmaceuticals Incorporated.
All other trademarks referenced herein are the property of their respective owners.
©2026 Vertex Pharmaceuticals Incorporated
For more information, go to www.alyftrek.com or call 1-877-752-5933. -
PRINCIPAL DISPLAY PANEL - 4 mg/20 mg/50 mg Tablet Wallet Carton
Rx Only
NDC: 51167-135-0184 tablets
4-wallets
(each containing
21 tablets)alyftrek™
(vanzacaftor/tezacaftor/deutivacaftor)
tablets4 mg/20 mg/50 mg
per tabletATTENTION PHARMACIST: Dispense enclosed Medication Guide to each patient.
Lift to open
-
PRINCIPAL DISPLAY PANEL - 10 mg/50 mg/125 mg Tablet Wallet Carton
Rx Only
NDC: 51167-121-0156 tablets
4-wallets
(each containing
14 tablets)alyftrek™
(vanzacaftor/tezacaftor/deutivacaftor)
tablets10 mg/50 mg/125 mg
per tabletATTENTION PHARMACIST: Dispense enclosed Medication Guide to each patient.
Lift to open
-
INGREDIENTS AND APPEARANCE
ALYFTREK
vanzacaftor, tezacaftor, and deutivacaftor tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 51167-135 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength vanzacaftor (UNII: COM1POP492) (vanzacaftor - UNII:COM1POP492) vanzacaftor 4 mg tezacaftor (UNII: 8RW88Y506K) (tezacaftor - UNII:8RW88Y506K) tezacaftor 20 mg deutivacaftor (UNII: SHA6U5FJZL) (deutivacaftor - UNII:SHA6U5FJZL) deutivacaftor 50 mg Inactive Ingredients Ingredient Name Strength HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) HYPROMELLOSE ACETATE SUCCINATE 06081224 (3 MPA.S) (UNII: 6N003M473W) Sodium lauryl sulfate (UNII: 368GB5141J) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) Microcrystalline cellulose (UNII: OP1R32D61U) Magnesium stearate (UNII: 70097M6I30) HYPROMELLOSE 2910 (15 MPA.S) (UNII: 36SFW2JZ0W) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) Titanium dioxide (UNII: 15FIX9V2JP) Talc (UNII: 7SEV7J4R1U) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color PURPLE Score no score Shape ROUND Size 7mm Flavor Imprint Code V4 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 51167-135-01 4 in 1 CARTON 12/20/2024 1 21 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA218730 12/20/2024 ALYFTREK
vanzacaftor, tezacaftor, and deutivacaftor tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 51167-121 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength vanzacaftor (UNII: COM1POP492) (vanzacaftor - UNII:COM1POP492) vanzacaftor 10 mg tezacaftor (UNII: 8RW88Y506K) (tezacaftor - UNII:8RW88Y506K) tezacaftor 50 mg deutivacaftor (UNII: SHA6U5FJZL) (deutivacaftor - UNII:SHA6U5FJZL) deutivacaftor 125 mg Inactive Ingredients Ingredient Name Strength HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) HYPROMELLOSE ACETATE SUCCINATE 06081224 (3 MPA.S) (UNII: 6N003M473W) Sodium lauryl sulfate (UNII: 368GB5141J) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) Microcrystalline cellulose (UNII: OP1R32D61U) Magnesium stearate (UNII: 70097M6I30) HYPROMELLOSE 2910 (15 MPA.S) (UNII: 36SFW2JZ0W) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) Titanium dioxide (UNII: 15FIX9V2JP) Talc (UNII: 7SEV7J4R1U) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color PURPLE Score no score Shape OVAL (Oblong) Size 15mm Flavor Imprint Code V10 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 51167-121-01 4 in 1 CARTON 12/20/2024 1 14 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA218730 12/20/2024 Labeler - Vertex Pharmaceuticals Incorporated (602478257)
Trademark Results [ALYFTREK]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ALYFTREK 98760764 not registered Live/Pending |
Vertex Pharmaceuticals Incorporated 2024-09-20 |
 ALYFTREK 98321063 not registered Live/Pending |
Vertex Pharmaceuticals Incorporated 2023-12-19 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.