ENHERTU- fam-trastuzumab deruxtecan-nxki injection, powder, lyophilized, for solution
Enhertu by
Drug Labeling and Warnings
Enhertu by is a Prescription medication manufactured, distributed, or labeled by Daiichi Sankyo Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ENHERTU safely and effectively. See full prescribing information for ENHERTU.
ENHERTU® (fam-trastuzumab deruxtecan-nxki) for injection, for intravenous use
Initial U.S. Approval: 2019WARNING: INTERSTITIAL LUNG DISEASE and EMBRYO-FETAL TOXICITY
See full prescribing information for complete boxed warning.
- Interstitial lung disease (ILD) and pneumonitis, including fatal cases, have been reported with ENHERTU. Monitor for and promptly investigate signs and symptoms including cough, dyspnea, fever, and other new or worsening respiratory symptoms. Permanently discontinue ENHERTU in all patients with Grade 2 or higher ILD/pneumonitis. Advise patients of the risk and to immediately report symptoms. (2.2, 5.1)
- Exposure to ENHERTU during pregnancy can cause embryo-fetal harm. Advise patients of these risks and the need for effective contraception. (5.4, 8.1, 8.3)
INDICATIONS AND USAGE
ENHERTU is a HER2-directed antibody and topoisomerase inhibitor conjugate indicated for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens in the metastatic setting. (1)
This indication is approved under accelerated approval based on tumor response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. (1, 14.1)
DOSAGE AND ADMINISTRATION
- Do not substitute ENHERTU for or with trastuzumab or ado-trastuzumab emtansine. (2.1, 2.3)
- For intravenous infusion only. Do not administer as an intravenous push or bolus. Do not use Sodium Chloride Injection, USP. (2.3)
- The recommended dosage of ENHERTU is 5.4 mg/kg given as an intravenous infusion once every 3 weeks (21-day cycle) until disease progression or unacceptable toxicity. (2.1, 2.2)
- Management of adverse reactions (ILD, neutropenia, or left ventricular dysfunction) may require temporary interruption, dose reduction, or discontinuation of ENHERTU. (2.2)
DOSAGE FORMS AND STRENGTHS
For injection: 100 mg lyophilized powder in a single-dose vial (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Neutropenia: Monitor complete blood counts prior to initiation of ENHERTU and prior to each dose, and as clinically indicated. Manage through treatment interruption or dose reduction. (2.2, 5.2)
- Left Ventricular Dysfunction: Assess LVEF prior to initiation of ENHERTU and at regular intervals during treatment as clinically indicated. Manage through treatment interruption or discontinuation. Permanently discontinue ENHERTU in patients with symptomatic congestive heart failure (CHF). (2.2, 5.3)
ADVERSE REACTIONS
The most common adverse reactions (≥20%) were nausea, fatigue, vomiting, alopecia, constipation, decreased appetite, anemia, neutropenia, diarrhea, leukopenia, cough, and thrombocytopenia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Daiichi Sankyo, Inc. at 1-877-437-7763 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: INTERSTITIAL LUNG DISEASE and EMBRYO-FETAL TOXICITY
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage and Schedules
2.2 Dose Modifications
2.3 Preparation for Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease/Pneumonitis
5.2 Neutropenia
5.3 Left Ventricular Dysfunction
5.4 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Metastatic Breast Cancer
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied/Storage
16.2 Special Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: INTERSTITIAL LUNG DISEASE and EMBRYO-FETAL TOXICITY
- Interstitial Lung Disease (ILD) and pneumonitis, including fatal cases, have been reported with ENHERTU. Monitor for and promptly investigate signs and symptoms including cough, dyspnea, fever, and other new or worsening respiratory symptoms. Permanently discontinue ENHERTU in all patients with Grade 2 or higher ILD/pneumonitis. Advise patients of the risk and the need to immediately report symptoms [see Dosage and Administration (2.2), Warnings and Precautions (5.1)].
- Embryo-Fetal Toxicity: Exposure to ENHERTU during pregnancy can cause embryo-fetal harm. Advise patients of these risks and the need for effective contraception [see Warnings and Precautions (5.4), Use in Specific Populations (8.1, 8.3)].
-
1 INDICATIONS AND USAGE
ENHERTU is indicated for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens in the metastatic setting.
This indication is approved under accelerated approval based on tumor response rate and duration of response [see Clinical Studies (14.1)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage and Schedules
Do not substitute ENHERTU for or with trastuzumab or ado-trastuzumab emtansine.
The recommended dosage of ENHERTU is 5.4 mg/kg given as an intravenous infusion once every 3 weeks (21-day cycle) until disease progression or unacceptable toxicity.
First infusion: Administer infusion over 90 minutes.
Subsequent infusions: Administer over 30 minutes if prior infusions were well tolerated.
Slow or interrupt the infusion rate if the patient develops infusion-related symptoms.
Permanently discontinue ENHERTU in case of severe infusion reactions.
2.2 Dose Modifications
Management of adverse reactions may require temporary interruption, dose reduction, or treatment discontinuation of ENHERTU as described in Tables 1 and 2.
Do not re-escalate the ENHERTU dose after a dose reduction is made.
If a planned dose is delayed or missed, administer as soon as possible; do not wait until the next planned cycle. Adjust the schedule of administration to maintain a 3-week interval between doses. Administer the infusion at the dose and rate the patient tolerated in the most recent infusion.
Table 1: Dose Reduction Schedule Dose Reduction Schedule
(Starting dose is 5.4 mg/kg.)Dose to be administered First dose reduction 4.4 mg/kg Second dose reduction 3.2 mg/kg Requirement for further dose reduction Discontinue treatment Table 2: Dose Modifications for Adverse Reactions Adverse Reaction Severity Treatment Modification Toxicity grades are in accordance with National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03 (NCI-CTCAE v.4.03). Interstitial Lung Disease (ILD)/pneumonitis Asymptomatic ILD/pneumonitis
(Grade 1)Interrupt ENHERTU until resolved to Grade 0, then: - if resolved in 28 days or less from date of onset, maintain dose.
- if resolved in greater than 28 days from date of onset, reduce dose one level (see Table 1).
- consider corticosteroid treatment as soon as ILD/pneumonitis is suspected [see Warnings and Precautions (5.1)].
Symptomatic ILD/pneumonitis
(Grade 2 or greater)- Permanently discontinue ENHERTU.
- Promptly initiate corticosteroid treatment as soon as ILD/pneumonitis is suspected [see Warnings and Precautions (5.1)].
Neutropenia Grade 3 (less than 1.0 to 0.5 × 109/L) - Interrupt ENHERTU until resolved to Grade 2 or less, then maintain dose.
Grade 4 (less than 0.5 × 109/L) - Interrupt ENHERTU until resolved to Grade 2 or less.
- Reduce dose by one level (see Table 1).
Febrile Neutropenia Absolute neutrophil count of less than 1.0 × 109/L and temperature greater than 38.3°C or a sustained temperature of 38°C or greater for more than one hour - Interrupt ENHERTU until resolved.
- Reduce dose by one level (see Table 1).
Left Ventricular Dysfunction LVEF greater than 45% and absolute decrease from baseline is 10% to 20% - Continue treatment with ENHERTU.
LVEF 40% to 45% And absolute decrease from baseline is less than 10% - Continue treatment with ENHERTU.
- Repeat LVEF assessment within 3 weeks.
And absolute decrease from baseline is 10% to 20% - Interrupt ENHERTU.
- Repeat LVEF assessment within 3 weeks.
- If LVEF has not recovered to within 10% from baseline, permanently discontinue ENHERTU.
- If LVEF recovers to within 10% from baseline, resume treatment with ENHERTU at the same dose.
LVEF less than 40% or absolute decrease from baseline is greater than 20% - Interrupt ENHERTU.
- Repeat LVEF assessment within 3 weeks.
- If LVEF of less than 40% or absolute decrease from baseline of greater than 20% is confirmed, permanently discontinue ENHERTU.
Symptomatic congestive heart failure (CHF) - Permanently discontinue ENHERTU.
2.3 Preparation for Administration
In order to prevent medication errors, check the vial labels to ensure that the drug being prepared and administered is ENHERTU (fam-trastuzumab deruxtecan-nxki) and not trastuzumab or ado-trastuzumab emtansine.
Reconstitute and further dilute ENHERTU prior to intravenous infusion. Use appropriate aseptic technique.
ENHERTU (fam-trastuzumab deruxtecan-nxki) is a cytotoxic drug. Follow applicable special handling and disposal procedures.1
Reconstitution
- Reconstitute immediately before dilution.
- More than one vial may be needed for a full dose. Calculate the dose (mg), the total volume of reconstituted ENHERTU solution required, and the number of vial(s) of ENHERTU needed [see Dosage and Administration (2.2)].
- Reconstitute each 100 mg vial by using a sterile syringe to slowly inject 5 mL of Sterile Water for Injection, USP into each vial to obtain a final concentration of 20 mg/mL.
- Swirl the vial gently until completely dissolved. Do not shake.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The solution should be clear and colorless to light yellow. Do not use if visible particles are observed or if the solution is cloudy or discolored.
- If not used immediately, store the reconstituted ENHERTU vials in a refrigerator at 2°C to 8°C (36°F to 46°F) for up to 24 hours from the time of reconstitution, protected from light. Do not freeze.
- The product does not contain a preservative. Discard unused ENHERTU after 24 hours refrigerated.
Dilution
- Dilute the calculated volume of reconstituted ENHERTU in an intravenous infusion bag containing 100 mL of 5% Dextrose Injection, USP. Do not use Sodium Chloride Injection, USP. ENHERTU is compatible with an infusion bag made of polyvinylchloride or polyolefin (copolymer of ethylene and polypropylene).
- Gently invert the infusion bag to thoroughly mix the solution. Do not shake.
- Cover the infusion bag to protect from light.
- If not used immediately, store at room temperature for up to 4 hours including preparation and infusion, or in a refrigerator at 2°C to 8°C (36°F to 46°F) for up to 24 hours, protected from light. Do not freeze.
- Discard any unused portion left in the vial.
Administration
- If the prepared infusion solution was stored refrigerated (2°C to 8°C [36°F to 46°F]), allow the solution to reach room temperature prior to administration.
- Administer ENHERTU as an intravenous infusion only with an infusion set made of polyolefin or polybutadiene and a 0.20 or 0.22 micron in-line polyethersulfone (PES) or polysulfone (PS) filter. Do not administer as an intravenous push or bolus.
- Do not mix ENHERTU with other drugs or administer other drugs through the same intravenous line.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease/Pneumonitis
Severe, life-threatening, or fatal interstitial lung disease (ILD), including pneumonitis, can occur in patients treated with ENHERTU [see Adverse Reactions (6.1)]. In clinical studies, of the 234 patients with unresectable or metastatic HER2-positive breast cancer treated with ENHERTU, ILD occurred in 9% of patients. Fatal outcomes due to ILD and/or pneumonitis occurred in 2.6% of patients treated with ENHERTU. Median time to first onset was 4.1 months (range: 1.2 to 8.3).
Advise patients to immediately report cough, dyspnea, fever, and/or any new or worsening respiratory symptoms. Monitor patients for signs and symptoms of ILD. Promptly investigate evidence of ILD. Evaluate patients with suspected ILD by radiographic imaging. Consider consultation with a pulmonologist. For asymptomatic (Grade 1) ILD, consider corticosteroid treatment (e.g., ≥0.5 mg/kg prednisolone or equivalent). Withhold ENHERTU until recovery [see Dosage and Administration (2.2)]. In cases of symptomatic ILD (Grade 2 or greater), promptly initiate corticosteroid treatment (e.g., ≥1 mg/kg prednisolone or equivalent). Upon improvement, follow by gradual taper (e.g., 4 weeks). Permanently discontinue ENHERTU in patients who are diagnosed with any symptomatic (Grade 2 or greater) ILD [see Dosage and Administration (2.2)].
5.2 Neutropenia
Severe neutropenia, including febrile neutropenia, can occur in patients treated with ENHERTU. Of the 234 patients with unresectable or metastatic HER2-positive breast cancer who received ENHERTU, a decrease in neutrophil count was reported in 30% of patients and 16% had Grade 3 or 4 events. Median time to first onset was 1.4 months (range: 0.3 to 18.2). Febrile neutropenia was reported in 1.7% of patients.
Monitor complete blood counts prior to initiation of ENHERTU and prior to each dose, and as clinically indicated. Based on the severity of neutropenia, ENHERTU may require dose interruption or reduction [see Dosage and Administration (2.2)].
5.3 Left Ventricular Dysfunction
Patients treated with ENHERTU may be at increased risk of developing left ventricular dysfunction. Left ventricular ejection fraction (LVEF) decrease has been observed with anti-HER2 therapies, including ENHERTU. In the 234 patients with unresectable or metastatic HER2-positive breast cancer who received ENHERTU, two cases (0.9%) of asymptomatic LVEF decrease were reported. Treatment with ENHERTU has not been studied in patients with a history of clinically significant cardiac disease or LVEF less than 50% prior to initiation of treatment.
Assess LVEF prior to initiation of ENHERTU and at regular intervals during treatment as clinically indicated. Manage LVEF decrease through treatment interruption. Permanently discontinue ENHERTU if LVEF of less than 40% or absolute decrease from baseline of greater than 20% is confirmed. Permanently discontinue ENHERTU in patients with symptomatic congestive heart failure (CHF) [see Dosage and Administration (2.2)].
5.4 Embryo-Fetal Toxicity
Based on its mechanism of action, ENHERTU can cause fetal harm when administered to a pregnant woman. In postmarketing reports, use of a HER2-directed antibody during pregnancy resulted in cases of oligohydramnios manifesting as fatal pulmonary hypoplasia, skeletal abnormalities, and neonatal death. Based on its mechanism of action, the topoisomerase inhibitor component of ENHERTU, DXd, can also cause embryo-fetal harm when administered to a pregnant woman because it is genotoxic and targets actively dividing cells [see Use in Specific Populations (8.1), Clinical Pharmacology (12.1), Nonclinical Toxicology (13.1)]. Advise patients of the potential risks to a fetus.
Verify the pregnancy status of females of reproductive potential prior to the initiation of ENHERTU. Advise females of reproductive potential to use effective contraception during treatment and for at least 7 months following the last dose of ENHERTU. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with ENHERTU and for at least 4 months after the last dose of ENHERTU [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Interstitial Lung Disease / Pneumonitis [see Warnings and Precautions (5.1)]
- Neutropenia [see Warnings and Precautions (5.2)]
- Left Ventricular Dysfunction [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of ENHERTU was evaluated in a pooled analysis of 234 patients with unresectable or metastatic HER2-positive breast cancer who received at least one dose of ENHERTU 5.4 mg/kg in DESTINY-Breast01 and Study DS8201-A-J101 (NCT02564900). ENHERTU was administered by intravenous infusion once every three weeks. The median duration of treatment was 7 months (range: 0.7 to 31).
In the pooled 234 patients, the median age was 56 years (range: 28-96), 74% of patients were <65 years, 99.6% of patients were female, and the majority were White (51%) or Asian (42%). Patients had an ECOG performance status of 0 (58%) or 1 (42%) at baseline. Ninety-four percent had visceral disease, 31% had bone metastases, and 13% had brain metastases.
Serious adverse reactions occurred in 20% of patients receiving ENHERTU. Serious adverse reactions in >1% of patients who received ENHERTU were interstitial lung disease, pneumonia, vomiting, nausea, cellulitis, hypokalemia, and intestinal obstruction. Fatalities due to adverse reactions occurred in 4.3% of patients including interstitial lung disease (2.6%), and the following events occurred in one patient each (0.4%): acute hepatic failure/acute kidney injury, general physical health deterioration, pneumonia, and hemorrhagic shock.
ENHERTU was permanently discontinued in 9% of patients, of which ILD accounted for 6%. Dose interruptions due to adverse reactions occurred in 33% of patients treated with ENHERTU. The most frequent adverse reactions (>2%) associated with dose interruption were neutropenia, anemia, thrombocytopenia, leukopenia, upper respiratory tract infection, fatigue, nausea, and ILD. Dose reductions occurred in 18% of patients treated with ENHERTU. The most frequent adverse reactions (>2%) associated with dose reduction were fatigue, nausea, and neutropenia.
The most common adverse reactions (frequency ≥20%) were nausea, fatigue, vomiting, alopecia, constipation, decreased appetite, anemia, neutropenia, diarrhea, leukopenia, cough, and thrombocytopenia.
Tables 3 and 4 summarize common adverse reactions and laboratory abnormalities observed in ENHERTU-treated patients.
Table 3: Common Adverse Reactions (≥10% All Grades or ≥2% Grades 3 or 4) in Patients in DESTINY-Breast01 and Study DS8201-A-J101 Adverse Reactions ENHERTU 5.4 mg/kg
N=234All Grades
%Grades 3 or 4
%Events were graded using NCI-CTCAE version 4.03. N=number of patients exposed; PT = preferred term.
Percentages were calculated using the number of patients in the Safety Analysis Set as the denominator.- * Grouped term of abdominal pain includes PTs of abdominal discomfort, gastrointestinal pain, abdominal pain, abdominal pain lower, and abdominal pain upper.
- † Grouped term of stomatitis includes PTs of stomatitis, aphthous ulcer, mouth ulceration, oral mucosa erosion, and oral mucosa blistering. One Grade 1 event of aphthous ulcer was not included in the summary of grouped term stomatitis (from DESTINY-Breast01).
- ‡ Grouped term of fatigue includes PTs of fatigue and asthenia.
- § This Grade 3 event was reported by the investigator. Per NCI-CTCAE v.4.03, the highest NCI-CTCAE grade for alopecia is Grade 2.
- ¶ Grouped term of rash includes PTs of rash, rash pustular, rash maculo-papular.
- # Grouped term of anemia includes PTs of anemia, hemoglobin decreased, hematocrit decreased, and red blood cell count decreased.
- Þ Grouped term of neutropenia includes PTs of neutropenia and neutrophil count decreased.
- ß Grouped term of leukopenia includes PTs of leukopenia, lymphopenia, and white blood cell count decreased.
- à Grouped term of thrombocytopenia includes PTs of thrombocytopenia and platelet count decreased.
- è Interstitial lung disease includes events that were adjudicated as ILD: pneumonitis, interstitial lung disease, respiratory failure, organizing pneumonia, acute respiratory failure, lung infiltration, lymphangitis, alveolitis.
- ð All events had fatal outcomes (n=6).
- ø Grouped term of headache includes PTs headache, sinus headache, and migraine.
- ý Grouped term of upper respiratory tract infection includes PTs influenza, influenza like illness, upper respiratory tract infection.
- £ This Grade 4 event was reported by the investigator. Per NCI-CTCAE v.4.03, the highest NCI-CTCAE grade for dry eye is Grade 3.
Gastrointestinal Disorders Nausea 79 7 Vomiting 47 3.8 Constipation 35 0.9 Diarrhea 29 1.7 Abdominal pain* 19 1.3 Stomatitis† 14 0.9 Dyspepsia 12 0 General Disorders and Administration Site Conditions Fatigue‡ 59 6 Skin and Subcutaneous Tissue Disorders Alopecia 46 0.4§ Rash¶ 10 0 Metabolism and Nutrition Disorders Decreased appetite 32 1.3 Hypokalemia 12 3.4 Blood and Lymphatic System Disorders Anemia# 31 7 NeutropeniaÞ 29 16 Leukopeniaß 22 6 Thrombocytopeniaà 20 3.4 Respiratory, Thoracic and Mediastinal Disorders Cough 20 0 Dyspnea 13 1.3 Epistaxis 13 0 Interstitial lung diseaseè 9 2.6ð Nervous System Disorders Headacheø 19 0 Dizziness 10 0 Infections and Infestation Upper respiratory tract infectioný 15 0 Investigations Aspartate aminotransferase increased 14 0.9 Alanine aminotransferase increased 10 0.9 Eye Disorders Dry eye 11 0.4£ Other clinically relevant adverse reactions reported in less than 10% of patients were:
- Injury, Poisoning and Procedural Complications: infusion-related reactions (2.6%)
- Blood and Lymphatic System Disorders: febrile neutropenia (1.7%)
Table 4: Selected Laboratory Abnormalities in Patients with Unresectable or Metastatic HER2-positive Breast Cancer Treated with ENHERTU Laboratory Parameter ENHERTU 5.4 mg/kg
N = 234All Grades
%Grades 3 or 4
%Percentages were calculated using patients with worsening laboratory values from baseline and the number of patients with both baseline and post-treatment measurements as the denominator.
Frequencies were based on NCI-CTCAE v.4.03 grade-derived laboratory abnormalities.Hematology White blood cell count decreased 70 7 Hemoglobin decreased 70 7 Neutrophil count decreased 62 16 Platelet count decreased 37 3.4 Chemistry Aspartate aminotransferase increased 41 0.9 Alanine aminotransferase increased 38 0.4 Hypokalemia 26 3.0 6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparisons of the incidence of antibodies to ENHERTU in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
Treatment-induced anti-fam-trastuzumab deruxtecan-nxki antibodies (ADA) developed in 0.6% (4/640) patients who received ENHERTU across all doses. Due to the limited number of patients who tested positive for ADA, no conclusions can be drawn concerning a potential effect of immunogenicity on efficacy or safety. In addition, neutralizing activity of anti-ENHERTU antibodies has not been assessed.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, ENHERTU can cause fetal harm when administered to a pregnant woman. There are no available data on the use of ENHERTU in pregnant women. In postmarketing reports, use of a HER2-directed antibody during pregnancy resulted in cases of oligohydramnios manifesting as fatal pulmonary hypoplasia, skeletal abnormalities, and neonatal death [see Data]. Based on its mechanism of action, the topoisomerase inhibitor component of ENHERTU, DXd, can also cause embryo-fetal harm when administered to a pregnant woman because it is genotoxic and targets actively dividing cells [see Clinical Pharmacology (12.1), Nonclinical Toxicology (13.1)]. Advise patients of the potential risks to a fetus.
There are clinical considerations if ENHERTU is used in pregnant women, or if a patient becomes pregnant within 7 months following the last dose of ENHERTU [see Clinical Considerations].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Human Data
There are no available data on the use of ENHERTU in pregnant women. In postmarketing reports in pregnant women receiving a HER2-directed antibody, cases of oligohydramnios manifesting as fatal pulmonary hypoplasia, skeletal abnormalities, and neonatal death have been reported. These case reports described oligohydramnios in pregnant women who received a HER2-directed antibody either alone or in combination with chemotherapy. In some case reports, amniotic fluid index increased after use of a HER2-directed antibody was stopped.
8.2 Lactation
Risk Summary
There is no data regarding the presence of fam-trastuzumab deruxtecan-nxki in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment with ENHERTU and for 7 months after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status of females of reproductive potential prior to initiation of ENHERTU.
Contraception
Females
ENHERTU can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with ENHERTU and for at least 7 months following the last dose.
Males
Because of the potential for genotoxicity, advise male patients with female partners of reproductive potential to use effective contraception during treatment with ENHERTU and for at least 4 months following the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Based on findings in animal toxicity studies, ENHERTU may impair male reproductive function and fertility [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of ENHERTU have not been established in pediatric patients.
8.5 Geriatric Use
Of the 234 patients with HER2-positive breast cancer treated with ENHERTU 5.4 mg/kg, 26% were 65 years or older and 5% were 75 years or older. No overall differences in efficacy were observed between patients ≥65 years of age compared to younger patients. There was a higher incidence of Grade 3-4 adverse reactions observed in patients aged 65 years or older (53%) as compared to younger patients (42%).
8.6 Renal Impairment
No dose adjustment of ENHERTU is required in patients with mild (creatinine clearance (CLcr) ≥60 and <90 mL/min) or moderate (CLcr ≥30 and <60 mL/min) renal impairment [see Clinical Pharmacology (12.3)]. No data are available in patients with severe renal impairment.
8.7 Hepatic Impairment
No dose adjustment of ENHERTU is required in patients with mild (total bilirubin ≤ULN and any AST >ULN or total bilirubin >1 to 1.5 times ULN and any AST) or moderate (total bilirubin >1.5 to 3 times ULN and any AST) hepatic impairment. In patients with moderate hepatic impairment, due to potentially increased exposure, closely monitor for increased toxicities related to the topoisomerase inhibitor, DXd [see Dosage and Administration (2.2)]. No data are available in patients with severe (total bilirubin >3 to 10 times ULN and any AST) hepatic impairment [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
Fam-trastuzumab deruxtecan-nxki is a HER2-directed antibody and topoisomerase inhibitor conjugate. Fam-trastuzumab deruxtecan-nxki is an antibody-drug conjugate (ADC) composed of three components: 1) a humanized anti-HER2 IgG1 monoclonal antibody (mAb), covalently linked to 2) a topoisomerase inhibitor, via 3) a tetrapeptide-based cleavable linker. Deruxtecan is composed of a protease-cleavable maleimide tetrapeptide linker and the topoisomerase inhibitor, DXd, which is an exatecan derivative.
The antibody is produced in Chinese hamster ovary cells by recombinant DNA technology, and the topoisomerase inhibitor and linker are produced by chemical synthesis. Approximately 8 molecules of deruxtecan are attached to each antibody molecule. Fam-trastuzumab deruxtecan-nxki has the following structure:
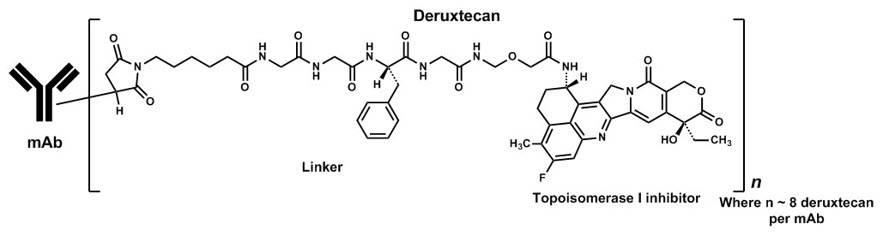
ENHERTU (fam-trastuzumab deruxtecan-nxki) is a sterile, white to yellowish white, preservative-free lyophilized powder in single-dose vials. Each vial delivers 100 mg of fam-trastuzumab deruxtecan-nxki, L-histidine (4.45 mg), L-histidine hydrochloride monohydrate (20.2 mg), polysorbate 80 (1.5 mg), and sucrose (450 mg). Following reconstitution with 5 mL of Sterile Water for Injection, USP, the resulting concentration of fam-trastuzumab deruxtecan-nxki is 20 mg/mL with a pH of 5.5. The resulting solution is administered by intravenous infusion following dilution.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Fam-trastuzumab deruxtecan-nxki is a HER2-directed antibody-drug conjugate. The antibody is a humanized anti-HER2 IgG1. The small molecule DXd, is a topoisomerase I inhibitor attached to the antibody by a cleavable linker. Following binding to HER2 on tumor cells, fam-trastuzumab deruxtecan-nxki undergoes internalization and intracellular linker cleavage by lysosomal enzymes. Upon release, the membrane-permeable DXd causes DNA damage and apoptotic cell death.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The administration of multiple doses of ENHERTU (6.4 mg/kg every 3 weeks, which is 1.2 times the recommended dosage) did not show large mean effect (i.e. >20 ms) on the QTc interval in an open label, single-arm study in 51 patients with HER2-expressing metastatic breast cancer.
12.3 Pharmacokinetics
The pharmacokinetics of fam-trastuzumab deruxtecan-nxki was evaluated in patients with cancer. Following a single dose, exposures (Cmax and AUC) of fam-trastuzumab deruxtecan-nxki and released topoisomerase inhibitor (DXd) increased proportionally over a dose range of 3.2 mg/kg to 8 mg/kg (approximately 0.6 to 1.5 times the recommended dose). At the recommended dosage of ENHERTU, the geometric mean (coefficient of variation [CV]%) Cmax of fam-trastuzumab deruxtecan-nxki and DXd were 122 µg/mL (20%) and 4.4 ng/mL (40%), respectively, and the AUC of fam-trastuzumab deruxtecan-nxki and DXd were 735 µg∙day/mL (31%) and 28 ng∙day/mL (38%), respectively, based on population pharmacokinetic analysis. Accumulation of fam-trastuzumab deruxtecan-nxki was approximately 35% at steady state (Cycle 3).
Distribution
Based on population pharmacokinetic analysis, the estimated volume of distribution of the central compartment (Vc) of fam-trastuzumab deruxtecan-nxki was 2.77 L.
For humans, DXd plasma protein binding is approximately 97% and the blood to plasma ratio is approximately 0.6, in vitro.
Elimination
The median elimination half-life (t1/2) of fam-trastuzumab deruxtecan-nxki was approximately 5.7 days. Based on population pharmacokinetic analysis, the estimated systemic clearance of fam-trastuzumab deruxtecan-nxki was 0.42 L/day.
The median apparent elimination half-life (t1/2) of DXd was approximately 5.8 days. Based on population pharmacokinetic analysis, the estimated apparent systemic clearance of DXd was 19.2 L/h.
Specific Populations
No clinically significant differences in the pharmacokinetics of fam-trastuzumab deruxtecan-nxki or DXd were observed for age (23-96 years), race (Asian [n=291] and non-Asian [n=221]), sex, body weight (34.6-125.4 kg), mild (total bilirubin ≤ULN and any AST >ULN or total bilirubin >1 to 1.5 times ULN and any AST, n=215) hepatic impairment, mild (creatinine clearance [CLcr] ≥60 and <90 mL/min, n=206) or moderate (CLcr ≥30 and <60 mL/min; n=58) renal impairment based on population pharmacokinetic analysis.
The pharmacokinetics of fam-trastuzumab deruxtecan-nxki or DXd in patients with moderate to severe hepatic impairment (total bilirubin >1.5 ULN with any AST) or severe renal impairment (CLcr <30 mL/min) is unknown.
Drug Interaction Studies
Clinical Studies
In Vitro Studies
Effects of DXd on CYP Enzymes: DXd does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A nor induce CYP1A2, CYP2B6, or CYP3A.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with fam-trastuzumab deruxtecan-nxki.
The topoisomerase inhibitor component of fam-trastuzumab deruxtecan-nxki, DXd, was clastogenic in both an in vivo rat bone marrow micronucleus assay and an in vitro Chinese hamster lung chromosome aberration assay and was not mutagenic in an in vitro bacterial reverse mutation assay.
Fertility studies have not been conducted with fam-trastuzumab deruxtecan-nxki. In a six-week repeat-dose toxicity study in rats, intravenous administration of fam-trastuzumab deruxtecan-nxki resulted in spermatid retention at 20 mg/kg and 60 mg/kg (approximately 4 and 9 times the human recommended dose of 5.4 mg/kg based on AUC, respectively). Decreased testes and epididymides weights, tubular atrophy/degeneration in testes, and reduced sperm count in epididymides were observed at a dose of 197 mg/kg (19 times the human recommended dose of 5.4 mg/kg based on AUC). In a three-month repeat-dose toxicity study in monkeys, intravenous administration of fam-trastuzumab deruxtecan-nxki resulted in decreased numbers of round spermatids in the testes at seminiferous tubule stages V to VI at ≥30 mg/kg (≥7 times the human recommended dose of 5.4 mg/kg based on AUC). Evidence of reversibility was observed in monkeys by the end of a three-month recovery period.
-
14 CLINICAL STUDIES
14.1 Metastatic Breast Cancer
The efficacy of ENHERTU was evaluated in study DESTINY-Breast01 (NCT03248492), a multicenter, single-arm, trial that enrolled 184 female patients with HER2-positive, unresectable and/or metastatic breast cancer who had received two or more prior anti-HER2 therapies. Patients were excluded for a history of treated ILD or current ILD at screening. Patients were also excluded for history of clinically significant cardiac disease, active brain metastases, and ECOG performance status >1. HER2 expression was based on archival tissue tested at a central laboratory prior to enrollment with HER2 positivity defined as HER2 IHC 3+ or ISH positive.
Patients received ENHERTU 5.4 mg/kg by intravenous infusion every 3 weeks until unacceptable toxicity or disease progression. Tumor imaging was obtained every 6 weeks and CT/MRI of the brain was mandatory for patients with brain metastases at baseline. The major efficacy outcomes were confirmed objective response rate (ORR) assessed by independent central review using RECIST v1.1 and duration of response (DOR).
The median age was 55 years (range: 28-96); 76% of patients were < 65 years. All 184 patients were female, and the majority were White (55%) or Asian (38%). Patients had an ECOG performance status of 0 (55%) or 1 (44%) at baseline. Ninety-two percent had visceral disease, 29% had bone metastases, and 13% had brain metastases. Fifty-three percent were hormone receptor positive. Sum of diameters of target lesions were < 5 cm in 42%, and ≥ 5 cm in 50% (not evaluable by central review in 8% of patients).
The median number of prior cancer regimens in the locally advanced/metastatic setting was 5 (range: 2-17).
All patients received prior trastuzumab, ado-trastuzumab emtansine, and 66% had prior pertuzumab.
Efficacy results are summarized in Table 5.
Table 5: Efficacy Results by Independent Central Review in DESTINY-Breast01 Efficacy Parameter DESTINY-Breast01
N=184ORR 95% CI calculated using Clopper-Pearson method - * DOR is based on median duration of follow-up of 11.1 months.
- † Median DOR based on Kaplan-Meier estimate; 95% CI calculated using Brookmeyer-Crowley method
Confirmed Objective Response Rate (95% CI) 60.3% (52.9, 67.4) Complete Response 4.3% Partial Response 56.0% Duration of Response*
Median, months (95% CI)†14.8
(13.8, 16.9) - 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied/Storage
ENHERTU (fam-trastuzumab deruxtecan-nxki) for injection is a white to yellowish white lyophilized powder supplied as:
Carton Contents NDC One 100 mg single-dose vial NDC: 65597-406-01 Store vials in a refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light until time of reconstitution. Do not freeze. Do not shake the reconstituted or diluted solution [see Dosage and Administration (2.3)].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Interstitial Lung Disease
- Inform patients of the risks of severe or fatal ILD. Advise patients to contact their healthcare provider immediately for any of the following: cough, shortness of breath, fever, or other new or worsening respiratory symptoms [see Warnings and Precautions (5.1)].
Neutropenia
- Advise patients of the possibility of developing neutropenia and to immediately contact their healthcare provider should they develop a fever, particularly in association with any signs of infection [see Warnings and Precautions (5.2)].
Left Ventricular Dysfunction
- Advise patients to contact their healthcare provider immediately for any of the following: new onset or worsening shortness of breath, cough, fatigue, swelling of ankles/legs, palpitations, sudden weight gain, dizziness, loss of consciousness [see Warnings and Precautions (5.3)].
Embryo-Fetal Toxicity
- Inform female patients of the potential risk to a fetus. Advise female patients to contact their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.4), Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective contraception during treatment with ENHERTU and for at least 7 months after the last dose [see Use in Specific Populations (8.3)].
- Advise male patients with female partners of reproductive potential to use effective contraception during treatment with ENHERTU and for at least 4 months after the last dose [see Use in Specific Populations (8.3)].
Lactation
- Advise women not to breastfeed during treatment and for 7 months after the last dose of ENHERTU [see Use in Specific Populations (8.2)].
Infertility
- Advise males of reproductive potential that ENHERTU may impair fertility [see Use in Specific Populations (8.3)].
-
SPL UNCLASSIFIED SECTION
Manufactured by:
Daiichi Sankyo, Inc., Basking Ridge, NJ 07920U.S. License No. 2128
Marketed by:
Daiichi Sankyo, Inc., Basking Ridge, NJ 07920 and AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850ENHERTU® is a registered trademark of Daiichi Sankyo Company, Ltd.
© 2019 Daiichi Sankyo Co., Ltd.USPI-ENH-C1-1219-r001
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Issued: 12/2019 Medication Guide
ENHERTU® (en-HER-too)
(fam-trastuzumab deruxtecan-nxki) for injectionWhat is the most important information I should know about ENHERTU?
ENHERTU can cause serious side effects, including:
Lung problems that may be severe, life-threatening or that may lead to death. If you develop lung problems your healthcare provider may treat you with corticosteroid medicines. Tell your healthcare provider right away if you get any of the following signs and symptoms:- cough
- trouble breathing or shortness of breath
- fever
- other new or worsening breathing symptoms (e.g., chest tightness, wheezing)
Low white blood cell count (neutropenia). Low white blood cell counts are common with ENHERTU and can sometimes be severe. Your healthcare provider will check your white blood cell counts before starting ENHERTU and before starting each dose. Tell your healthcare provider right away if you develop any signs or symptoms of an infection or have fever or chills during treatment with ENHERTU. Heart problems that may affect your heart's ability to pump blood. Your healthcare provider will check your heart function before starting treatment with ENHERTU. Tell your healthcare provider right away if you get any of the following signs and symptoms: - new or worsening shortness of breath
- coughing
- feeling tired
- swelling of your ankles or legs
- irregular heartbeat
- sudden weight gain
- dizziness or feeling light-headed
- loss of consciousness
Your healthcare provider will check you for these side effects during your treatment with ENHERTU. Your healthcare provider may reduce your dose, delay treatment or completely stop treatment with ENHERTU if you have severe side effects. Harm to your unborn baby. Tell your healthcare provider right away if you become pregnant or think you might be pregnant during treatment with ENHERTU. - If you are able to become pregnant, your healthcare provider should do a pregnancy test before you start treatment with ENHERTU.
- Females who are able to become pregnant should use effective birth control (contraception) during treatment with ENHERTU and for at least 7 months after the last dose.
- Males who have female partners that are able to become pregnant should use effective birth control (contraception) during treatment with ENHERTU and for at least 4 months after the last dose.
See "What are the possible side effects of ENHERTU?" for more information about side effects. What is ENHERTU?
ENHERTU is a prescription medicine used in adults to treat human epidermal growth factor receptor 2 (HER2)-positive breast cancer that cannot be removed by surgery or that has spread to other parts of your body (metastatic), and who have received two or more prior anti-HER2 breast cancer treatments.
It is not known if ENHERTU is safe and effective in children.Before you receive ENHERTU, tell your healthcare provider about all of your medical conditions, including if you: - have lung or breathing problems.
- have signs or symptoms of an infection.
- have or have had any heart problems.
- are breastfeeding or plan to breastfeed. It is not known if ENHERTU passes into your breast milk. Do not breastfeed during treatment with ENHERTU and for 7 months after the last dose.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. How will I receive ENHERTU? - You will receive ENHERTU into your vein through an intravenous (IV) line by your healthcare provider.
- ENHERTU is given 1 time every three weeks (21-day treatment cycle).
- Your healthcare provider will decide how many treatments you need.
- Your healthcare provider may slow down or temporarily stop your infusion of ENHERTU if you have an infusion-related reaction, or permanently stop ENHERTU if you have severe infusion reactions.
- If you miss a planned dose of ENHERTU, call your healthcare provider right away to schedule an appointment. Do not wait until the next planned treatment cycle.
What are the possible side effects of ENHERTU?
ENHERTU can cause serious side effects. See "What is the most important information I should know about ENHERTU?"
The most common side effects of ENHERTU include:- nausea
- feeling tired
- vomiting
- hair loss
- constipation
- decreased appetite
- low red blood cell counts
- low white blood cell counts
- diarrhea
- cough
- low platelet counts
ENHERTU may cause fertility problems in males, which may affect the ability to father children. Talk to your healthcare provider if you have concerns about fertility.
These are not all of the possible side effects of ENHERTU. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of ENHERTU.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about ENHERTU that is written for healthcare professionals.What are the ingredients in ENHERTU?
Active Ingredient: fam-trastuzumab deruxtecan-nxki.
Inactive Ingredient: L-histidine, L-histidine hydrochloride monohydrate, polysorbate 80, and sucrose.Manufactured by: Daiichi Sankyo, Inc., Basking Ridge, NJ 07920
U.S. License No. 2128
Marketed by: Daiichi Sankyo, Inc., Basking Ridge, NJ 07920 and AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
ENHERTU® is a registered trademark of Daiichi Sankyo Company, Ltd.
© 2019 Daiichi Sankyo Co., Ltd.
USMG-ENH-C1-1219-r001
For more information, call 1-877-437-7763 or go to http://www.ENHERTU.com. -
PRINCIPAL DISPLAY PANEL - 100 mg Vial Carton
NDC: 65597-406-01
Rx onlyENHERTU®
(fam-trastuzumab deruxtecan-nxki)For Injection
100 mg per vial
For Intravenous Infusion Only
Dispense the enclosed Medication Guide to each patient.
Reconstitute and Dilute prior to administration
Single-Dose Vial
Discard Unused PortionCAUTION: Cytotoxic Agent
KEEP REFRIGERATED
1 vial
Daiichi-Sankyo
AstraZeneca
-
INGREDIENTS AND APPEARANCE
ENHERTU
fam-trastuzumab deruxtecan-nxki injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 65597-406 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Trastuzumab Deruxtecan (UNII: 5384HK7574) (Trastuzumab Deruxtecan - UNII:5384HK7574) Trastuzumab Deruxtecan 100 mg in 5 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) HISTIDINE MONOHYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) SUCROSE (UNII: C151H8M554) POLYSORBATE 80 (UNII: 6OZP39ZG8H) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 65597-406-01 1 in 1 CARTON 12/20/2019 1 5 mL in 1 VIAL, SINGLE-DOSE; Type 6: Drug/Biologic Combination Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761139 12/20/2019 Labeler - Daiichi Sankyo Inc. (068605067)
Trademark Results [Enhertu]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ENHERTU 87675518 5640578 Live/Registered |
DAIICHI SANKYO COMPANY, LIMITED 2017-11-07 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.