ISENTRESS- raltegravir tablet, film coated
ISENTRESS by
Drug Labeling and Warnings
ISENTRESS by is a Prescription medication manufactured, distributed, or labeled by A-S Medication Solutions. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ISENTRESS safely and effectively. See full prescribing information for ISENTRESS.
ISENTRESS® (raltegravir) film-coated tablets, for oral use
ISENTRESS® HD (raltegravir) film-coated tablets, for oral use
ISENTRESS® (raltegravir) chewable tablets, for oral use
ISENTRESS® (raltegravir) for oral suspension
Initial U.S. Approval: 2007INDICATIONS AND USAGE
Adult Patients:
ISENTRESS and ISENTRESS HD are human immunodeficiency virus integrase strand transfer inhibitors (HIV-1 INSTI) indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in adult patients (1).
Pediatric Patients:
ISENTRESS is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in pediatric patients weighing at least 2 kg (1).
ISENTRESS HD is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in pediatric patients weighing at least 40 kg (1).
DOSAGE AND ADMINISTRATION
ISENTRESS and ISENTRESS HD can be administered with or without food (2.1).
Do not substitute ISENTRESS chewable tablets or ISENTRESS for oral suspension for the ISENTRESS 400 mg or 600 mg film-coated tablet.
See specific dosing guidance for chewable tablets and the formulation for oral suspension (2.1).
Adults
- Treatment-naïve patients or patients who are virologically suppressed on an initial regimen of ISENTRESS 400 mg twice daily:
- 1200 mg (2 × 600 mg) film-coated tablet orally, once daily or
- 400 mg film-coated tablet orally, twice daily (2.2).
- Treatment experienced patients:
- 400 mg film-coated tablet orally, twice daily (2.2).
- During coadministration with rifampin in adults, 800 mg (2 × 400 mg) twice daily (2.2).
Pediatrics
- If weighing at least 40 kg, and either
- treatment-naïve patients or
- patients who are virologically suppressed on an initial regimen of ISENTRESS 400 mg twice daily:
- 1200 mg (2 × 600 mg) film-coated tablet orally, once daily or
- 400 mg film-coated tablet orally, twice daily or
- 300 mg chewable tablets, twice daily (2.3).
- If weighing at least 25 kg: One 400 mg film-coated tablet orally, twice daily. If unable to swallow a tablet, consider the chewable tablet, as specified in Table 2 (2.3).
- If weighing at least 3 kg to less than 25 kg: Weight based dosing, as specified in Table 4. For patients weighing between 11 and 20 kg, either the chewable tablet or the formulation for oral suspension can be used, as specified in Table 4 (2.3).
- For neonates (birth to 4 weeks [28 days] of age): Weight-based dosing of the oral suspension as specified in Table 5 (2.3).
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
None (4).
WARNINGS AND PRECAUTIONS
- Severe, potentially life-threatening and fatal skin reactions have been reported. This includes cases of Stevens-Johnson syndrome, hypersensitivity reaction and toxic epidermal necrolysis. Immediately discontinue treatment with ISENTRESS or ISENTRESS HD and other suspect agents if severe hypersensitivity, severe rash, or rash with systemic symptoms or liver aminotransferase elevations develops and monitor clinical status, including liver aminotransferases closely (5.1).
- Monitor for Immune Reconstitution Syndrome (5.2).
- Inform patients with phenylketonuria that the 100 mg and 25 mg chewable tablets contain phenylalanine (5.3).
ADVERSE REACTIONS
- The most common adverse reactions of moderate to severe intensity (≥2%) are insomnia, headache, dizziness, nausea and fatigue (6.1).
- Creatine kinase elevations were observed in subjects who received ISENTRESS or ISENTRESS HD. Myopathy and rhabdomyolysis have been reported. Use with caution in patients at increased risk of myopathy or rhabdomyolysis, such as patients receiving concomitant medications known to cause these conditions and patients with a history of rhabdomyolysis, myopathy or increased serum creatine kinase (6.2).
To report SUSPECTED ADVERSE REACTIONS, contact Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., at 1-877-888-4231 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Coadministration of ISENTRESS or ISENTRESS HD and other drugs may alter the plasma concentration of raltegravir. The potential for drug-drug interactions must be considered prior to and during therapy (7).
- Coadministration of ISENTRESS or ISENTRESS HD with drugs that are strong inducers of UGT1A1, such as rifampin, may result in reduced plasma concentrations of raltegravir (2.1, 7.1).
USE IN SPECIFIC POPULATIONS
Lactation: Women infected with HIV should be instructed not to breastfeed due to the potential for HIV transmission (8.2).
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2019
- Treatment-naïve patients or patients who are virologically suppressed on an initial regimen of ISENTRESS 400 mg twice daily:
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Recommendations
2.2 Adults
2.3 Pediatrics
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Severe Skin and Hypersensitivity Reactions
5.2 Immune Reconstitution Syndrome
5.3 Phenylketonurics
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Agents on the Pharmacokinetics of Raltegravir
7.2 Drugs without Clinically Significant Interactions with ISENTRESS or ISENTRESS HD
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Use in Patients with Hepatic Impairment
8.7 Use in Patients with Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Description of Clinical Studies
14.2 Treatment-Naïve Adult Subjects
14.3 Treatment-Experienced Adult Subjects
14.4 Pediatric Subjects
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Adult Patients:
ISENTRESS® and ISENTRESS® HD are indicated in combination with other antiretroviral agents for the treatment of human immunodeficiency virus (HIV-1) infection in adult patients.
Pediatric Patients:
ISENTRESS is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in pediatric patients weighing at least 2 kg.
ISENTRESS HD is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in pediatric patients weighing at least 40 kg.
-
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Recommendations
- Because the formulations have different pharmacokinetic profiles, do not substitute ISENTRESS chewable tablets or ISENTRESS for oral suspension for the ISENTRESS 400 mg film-coated tablet or the ISENTRESS HD 600 mg film-coated tablet. See specific dosing guidance for chewable tablets and the formulation for oral suspension.
- Because the extent to which ISENTRESS may be dialyzable is unknown, dosing before a dialysis session should be avoided [see Clinical Pharmacology (12.3)].
- ISENTRESS film-coated tablets must be swallowed whole.
- ISENTRESS chewable tablets may be chewed or swallowed whole. Maximum daily dose is 300 mg taken by mouth twice daily.
- ISENTRESS for oral suspension:
- Using the provided mixing cup, combine 10 mL of water and the entire contents of one packet of ISENTRESS for oral suspension and mix. Each single-use packet for oral suspension contains 100 mg of raltegravir which is suspended in 10 mL of water giving a final concentration of 10 mg per mL. Maximum daily dose is 100 mg taken by mouth twice daily.
- Gently swirl the mixing cup for 45 seconds in a circular motion to mix the powder into a uniform suspension. Do not shake.
- Once mixed, measure the prescribed dose volume of suspension with a syringe and administer the dose orally. The dose should be administered orally within 30 minutes of mixing.
- Discard any remaining suspension into the trash.
- For more details on preparation and administration of the suspension, see Instructions for Use.
2.2 Adults
The recommended adult dosage of ISENTRESS film-coated tablets is displayed in Table 1. ISENTRESS and ISENTRESS HD should be taken by mouth and may be taken with or without food [see Clinical Pharmacology (12.3)].
Table 1: Dosing Recommendations for ISENTRESS and ISENTRESS HD in Adult Patients Population Recommended Dose Treatment-naïve patients or patients who are virologically suppressed on an initial regimen of ISENTRESS 400 mg twice daily 1200 mg (2 × 600 mg) once daily or 400 mg twice daily Treatment-experienced 400 mg twice daily Treatment-naïve or treatment-experienced when coadministered with rifampin [see Drug Interactions (7.1)] 800 mg (2 × 400 mg) twice daily 2.3 Pediatrics
The recommended pediatric dosage of ISENTRESS is displayed in Table 2. ISENTRESS film-coated tablets, chewable tablets and for oral suspension should be taken by mouth and may be taken with or without food [see Clinical Pharmacology (12.3)].
Table 2: Dosing Recommendations for ISENTRESS and ISENTRESS HD in Pediatric Patients Recommended Pediatric Dosage and Formulation Population/Weight Film-Coated Tablets 400 mg Film-Coated Tablets 600 mg Chewable Tablets 100 mg and 25 mg For Oral Suspension 100 mg - * If able to swallow a tablet
If at least 40 kg and either - treatment-naïve or
- virologically suppressed on an initial regimen of ISENTRESS 400 mg twice daily
400 mg twice daily 1200 mg (2 × 600 mg) once daily 300 mg twice daily (see Table 3) NA If at least 25 kg 400 mg twice daily* NA Weight-based dosing twice daily (see Table 3) NA If at least 4 weeks of age and weighing 3 kg to less than 25 kg NA NA Weight-based dosing twice daily (see Table 4) Weight-based dosing twice daily up to 20 kg (see Table 4) From birth to 4 weeks (28 days) weighing at least 2 kg NA NA NA Weight-based dosing once daily or twice daily (see Table 5) Table 3: Alternative Dosage* with ISENTRESS Chewable Tablets for Pediatric Patients Weighing at Least 25 kg Body Weight
(kg)Dose Number of Chewable Tablets - * The weight-based dosing recommendation for the chewable tablet is based on approximately 6 mg/kg/dose twice daily [see Clinical Pharmacology (12.3)].
- † The 100 mg chewable tablet can be divided into equal halves.
25 to less than 28 150 mg twice daily 1.5 × 100 mg† twice daily 28 to less than 40 200 mg twice daily 2 × 100 mg twice daily At least 40 300 mg twice daily 3 × 100 mg twice daily Table 4: Recommended Dosage* for ISENTRESS For Oral Suspension and Chewable Tablets in Pediatric Patients at Least 4 Weeks of Age and Weighing at Least 3 kg and Less than 25 kg Body Weight
(kg)Volume (Dose) of Suspension to be Administered Number of Chewable Tablets† - * The weight-based dosing recommendation for the chewable tablet and oral suspension is based on approximately 6 mg/kg/dose twice daily [see Clinical Pharmacology (12.3)].
- † The chewable tablets are available as 25 mg and 100 mg tablets.
- ‡ For weight between 11 and 20 kg, either formulation can be used.
- § The 100 mg chewable tablet can be divided into equal halves.
3 to less than 4 2.5 mL (25 mg) twice daily 4 to less than 6 3 mL (30 mg) twice daily 6 to less than 8 4 mL (40 mg) twice daily 8 to less than 11 6 mL (60 mg) twice daily 11 to less than 14‡ 8 mL (80 mg) twice daily 3 × 25 mg twice daily 14 to less than 20‡ 10 mL (100 mg) twice daily 1 × 100 mg twice daily 20 to less than 25 1.5 × 100 mg§ twice daily - For full-term neonates (birth to 4 weeks [28 days] of age): Weight-based dosing of the oral suspension as specified in Table 5.
- No data are available in pre-term neonates. The use of ISENTRESS is not recommended in pre-term neonates.
Table 5: Recommended Dose for ISENTRESS For Oral Suspension in Full-Term Neonates (Birth to 4 Weeks [28 days] of Age) Body Weight
(kg)Volume (Dose) of Suspension to be Administered Note: If the mother has taken ISENTRESS or ISENTRESS HD 2-24 hours before delivery, the neonate's first dose should be given between 24-48 hours after birth. - * The dosing recommendations are based on approximately 1.5 mg/kg/dose.
- † The dosing recommendations are based on approximately 3 mg/kg/dose.
Birth to 1 Week - Once daily dosing* 2 to less than 3 0.4 mL (4 mg) once daily 3 to less than 4 0.5 mL (5 mg) once daily 4 to less than 5 0.7 mL (7 mg) once daily 1 to 4 Weeks - Twice daily dosing † 2 to less than 3 0.8 mL (8 mg) twice daily 3 to less than 4 1 mL (10 mg) twice daily 4 to less than 5 1.5 mL (15 mg) twice daily -
3 DOSAGE FORMS AND STRENGTHS
- Film-coated Tablets
- 400 mg pink, oval-shaped, film-coated tablets with "227" on one side (ISENTRESS).
- 600 mg yellow, oval-shaped, film-coated tablets with Merck logo and "242" on one side and plain on the other side (ISENTRESS HD).
- Chewable Tablets
- 100 mg pale orange, oval-shaped, orange-banana flavored, chewable tablets scored on both sides and imprinted on one face with the Merck logo and "477" on opposite sides of the score.
- 25 mg pale yellow, round, orange-banana flavored, chewable tablets with the Merck logo on one side and "473" on the other side.
- For Oral Suspension
- 100 mg white to off-white, banana flavored, granular powder that may contain yellow or beige to tan particles in a child resistant single-use foil packet.
- Film-coated Tablets
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Severe Skin and Hypersensitivity Reactions
Severe, potentially life-threatening, and fatal skin reactions have been reported. These include cases of Stevens-Johnson syndrome and toxic epidermal necrolysis. Hypersensitivity reactions have also been reported and were characterized by rash, constitutional findings, and sometimes, organ dysfunction, including hepatic failure. Discontinue ISENTRESS or ISENTRESS HD and other suspect agents immediately if signs or symptoms of severe skin reactions or hypersensitivity reactions develop (including, but not limited to, severe rash or rash accompanied by fever, general malaise, fatigue, muscle or joint aches, blisters, oral lesions, conjunctivitis, facial edema, hepatitis, eosinophilia, angioedema). Clinical status including liver aminotransferases should be monitored and appropriate therapy initiated. Delay in stopping ISENTRESS or ISENTRESS HD treatment or other suspect agents after the onset of severe rash may result in a life-threatening reaction.
5.2 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including ISENTRESS. During the initial phase of combination antiretroviral treatment, patients whose immune systems respond may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jiroveci pneumonia, tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
5.3 Phenylketonurics
ISENTRESS Chewable Tablets contain phenylalanine, a component of aspartame. Each 25 mg ISENTRESS Chewable Tablet contains approximately 0.05 mg phenylalanine. Each 100 mg ISENTRESS Chewable Tablet contains approximately 0.10 mg phenylalanine. Phenylalanine can be harmful to patients with phenylketonuria.
-
6 ADVERSE REACTIONS
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
6.1 Clinical Trials Experience
Treatment-Naïve Adults
The safety of ISENTRESS was evaluated in HIV-infected treatment-naïve subjects in 2 Phase III studies: STARTMRK evaluated ISENTRESS 400 mg twice daily versus efavirenz, both in combination with emtricitabine (+) tenofovir disoproxil fumarate (TDF), and ONCEMRK evaluated ISENTRESS HD 1200 mg (2 × 600 mg) once daily versus ISENTRESS 400 mg twice daily, both in combination with emtricitabine (+) tenofovir disoproxil fumarate. Safety data from these two studies are presented side-by-side in Tables 5 and 6 to simplify presentation; direct comparisons across trials should not be made due to differing duration of follow-up and study design.
STARTMRK (ISENTRESS 400 mg twice daily)
In STARTMRK, subjects received ISENTRESS 400 mg twice daily (N=281) or efavirenz (EFV) 600 mg at bedtime (N=282) both in combination with emtricitabine (+) tenofovir disoproxil fumarate, (N=282). During double-blind treatment, the total follow-up for subjects receiving ISENTRESS 400 mg twice daily + emtricitabine (+) tenofovir disoproxil fumarate was 1104 patient-years and 1036 patient-years for subjects receiving efavirenz 600 mg at bedtime + emtricitabine (+) tenofovir disoproxil fumarate.
In STARTMRK, the rate of discontinuation of therapy due to adverse events through Week 240 was 5% in subjects receiving ISENTRESS + emtricitabine (+) tenofovir disoproxil fumarate and 10% in subjects receiving efavirenz + emtricitabine (+) tenofovir disoproxil fumarate.
ONCEMRK (ISENTRESS HD 1200 mg [2 × 600 mg] once daily)
In ONCEMRK, subjects received ISENTRESS HD 1200 mg once daily (n=531) or ISENTRESS 400 mg twice daily (n=266) both in combination with emtricitabine (+) tenofovir disoproxil fumarate. During double-blind treatment, the total follow-up for subjects with ISENTRESS HD 1200 mg once daily was 913 patient-years and for ISENTRESS 400 mg twice daily was 450 patient-years.
In ONCEMRK, the rate of discontinuation of therapy due to adverse events through Week 96 was 1% in subjects receiving ISENTRESS HD 1200 mg (2 × 600 mg) once daily and 2% in subjects receiving ISENTRESS 400 mg twice daily.
Clinical adverse reactions of moderate to severe intensity occurring in ≥2% of treatment-naïve subjects treated with ISENTRESS 400 mg twice daily or efavirenz in STARTMRK through Week 240 or ISENTRESS HD 1200 mg once daily or ISENTRESS 400 mg twice daily in ONCEMRK through Week 96 are presented in Table 6.
In STARTMRK, clinical adverse reactions of all intensities (mild, moderate and severe) occurring in ≥2% of subjects on ISENTRESS 400 mg twice daily through Week 240 also include diarrhea, flatulence, asthenia, decreased appetite, abnormal dreams, depression and nightmare. In ONCEMRK, clinical adverse reactions of all intensities (mild, moderate and severe) occurring in ≥2% of subjects on ISENTRESS HD or ISENTRESS 400 mg twice daily through Week 96 also include abdominal pain, diarrhea, vomiting, and decreased appetite.
Table 6: Adverse Reactions* of Moderate to Severe Intensity† Occurring in ≥2% of Treatment-Naïve Adult Subjects Receiving ISENTRESS and ISENTRESS HD System Organ Class, Preferred Term STARTMRK
Week 240ONCEMRK
Week 96ISENTRESS 400 mg Twice Daily
(N= 281)Efavirenz 600 mg At Bedtime
(N= 282)ISENTRESS HD 1200 mg Once Daily
(N=531)ISENTRESS 400 mg Twice Daily
(N=266)Note: ISENTRESS BID, ISENTRESS HD and efavirenz were administered with emtricitabine (+) tenofovir disoproxil fumarate N= total number of subjects per treatment group - * Includes adverse experiences considered by investigators to be at least possibly, probably, or definitely related to the drug.
- † Intensities are defined as follows: Moderate (discomfort enough to cause interference with usual activity); Severe (incapacitating with inability to work or do usual activity).
Headache 4% 5% 1% <1% Insomnia 4% 4% <1% <1% Nausea 3% 4% 1% 0% Dizziness 2% 6% <1% 0% Fatigue 2% 3% 0% 0% Laboratory Abnormalities
The percentages of adult subjects with selected Grade 2 to 4 laboratory abnormalities (that represent a worsening Grade from baseline) who were treated with ISENTRESS 400 mg twice daily or efavirenz in STARTMRK or ISENTRESS HD 1200 mg once daily or ISENTRESS 400 mg twice daily in ONCEMRK are presented in Table 7.
Table 7: Selected Grade 2 to 4 Laboratory Abnormalities Reported in Treatment-Naïve Subjects STARTMRK
Week 240ONCEMRK
Week 96Laboratory Parameter Preferred Term (Unit) Limit ISENTRESS
400 mg
Twice Daily
(N = 281)Efavirenz
600 mg
At Bedtime
(N = 282)ISENTRESS HD
1200 mg
Once Daily
(N=531)ISENTRESS
400 mg
Twice Daily
(N=266)ULN = Upper limit of normal range Notes: ISENTRESS BID, ISENTRESS HD and Efavirenz were administered with emtricitabine (+) tenofovir disoproxil fumarate - * Test not done in ONCEMRK
- † Test not done in STARTMRK
Hematology Absolute neutrophil count (103/µL) Grade 2 0.75 - 0.999 3% 5% 2% 1% Grade 3 0.50 - 0.749 3% 1% 1% 1% Grade 4 <0.50 1% 1% <1% 0% Hemoglobin (gm/dL) Grade 2 7.5 - 8.4 1% 1% 0% 0% Grade 3 6.5 - 7.4 1% 1% 0% 0% Grade 4 <6.5 <1% 0% 0% 0% Platelet count (103/µL) Grade 2 50 - 99.999 1% 0% 1% <1% Grade 3 25 - 49.999 <1% <1% 0% 0% Grade 4 <25 0% 0% 0% <1% Blood chemistry Fasting (non-random) serum glucose test (mg/dL)* Grade 2 126 - 250 7% 6% - - Grade 3 251 - 500 2% 1% - - Grade 4 >500 0% 0% - - Total serum bilirubin Grade 2 1.6 - 2.5 × ULN 5% <1% 3% 2% Grade 3 2.6 - 5.0 × ULN 1% 0% 1% <1% Grade 4 >5.0 × ULN <1% 0% <1% 0% Creatinine Grade 2 1.4-1.8 × ULN 1% 1% 0% <1% Grade 3 1.9-3.4 × ULN 0% <1% 0% 0% Grade 4 ≥3.5 × ULN 0% 0% 0% 0% Serum aspartate aminotransferase Grade 2 2.6 - 5.0 × ULN 8% 10% 5% 3% Grade 3 5.1 - 10.0 × ULN 5% 3% 2% <1% Grade 4 >10.0 × ULN 1% <1% 1% <1% Serum alanine aminotransferase Grade 2 2.6 - 5.0 × ULN 11% 12% 4% 2% Grade 3 5.1 - 10.0 × ULN 2% 2% 1% <1% Grade 4 >10.0 × ULN 2% 1% 1% <1% Serum alkaline phosphatase Grade 2 2.6 - 5.0 × ULN 1% 3% 1% 0% Grade 3 5.1 - 10.0 × ULN 0% 1% <1% 0% Grade 4 >10.0 × ULN <1% <1% 0% 0% Lipase† Grade 2 1.6-3.0 x ULN - - 7% 5% Grade 3 3.1-5.0 × ULN - - 2% 1% Grade 4 >5.0 × ULN - - 2% 1% Creatine kinase† Grade 2 6.0-9.9 × ULN - - 4% 5% Grade 3 10.0-19.9 × ULN - - 3% 3% Grade 4 ≥20.0 × ULN - - 3% 2% Lipids, Change from Baseline
Changes from baseline in fasting lipids are shown in Table 8.
Table 8: Lipid Values, Mean Change from Baseline, STARTMRK Study Laboratory Parameter Preferred Term ISENTRESS 400 mg
Twice Daily + Emtricitabine (+) Tenofovir Disoproxil Fumarate
N = 207Efavirenz 600 mg
At Bedtime + Emtricitabine (+) Tenofovir Disoproxil Fumarate
N = 187Change from Baseline at
Week 240Change from Baseline at
Week 240Baseline
MeanWeek 240
MeanMean Change Baseline
MeanWeek 240
MeanMean Change (mg/dL) (mg/dL) (mg/dL) (mg/dL) (mg/dL) (mg/dL) Notes:
N = total number of subjects per treatment group with at least one lipid test result available. The analysis is based on all available data.
If subjects initiated or increased serum lipid-reducing agents, the last available lipid values prior to the change in therapy were used in the analysis. If the missing data was due to other reasons, subjects were censored thereafter for the analysis. At baseline, serum lipid-reducing agents were used in 5% of subjects in the group receiving ISENTRESS and 3% in the efavirenz group. Through Week 240, serum lipid-reducing agents were used in 9% of subjects in the group receiving ISENTRESS and 15% in the efavirenz group.- * Fasting (non-random) laboratory tests at Week 240.
LDL-Cholesterol* 96 106 10 93 118 25 HDL-Cholesterol* 38 44 6 38 51 13 Total Cholesterol* 159 175 16 157 201 44 Triglyceride* 128 130 2 141 178 37 Treatment-Experienced Adults
The safety assessment of ISENTRESS in treatment-experienced subjects is based on the pooled safety data from the randomized, double-blind, placebo-controlled trials, BENCHMRK 1 and BENCHMRK 2 in antiretroviral treatment-experienced HIV-1 infected adult subjects. A total of 462 subjects received the recommended dose of ISENTRESS 400 mg twice daily in combination with optimized background therapy (OBT) compared to 237 subjects taking placebo in combination with OBT. The median duration of therapy in these trials was 96 weeks for subjects receiving ISENTRESS and 38 weeks for subjects receiving placebo. The total exposure to ISENTRESS was 708 patient-years versus 244 patient-years on placebo. The rates of discontinuation due to adverse events were 4% in subjects receiving ISENTRESS and 5% in subjects receiving placebo.
Clinical ADRs were considered by investigators to be causally related to ISENTRESS + OBT or placebo + OBT. Clinical ADRs of moderate to severe intensity occurring in ≥2% of subjects treated with ISENTRESS and occurring at a higher rate compared to placebo are presented in Table 9.
Table 9: Adverse Drug Reactions* of Moderate to Severe Intensity† Occurring in ≥2% of Treatment-Experienced Adult Subjects Receiving ISENTRESS and at a Higher Rate Compared to Placebo (96 Week Analysis) System Organ Class, Adverse Reactions Randomized Studies BENCHMRK 1 and BENCHMRK 2 ISENTRESS 400 mg Twice Daily + OBT
(n = 462)Placebo + OBT
(n = 237)Nervous System Disorders n=total number of subjects per treatment group. - * Includes adverse reactions at least possibly, probably, or definitely related to the drug.
- † Intensities are defined as follows: Moderate (discomfort enough to cause interference with usual activity); Severe (incapacitating with inability to work or do usual activity).
Headache 2% <1% Laboratory Abnormalities
The percentages of adult subjects treated with ISENTRESS 400 mg twice daily or placebo in Studies BENCHMRK 1 and BENCHMRK 2 with selected Grade 2 to 4 laboratory abnormalities representing a worsening Grade from baseline are presented in Table 10.
Table 10: Selected Grade 2 to 4 Laboratory Abnormalities Reported in Treatment-Experienced Subjects (96 Week Analysis) Randomized Studies BENCHMRK 1 and BENCHMRK 2 Laboratory Parameter Preferred Term
(Unit)Limit ISENTRESS 400 mg Twice Daily + OBT
(N = 462)Placebo + OBT
(N = 237)ULN = Upper limit of normal range Hematology Absolute neutrophil count (103/µL) Grade 2 0.75 - 0.999 4% 5% Grade 3 0.50 - 0.749 3% 3% Grade 4 <0.50 1% <1% Hemoglobin (gm/dL) Grade 2 7.5 - 8.4 1% 3% Grade 3 6.5 - 7.4 1% 1% Grade 4 <6.5 <1% 0% Platelet count (103/µL) Grade 2 50 - 99.999 3% 5% Grade 3 25 - 49.999 1% <1% Grade 4 <25 1% <1% Blood chemistry Fasting (non-random) serum glucose test (mg/dL) Grade 2 126 - 250 10% 7% Grade 3 251 - 500 3% 1% Grade 4 >500 0% 0% Total serum bilirubin Grade 2 1.6 - 2.5 × ULN 6% 3% Grade 3 2.6 - 5.0 × ULN 3% 3% Grade 4 >5.0 × ULN 1% 0% Serum aspartate aminotransferase Grade 2 2.6 - 5.0 × ULN 9% 7% Grade 3 5.1 - 10.0 × ULN 4% 3% Grade 4 >10.0 × ULN 1% 1% Serum alanine aminotransferase Grade 2 2.6 - 5.0 × ULN 9% 9% Grade 3 5.1 - 10.0 × ULN 4% 2% Grade 4 >10.0 × ULN 1% 2% Serum alkaline phosphatase Grade 2 2.6 - 5.0 × ULN 2% <1% Grade 3 5.1 - 10.0 × ULN <1% 1% Grade 4 >10.0 × ULN 1% <1% Serum pancreatic amylase test Grade 2 1.6 - 2.0 × ULN 2% 1% Grade 3 2.1 - 5.0 × ULN 4% 3% Grade 4 >5.0 × ULN <1% <1% Serum lipase test Grade 2 1.6 - 3.0 × ULN 5% 4% Grade 3 3.1 - 5.0 × ULN 2% 1% Grade 4 >5.0 × ULN 0% 0% Serum creatine kinase Grade 2 6.0 - 9.9 × ULN 2% 2% Grade 3 10.0 - 19.9 × ULN 4% 3% Grade 4 ≥20.0 × ULN 3% 1% Less Common Adverse Reactions Observed in Treatment-Naïve and Treatment-Experienced Studies
The following ADRs occurred in <2% of treatment-naïve or treatment-experienced subjects receiving ISENTRESS or ISENTRESS HD in a combination regimen. These events have been included because of their seriousness, increased frequency compared with efavirenz or placebo, or investigator's assessment of potential causal relationship.
Gastrointestinal Disorders: abdominal pain, gastritis, dyspepsia, vomiting
General Disorders and Administration Site Conditions: asthenia
Hepatobiliary Disorders: hepatitis
Immune System Disorders: hypersensitivity
Infections and Infestations: genital herpes, herpes zoster
Psychiatric Disorders: depression (particularly in subjects with a pre-existing history of psychiatric illness), including suicidal ideation and behaviors
Renal and Urinary Disorders: nephrolithiasis, renal failure
Selected Adverse Events - Adults
In studies of ISENTRESS 400 mg twice daily, cancers were reported in treatment-experienced subjects who initiated ISENTRESS or placebo, both with OBT, and in treatment-naïve subjects who initiated ISENTRESS or efavirenz, both with emtricitabine (+) tenofovir disoproxil fumarate; several were recurrent. The types and rates of specific cancers were those expected in a highly immunodeficient population (many had CD4+ counts below 50 cells/mm3 and most had prior AIDS diagnoses). The risk of developing cancer in these studies was similar in the group receiving ISENTRESS and the group receiving the comparator.
Grade 2-4 creatine kinase laboratory abnormalities were observed in subjects treated with ISENTRESS and ISENTRESS HD (see Tables 6 and 8). Myopathy and rhabdomyolysis have been reported with ISENTRESS. Use with caution in patients at increased risk of myopathy or rhabdomyolysis, such as patients receiving concomitant medications known to cause these conditions and patients with a history of rhabdomyolysis, myopathy or increased serum creatine kinase.
Rash occurred more commonly in treatment-experienced subjects receiving regimens containing ISENTRESS + darunavir/ritonavir compared to subjects receiving ISENTRESS without darunavir/ritonavir or darunavir/ritonavir without ISENTRESS. However, rash that was considered drug related occurred at similar rates for all three groups. These rashes were mild to moderate in severity and did not limit therapy; there were no discontinuations due to rash.
Patients with Co-existing Conditions - Adults
Patients Co-infected with Hepatitis B and/or Hepatitis C Virus
In Phase III studies of ISENTRESS, patients with chronic (but not acute) active hepatitis B and/or hepatitis C virus co-infection were permitted to enroll provided that baseline liver function tests did not exceed 5 times the upper limit of normal (ULN). In the treatment-experienced studies, BENCHMRK 1 and BENCHMRK 2, 16% of all patients (114/699) were co-infected; in the treatment-naïve studies, STARTMRK and ONCEMRK, 6% (34/563) and 3% (23/797), respectively, were co-infected. In general the safety profile of ISENTRESS in subjects with hepatitis B and/or hepatitis C virus co-infection was similar to that in subjects without hepatitis B and/or hepatitis C virus co-infection, although the rates of AST and ALT abnormalities were higher in the subgroup with hepatitis B and/or hepatitis C virus co-infection for all treatment groups.
At 96 weeks, in treatment-experienced subjects receiving ISENTRESS 400 mg twice daily, Grade 2 or higher laboratory abnormalities that represent a worsening Grade from baseline of AST, ALT or total bilirubin occurred in 29%, 34% and 13%, respectively, of co-infected subjects treated with ISENTRESS as compared to 11%, 10% and 9% of all other subjects treated with ISENTRESS. At 240 weeks, in treatment-naïve subjects receiving ISENTRESS 400 mg twice daily, Grade 2 or higher laboratory abnormalities that represent a worsening Grade from baseline of AST, ALT or total bilirubin occurred in 22%, 44% and 17%, respectively, of co-infected subjects treated with ISENTRESS as compared to 13%, 13% and 5% of all other subjects treated with ISENTRESS.
At 96 weeks, in treatment-naïve subjects receiving ISENTRESS HD 1200 mg (2 × 600 mg) once daily, Grade 2 or higher laboratory abnormalities that represent a worsening Grade from baseline of AST, ALT or total bilirubin occurred in 27%, 40% and 13%, respectively, of co-infected subjects treated with ISENTRESS HD 1200 mg once daily as compared to 7%, 5% and 3% of all other subjects treated with ISENTRESS HD 1200 mg once daily.
Pediatrics
2 to 18 Years of Age
ISENTRESS has been studied in 126 antiretroviral treatment-experienced HIV-1 infected children and adolescents 2 to 18 years of age, in combination with other antiretroviral agents in IMPAACT P1066 [see Use in Specific Populations (8.4) and Clinical Studies (14.4)]. Of the 126 patients, 96 received the recommended dose of ISENTRESS.
In these 96 children and adolescents, frequency, type and severity of drug related adverse reactions through Week 24 were comparable to those observed in adults.
One patient experienced drug related clinical adverse reactions of Grade 3 psychomotor hyperactivity, abnormal behavior and insomnia; one patient experienced a Grade 2 serious drug related allergic rash.
One patient experienced drug related laboratory abnormalities, Grade 4 AST and Grade 3 ALT, which were considered serious.
4 Weeks to Less than 2 Years of Age
ISENTRESS has also been studied in 26 HIV-1 infected infants and toddlers 4 weeks to less than 2 years of age, in combination with other antiretroviral agents in IMPAACT P1066 [see Use in Specific Populations (8.4) and Clinical Studies (14.4)].
In these 26 infants and toddlers, the frequency, type and severity of drug-related adverse reactions through Week 48 were comparable to those observed in adults.
One patient experienced a Grade 3 serious drug-related allergic rash that resulted in treatment discontinuation.
HIV-1 Exposed Neonates
Forty-two neonates were treated with ISENTRESS for up to 6 weeks from birth, and followed for a total of 24 weeks in IMPAACT P1110 [see Use in Specific Populations (8.4)]. There were no drug related clinical adverse reactions and three drug-related laboratory adverse reactions (one case of transient Grade 4 neutropenia in a subject receiving zidovudine-containing regimen for prevention of mother to child transmission (PMTCT), and two bilirubin elevations (one each, Grade 1 and Grade 2) considered non-serious and not requiring specific therapy). The safety profile in neonates was generally similar to that observed in older patients treated with ISENTRESS. No clinically meaningful differences in the adverse event profile of neonates were observed when compared to adults.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of ISENTRESS. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System Disorders: thrombocytopenia
Gastrointestinal Disorders: diarrhea
Hepatobiliary Disorders: hepatic failure (with and without associated hypersensitivity) in patients with underlying liver disease and/or concomitant medications
Musculoskeletal and Connective Tissue Disorders: rhabdomyolysis
Nervous System Disorders: cerebellar ataxia
Psychiatric Disorders: anxiety, paranoia
-
7 DRUG INTERACTIONS
7.1 Effect of Other Agents on the Pharmacokinetics of Raltegravir
Raltegravir is not a substrate of cytochrome P450 (CYP) enzymes. Based on in vivo and in vitro studies, raltegravir is eliminated mainly by metabolism via a UGT1A1-mediated glucuronidation pathway. Coadministration of ISENTRESS with drugs that inhibit UGT1A1 may increase plasma levels of raltegravir and coadministration of ISENTRESS with drugs that induce UGT1A1, such as rifampin, may reduce plasma levels of raltegravir (see Table 11).
Selected drug interactions are presented in Table 11 [see Clinical Pharmacology (12.3)]. In some cases, recommendations differ for ISENTRESS versus ISENTRESS HD.
Table 11: Selected Drug Interactions in Adults Concomitant Drug Class:
Drug NameEffect on Concentration of Raltegravir Clinical Comment for ISENTRESS Clinical Comment for ISENTRESS HD Metal-Containing Antacids Aluminum and/or magnesium-containing antacids ↓ Coadministration or staggered administration is not recommended. Calcium carbonate antacid ↓ No dose adjustment Co-administration is not recommended Other Agents Rifampin ↓ The recommended dosage is 800 mg twice daily during coadministration with rifampin. There are no data to guide co-administration of ISENTRESS with rifampin in patients below 18 years of age [see Dosage and Administration (2.1)]. Coadministration is not recommended. Tipranavir/ritonavir No dose adjustment Coadministration is not recommended Etravirine ↓ No dose adjustment Coadministration is not recommended. Strong inducers of drug metabolizing enzymes not mentioned above e.g., Carbamazepine
Phenobarbital
Phenytoin↓↔ The impact of other strong inducers of drug metabolizing enzymes on raltegravir is unknown. Coadministration is not recommended. 7.2 Drugs without Clinically Significant Interactions with ISENTRESS or ISENTRESS HD
ISENTRESS
In drug interaction studies performed using ISENTRESS film-coated tablets 400 mg twice daily dose, raltegravir did not have a clinically meaningful effect on the pharmacokinetics of the following: ethinyl estradiol/norgestimate, methadone, midazolam, lamivudine, tenofovir disoproxil fumarate, etravirine, darunavir/ritonavir, or boceprevir. Moreover, atazanavir, atazanavir/ritonavir, boceprevir, calcium carbonate antacids, darunavir/ritonavir, efavirenz, etravirine, omeprazole, or tipranavir/ritonavir did not have a clinically meaningful effect on the pharmacokinetics of 400 mg twice daily raltegravir. No dose adjustment is required when ISENTRESS 400 mg twice daily is coadministered with these drugs.
There is no predicted pharmacokinetic drug interaction between ISENTRESS and tenofovir alafenamide.
ISENTRESS HD
In drug interaction studies, efavirenz did not have a clinically meaningful effect on the pharmacokinetics of ISENTRESS HD 1200 mg (2 × 600 mg) once daily. No dose adjustment is recommended when ISENTRESS HD 1200 mg once daily is coadministered with atazanavir, atazanavir/ritonavir, hormonal contraceptives, methadone, lamivudine, tenofovir disoproxil fumarate, darunavir/ritonavir, boceprevir, efavirenz and omeprazole.
There is no predicted pharmacokinetic drug interaction between ISENTRESS HD and tenofovir alafenamide.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
Available data from the APR show no difference in the rate of overall birth defects for raltegravir compared to the background rate for major birth defects of 2.7% in the U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP) (see Data). The rate of miscarriage is not reported in the APR. The estimated background rate of miscarriage in clinically recognized pregnancies in the U.S. general population is 15-20%. The background risk for major birth defects and miscarriage for the indicated population is unknown. Methodological limitations of the APR include the use of MACDP as the external comparator group. The MACDP population is not disease-specific, evaluates women and infants from a limited geographic area, and does not include outcomes for births that occurred at <20 weeks gestation (see Data).
In animal reproduction studies in rats and rabbits, no evidence of adverse developmental outcomes was observed with oral administration of raltegravir during organogenesis at doses that produced exposures up to approximately 4 times the maximal recommended human dose (MRHD) of 1200 mg (see Data).
Data
Human Data
Based on prospective reports from the APR of over 500 exposures to raltegravir during pregnancy resulting in live births (including over 250 exposures in the first trimester), there was no difference between the overall risk of birth defects for raltegravir compared with the background birth defect rate of 2.7% in the U.S. reference population of the MACDP. The prevalence of defects in live births was 2.9% (95% CI: 1.2% to 5.6%) following first trimester exposure to raltegravir-containing regimens and 3.6% (95% CI: 1.6% to 6.6%) following second and third trimester exposure to raltegravir-containing regimens.
Animal Data
In a combined embryo/fetal and pre/postnatal development study, raltegravir was administered orally to rats at doses of 100, 300, 600 mg/kg/day on gestation day 6 to 20 or from gestation day 6 to lactation day 20. No effects on pre/postnatal development were observed up to the highest dose tested. Embryo-fetal findings were limited to an increase in the incidence of supernumerary ribs in the 600 mg/kg/day group. Systemic exposure (AUC) at 600 mg/kg/day was approximately 3 times higher than exposure at the MRHD of 1200 mg.
In pregnant rabbits, raltegravir was administered orally at doses of 100, 500, or 1000 mg/kg/day during the gestation days 7 to 20. No embryo/fetal effects were noted up to the highest dose of 1000 mg/kg/day. Systemic exposure (AUC) at 1000 mg/kg/day was approximately 4 times higher than exposures at the MRHD of 1200 mg. In both species, raltegravir has been shown to cross the placenta, with fetal plasma concentrations observed in rats approximately 1.5 to 2.5 times greater than in maternal plasma and fetal plasma concentrations in rabbits approximately 2% that of maternal concentrations on gestation day 20.
8.2 Lactation
Risk Summary
The Centers for Disease Control and Prevention recommend that HIV-1 infected mothers in the United States not breastfeed their infants to avoid risking postnatal transmission of HIV-1 infection. There are no data on the presence of raltegravir in human milk, the effects on the breastfed infant, or the effects on milk production. When administered to lactating rats, raltegravir was present in milk [see Data]. Because of the potential for: 1) HIV transmission (in HIV-negative infants), 2) developing viral resistance (in HIV-positive infants), and 3) adverse reactions in a breastfed infant, instruct mothers not to breastfeed if they are receiving ISENTRESS/ISENTRESS HD.
8.4 Pediatric Use
ISENTRESS
HIV-1 Infected Children
The safety, tolerability, pharmacokinetic profile, and efficacy of twice daily ISENTRESS were evaluated in HIV-1 infected infants, children and adolescents 4 weeks to 18 years of age in an open-label, multicenter clinical trial, IMPAACT P1066 [see Dosage and Administration (2.3), Clinical Pharmacology (12.3) and Clinical Studies (14.4)]. The safety profile was comparable to that observed in adults [see Adverse Reactions (6.1)].
HIV-1 Exposed Neonates
The safety and pharmacokinetics of ISENTRESS for oral suspension were evaluated in 42 full-term HIV-1 exposed neonates at high risk of acquiring HIV-1 infection in a Phase 1, open-label, multicenter clinical study, IMPAACT P1110. Cohort 1 neonates received 2 single doses of ISENTRESS for oral suspension: the first within 48 hours of birth and the second at 7 to 10 days of age. Cohort 2 neonates received daily dosing of ISENTRESS for oral suspension for 6 weeks: 1.5 mg/kg once daily starting within 48 hours of birth through Day 7 (week 1); 3 mg/kg twice daily on Days 8 to 28 of age (weeks 2 to 4); and 6 mg/kg twice daily on Days 29 to 42 of age (weeks 5 and 6). Sixteen neonates were enrolled in Cohort 1 (10 were exposed and 6 were unexposed to raltegravir in utero) and 26 in Cohort 2 (all unexposed to raltegravir in utero); all infants received a standard of care antiretroviral drug regimen for prevention of mother to child transmission. All enrolled neonates were followed for safety for a duration of 24 weeks. The 42 infants were 52% male, 69% Black and 12% Caucasian. HIV-1 status was assessed by nucleic acid test at birth, week 6 and week 24; all patients were HIV-1 negative at completion of the study. The safety profile was comparable to that observed in adults [see Adverse Reactions (6.1)].
ISENTRESS is not recommended in pre-term neonates or in pediatric patients weighing less than 2 kg.
ISENTRESS HD
ISENTRESS HD once daily has not been studied in pediatric patients. However population PK modeling and simulation support the use of 1200 mg (2 × 600 mg) once daily in pediatric patients weighing at least 40 kg [see Clinical Pharmacology (12.3)].
8.5 Geriatric Use
Clinical studies of ISENTRESS/ISENTRESS HD did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger subjects. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Use in Patients with Hepatic Impairment
No dosage adjustment of ISENTRESS is necessary for patients with mild to moderate (Child-Pugh A and B) hepatic impairment. No hepatic impairment study has been conducted with ISENTRESS HD and therefore administration in subjects with hepatic impairment is not recommended. The effect of severe hepatic impairment on the pharmacokinetics of raltegravir has not been studied [see Clinical Pharmacology (12.3)].
8.7 Use in Patients with Renal Impairment
No dosage adjustment of ISENTRESS or ISENTRESS HD is necessary in patients with any degree of renal impairment [see Clinical Pharmacology (12.3)]. The extent to which ISENTRESS may be dialyzable is unknown.
-
10 OVERDOSAGE
In the event of an overdose, it is reasonable to employ the standard supportive measures, e.g., remove unabsorbed material from the gastrointestinal tract, employ clinical monitoring (including obtaining an electrocardiogram), and institute supportive therapy if required. The extent to which ISENTRESS may be dialyzable is unknown.
-
11 DESCRIPTION
ISENTRESS contains raltegravir potassium, a human immunodeficiency virus integrase strand transfer inhibitor. The chemical name for raltegravir potassium is N-[(4-Fluorophenyl) methyl]-1,6-dihydro-5-hydroxy-1-methyl-2-[1-methyl-1-[[(5-methyl-1,3,4-oxadiazol-2-yl)carbonyl]amino]ethyl]-6-oxo-4-pyrimidinecarboxamide monopotassium salt.
The empirical formula is C20H20FKN6O5 and the molecular weight is 482.51. The structural formula is:
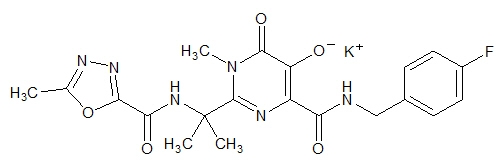
Raltegravir potassium is a white to off-white powder. It is soluble in water, slightly soluble in methanol, very slightly soluble in ethanol and acetonitrile and insoluble in isopropanol.
Each 400 mg film-coated tablet of ISENTRESS for oral administration contains 434.4 mg of raltegravir (as potassium salt), equivalent to 400 mg of raltegravir free phenol and the following inactive ingredients: calcium phosphate dibasic anhydrous, hypromellose 2208, lactose monohydrate, magnesium stearate, microcrystalline cellulose, poloxamer 407 (contains 0.01% butylated hydroxytoluene as antioxidant), sodium stearyl fumarate. In addition, the film coating contains the following inactive ingredients: black iron oxide, polyethylene glycol 3350, polyvinyl alcohol, red iron oxide, talc and titanium dioxide.
Each 600 mg film-coated tablet of ISENTRESS HD for oral administration contains 651.6 mg of raltegravir (as potassium salt), equivalent to 600 mg of raltegravir free phenol and the following inactive ingredients: croscarmellose sodium, hypromellose 2910, magnesium stearate, microcrystalline cellulose. The film coating contains the following inactive ingredients: ferrosoferric oxide, hypromellose 2910, iron oxide yellow, lactose monohydrate, triacetin and titanium dioxide. The tablet may also contain trace amount of carnauba wax.
Each 100 mg chewable tablet of ISENTRESS for oral administration contains 108.6 mg of raltegravir (as potassium salt), equivalent to 100 mg of raltegravir free phenol and the following inactive ingredients: ammonium hydroxide, crospovidone, ethylcellulose 20 cP, fructose, hydroxypropyl cellulose, hypromellose 2910/6cP, magnesium stearate, mannitol, medium chain triglycerides, monoammonium glycyrrhizinate, natural and artificial flavors (orange, banana, and masking that contains aspartame), oleic acid, PEG 400, red iron oxide, saccharin sodium, sodium citrate dihydrate, sodium stearyl fumarate, sorbitol, sucralose and yellow iron oxide.
Each 25 mg chewable tablet of ISENTRESS for oral administration contains 27.16 mg of raltegravir (as potassium salt), equivalent to 25 mg of raltegravir free phenol and the following inactive ingredients: ammonium hydroxide, crospovidone, ethylcellulose 20 cP, fructose, hydroxypropyl cellulose, hypromellose 2910/6cP, magnesium stearate, mannitol, medium chain triglycerides, monoammonium glycyrrhizinate, natural and artificial flavors (orange, banana, and masking that contains aspartame), oleic acid, PEG 400, saccharin sodium, sodium citrate dihydrate, sodium stearyl fumarate, sorbitol, sucralose and yellow iron oxide.
Each packet of ISENTRESS for oral suspension 100 mg, contains 108.6 mg of raltegravir (as potassium salt), equivalent to 100 mg of raltegravir free phenol and the following inactive ingredients: ammonium hydroxide, banana with other natural flavors, carboxymethylcellulose sodium, crospovidone, ethylcellulose 20 cP, fructose, hydroxypropyl cellulose, hypromellose 2910/6cP, macrogol/PEG 400, magnesium stearate, maltodextrin, mannitol, medium chain triglycerides, microcrystalline cellulose, monoammonium glycyrrhizinate, oleic acid, sorbitol, sucralose and sucrose.
-
12 CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
In a monotherapy study raltegravir (400 mg twice daily) demonstrated rapid antiviral activity with mean viral load reduction of 1.66 log10 copies/mL by Day 10.
12.3 Pharmacokinetics
Adults
Absorption
Raltegravir, given 400 mg twice daily, is absorbed with a Tmax of approximately 3 hours postdose in the fasted state in healthy subjects. Raltegravir 1200 mg once daily is rapidly absorbed with median Tmax of ~1.5 to 2 hours in the fasted state.
Raltegravir increases dose proportionally (AUC and Cmax) or slightly less than dose proportionally (C12hr) over the dose range 100 mg to 1600 mg.
The absolute bioavailability of raltegravir has not been established. The chewable tablet and oral suspension have higher oral bioavailability compared to the 400 mg film-coated tablet.
Relative to the raltegravir 400 mg formulation, the raltegravir 600 mg formulation has higher relative bioavailability.
Steady-state is generally reached in 2 days, with little to no accumulation with multiple dose administration for the 400 mg twice daily and 1200 once daily formulation.
Effect of Food on Oral Absorption
The food effect of various formulations are presented in Table 12.
Table 12: Effect of Food on the Pharmacokinetics of Raltegravir Formulations PK parameter ratio (fed/fasted) Formulation Meal Type AUC Ratio
(90% CI)Cmax Ratio
(90% CI)Cmin Ratio
(90% CI)Low-fat meal: 300 Kcal, 2.5 g fat Moderate-fat meal: 600 Kcal, 21 g fat High-fat meal: 825 Kcal, 52 g fat 400 mg twice daily Low Fat 0.54 (0.41-0.71) 0.48 (0.35-0.67) 0.86 (0.54-1.36) Moderate Fat 1.13 (0.85-1.49) 1.05 (0.75-1.46) 1.66 (1.04-2.64) High Fat 2.11 (1.60-2.80) 1.96 (1.41-2.73) 4.13 (2.60-6.57) 1200 mg once daily Low Fat 0.58 (0.46-0.74) 0.48 (0.37-0.62) 0.84 (0.63-1.10) High Fat 1.02 (0.86-1.21) 0.72 (0.58-0.90) 0.88 (0.66-1.18) Chewable tablet High Fat 0.94 (0.78-1.14) 0.38 (0.28-0.52) 2.88 (2.21-3.75) Oral suspension The effect of food on oral suspension was not studied. Distribution
Raltegravir is approximately 83% bound to human plasma protein over the concentration range of 2 to 10 µM.
In one study of HIV-1 infected subjects who received raltegravir 400 mg twice daily, raltegravir was measured in the cerebrospinal fluid. In the study (n=18), the median cerebrospinal fluid concentration was 5.8% (range 1 to 53.5%) of the corresponding plasma concentration. This median proportion was approximately 3-fold lower than the free fraction of raltegravir in plasma. The clinical relevance of this finding is unknown.
Metabolism and Excretion
The apparent terminal half-life of raltegravir is approximately 9 hours, with a shorter α-phase half-life (~1 hour) accounting for much of the AUC. Following administration of an oral dose of radiolabeled raltegravir, approximately 51 and 32% of the dose was excreted in feces and urine, respectively. In feces, only raltegravir was present, most of which is likely derived from hydrolysis of raltegravir-glucuronide secreted in bile as observed in preclinical species. Two components, namely raltegravir and raltegravir-glucuronide, were detected in urine and accounted for approximately 9 and 23% of the dose, respectively. The major circulating entity was raltegravir and represented approximately 70% of the total radioactivity; the remaining radioactivity in plasma was accounted for by raltegravir-glucuronide. The major mechanism of clearance of raltegravir in humans is UGT1A1-mediated glucuronidation.
Table 13: Multiple-Dose Pharmacokinetic Parameters of Raltegravir Following the Administration of 400 mg Twice Daily and 1200 mg Once Daily in HIV-infected Subjects Parameter 400 mg BID
Geometric Mean (%CV)
N=61200 mg QD
Geometric Mean (%CV)
N=524AUC (µM∙hr) AUC0-12= 14.3 (88.6) AUC0-24 = 55.3 (41.5) Cmax (µM) 4.5 (128) 15.7 (45.8) Cmin (nM) C12 = 142 (63.8) C24 = 107 (97.5) Special Populations
Pediatric
ISENTRESS
Two pediatric formulations were evaluated in healthy adult volunteers, where the chewable tablet and oral suspension were compared to the 400 mg tablet. The chewable tablet and oral suspension demonstrated higher oral bioavailability, thus higher AUC, compared to the 400 mg tablet. In the same study, the oral suspension resulted in higher oral bioavailability compared to the chewable tablet. These observations resulted in proposed pediatric doses targeting 6 mg/kg/dose for the chewable tablets and oral suspension. As displayed in Table 13, the doses recommended for HIV-infected infants, children and adolescents 4 weeks to 18 years of age [see Dosage and Administration (2.3)] resulted in a pharmacokinetic profile of raltegravir similar to that observed in adults receiving 400 mg twice daily.
Overall, dosing in pediatric patients achieved exposures (Ctrough) above 45 nM in the majority of subjects, but some differences in exposures between formulations were observed. Pediatric patients above 25 kg administered the chewable tablets had lower trough concentrations (113 nM) compared to pediatric patients above 25 kg administered the 400 mg tablet formulation (233 nM) [see Clinical Studies (14.4)]. As a result, the 400 mg film-coated tablet is the recommended dose in patients weighing at least 25 kg; however, the chewable tablet offers an alternative regimen in patients weighing at least 25 kg who are unable to swallow the film-coated tablet [see Dosage and Administration (2.3)]. In addition, pediatric patients weighing 11 to 25 kg who were administered the chewable tablets had the lowest trough concentrations (82 nM) compared to all other pediatric subgroups.
Table 14: Raltegravir Steady State Pharmacokinetic Parameters in Pediatric Patients Following Administration of Recommended Twice-Daily Doses Body Weight Formulation Dose N* Geometric Mean
(%CV†)
AUC0-12hr (µM∙hr)Geometric Mean
(%CV†)
C12hr (nM)- * Number of patients with intensive pharmacokinetic (PK) results at the final recommended dose.
- † Geometric coefficient of variation.
≥25 kg Film-coated tablet 400 mg twice daily 18 14.1 (121%) 233 (157%) ≥25 kg Chewable tablet Weight based dosing, see Table 3 9 22.1 (36%) 113 (80%) 11 to less than 25 kg Chewable tablet Weight based dosing, see Table 4 13 18.6 (68%) 82 (123%) 3 to less than 20 kg Oral suspension Weight based dosing, see Table 4 19 24.5 (43%) 113 (69%) Elimination of raltegravir in vivo in human is primarily through the UGT1A1-mediated glucuronidation pathway. UGT1A1 catalytic activity is negligible at birth and matures after birth. The dose recommended for neonates less than 4 weeks of age takes into consideration the rapidly increasing UGT1A1 activity and drug clearance from birth to 4 weeks of age. Table 15 displays pharmacokinetic parameters for neonates receiving the granules for oral suspension at the recommended dose [see Dosage and Administration (2.3)].
Table 15: Raltegravir Pharmacokinetic Parameters from IMPAACT P1110 Following Age and Weight Based Dosing of Oral Suspension Age (hours/days) at PK Sampling Dose (See Table 5) N* Geometric Mean
(%CV†)
AUC (μM∙hr )Geometric Mean
(%CV†)
Ctrough (nM)- * Number of patients with intensive pharmacokinetic (PK) results at the final recommended dose.
- † Geometric coefficient of variation.
- ‡ AUC0-24hr (N = 24) and C24hr
- § AUC0-12hr and C12hr
Birth – 48 hours 1.5 mg/kg once daily 25 85.9 (38.4%)‡ 2132.9 (64.2%)‡ 15 to 18 days 3.0 mg/kg twice daily 23 32.2 (43.3%)§ 1255.5 (83.7%)§ Age/Race/Gender
There is no clinically meaningful effect of age (18 years and older), race, or gender on the pharmacokinetics of raltegravir.
Hepatic Impairment
Raltegravir is eliminated primarily by glucuronidation in the liver. The pharmacokinetics of a single 400-mg dose of raltegravir were not altered in patients with moderate (Child-Pugh Score 7 to 9) hepatic impairment.
No hepatic impairment study has been conducted with ISENTRESS HD 1200 mg (2 × 600 mg) once daily.The effect of severe hepatic impairment on the pharmacokinetics of raltegravir has not been studied.
Renal Impairment
Renal clearance of unchanged drug is a minor pathway of elimination. The pharmacokinetics of a single 400-mg dose of raltegravir were not altered in patients with severe (24-hour creatinine clearance of <30 mL/min/1.73 m2) renal impairment.
No renal impairment study was conducted with ISENTRESS HD 1200 mg (2 × 600 mg) once daily.
The extent to which ISENTRESS may be dialyzable is unknown.
Drug Interactions
In vitro, raltegravir does not inhibit (IC50>100 µM) CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 or CYP3A. In vivo, raltegravir does not inhibit CYP3A4. Moreover, in vitro, raltegravir did not induce CYP1A2, CYP2B6 or CYP3A4. Similarly, raltegravir is not an inhibitor (IC50>50 µM) of UGT1A1 or UGT2B7, and raltegravir does not inhibit P-glycoprotein-mediated transport.
Raltegravir drug interaction study results are shown in Tables 16 and 17. For information regarding clinical recommendations [see Drug Interactions (7)].
Table 16: Effect of Other Agents on the Pharmacokinetics of Raltegravir in Adults Coadministered Drug Coadministered Drug Dose/Schedule Raltegravir
Dose/ScheduleRatio (90% Confidence Interval) of Raltegravir Pharmacokinetic Parameters with/without Coadministered Drug;
No Effect = 1.00n Cmax AUC Cmin - * Study conducted in HIV-infected subjects.
aluminum and magnesium hydroxide antacid* 20 mL single dose given with raltegravir 400 mg twice daily 25 0.56
(0.42, 0.73)0.51
(0.40, 0.65)0.37
(0.29, 0.48)20 mL single dose given 2 hours before raltegravir 23 0.49
(0.33, 0.71)0.49
(0.35, 0.67)0.44
(0.34, 0.55)20 mL single dose given 2 hours after raltegravir 23 0.78
(0.53, 1.13)0.70
(0.50, 0.96)0.43
(0.34, 0.55)20 mL single dose given 4 hours before raltegravir 17 0.78
(0.55, 1.10)0.81
(0.63, 1.05)0.40
(0.31, 0.52)20 mL single dose given 4 hours after raltegravir 18 0.70
(0.48, 1.04)0.68
(0.50, 0.92)0.38
(0.30, 0.49)20 mL single dose given 6 hours before raltegravir 16 0.90
(0.58, 1.40)0.87
(0.64, 1.18)0.50
(0.39, 0.65)20 mL single dose given 6 hours after raltegravir 16 0.90
(0.58, 1.41)0.89
(0.64, 1.22)0.51
(0.40, 0.64)aluminum and magnesium hydroxide antacid* 20 mL single dose given 12 hours after raltegravir 1200 mg single dose 19 0.86
(0.65, 1.15)0.86
(0.73, 1.03)0.42
(0.34, 0.52)atazanavir 400 mg daily 100 mg single dose 10 1.53 (1.11, 2.12) 1.72 (1.47, 2.02) 1.95 (1.30, 2.92) atazanavir 400 mg daily 1200 mg single dose 14 1.16 (1.01, 1.33) 1.67 (1.34, 2.10) 1.26 (1.08, 1.46) atazanavir/ritonavir 300 mg/100 mg daily 400 mg twice daily 10 1.24 (0.87, 1.77) 1.41 (1.12, 1.78) 1.77 (1.39, 2.25) boceprevir 800 mg three times daily 400 mg single dose 22 1.11
(0.91-1.36)1.04
(0.88-1.22)0.75
(0.45-1.23)calcium carbonate antacid* 3000 mg single dose given with raltegravir 400 mg twice daily 24 0.48
(0.36, 0.63)0.45
(0.35, 0.57)0.68
(0.53, 0.87)calcium carbonate antacid* 3000 mg single dose given with raltegravir 1200 mg single dose 19 0.26
(0.21, 0.32)0.28
(0.24, 0.32)0.52
(0.45, 0.61)3000 mg single dose given 12 hours after raltegravir 0.98
(0.81, 1.17)0.90
(0.80, 1.03)0.43
(0.36, 0.51)efavirenz 600 mg daily 400 mg single dose 9 0.64
(0.41, 0.98)0.64
(0.52, 0.80)0.79
(0.49, 1.28)efavirenz 600 mg daily 1200 mg single dose 21 0.91
(0.70, 1.17)0.86
(0.73, 1.01)0.94
(0.76, 1.17)etravirine 200 mg twice daily 400 mg twice daily 19 0.89
(0.68, 1.15)0.90
(0.68, 1.18)0.66
(0.34, 1.26)omeprazole* 20 mg daily 400 mg twice daily 18 1.51
(0.98, 2.35)1.37
(0.99, 1.89)1.24
(0.95, 1.62)rifampin 600 mg daily 400 mg single dose 9 0.62
(0.37, 1.04)0.60
(0.39, 0.91)0.39
(0.30, 0.51)rifampin 600 mg daily 400 mg twice daily when administered alone; 800 mg twice daily when administered with rifampin 14 1.62
(1.12, 2.33)1.27
(0.94, 1.71)0.47
(0.36, 0.61)ritonavir 100 mg twice daily 400 mg single dose 10 0.76 (0.55, 1.04) 0.84 (0.70, 1.01) 0.99 (0.70, 1.40) tenofovir disoproxil fumarate 300 mg daily 400 mg twice daily 9 1.64
(1.16, 2.32)1.49
(1.15, 1.94)1.03
(0.73, 1.45)tipranavir/ritonavir 500 mg/200 mg twice daily 400 mg twice daily 15
(14 for Cmin)0.82
(0.46, 1.46)0.76
(0.49, 1.19)0.45
(0.31, 0.66)Table 17: Effect of Raltegravir on the Pharmacokinetics of Other Agents in Adults Substrate Drug Raltegravir
Dose/ScheduleRatio (90% Confidence Interval) of Substrate Pharmacokinetic Parameters with/without Coadministered Drug;
No Effect = 1.00n Cmax AUC Cmin Tenofovir disoproxil fumarate 300 mg 400 mg 9 0.77 (0.69, 0.85) 0.90 (0.82, 0.99) C24hr
0.87 (0.74, 1.02)Etravirine 200 mg 400 mg 19 1.04 (0.97, 1.12) 1.10 (1.03, 1.16) 1.17 (1.10, 1.26) In drug interaction studies, there was no effect of raltegravir on the PK of ethinyl estradiol, methadone, midazolam or boceprevir.
12.4 Microbiology
Mechanism of Action
Raltegravir inhibits the catalytic activity of HIV-1 integrase, an HIV-1 encoded enzyme that is required for viral replication. Inhibition of integrase prevents the covalent insertion, or integration, of unintegrated linear HIV-1 DNA into the host cell genome preventing the formation of the HIV-1 provirus. The provirus is required to direct the production of progeny virus, so inhibiting integration prevents propagation of the viral infection. Raltegravir did not significantly inhibit human phosphoryltransferases including DNA polymerases α, β, and γ.
Antiviral Activity in Cell Culture
Raltegravir at concentrations of 31 ± 20 nM resulted in 95% inhibition (EC95) of viral spread (relative to an untreated virus-infected culture) in human T-lymphoid cell cultures infected with the cell-line adapted HIV-1 variant H9IIIB. In addition, 5 clinical isolates of HIV-1 subtype B had EC95 values ranging from 9 to 19 nM in cultures of mitogen-activated human peripheral blood mononuclear cells. In a single-cycle infection assay, raltegravir inhibited infection of 23 HIV-1 isolates representing 5 non-B subtypes (A, C, D, F, and G) and 5 circulating recombinant forms (AE, AG, BF, BG, and cpx) with EC50 values ranging from 5 to 12 nM. Raltegravir also inhibited replication of an HIV-2 isolate when tested in CEMx174 cells (EC95 value = 6 nM). No antagonism was observed when human T-lymphoid cells infected with the H9IIIB variant of HIV-1 were incubated with raltegravir in combination with non-nucleoside reverse transcriptase inhibitors (delavirdine, efavirenz, or nevirapine); nucleoside analog reverse transcriptase inhibitors (abacavir, didanosine, lamivudine, stavudine, tenofovir, or zidovudine); protease inhibitors (amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, or saquinavir); or the entry inhibitor enfuvirtide.
Resistance
The mutations observed in the HIV-1 integrase coding sequence that contributed to raltegravir resistance (evolved either in cell culture or in subjects treated with raltegravir) generally included an amino acid substitution at either Y143 (changed to C, H, or R) or Q148 (changed to H, K, or R) or N155 (changed to H) plus one or more additional substitutions (i.e., L74M, E92Q, Q95K/R, T97A, E138A/K, G140A/S, V151I, G163R, H183P, Y226C/D/F/H, S230R, and D232N). E92Q, T97A, and F121C are occasionally seen in the absence of substitutions at Y143, Q148, or N155 in raltegravir-treatment failure subjects.
Treatment-Naïve Adult Subjects: By Week 240 in the STARTMRK trial, the primary raltegravir resistance-associated substitutions were observed in 4 (2 with Y143H/R and 2 with Q148H/R) of the 12 virologic failure subjects with evaluable genotypic data from paired baseline and raltegravir treatment-failure isolates. By Week 96 in the ONCEMRK trial, primary raltegravir resistance substitutions were observed in on treatment isolates obtained from 4 (3 with N155H and 1 with E92Q) of 14 virologic failure subjects with evaluable genotypic data in the 1,200 mg QD arm and 2 (1 with N155H and 1 with T97A) of 6 virologic failure subjects in the 400 mg BID arm. Additional integrase substitutions observed included L74M, Q95K, V151I, E170A, I203M and D232N. These resistant isolates exhibited 6.2- to 19-fold reductions in susceptibility to raltegravir. Overall, at Week 96, detection of raltegravir resistance was not different between the QD and BID arms in subjects who were failing treatment and had resistance data evaluable (28.6% versus 33.3%, respectively).
Treatment-Experienced Adult Subjects: By Week 96 in the BENCHMRK trials, at least one of the primary raltegravir resistance-associated substitutions, Y143C/H/R, Q148H/K/R, and N155H, was observed in 76 of the 112 virologic failure subjects with evaluable genotypic data from paired baseline and raltegravir treatment-failure isolates. The emergence of the primary raltegravir resistance-associated substitutions was observed cumulatively in 70 subjects by Week 48 and 78 subjects by Week 96, 15.2% and 17% of the raltegravir recipients, respectively. Some (n=58) of those HIV-1 isolates harboring one or more of the primary raltegravir resistance-associated substitutions were evaluated for raltegravir susceptibility yielding a median decrease of 26.3-fold (mean 48.9 ± 44.8-fold decrease, ranging from 0.8- to 159-fold) compared to the wild-type reference.
Cross Resistance
Cross resistance has been observed among HIV-1 integrase strand transfer inhibitors (INSTIs). Amino acid substitutions in HIV-1 integrase conferring resistance to raltegravir generally also confer resistance to elvitegravir. Substitutions at amino acid Y143 confer greater reductions in susceptibility to raltegravir than to elvitegravir, and the E92Q substitution confers greater reductions in susceptibility to elvitegravir than to raltegravir. Viruses harboring a substitution at amino acid Q148, along with one or more other raltegravir resistance substitutions, may also have clinically significant resistance to dolutegravir.
12.5 Pharmacogenomics
UGT1A1 Polymorphism
There is no evidence that common UGT1A1 polymorphisms alter raltegravir pharmacokinetics to a clinically meaningful extent. In a comparison of 30 adult subjects with *28/*28 genotype (associated with reduced activity of UGT1A1) to 27 adult subjects with wild-type genotype, the geometric mean ratio (90% CI) of AUC was 1.41 (0.96, 2.09).
In the neonatal study IMPAACT P1110, there was no association between apparent clearance (CL/F) of raltegravir and UGT 1A1 genotype polymorphisms.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies of raltegravir in mice did not show any carcinogenic potential. At the highest dose levels, 400 mg/kg/day in females and 250 mg/kg/day in males, systemic exposure was 1.8-fold (females) or 1.2-fold (males) greater than the AUC (54 µM∙hr) at the 400-mg twice daily human dose. Treatment-related squamous cell carcinoma of nose/nasopharynx was observed in female rats dosed with 600 mg/kg/day raltegravir for 104 weeks. These tumors were possibly the result of local irritation and inflammation due to local deposition and/or aspiration of drug in the mucosa of the nose/nasopharynx during dosing. No tumors of the nose/nasopharynx were observed in rats dosed with 150 mg/kg/day (males) and 50 mg/kg/day (females) and the systemic exposure in rats was 1.7-fold (males) to 1.4-fold (females) greater than the AUC (54 µM∙hr) at the 400-mg twice daily human dose.
No evidence of mutagenicity or genotoxicity was observed in in vitro microbial mutagenesis (Ames) tests, in vitro alkaline elution assays for DNA breakage, and in vitro and in vivo chromosomal aberration studies.
No effect on fertility was seen in male and female rats at doses up to 600 mg/kg/day which resulted in a 3-fold exposure above the exposure at the recommended human dose.
-
14 CLINICAL STUDIES
14.1 Description of Clinical Studies
The evidence of durable efficacy of ISENTRESS 400 mg twice daily is based on the analyses of 240-week data from a randomized, double-blind, active-controlled trial, STARTMRK evaluating ISENTRESS 400 mg twice daily in antiretroviral treatment-naïve HIV-1 infected adult subjects, the analysis of 96-week data from a randomized, double-blind, active-control trial, ONCEMRK evaluating ISENTRESS HD 1200 mg (2 × 600 mg) once daily in treatment-naïve adult subjects, and 96-week data from 2 randomized, double-blind, placebo-controlled studies, BENCHMRK 1 and BENCHMRK 2, evaluating ISENTRESS 400 mg twice daily in antiretroviral treatment-experienced HIV-1 infected adult subjects. See Table 18.
Table 18: Trials Conducted with ISENTRESS and ISENTRESS HD in Subjects with HIV-1 Infection Trial Study Type Population Study Arms (N) Dose/Formulation Timepoint STARTMRK Randomized, double-blind, active-controlled Treatment-Naïve Adults ISENTRESS 400 mg Twice Daily (281)
Efavirenz 600 mg At Bedtime (282)
Both in combination with emtricitabine (+) tenofovir disoproxil fumarate400 mg film-coated tablet Week 240 ONCEMRK Randomized, double-blind, active-controlled Treatment-Naïve Adults ISENTRESS HD 1200 mg Once Daily (531) 600 mg film-coated tablet Week 96 ISENTRESS 400 mg Twice Daily (266)
Both in combination with emtricitabine (+) tenofovir disoproxil fumarate400 mg film-coated tablet BENCHMRK 1 Randomized, double-blind, placebo-controlled Treatment-Experienced Adults ISENTRESS 400 mg Twice Daily (232)
Placebo (118)
Both in combination with optimized background therapy400 mg film-coated tablet Week 240 (Week 156 on double-blind plus Week 84 on open-label) BENCHMRK 2 Randomized, double-blind, placebo-controlled Treatment-Experienced Adults ISENTRESS 400 mg Twice Daily (230)
Placebo (119)
Both in combination with optimized background therapy400 mg film-coated tablet Week 240 (Week 156 on double-blind plus Week 84 on open-label) IMPAACT P1066 Open-label, non-comparative Pediatric Patients – 4 weeks to 18 years of age
(Treatment-Experienced or Failed Prior PMTCT)ISENTRESS 400 mg tablet Twice Daily – 12 to 18 years or 6 to <12 years and ≥25 kg (87)
ISENTRESS chewable tablet- Weight-Based Dose to Approximate 6 mg/kg Twice Daily – 2 to <12 years (39)
ISENTRESS for oral suspension- Weight-Based Dose to Approximate 6 mg/kg Twice Daily – 4 weeks to <2 years (26)
In combination with optimized background therapy400 mg film-coated tablet
25 mg and 100 mg chewable tablet
100 mg sachet for oral suspensionWeek 240 14.2 Treatment-Naïve Adult Subjects
STARTMRK (ISENTRESS 400 mg twice daily)
STARTMRK is a Phase 3 randomized, international, double-blind, active-controlled trial to evaluate the safety and efficacy of ISENTRESS 400 mg twice daily versus efavirenz 600 mg at bedtime both with emtricitabine (+) tenofovir disoproxil fumarate in treatment-naïve HIV-1-infected subjects with HIV-1 RNA >5000 copies/mL. Randomization was stratified by screening HIV-1 RNA level (≤50,000 copies/mL; or >50,000 copies/mL) and by hepatitis status. In STARTMRK, 563 subjects were randomized and received at least 1 dose of either raltegravir 400 mg twice daily or efavirenz 600 mg at bedtime, both in combination with emtricitabine (+) tenofovir disoproxil fumarate. There were 563 subjects included in the efficacy and safety analyses. At baseline, the median age of subjects was 37 years (range 19-71), 19% female, 58% non-white, 6% had hepatitis B and/or C virus co-infection, 20% were CDC Class C (AIDS), 53% had HIV-1 RNA greater than 100,000 copies per mL, and 47% had CD4+ cell count less than 200 cells per mm3; the frequencies of these baseline characteristics were similar between treatment groups.
ONCEMRK (ISENTRESS HD 1200 mg [2 × 600 mg] once daily)
ONCEMRK is a Phase 3 randomized, international, double-blind, active-controlled trial to evaluate the safety and efficacy of ISENTRESS HD 1200 mg (2 × 600 mg) once daily versus ISENTRESS 400 mg twice daily, both in combination with emtricitabine (+) tenofovir disoproxil fumarate, in treatment-naïve HIV-1-infected subjects with HIV-1 RNA ≥1000 copies/mL. Randomization was stratified by screening HIV-1 RNA level (≤100,000 or >100,000 copies/mL) and by hepatitis B and C infection status.
In ONCEMRK, 797 subjects were randomized and received at least 1 dose of either raltegravir 1200 mg once daily or raltegravir 400 mg twice daily, both in combination with emtricitabine (+) tenofovir disoproxil fumarate. There were 797 subjects included in the efficacy and safety analyses. At baseline, the median age of subjects was 34 years (range 18-84), 15% female, 41% non-white, 3% had hepatitis B and/or C virus co-infection, 13% were CDC Class C (AIDS), 28% had HIV-1 RNA greater than 100,000 copies per mL, and 13% had CD4+ cell count less than 200 cells per mm3; the frequencies of these baseline characteristics were similar between treatment groups.
Table 19 shows the virologic outcomes in both studies. Side-by-side tabulation is to simplify presentation; direct comparisons across trials should not be made due to differing duration of follow-up.
Table 19: Virologic Outcomes of Randomized Treatment in STARTMRK and ONCEMRK (Snapshot Algorithm) in HIV Treatment-Naïve Adults STARTMRK
Week 240ONCEMRK
Week 96ISENTRESS
400 mg Twice Daily
(N=281)Efavirenz
600 mg At Bedtime
(N=282)ISENTRESS HD
1200 mg Once Daily
(N=531)ISENTRESS
400 mg Twice Daily
(N=266)Notes: ISENTRESS BID, ISENTRESS HD and Efavirenz were administered with emtricitabine (+) tenofovir disoproxil fumarate - * Lower Limit of Quantitation: STARTMRK <50 copies/mL; ONCEMRK < 40 copies/mL.
- † Includes subjects who discontinued because of adverse event (AE) or death at any time point from Day 1 through the time window if this resulted in no virologic data on treatment during the specified window.
- ‡ Other Reasons includes: lost to follow-up, moved, non-compliance with study drug, physician decision, pregnancy, withdrawal by subject.
HIV RNA < Lower Limit of Quantitation* 66% 60% 82% 80% Treatment Difference 6.6% (95% CI: -1.4%, 14.5%) 1.4% (95% CI: -4.4%, 7.3%) HIV RNA ≥ Lower Limit of Quantitation 8% 15% 9% 8% No Virologic Data at Analysis Timepoint 26% 26% 9% 12% Reasons Discontinued trial due to AE or Death† 5% 10% 1% 3% Discontinued trial for Other Reasons‡ 15% 14% 7% 8% On trial but missing data at timepoint 6% 2% 1% 2% In the ONCEMRK trial, ISENTRESS HD 1200 mg (2 × 600 mg) once daily demonstrated consistent virologic and immunologic efficacy relative to ISENTRESS 400 mg twice daily, both in combination with emtricitabine (+) tenofovir disoproxil fumarate, across demographic and baseline prognostic factors, including: baseline HIV RNA levels >100,000 copies/mL and demographic groups (including age, gender, race, ethnicity and region), concomitant proton pump inhibitors/H2 blockers use and viral subtypes (comparing non-clade B as a group to clade B).
Consistent efficacy in subjects receiving ISENTRESS HD 1200 mg (2 × 600 mg) once daily was observed across HIV subtypes with 80.6% (270/335) and 83.5% (162/194) of subjects with B and non-B subtypes respectively, achieving HIV RNA <40 copies/mL at week 96 (Snapshot approach).
14.3 Treatment-Experienced Adult Subjects
BENCHMRK 1 and BENCHMRK 2 are Phase 3 studies to evaluate the safety and antiretroviral activity of ISENTRESS 400 mg twice daily in combination with an optimized background therapy (OBT), versus OBT alone, in HIV-1-infected subjects, 16 years or older, with documented resistance to at least 1 drug in each of 3 classes (NNRTIs, NRTIs, PIs) of antiretroviral therapies. Randomization was stratified by degree of resistance to PI (1PI vs. >1PI) and the use of enfuvirtide in the OBT. Prior to randomization, OBT was selected by the investigator based on genotypic/phenotypic resistance testing and prior ART history.
Table 20 shows the demographic characteristics of subjects in the group receiving ISENTRESS 400 mg twice daily and subjects in the placebo group.
Table 20: Trials BENCHMRK 1 and BENCHMRK 2 Baseline Characteristics Randomized Studies
BENCHMRK 1 and BENCHMRK 2ISENTRESS 400 mg Twice Daily + OBT
(N = 462)Placebo + OBT
(N = 237)- * Hepatitis B virus surface antigen positive or hepatitis C virus antibody positive.
Gender Male 88% 89% Female 12% 11% Race White 65% 73% Black 14% 11% Asian 3% 3% Hispanic 11% 8% Others 6% 5% Age (years) Median (min, max) 45 (16 to 74) 45 (17 to 70) CD4+ Cell Count Median (min, max), cells/mm3 119 (1 to 792) 123 (0 to 759) ≤50 cells/mm3 32% 33% >50 and ≤200 cells/mm3 37% 36% Plasma HIV-1 RNA Median (min, max), log10 copies/mL 4.8 (2 to 6) 4.7 (2 to 6) >100,000 copies/mL 36% 33% History of AIDS Yes 92% 91% Prior Use of ART, Median (1st Quartile, 3rd Quartile) Years of ART Use 10 (7 to 12) 10 (8 to 12) Number of ART 12 (9 to 15) 12 (9 to 14) Hepatitis Co-infection* No Hepatitis B or C virus 83% 84% Hepatitis B virus only 8% 3% Hepatitis C virus only 8% 12% Co-infection of Hepatitis B and C virus 1% 1% Stratum Enfuvirtide in OBT 38% 38% Resistant to ≥2 PI 97% 95% Table 21 compares the characteristics of optimized background therapy at baseline in the group receiving ISENTRESS 400 mg twice daily and subjects in the control group.
Table 21: Trials BENCHMRK 1 and BENCHMRK 2 Characteristics of Optimized Background Therapy at Baseline Randomized Studies
BENCHMRK 1 and BENCHMRK 2ISENTRESS 400 mg Twice Daily + OBT
(N = 462)Placebo + OBT
(N = 237)- * Darunavir use in OBT in darunavir-naïve subjects was counted as one active PI.
- † The Phenotypic Sensitivity Score (PSS) and the Genotypic Sensitivity Score (GSS) were defined as the total oral ARTs in OBT to which a subject's viral isolate showed phenotypic sensitivity and genotypic sensitivity, respectively, based upon phenotypic and genotypic resistance tests. Enfuvirtide use in OBT in enfuvirtide-naïve subjects was counted as one active drug in OBT in the GSS and PSS. Similarly, darunavir use in OBT in darunavir-naïve subjects was counted as one active drug in OBT.
Number of ARTs in OBT Median (min, max) 4 (1 to 7) 4 (2 to 7) Number of Active PI in OBT by Phenotypic Resistance Test* 0 36% 41% 1 or more 60% 58% Phenotypic Sensitivity Score (PSS)† 0 15% 18% 1 31% 30% 2 31% 28% 3 or more 18% 20% Genotypic Sensitivity Score (GSS)† 0 25% 27% 1 38% 40% 2 24% 21% 3 or more 11% 10% Week 96 outcomes for the 699 subjects randomized and treated with the recommended dose of ISENTRESS 400 mg twice daily or placebo in the pooled BENCHMRK 1 and 2 studies are shown in Table 22.
Table 22: Virologic Outcomes of Randomized Treatment of BENCHMRK 1 and BENCHMRK 2 Trials at 96 Weeks (Pooled Analysis) ISENTRESS 400 mg Twice Daily + OBT
(N = 462)Placebo + OBT
(N = 237)- * Includes subjects who switched to open-label raltegravir after Week 16 due to the protocol-defined virologic failure, subjects who discontinued prior to Week 96 for lack of efficacy, subjects changed OBT due to lack of efficacy prior to Week 96, or subjects who were ≥50 copies in the 96 week window.
- † Includes subjects who discontinued due to AE or death at any time point from Day 1 through the Week 96 window if this resulted in no virologic data on treatment during the Week 96 window.
- ‡ Other includes: withdrew consent, loss to follow-up, moved etc., if the viral load at the time of discontinuation was <50 copies/mL.
Subjects with HIV-1 RNA less than 50 copies/mL 55% 27% Virologic Failure* 35% 66% No virologic data at Week 96 Window Reasons Discontinued study due to AE or death† 3% 3% Discontinued study for other reasons‡ 4% 4% Missing data during window but on study 4% <1% The mean changes in CD4 count from baseline were 118 cells/mm3 in the group receiving ISENTRESS 400 mg twice daily and 47 cells/mm3 for the control group.
Treatment-emergent CDC Category C events occurred in 4% of the group receiving ISENTRESS 400 mg twice daily and 5% of the control group.
Virologic responses at Week 96 by baseline genotypic and phenotypic sensitivity score are shown in Table 23.
Table 23: Virologic Response at 96 Week Window by Baseline Genotypic/Phenotypic Sensitivity Score Percent with HIV-1 RNA <50 copies/mL At Week 96 n ISENTRESS 400 mg Twice Daily + OBT
(N = 462)n Placebo + OBT
(N = 237)- * The Phenotypic Sensitivity Score (PSS) and the Genotypic Sensitivity Score (GSS) were defined as the total oral ARTs in OBT to which a subject's viral isolate showed phenotypic sensitivity and genotypic sensitivity, respectively, based upon phenotypic and genotypic resistance tests. Enfuvirtide use in OBT in enfuvirtide-naïve subjects was counted as one active drug in OBT in the GSS and PSS. Similarly, darunavir use in OBT in darunavir-naïve subjects was counted as one active drug in OBT.
Phenotypic Sensitivity Score (PSS)* 0 67 43 43 5 1 144 58 71 23 2 142 61 66 32 3 or more 85 48 48 42 Genotypic Sensitivity Score (GSS)* 0 116 39 65 5 1 177 62 95 26 2 111 61 49 53 3 or more 51 49 23 35 Switch of Suppressed Subjects from Lopinavir (+) Ritonavir to Raltegravir
The SWITCHMRK 1 & 2 Phase 3 studies evaluated HIV-1 infected subjects receiving suppressive therapy (HIV-1 RNA <50 copies/mL on a stable regimen of lopinavir 200 mg (+) ritonavir 50 mg 2 tablets twice daily plus at least 2 nucleoside reverse transcriptase inhibitors for >3 months) and randomized them 1:1 to either continue lopinavir (+) ritonavir (n=174 and n=178, SWITCHMRK 1 & 2, respectively) or replace lopinavir (+) ritonavir with ISENTRESS 400 mg twice daily (n=174 and n=176, respectively). The primary virology endpoint was the proportion of subjects with HIV-1 RNA less than 50 copies/mL at Week 24 with a prespecified non-inferiority margin of -12% for each study; and the frequency of adverse events up to 24 weeks.
Subjects with a prior history of virological failure were not excluded and the number of previous antiretroviral therapies was not limited.
These studies were terminated after the primary efficacy analysis at Week 24 because they each failed to demonstrate non-inferiority of switching to ISENTRESS versus continuing on lopinavir (+) ritonavir. In the combined analysis of these studies at Week 24, suppression of HIV-1 RNA to less than 50 copies/mL was maintained in 82.3% of the ISENTRESS group versus 90.3% of the lopinavir (+) ritonavir group. Clinical and laboratory adverse events occurred at similar frequencies in the treatment groups.
14.4 Pediatric Subjects
2 to 18 Years of Age
IMPAACT P1066 is a Phase I/II open label multicenter trial to evaluate the pharmacokinetic profile, safety, tolerability, and efficacy of raltegravir in HIV infected children. This study enrolled 126 treatment experienced children and adolescents 2 to 18 years of age. Subjects were stratified by age, enrolling adolescents first and then successively younger children. Subjects were enrolled into cohorts according to age and received the following formulations: Cohort I (12 to less than 18 years old), 400 mg film-coated tablet; Cohort IIa (6 to less than 12 years old), 400 mg film-coated tablet; Cohort IIb (6 to less than 12 years old), chewable tablet; Cohort III (2 to less than 6 years), chewable tablet. Raltegravir was administered with an optimized background regimen.
The initial dose finding stage included intensive pharmacokinetic evaluation. Dose selection was based upon achieving similar raltegravir plasma exposure and trough concentration as seen in adults, and acceptable short term safety. After dose selection, additional subjects were enrolled for evaluation of long term safety, tolerability and efficacy. Of the 126 subjects, 96 received the recommended dose of ISENTRESS [see Dosage and Administration (2.3)].
These 96 subjects had a median age of 13 (range 2 to 18) years, were 51% Female, 34% Caucasian, and 59% Black. At baseline, mean plasma HIV-1 RNA was 4.3 log10 copies/mL, median CD4 cell count was 481 cells/mm3 (range: 0 – 2361) and median CD4% was 23.3% (range: 0 – 44). Overall, 8% had baseline plasma HIV-1 RNA >100,000 copies/mL and 59% had a CDC HIV clinical classification of category B or C. Most subjects had previously used at least one NNRTI (78%) or one PI (83%).
Ninety-three (97%) subjects 2 to 18 years of age completed 24 weeks of treatment (3 discontinued due to non-compliance). At Week 24, 54% achieved HIV RNA <50 copies/mL; 66% achieved HIV RNA <400 copies/mL. The mean CD4 count (percent) increase from baseline to Week 24 was 119 cells/mm3 (3.8%).
4 Weeks to Less Than 2 Years of Age
IMPAACT P1066 also enrolled HIV-infected, infants and toddlers 4 weeks to less than 2 years of age (Cohorts IV and V) who had received prior antiretroviral therapy either as prophylaxis for prevention of mother-to-child transmission (PMTCT) and/or as combination antiretroviral therapy for treatment of HIV infection. Raltegravir was administered as an oral suspension without regard to food in combination with an optimized background regimen.
The 26 subjects had a median age of 28 weeks (range: 4 -100), were 35% female, 85% Black and 8% Caucasian. At baseline, mean plasma HIV-1 RNA was 5.7 log10 copies/mL (range: 3.1 – 7), median CD4 cell count was 1400 cells/mm3 (range: 131 – 3648) and median CD4% was 18.6% (range: 3.3 – 39.3). Overall, 69% had baseline plasma HIV-1 RNA exceeding 100,000 copies/mL and 23% had a CDC HIV clinical classification of category B or C. None of the 26 subjects were completely treatment naïve. All infants under 6 months of age had received nevirapine or zidovudine for prevention of mother-to-infant transmission, and 43% of subjects greater than 6 months of age had received two or more antiretrovirals.
Of the 26 treated subjects, 23 subjects were included in the Week 24 and 48 efficacy analyses, respectively. All 26 treated subjects were included for safety analyses.
At Week 24, 39% achieved HIV RNA <50 copies/mL and 61% achieved HIV RNA <400 copies/mL. The mean CD4 count (percent) increase from baseline to Week 24 was 500 cells/mm3 (7.5%).
At Week 48, 44% achieved HIV RNA <50 copies/mL and 61% achieved HIV RNA <400 copies/mL. The mean CD4 count (percent) increase from baseline to Week 48 was 492 cells/mm3 (7.8%).
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Severe and Potentially Life-threatening Rash
Inform patients that severe and potentially life-threatening rash has been reported. Advise patients to immediately contact their healthcare provider if they develop rash. Instruct patients to immediately stop taking ISENTRESS or ISENTRESS HD and other suspect agents, and seek medical attention if they develop a rash associated with any of the following symptoms as it may be a sign of a more serious reaction such as Stevens-Johnson syndrome, toxic epidermal necrolysis or severe hypersensitivity: fever, generally ill feeling, extreme tiredness, muscle or joint aches, blisters, oral lesions, eye inflammation, facial swelling, swelling of the eyes, lips, mouth, breathing difficulty, and/or signs and symptoms of liver problems (e.g., yellowing of the skin or whites of the eyes, dark or tea colored urine, pale colored stools/bowel movements, nausea, vomiting, loss of appetite, or pain, aching or sensitivity on the right side below the ribs). Inform patients that if severe rash occurs, their physician will closely monitor them, order laboratory tests and initiate appropriate therapy.
Immune Reconstitution Syndrome
Advise patients to inform their healthcare provider immediately of any symptoms of infection, as in some patients with advanced HIV infection (AIDS), signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started [see Warnings and Precautions (5.2)].
Rhabdomyolysis
Before patients begin ISENTRESS or ISENTRESS HD, ask them if they have a history of rhabdomyolysis, myopathy or increased creatine kinase or if they are taking medications known to cause these conditions such as statins, fenofibrate, gemfibrozil or zidovudine [see Adverse Reactions (6.1)].
Instruct patients to immediately report to their healthcare provider any unexplained muscle pain, tenderness, or weakness while taking ISENTRESS.
Phenylketonuria
Alert patients with phenylketonuria that ISENTRESS Chewable Tablets contain phenylalanine [see Warnings and Precautions (5.3)].
Drug Interactions
Inform patients that ISENTRESS or ISENTRESS HD may interact with some drugs and ask them to identify all their current medications including over-the-counter agents [see Drug Interactions (7.2)].
Missed Dosage
Inform patients that it is important to take ISENTRESS or ISENTRESS HD on a regular dosing schedule as instructed by their healthcare provider and to avoid missing doses as it can result in development of resistance [see Dosage and Administration (2)].
Important Administration Instructions
Pregnancy Registry
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ISENTRESS or ISENTRESS HD during pregnancy [see Use in Specific Populations (8.1)].
Lactation
Instruct women with HIV-1 infection not to breastfeed because HIV-1 can be passed to the baby in the breast milk [see Use in Specific Populations (8.2)].
-
SPL UNCLASSIFIED SECTION
Distributed by:
Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
Whitehouse Station, NJ 08889, USAFor patent information: www.merck.com/product/patent/home.html
Copyright © 2014-2019 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
All rights reserved.uspi-mk0518-mf-1901r034
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION This Patient Information has been approved by the U.S. Food and Drug Administration. Revised November 2017 ISENTRESS® (eye sen tris)
(raltegravir)
film-coated tabletsISENTRESS® (eye sen tris)
(raltegravir)
chewable tabletsISENTRESS® HD (eye sen tris HD)
(raltegravir)
film-coated tabletsISENTRESS® (eye sen tris)
(raltegravir)
for oral suspensionWhat are ISENTRESS and ISENTRESS HD? ISENTRESS is a prescription HIV medicine used with other antiretroviral medicines to treat Human Immunodeficiency Virus-1 (HIV-1) infection in adults, and in children weighing at least 4.4 pounds (2 kg). HIV is the virus that causes AIDS (Acquired Immune Deficiency Syndrome). ISENTRESS HD is a prescription HIV medicine used with other antiretroviral medicines to treat HIV-1 infection in adults, and in children weighing at least 88 pounds (40 kg).
ISENTRESS should not be used in children who weigh less than 4.4 pounds (2 kg).Before you take ISENTRESS or ISENTRESS HD, tell your doctor about all of your medical conditions, including if you: - have liver problems
- have a history of a muscle disorder called rhabdomyolysis or myopathy
- have increased levels of creatine kinase in your blood
- have phenylketonuria (PKU). ISENTRESS chewable tablets contain phenylalanine as part of the artificial sweetener, aspartame. The artificial sweetener may be harmful to people with PKU.
- receive kidney dialysis treatment
- are pregnant or plan to become pregnant. It is not known if ISENTRESS or ISENTRESS HD can harm your unborn baby.
Pregnancy Registry: There is a pregnancy registry for women who take antiretroviral medicines during pregnancy. The purpose of this registry is to collect information about the health of you and your baby. Talk to your doctor about how you can take part in this registry. - are breastfeeding or plan to breastfeed. Do not breastfeed if you take ISENTRESS or ISENTRESS HD.
- You should not breastfeed if you have HIV-1 because of the risk of passing HIV-1 to your baby.
- It is not known if ISENTRESS or ISENTRESS HD can pass into your breast milk.
- Talk with your doctor about the best way to feed your baby.
Tell your doctor about all the medicines you take, including, prescription and over-the-counter medicines, vitamins, and herbal supplements. Some medicines interact with ISENTRESS and ISENTRESS HD. - Keep a list of your medicines to show your doctor and pharmacist.
- You can ask your doctor or pharmacist for a list of medicines that interact with ISENTRESS and ISENTRESS HD.
- Do not start taking a new medicine without telling your doctor. Your doctor can tell you if it is safe to take ISENTRESS or ISENTRESS HD with other medicines.
How should I take ISENTRESS or ISENTRESS HD? - Take ISENTRESS or ISENTRESS HD exactly as prescribed by your doctor.
- Do not change your dose of ISENTRESS or ISENTRESS HD or stop your treatment without talking with your doctor first.
- Stay under the care of your doctor during treatment with ISENTRESS or ISENTRESS HD.
- ISENTRESS film-coated tablets and ISENTRESS HD film-coated tablets must be swallowed whole.
- ISENTRESS chewable tablets may be chewed or swallowed whole.
- Do not switch between the film-coated tablet, the chewable tablet, or the oral suspension without talking with your doctor first.
- Do not switch between the ISENTRESS 400 mg film-coated tablet and the ISENTRESS HD 600 mg film-coated tablet if your prescribed dose is 1200 mg.
- Do not run out of ISENTRESS or ISENTRESS HD. The virus in your blood may increase and the virus may become harder to treat. Get a refill of your ISENTRESS or ISENTRESS HD from your doctor or pharmacy before you run out.
- Take ISENTRESS or ISENTRESS HD on a regular dosing schedule as instructed by your doctor. Do not miss doses.
- If you take too much ISENTRESS or ISENTRESS HD, call your doctor or go to the nearest hospital emergency room right away.
If ISENTRESS for oral suspension is prescribed for your child, be sure to read the following information:
- Before giving the first dose of ISENTRESS for oral suspension, read the Instructions for Use booklet that comes with ISENTRESS for oral suspension for information about the correct way to mix and give a dose of ISENTRESS for oral suspension to your child. Keep the booklet and follow it each time you prepare the medicine. Bring this booklet to your child's appointments.
- Make sure your doctor shows you how to mix and give the right dose of ISENTRESS for oral suspension to your child. If you have questions about how to mix or give ISENTRESS for oral suspension, talk with your doctor or pharmacist.
- Give the dose of ISENTRESS for oral suspension within 30 minutes of mixing.
- If your child does not take all of the prescribed dose or spits some of it out, call your doctor to find out what to do.
- Your child's dose will change over time. Make sure you follow your doctor's instructions. Your doctor will tell you if and when to stop giving ISENTRESS to your child.
What are the possible side effects of ISENTRESS or ISENTRESS HD? ISENTRESS and ISENTRESS HD can cause serious side effects including: -
Severe skin reactions and allergic reactions. Some people who take ISENTRESS or ISENTRESS HD develop severe skin reactions and allergic reactions that can be serious, and may be life-threatening or lead to death.
- If you develop a rash, call your doctor right away.
- If you develop a rash with any of the following symptoms, stop using ISENTRESS or ISENTRESS HD and call your doctor or get medical help right away:
- fever
- generally ill feeling
- extreme tiredness
- muscle or joint aches
- blisters or sores in mouth
- blisters or peeling of the skin
- redness or swelling of the eyes
- swelling of the mouth, lips, or face
- problems breathing
Sometimes allergic reactions can affect body organs, such as your liver. Call your doctor right away if you have any of the following signs or symptoms of liver problems: - yellowing of your skin or whites of your eyes
- dark or tea colored urine
- pale colored stools (bowel movements)
- nausea or vomiting
- loss of appetite
- pain, aching, or tenderness on the right side of your stomach area
- Changes in your immune system (Immune Reconstitution Syndrome) can happen when you start taking HIV-1 medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your doctor right away if you start having new symptoms after starting your HIV-1 medicine.
The most common side effects of ISENTRESS and ISENTRESS HD include: - trouble sleeping
- headache
- dizziness
- nausea
- tiredness
Less common side effects of ISENTRESS and ISENTRESS HD include: - depression
- hepatitis
- genital herpes
- herpes zoster including shingles
- kidney failure
- kidney stones
- indigestion or stomach area pain
- vomiting
- suicidal thoughts and actions
- weakness
Tell your doctor right away if you get unexplained muscle pain, tenderness, or weakness during treatment with ISENTRESS or ISENTRESS HD. These may be signs of a rare serious muscle problem that can lead to kidney problems. These are not all the possible side effects of ISENTRESS and ISENTRESS HD. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store ISENTRESS and ISENTRESS HD? ISENTRESS and ISENTRESS HD film-coated tablets: - Store ISENTRESS and ISENTRESS HD film-coated tablets at room temperature between 68°F to 77°F (20°C to 25°C).
- Store ISENTRESS and ISENTRESS HD film-coated tablets in the original package with the bottle tightly closed.
- Keep the drying agent (desiccant) in the ISENTRESS and ISENTRESS HD bottle to protect from moisture.
ISENTRESS chewable tablets: - Store ISENTRESS chewable tablets at room temperature between 68°F to 77°F (20°C to 25°C).
- Store ISENTRESS chewable tablets in the original package with the bottle tightly closed.
- Keep the drying agent (desiccant) in the bottle to protect from moisture.
ISENTRESS for oral suspension: - Store ISENTRESS for oral suspension at room temperature between 68°F to 77°F (20°C to 25°C).
- Store in the original container. Do not open the foil packet until ready for use.
Keep ISENTRESS and all medicines out of the reach of children. General information about the safe and effective use of ISENTRESS and ISENTRESS HD Medicines are sometimes prescribed for purposes other than those listed in a Patient Information Leaflet. Do not use ISENTRESS or ISENTRESS HD for a condition for which it was not prescribed. Do not give ISENTRESS or ISENTRESS HD to other people, even if they have the same symptoms that you have. It may harm them. You can ask your doctor or pharmacist for information about ISENTRESS or ISENTRESS HD that is written for health professionals. What are the ingredients in ISENTRESS and ISENTRESS HD? ISENTRESS 400 mg film-coated tablets: Active ingredient: raltegravir Inactive ingredients: calcium phosphate dibasic anhydrous, hypromellose 2208, lactose monohydrate, magnesium stearate, microcrystalline cellulose, poloxamer 407 (contains 0.01% butylated hydroxytoluene as antioxidant), sodium stearyl fumarate. The film coating contains: black iron oxide, polyethylene glycol 3350, polyvinyl alcohol, red iron oxide, talc and titanium dioxide. ISENTRESS HD 600 mg film-coated tablets: Active ingredient: raltegravir Inactive ingredients: croscarmellose sodium, hypromellose 2910, magnesium stearate, microcrystalline cellulose. The film coating contains: ferrosoferric oxide, hypromellose 2910, iron oxide yellow, lactose monohydrate, triacetin and titanium dioxide. The tablet may also contain trace amount of carnauba wax. ISENTRESS chewable tablets: Active ingredient: raltegravir Inactive ingredients: ammonium hydroxide, crospovidone, ethylcellulose 20 cP, fructose, hydroxypropyl cellulose, hypromellose 2910/6cP, magnesium stearate, mannitol, medium chain triglycerides, monoammonium glycyrrhizinate, natural and artificial flavors (orange, banana, and masking that contains aspartame), oleic acid, PEG 400, saccharin sodium, sodium citrate dihydrate, sodium stearyl fumarate, sorbitol, sucralose and yellow iron oxide. The 100 mg chewable tablet also contains red iron oxide. ISENTRESS for oral suspension: Active ingredient: raltegravir Inactive ingredients: ammonium hydroxide, banana with other natural flavors, carboxymethylcellulose sodium, crospovidone, ethylcellulose 20 cP, fructose, hydroxypropyl cellulose, hypromellose 2910/6cP, macrogol/PEG 400, magnesium stearate, maltodextrin, mannitol, medium chain triglycerides, microcrystalline cellulose, monoammonium glycyrrhizinate, oleic acid, sorbitol, sucralose and sucrose. Distributed by: Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. Whitehouse Station, NJ 08889, USA For patent information: www.merck.com/product/patent/home.html Copyright © 2007-2017 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. All rights reserved. usppi-mk0518-mf-1711r028
For more information go to www.ISENTRESS.com or call 1-800-622-4477. -
INSTRUCTIONS FOR USE
Bring this booklet to your child's appointments.
ISENTRESS (raltegravir)
for oral suspensionInstructions for Use
for babies and toddlers
Before You Start
Note: Make sure your doctor shows you how to prepare and give ISENTRESS for oral suspension. - Be sure you understand these instructions before you start. Call your doctor if you have any questions.
- It is very important that you measure the water and ISENTRESS carefully using the correct syringe.
- Before you give ISENTRESS to your child, check the expiration date. The expiration date is printed on the box and the ISENTRESS packets.
- Do not open the ISENTRESS packets until you are ready to mix a dose.
- The amount of ISENTRESS depends on your child's age and weight, so it will change over time.
Your doctor will tell you the right dose at each check-up after weighing your child.
Be sure to keep your doctor's appointments so you get new dosing information as your child grows.
During your child's first week of life, you will give ISENTRESS 1 time a day. After the first week of life, you will give it 2 times a day. - This booklet tells you how to:
- Mix ISENTRESS into a liquid form
- Measure the right dose using a syringe
- Give mixed ISENTRESS to your child
- Clean up
Kit Contents
- Prescription (on box)

- 2 mixing cups
- Instructions for Use (this booklet)
- Prescribing Information
- 6 syringes
- 60 packets of ISENTRESS


2 blue (10mL) syringes 2 green (3mL) syringes 2 white (1mL) syringes The kit has an extra cup and set of syringes in case one is lost or damaged.
Do not use any damaged cups or syringes.
Step 1. Get ready
- Put your child in a safe place. You will need both hands to prepare ISENTRESS.
- Wash your hands with soap and water.
- Take out what you need from the kit as shown below to make 1 dose and place on a clean surface:

1 mixing cup
(Using the tab on the mixing cup, pull open the lid)1 packet of ISENTRESS a clean glass 3 syringes
(Have one of each size ready, but you will only need 1 or 2, depending on the prescribed dose)Step 2. Fill a clean glass with water
Fill a clean glass with room-temperature drinking water from your sink or use bottled water. 
Step 3. Fill the blue syringe with water
Push the plunger of the blue syringe into the syringe as far as it goes. 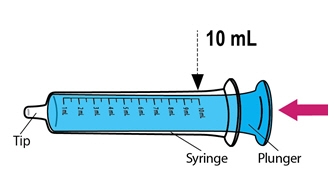
Put the tip of the syringe into the glass of water. Pull back the plunger to draw up water into the syringe. Stop when you get to the 10mL mark. 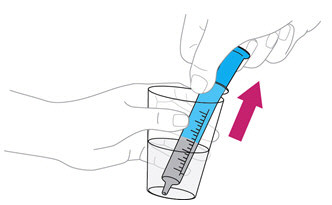
Step 4. Check for air bubbles
Hold the syringe with tip up. Tap it with your finger to move any air bubbles up towards the tip.
Slowly push up on the plunger to make the air come out. You may see some drops of water come out.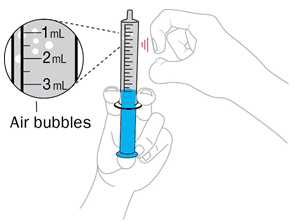
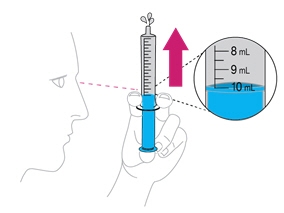
Re-check the amount of water in the syringe. If it is less than 10mL, put the tip back into the water and pull back on the plunger until you get to the 10mL mark.Step 5. Add the 10mL of water to the mixing cup
Add the 10 mL of water from the syringe to the mixing cup by pushing all the way down on the plunger.
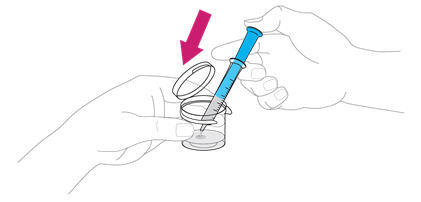
Step 6. Add ISENTRESS to the mixing cup
Note before adding ISENTRESS:
Make sure you and your child are ready! After mixing ISENTRESS, use it within 30 minutes. Throw away any leftover ISENTRESS after you have given the dose to your child.
Take 1 packet of ISENTRESS and shake the powder to the bottom of the packet. Tear or cut open the packet at the dotted line and add all of the powder to the water in the mixing cup. Make sure the packet is completely empty. 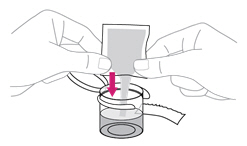
Step 7. Mix ISENTRESS and water
Snap the lid of the mixing cup shut.
Gently swirl the mixing cup for 45 seconds in a circular motion to mix the powder and water. Use a clock or timer to time for 45 seconds. Do not shake the mixture.
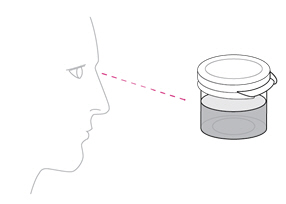
Check to make sure the powder is mixed. If it is not mixed, swirl it some more. The mixture should look cloudy. Step 8. Check your prescription
Find the dose amount in 'mL' prescribed by your doctor. This is written on the prescription label on the box from your pharmacy.
Remember that the dose may change each time you go to the doctor, so make sure you check the prescribed dose each time. Be sure to go to all of your doctor's appointments so your child gets the right dose!
Step 9. Choose the syringe you need
Your doctor will prescribe the dose in milliliters (mL). Choose the right syringe for your child's dose:
WHITE
(1mL) for 1mL
or less
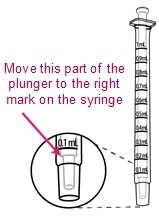
GREEN
(3mL)
for 1.5mL
to 3mL
BLUE
(10mL)
for 3.5mL
to 10mL
Then find the mL mark on the syringe that matches your child's dose.
Step 10. Measure ISENTRESS
Push the plunger into the barrel of the syringe as far as it goes. 
Put the tip of the syringe into the cup of the mixed ISENTRESS and pull back on the plunger.
Stop when you get to the line that matches your child's prescribed dose.
IMPORTANT: - Your child's dose may be different from the one shown in this figure.
- There will usually be some leftover mixed ISENTRESS in the mixing cup.
Step 11. Check for air bubbles
Hold the syringe with tip up. Tap it with your finger to move any air bubbles up towards the tip.
Slowly push the plunger to make the air come out. You may see some drops of medicine come out.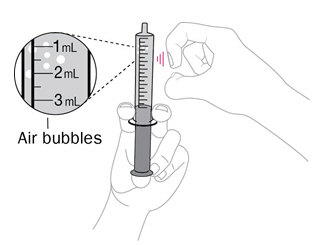
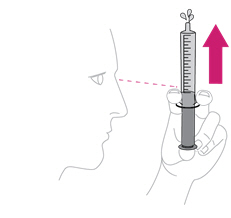
Re-check the amount of ISENTRESS in the syringe. If it is less than the prescribed dose, put the tip back into the cup of mixed ISENTRESS and pull back on the plunger until you get to the right dose mark. Step 12. Give ISENTRESS to your child
Place the tip of the syringe inside your child's mouth so that it touches either the right or left cheek. 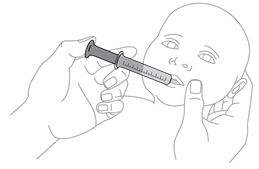
Slowly push in the plunger to give the dose of mixed ISENTRESS to your child. If your child fusses, take the tip of the syringe out of the mouth and try again. It is important that your child takes all of the prescribed dose (a little left in the syringe tip is OK).
IMPORTANT:
If your child does not take all of the prescribed dose or spits some of it out, call your doctor to find out what to do.Step 13. Clean up
Pour the leftover mixed ISENTRESS into the trash.
Do not pour it into the sink.Pull the plungers out of any syringes you used.
Hand wash the syringes, plungers, and mixing cup with warm water and dish soap. Do not wash in the dishwasher.
Rinse with water and let air dry.
Put everything in a clean, dry place.
How should I store ISENTRESS?
Store the ISENTRESS for oral suspension kit at room temperature between 68°F to 77°F (20°C to 25°C).
Store in the original container.
Keep ISENTRESS and all medicines out of the reach of children.
Be sure to keep your doctor's appointments so you always know how much ISENTRESS to give to your child. For more information, go to www.ISENTRESS.com or call 1-800-622-4477.
-
SPL UNCLASSIFIED SECTION
Distributed by:
Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
Whitehouse Station, NJ, USAFor patent information: www.merck.com/product/patent/home.html
Copyright © 2013-2017 Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.
All rights reserved.ifu-mk0518-mf-1711r001
Revised November/2017 - RALTEGRAVIR
-
INGREDIENTS AND APPEARANCE
ISENTRESS
raltegravir tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50090-1085(NDC:0006-0227) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RALTEGRAVIR POTASSIUM (UNII: 43Y000U234) (RALTEGRAVIR - UNII:22VKV8053U) RALTEGRAVIR 400 mg Inactive Ingredients Ingredient Name Strength FERROSOFERRIC OXIDE (UNII: XM0M87F357) BUTYLATED HYDROXYTOLUENE (UNII: 1P9D0Z171K) ANHYDROUS DIBASIC CALCIUM PHOSPHATE (UNII: L11K75P92J) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POLOXAMER 407 (UNII: TUF2IVW3M2) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) FERRIC OXIDE RED (UNII: 1K09F3G675) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color PINK (pink) Score no score Shape OVAL (oval-shaped) Size 16mm Flavor Imprint Code 227 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50090-1085-0 60 in 1 BOTTLE; Type 0: Not a Combination Product 11/28/2014 2 NDC: 50090-1085-1 8 in 1 BOTTLE; Type 0: Not a Combination Product 11/28/2014 3 NDC: 50090-1085-2 6 in 1 BOTTLE; Type 0: Not a Combination Product 03/16/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022145 10/12/2007 Labeler - A-S Medication Solutions (830016429) Establishment Name Address ID/FEI Business Operations A-S Medication Solutions 830016429 RELABEL(50090-1085) , REPACK(50090-1085)

Trademark Results [ISENTRESS]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ISENTRESS 78866853 3357758 Live/Registered |
MERCK SHARP & DOHME CORP. 2006-04-21 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.