NEXVIAZYME- avalglucosidase alfa-ngpt injection, powder, lyophilized, for solution
Nexviazyme by
Drug Labeling and Warnings
Nexviazyme by is a Prescription medication manufactured, distributed, or labeled by Genzyme Corporation, Genzyme Flanders, Genzyme Ireland Limited, Eurofins Biopharma Product Testing Ireland Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NEXVIAZYME® safely and effectively. See full prescribing information for NEXVIAZYME.
NEXVIAZYME (avalglucosidase alfa-ngpt) for injection, for intravenous use
Initial U.S. Approval: 2021WARNING: SEVERE HYPERSENSITIVITY REACTIONS, INFUSION-ASSOCIATED REACTIONS, and RISK OF ACUTE CARDIORESPIRATORY FAILURE IN SUSCEPTIBLE PATIENTS
See full prescribing information for complete boxed warning.
Hypersensitivity Reactions Including Anaphylaxis
- Appropriate medical monitoring and support measures, including cardiopulmonary resuscitation equipment, should be readily available. If a severe hypersensitivity reaction occurs, discontinue NEXVIAZYME immediately and initiate appropriate medical treatment. (5.1)
Infusion-Associated Reactions (IARs)
- If severe IARs occur, consider immediate discontinuation and initiation of appropriate medical treatment. (5.2)
Risk of Acute Cardiorespiratory Failure in Susceptible Patients
- Patients susceptible to fluid volume overload, or those with acute underlying respiratory illness or compromised cardiac or respiratory function, may be at risk of serious exacerbation of their cardiac or respiratory status during NEXVIAZYME infusion. (5.3)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
NEXVIAZYME is a hydrolytic lysosomal glycogen-specific enzyme indicated for the treatment of patients 1 year of age and older with late-onset Pompe disease (lysosomal acid alpha-glucosidase [GAA] deficiency). (1)
DOSAGE AND ADMINISTRATION
- Consider administering antihistamines, antipyretics, and/or corticosteroids prior to NEXVIAZYME administration to reduce the risk of IARs. (2.1)
- NEXVIAZYME is administered as intravenous infusion. For patients weighing (2.2):
- ≥30 kg, the recommended dosage is 20 mg/kg (of actual body weight) every two weeks.
- <30 kg, the recommended dosage is 40 mg/kg (of actual body weight) every two weeks.
- See the full prescribing information for dosage modifications due to hypersensitivity reactions or IARs. (2.3)
- Must be reconstituted and diluted prior to use. (2.4)
- For instructions on storage and administration, see full prescribing information. (2.5, 2.6)
DOSAGE FORMS AND STRENGTHS
For injection: 100 mg of avalglucosidase alfa-ngpt as a lyophilized powder in a single-dose vial for reconstitution. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
See boxed warning. (5.1, 5.2, 5.3)
ADVERSE REACTIONS
The most common adverse reactions (>5%) were headache, fatigue, diarrhea, nausea, arthralgia, dizziness, myalgia, pruritus, vomiting, dyspnea, erythema, paresthesia and urticaria. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genzyme at 1-800-633-1610, option 1 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 9/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SEVERE HYPERSENSITIVITY REACTIONS, INFUSION-ASSOCIATED REACTIONS, and RISK OF ACUTE CARDIORESPIRATORY FAILURE IN SUSCEPTIBLE PATIENTS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommendations Prior to NEXVIAZYME Treatment
2.2 Recommended Dosage and Administration
2.3 Administration Modifications Due to Hypersensitivity Reactions and/or Infusion-Associated Reactions
2.4 Reconstitution and Dilution Instructions
2.5 Storage Instructions for the Reconstituted and Diluted Product
2.6 Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions Including Anaphylaxis
5.2 Infusion-Associated Reactions
5.3 Risk of Acute Cardiorespiratory Failure in Susceptible Patients
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Clinical Trial in Patients with Late-Onset Pompe Disease
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SEVERE HYPERSENSITIVITY REACTIONS, INFUSION-ASSOCIATED REACTIONS, and RISK OF ACUTE CARDIORESPIRATORY FAILURE IN SUSCEPTIBLE PATIENTS
Hypersensitivity Reactions Including Anaphylaxis
Patients treated with NEXVIAZYME have experienced life-threatening hypersensitivity reactions, including anaphylaxis. Appropriate medical monitoring and support measures, including cardiopulmonary resuscitation equipment, should be readily available during NEXVIAZYME administration. If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue NEXVIAZYME immediately and initiate appropriate medical treatment. In patients with severe hypersensitivity reactions, a desensitization procedure to NEXVIAZYME may be considered [see Warnings and Precautions (5.1)].
Infusion-Associated Reactions (IARs)
Patients treated with NEXVIAZYME have experienced severe IARs. If severe IARs occur, consider immediate discontinuation of NEXVIAZYME, initiation of appropriate medical treatment, and the benefits and risks of readministering NEXVIAZYME following severe IARs. Patients with an acute underlying illness at the time of NEXVIAZYME infusion may be at greater risk for IARs. Patients with advanced Pompe disease may have compromised cardiac and respiratory function, which may predispose them to a higher risk of severe complications from IARs [see Warnings and Precautions (5.2)].
Risk of Acute Cardiorespiratory Failure in Susceptible Patients
Patients susceptible to fluid volume overload, or those with acute underlying respiratory illness or compromised cardiac or respiratory function for whom fluid restriction is indicated may be at risk of serious exacerbation of their cardiac or respiratory status during NEXVIAZYME infusion. More frequent monitoring of vitals should be performed during NEXVIAZYME infusion in such patients [see Warnings and Precautions (5.3)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommendations Prior to NEXVIAZYME Treatment
- Prior to NEXVIAZYME administration, consider pretreating with antihistamines, antipyretics, and/or corticosteroids [see Warnings and Precautions (5.1, 5.2)].
- NEXVIAZYME must be reconstituted and diluted prior to use [see Dosage and Administration (2.4)].
- Appropriate medical monitoring and support measures, including cardiopulmonary resuscitation equipment, should be readily available during NEXVIAZYME administration.
2.2 Recommended Dosage and Administration
- NEXVIAZYME is administered as intravenous infusion. For patients weighing:
- 30 kg or more- the recommended dosage is 20 mg/kg (of actual body weight) every two weeks [see Dosage and Administration (2.6)]
- Less than 30 kg- the recommended dosage is 40 mg/kg (of actual body weight) every two weeks [see Dosage and Administration (2.6)]
- The initial recommended infusion rate is 1 mg/kg/hour. Gradually increase the infusion rate every 30 minutes if there are no signs of infusion-associated reactions (IARs) [see Dosage and Administration (2.6)]. If one or more doses are missed, restart NEXVIAZYME treatment as soon as possible, maintaining the 2 week interval between infusions thereafter.
2.3 Administration Modifications Due to Hypersensitivity Reactions and/or Infusion-Associated Reactions
- In the event of a severe hypersensitivity reaction (e.g., anaphylaxis) or severe infusion-associated reaction (IAR), immediately discontinue NEXVIAZYME administration and initiate appropriate medical treatment. For additional recommendations in the event of a severe hypersensitivity reaction or IAR, [see Warnings and Precautions (5.1, 5.2)].
- In the event of a mild to moderate hypersensitivity reaction or a mild to moderate IAR, consider temporarily holding the infusion for 30 minutes or slowing the infusion rate by 50% [see Dosage and Administration (2.6)] and initiating appropriate medical treatment [see Warnings and Precautions (5.1, 5.2)].
- If symptoms persist for longer than 30 minutes despite holding or slowing the infusion, stop the infusion and monitor the patient. Consider re-initiating the infusion on the same day when symptoms subside at 50% of the rate at which the reaction occurred with appropriate pretreatment.
- If symptoms subside after holding the infusion, resume infusion at 50% of the rate at which the reaction occurred, and subsequently increase the infusion rate every 15 to 30 minutes by 50% as tolerated. Alternatively, if symptoms subside after slowing the infusion, complete the infusion at the reduced rate as tolerated.
- Starting with the next infusion, increase the infusion rate until the infusion rate at which the reaction occurred is reached. Consider continuing to increase the infusion rate in a stepwise manner until reaching the recommended infusion rate. Closely monitor the patient.
2.4 Reconstitution and Dilution Instructions
Reconstitute and dilute NEXVIAZYME in the following manner. Use aseptic technique during preparation.
Reconstitute the Lyophilized Powder
- Determine the number of NEXVIAZYME vials to be reconstituted based on actual body weight in kg and the recommended dose [see Dosage and Administration (2.2)]. Round the number of vials up to the next whole number.
- Remove the required number of NEXVIAZYME vials from the refrigerator and allow the vials to sit for 30 minutes at room temperature 20°C to 25°C (68°F to 77°F) before use.
- Reconstitute each vial by injecting 10 mL of Sterile Water for Injection, down the inside wall of each vial. Avoid adding the Sterile Water for Injection to the vial forcefully or directly onto the lyophilized powder to minimize foaming.
- Gently tilt and roll each vial to enhance the dissolution process. Do not invert, swirl, or shake the vial. Allow the solution to become dissolved. Each vial will yield a concentration of 100 mg/10 mL (10 mg/mL) of avalglucosidase alfa-ngpt.
- Visually inspect the reconstituted solution in the vials for particulate matter and discoloration. The reconstituted solution should be clear, colorless to pale-yellow. Discard if particles are present or the solution is discolored.
Dilute the Reconstituted Solution
- Select an appropriate size 5% Dextrose Injection infusion bag and prepare by removing a volume equal to the required NEXVIAZYME volume and any overfill to achieve a fixed total volume per Table 1 based on actual body weight.
- Slowly withdraw the required volume of reconstituted solution from the NEXVIAZYME vial(s). Discard any unused reconstituted solution remaining in the vial.
- Gently inject the NEXVIAZYME reconstituted solution into the port of the 5% Dextrose Injection bag. Avoid foaming or agitation of the infusion bag and avoid introducing air into the infusion bag.
- Gently invert the infusion bag to mix the solution. Do not shake. After dilution, the solution will have a final concentration of 0.5 to 4 mg/mL of avalglucosidase alfa-ngpt.
Administer the diluted solution without delay. The recommended infusion duration is between 4 to 7 hours [see Dosage and Administration (2.6)]. Discard any unused diluted solution after 9 hours.
Table 1: Projected Intravenous Infusion Volume for NEXVIAZYME Administration According to Actual Body Weight Patient Actual Body Weight Range (kg) Total Infusion Volume (mL) for 20 mg/kg Total Infusion Volume (mL) for 40 mg/kg 5 to 9.9 kg N/A 100 mL 10 to 19.9 kg N/A 200 mL 20 to 29.9 kg N/A 300 mL 30 to 34.9 kg 200 mL N/A 35 to 49.9 kg 250 mL N/A 50 to 59.9 kg 300 mL N/A 60 to 99.9 kg 500 mL N/A 100 to 119.9 kg 600 mL N/A 120 to 140 kg 700 mL N/A 2.5 Storage Instructions for the Reconstituted and Diluted Product
Storage of the Reconstituted Solution
- Dilute the reconstituted solution without delay. If the reconstituted solution is not diluted immediately, refrigerate at 2°C to 8°C (36°F to 46°F) for up to 24 hours.
- Do not freeze.
Storage of the Diluted Solution
- If the diluted solution is not used immediately, refrigerate the diluted solution at 2°C to 8°C (36°F to 46°F) for up to 24 hours.
- The diluted solution must be infused within 9 hours after removal from the refrigerator, inclusive of infusion time, or discarded.
- Once the diluted solution is removed from the refrigerator, it must not be restored back into the refrigerator.
- Do not freeze.
2.6 Administration Instructions
- If the diluted solution was refrigerated, allow solution to equilibrate to room temperature for 30 minutes prior to infusion.
- It is recommended to use an in-line, low protein-binding, 0.2 micron filter during administration.
- Administer the infusion incrementally, as determined by the patient's response and comfort.
When the recommended dose is 20 mg/kg- Initial and Subsequent Infusions: The initial recommended infusion rate is 1 mg/kg/hour (see Table 2). If there are no signs of hypersensitivity or infusion-associated reactions (IARs), gradually increase the infusion rate every 30 minutes in each of the following three steps: 3 mg/kg/hour, 5 mg/kg/hour, and then 7 mg/kg/hour; then, maintain the infusion rate at 7 mg/kg/hour until the infusion is complete. The approximate total infusion duration is 4 to 5 hours.
Table 2: Recommended Infusion Rates at 20 mg/kg Dose Dose Step 1 Step 2 Step 3 Step 4 Step 5 Start infusion at step 1 and in absence of infusion-associated reaction increase infusion rate sequentially per the steps of infusion every 30 minutes until completion (total time approximately 4 to 5 hours). 20 mg/kg 1 mg/kg/hour 3 mg/kg/hour 5 mg/kg/hour 7 mg/kg/hour Continue 7 mg/kg/hour
When the recommended dose is 40 mg/kg- Initial Infusion: The initial recommended infusion rate is 1 mg/kg/hour (see Table 3). If there are no signs of hypersensitivity or IARs, gradually increase the infusion rate every 30 minutes in each of the following three steps: 3 mg/kg/hour, 5 mg/kg/hour, and then 7 mg/kg/hour; then, maintain the infusion rate at 7 mg/kg/hour until the infusion is complete (4-step process). The approximate total infusion duration is 7 hours.
- Subsequent Infusions: The initial recommended infusion rate is 1 mg/kg/hour (see Table 3) with gradual increase in infusion rate every 30 minutes if there are no signs of hypersensitivity or IARs. The process may use either the above 4-step process or the following 5-step process: 3 mg/kg/hour, 6 mg/kg/hour, 8 mg/kg/hour, and then 10 mg/kg/hour; then, maintain the infusion rate at 10 mg/kg/hour until the infusion is complete. The approximate total 5-step infusion duration is 5 hours.
Table 3: Recommended Infusion Rates at 40 mg/kg Dose Dose Step 1 Step 2 Step 3 Step 4 Step 5 Start infusion at step 1 and in absence of infusion-associated reaction increase infusion rate sequentially per the steps of infusion every 30 minutes until completion. Total time for initial infusion approximately 7 hours and can optionally increase rate of subsequent infusions to decrease total duration to 5 hours. 40 mg/kg Initial infusion rate 1 mg/kg/hour 3 mg/kg/hour 5 mg/kg/hour 7 mg/kg/hour Continue 7 mg/kg/hour Subsequent infusions (optional) 1 mg/kg/hour 3 mg/kg/hour 6 mg/kg/hour 8 mg/kg/hour Continue 10 mg/kg/hour - After the infusion is complete, flush the intravenous line with 5% Dextrose Injection.
- Do not infuse NEXVIAZYME in the same intravenous line with other products.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions Including Anaphylaxis
Life-threatening hypersensitivity reactions, including anaphylaxis, have been reported in NEXVIAZYME-treated patients. In NEXVIAZYME clinical studies, 67 (48%) NEXVIAZYME-treated patients experienced hypersensitivity reactions, including 6 (4%) patients who reported severe hypersensitivity reactions and 3 (2%) patients who experienced anaphylaxis; 2 (1%) patients who experienced anaphylaxis discontinued from the study. Some of the hypersensitivity reactions were IgE mediated. Symptoms of severe hypersensitivity reactions (e.g., anaphylaxis) included chest discomfort, erythema, generalized edema, hypotension, hypoxia, rash, respiratory distress, tongue edema, and urticaria.
Prior to NEXVIAZYME administration, consider pretreating with antihistamines, antipyretics, and/or corticosteroids. Appropriate medical monitoring and support measures, including cardiopulmonary resuscitation equipment, should be readily available during NEXVIAZYME administration.
- If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue NEXVIAZYME immediately and initiate appropriate medical treatment. Consider the risks and benefits of re-administering NEXVIAZYME following severe hypersensitivity reactions (including anaphylaxis). Patients may be rechallenged using slower infusion rates at a dosage lower than the recommended dosage. In patients with severe hypersensitivity reaction, desensitization measures to NEXVIAZYME may be considered. If the decision is made to readminister NEXVIAZYME, ensure the patient tolerates the infusion. Once the patient tolerates the infusion, the dosage (dose and/or the rate) may be increased to reach the recommended dosage.
- If a mild or moderate hypersensitivity reaction occurs, consider temporarily holding the infusion or slowing the infusion rate [see Dosage and Administration (2.3)].
5.2 Infusion-Associated Reactions
In clinical studies, IARs were reported to occur at any time during and/or within a few hours after the NEXVIAZYME infusion and were more likely to occur with higher infusion rates. IARs were reported in 48 (34%) NEXVIAZYME-treated patients in all clinical studies. In these studies, 5 (4%) NEXVIAZYME-treated patients reported 10 severe IARs including symptoms of chest discomfort, decreased or increased blood pressure, dysphagia, erythema, generalized edema, hypoxia, nausea, respiratory distress, tongue edema, and urticaria. The majority of IARs were assessed as mild to moderate. IARs that led to treatment discontinuation were chest discomfort, cough, dizziness, erythema, flushing, nausea, ocular hyperemia, respiratory distress, and urticaria. Increased incidence of IARs was observed in patients with higher ADA titers [see Adverse Reactions (6.1)].
Prior to NEXVIAZYME administration, consider pretreating with antihistamines, antipyretics, and/or corticosteroids to reduce the risk of IARs. However, IARs may still occur in patients after receiving pretreatment.
If a severe IAR occurs, discontinue NEXVIAZYME immediately and initiate appropriate medical treatment. Consider the benefits and risks of readministering NEXVIAZYME following a severe IAR. Patients may be rechallenged using slower infusion rates at a dosage lower than the recommended dosage. Once the patient tolerates the infusion, the dosage (dose and/or the rate) may be increased to reach the recommended dosage.
If a mild or moderate IAR occurs, consider temporarily holding the infusion or slowing the infusion rate [see Dosage and Administration (2.3)].
Patients with an acute underlying illness at the time of NEXVIAZYME infusion appear to be at greater risk for IARs. Patients with advanced Pompe disease may have compromised cardiac and respiratory function, which may predispose them to a higher risk of severe complications from IARs.
5.3 Risk of Acute Cardiorespiratory Failure in Susceptible Patients
Patients susceptible to fluid volume overload, or those with acute underlying respiratory illness or compromised cardiac or respiratory function for whom fluid restriction is indicated may be at risk of serious exacerbation of their cardiac or respiratory status during the NEXVIAZYME infusion. More frequent monitoring of vitals should be performed during NEXVIAZYME infusion in these patients. Some patients may require prolonged observation times.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in greater detail in other sections of the labeling:
- Hypersensitivity Reactions Including Anaphylaxis [see Warnings and Precautions (5.1)]
- Infusion-Associated Reactions (IARs) [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions from Clinical Trials in the Pompe Disease Population
The pooled safety analysis from 4 clinical trials in the Pompe disease population (including Study 1) with a mean exposure of 26 months and up to 85 months of treatment included a total of 141 NEXVIAZYME-treated patients (118 adult and 23 pediatric patients [see Clinical Studies (14.1)].
Adverse reactions were similar across both adult and pediatric populations.
Serious adverse reactions reported in 2 or more NEXVIAZYME-treated patients were respiratory distress, chills, and pyrexia.
A total of 4 NEXVIAZYME-treated patients in clinical trials permanently discontinued NEXVIAZYME due to adverse reactions, including 3 patients who discontinued the treatment because of a serious adverse reaction.
The most frequently reported adverse reactions (>5%) in the pooled safety population were abdominal pain, arthralgia, chills, diarrhea, dizziness, dyspnea, erythema, fatigue, flushing, headache, hypertension, hypotension, myalgia, nausea, pruritus, pyrexia, rash, vomiting, and urticaria.
Hypersensitivity reactions were reported in 67 (48%) NEXVIAZYME-treated patients, including 6 (4%) patients who reported severe hypersensitivity reactions. Symptoms of severe hypersensitivity reactions (e.g., anaphylaxis) included chest discomfort, erythema, generalized edema, hypotension, hypoxia, rash, respiratory distress, tongue edema, and urticaria.
IARs were reported in 48 (34%) NEXVIAZYME-treated patients. Six (4%) NEXVIAZYME-treated patients reported 13 severe IARs including symptoms of chest discomfort, decreased or increased blood pressure, dysphagia, erythema, generalized edema, hypoxia, nausea, respiratory distress, tongue edema, and urticaria. IARs reported in more than 1 patient included chest discomfort, cyanosis, decreased or increased blood pressure, diarrhea, dizziness, erythema, fatigue, feeling hot or cold, generalized edema, headache, hyperhidrosis, hypoxia, influenza-like illness, nausea, pain, pruritus, pyrexia, rash, respiratory distress, tachycardia, throat irritation, tongue edema, tremor, urticaria, and vomiting.
Adverse Reactions from Clinical Trials in Late-Onset Pompe Disease (LOPD)
In Study 1, 100 patients aged 16 to 78 years of age with LOPD (naïve to enzyme replacement therapy) were treated with either 20 mg/kg of NEXVIAZYME (n=51) or 20 mg/kg of alglucosidase alfa (n=49) given every other week as an intravenous infusion for 49 weeks followed by an open-label extension period [see Clinical Studies (14.1)].
During the double-blind active-controlled period of 49 weeks, serious adverse reactions were reported in 1 (2%) patient treated with NEXVIAZYME and in 3 (6%) patients treated with alglucosidase alfa. The most frequently reported adverse reactions in (>5%) NEXVIAZYME-treated patients were headache, fatigue, diarrhea, nausea, arthralgia, dizziness, myalgia, pruritus, vomiting, dyspnea, erythema, paresthesia, and urticaria.
IARs were reported in 13 (25%) of the NEXVIAZYME-treated patients. IARs reported in more than 1 patient on NEXVIAZYME were mild to moderate and included headache, diarrhea, pruritus, urticaria, and rash. None of them were severe IARs. IARs were reported in 16 (33%) patients treated with alglucosidase alfa. IARs reported in more than 1 patient on alglucosidase alfa were mild to severe and included dizziness, flushing, dyspnea, nausea, pruritis, rash, erythema, chills, and feeling hot. Severe IARs were reported in 2 patients treated with alglucosidase alfa.
Table 4 summarizes the adverse reactions that occurred in at least 3 NEXVIAZYME-treated patients (≥6%) in Study 1. Study 1 was not designed to demonstrate a statistically significant difference in the incidence of adverse reactions in the NEXVIAZYME and the alglucosidase alfa treatment groups.
Table 4: Adverse Reactions Reported in at Least 6% of NEXVIAZYME-Treated Patients with LOPD in Study 1 Adverse Reaction NEXVIAZYME
(N=51)
n (%)Alglucosidase Alfa
(N=49)
n (%)Headache 11 (22%) 16 (33%) Fatigue 9 (18%) 7 (14%) Diarrhea 6 (12%) 8 (16%) Nausea 6 (12%) 7 (14%) Arthralgia 5 (10%) 8 (16%) Dizziness 5 (10%) 4 (8%) Myalgia 5 (10%) 7 (14%) Pruritus 4 (8%) 4 (8%) Vomiting 4 (8%) 3 (6%) Dyspnea 3 (6%) 4 (8%) Erythema 3 (6%) 3 (6%) Paresthesia 3 (6%) 2 (4%) Urticaria 3 (6%) 1 (2%) Immunogenicity: Anti-Drug Antibody-Associated Adverse Reactions
In enzyme replacement therapy (ERT)-naïve NEXVIAZYME-treated patients (mean of 47 months, up to 100 months of treatment), the incidence of [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.6)] IARs was 69% (9/13) in patients with an anti-drug antibody (ADA) peak titer ≥12,800, compared with incidences of 27% (12/44) in those with ADA peak titer <12,800 and 33% (1/3) in those who were ADA-negative.
In ERT-experienced adult patients, the incidence of IARs and hypersensitivity reactions were higher in patients who developed ADA compared to patients who were ADA-negative [see Clinical Pharmacology (12.6)].
In adults, one ERT-naïve patient (ADA peak titer 3,200) and 2 ERT-experienced patients (peak ADA titers; 800 and 12,800, respectively) developed anaphylaxis. One ERT-experienced pediatric patient (peak ADA titer 6,400) developed anaphylaxis [see Warnings and Precautions (5.1)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from case reports of NEXVIAZYME use in pregnant women are insufficient to evaluate for a drug associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. However, available data from postmarketing reports and published case reports on alglucosidase alfa (another hydrolytic lysosomal glycogen-specific enzyme replacement therapy) use in pregnant women have not identified a drug-associated risk of adverse pregnancy outcomes. The continuation of treatment for Pompe disease during pregnancy should be individualized to the pregnant woman. Untreated Pompe disease may result in worsening disease symptoms in pregnant women (see Clinical Considerations).
Embryo-fetal toxicity studies performed in pregnant mice resulted in maternal toxicity related to an immunologic response (including an anaphylactoid response) and embryo-fetal loss at 17 times the human steady-state AUC at the recommended biweekly dose of 20 mg/kg for LOPD patients weighing ≥30 kg or 10 times the human steady-state AUC at the recommended biweekly dose of 40 mg/kg for LOPD patients weighing <30 kg. Avalglucosidase alfa-ngpt did not cross the placenta in mice, therefore, the adverse effects were likely related to the immunologic response in the mothers. Embryo-fetal toxicity studies performed in pregnant rabbits showed no adverse effects on the fetuses at exposure up to 91 times the human steady-state AUC at the recommended biweekly dosage of 20 mg/kg for LOPD patients weighing ≥30 kg or 50 times the human steady-state AUC at the recommended biweekly dose of 40 mg/kg for LOPD patients weighing <30 kg (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, miscarriage, or other adverse outcomes. In the U.S. general population, the background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Pregnant women exposed to NEXVIAZYME, or their healthcare providers, should report NEXVIAZYME exposure by calling 1-800-633-1610, option 1.
Data
Animal data
The majority of reproductive toxicity studies in mice included the pretreatment with diphenhydramine (DPH) to prevent or minimize hypersensitivity reactions. The effects of NEXVIAZYME were evaluated based on comparison with a control group treated with DPH alone. Rabbits tested in reproductive toxicity studies were not pretreated with DPH because hypersensitivity reactions were not observed.
Embryo-fetal toxicity studies performed in pregnant mice at doses of 0, 10, 20, or 50 mg/kg/day administered intravenously once daily on gestational days 6 through 15 resulted in an immunologic response, including an anaphylactoid response, in some dams at the highest dose of 50 mg/kg/day (17 times the human steady-state AUC at the recommended biweekly dose of 20 mg/kg for LOPD patients weighing ≥30 kg or 10 times the human steady-state AUC at the recommended biweekly dose of 40 mg/kg for LOPD patients weighing <30 kg). Increased post-implantation loss and mean number of late resorptions were observed in this group. Placental transfer studies determined that avalglucosidase alfa-ngpt was not transported from the maternal to the fetal circulation in mice, suggesting that the embryo-fetal effects were due to maternal toxicity relating to the immunologic response. The maternal no observed adverse effect level (NOAEL) was 50 mg/kg/day intravenously (17 times the human AUC) and the developmental NOAEL was 20 mg/kg/day intravenously (4.8 times the human AUC).
Embryo-fetal toxicity studies performed in rabbits at doses of 0, 30, 60, and 100 mg/kg/day administered intravenously once daily on gestational days 6 through 19 resulted in no adverse effects in the fetuses at the highest dose (100 mg/kg/day; 91 times the human steady-state AUC at the recommended biweekly dosage of 20 mg/kg for LOPD patients weighing ≥30 kg or 50 times the human steady-state AUC at the recommended biweekly dose of 40 mg/kg for LOPD patients weighing <30 kg). Furthermore, the administration of NEXVIAZYME intravenously every other day in mice from gestational day 6 through postpartum day 20 did not produce adverse effects in the offspring at the highest dose of 50 mg/kg (maternal exposure not evaluated).
8.2 Lactation
Risk Summary
There are no data on the presence of avalglucosidase alfa-ngpt in either human or animal milk, the effects on the breastfed infant, or the effects on milk production. Available published literature suggests the presence of alglucosidase alfa (another hydrolytic lysosomal glycogen-specific enzyme replacement therapy) in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for NEXVIAZYME and any potential adverse effects on the breastfed infant from NEXVIAZYME or from the underlying maternal condition.
Lactating women exposed to NEXVIAZYME, or their healthcare providers, should report NEXVIAZYME exposure by calling 1-800-633-1610, option 1.
8.4 Pediatric Use
The safety and effectiveness of NEXVIAZYME for the treatment of late-onset Pompe disease have been established in pediatric patients aged 1 year and older. Use of NEXVIAZYME for this indication is supported by evidence from two clinical studies which included adults with LOPD, and 1 pediatric patient with LOPD (16 years of age) and from safety experience in 19 pediatric patients with infantile-onset Pompe disease (IOPD) (1 to 12 years of age) treated with NEXVIAZYME [see Clinical Studies (14.1)]. NEXVIAZYME is not approved for the treatment of IOPD.
The safety profile of NEXVIAZYME in pediatric patients 1 to 12 years old with Pompe disease was similar to the safety profile of NEXVIAZYME in older pediatric and adult patients with LOPD. The safety and effectiveness of NEXVIAZYME have not been established in pediatric patients younger than 1 year of age.
8.5 Geriatric Use
Clinical studies with NEXVIAZYME included 13 patients 65 to 74 years of age and 4 patients 75 years of age and older. The recommended dosage in geriatric patients is the same as the recommended dosage in younger adult patients [see Adverse Reactions (6.1)].
-
11 DESCRIPTION
Avalglucosidase alfa-ngpt is a hydrolytic lysosomal glycogen-specific recombinant human α-glucosidase enzyme conjugated with multiple synthetic bis-mannose-6-phosphate (bis-M6P)-tetra-mannose glycans resulting in approximately 15 moles of M6P per mole of enzyme (15 M6P) and is produced in Chinese hamster ovary cells (CHO). Avalglucosidase alfa-ngpt has a molecular weight of approximately 124 kDa.
NEXVIAZYME (avalglucosidase alfa-ngpt) for injection is a sterile white to pale-yellow lyophilized powder for intravenous use after reconstitution and dilution. Each single-dose vial contains 100 mg of avalglucosidase alfa-ngpt, glycine (200 mg), L-Histidine (10.7 mg), L-Histidine HCl monohydrate (6.5 mg), mannitol (200 mg), and polysorbate 80 (1 mg). After reconstitution with 10 mL of Sterile Water for Injection, USP, the resultant concentration is 100 mg/10 mL (10 mg/mL) with a pH of approximately 6.2.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Pompe disease (also known as glycogen storage disease type II, acid maltase deficiency, and glycogenosis type II) is an inherited disorder of glycogen metabolism caused by a deficiency of the lysosomal enzyme acid α-glucosidase (GAA) that degrades glycogen to glucose in the lysosome. GAA deficiency results in intralysosomal accumulation of glycogen in various tissues.
Avalglucosidase alfa-ngpt provides an exogenous source of GAA. The M6P on avalglucosidase alfa-ngpt mediates binding to M6P receptors on the cell surface with high affinity. After binding, it is internalized and transported into lysosomes where it undergoes proteolytic cleavage that results in increased GAA enzymatic activity. Avalglucosidase alfa-ngpt then exerts enzymatic activity in cleaving glycogen.
12.2 Pharmacodynamics
In patients with Pompe disease, excess of glycogen is degraded to hexose tetrasaccharide (Hex4) which is then excreted in urine. The urinary Hex4 assay measures the major component, glucose tetrasaccharide (Glc4). Treatment with NEXVIAZYME resulted in reductions of urinary Glc4 concentrations (normalized by urine creatinine and reported as mmol Glc4/mol creatinine) in patients with Pompe disease.
In ERT-naïve LOPD patients in Study 1, the baseline mean (SD) urinary Glc4 concentration was 12.7 mmol/mol (10.10) and 8.7 mmol/mol (5.04) in NEXVIAZYME and alglucosidase alfa treatment groups, respectively [see Clinical Studies (14.1)]. At Week 145, the mean urinary Glc4 concentration was 4.32 mmol/mol (4.28) in patients who continued with NEXVIAZYME and 5.25 mmol/mol (7.48) in patients who switched from alglucosidase alfa to NEXVIAZYME.
For patients who started on NEXVIAZYME, the mean percentage (SD) change in urinary Glc4 concentration from baseline was -54% (24), at Week 49 and -53% (73) at Week 145. For patients who started on alglucosidase alfa and switched to NEXVIAZYME at Week 49, the mean percentage (SD) change in urinary Glc4 concentration from baseline was -11% (32) at Week 49, and -48% (42) at Week 145.
12.3 Pharmacokinetics
The avalglucosidase alfa-ngpt exposure increases in an approximately proportional manner with increasing doses over a range from 5 to 20 mg/kg (0.25 to 1 time the approved recommended dosage in LOPD patients weighing greater than or equal to 30 kg or 0.125 to 0.5 times the approved recommended dosage in LOPD patients weighing less than 30 kg). No accumulation was observed following every two weeks dosing. Following intravenous infusion of 20 mg/kg of NEXVIAZYME every two weeks in LOPD patients weighing greater than or equal to 30 kg, the mean ± SD plasma Cmax of avalglucosidase alfa-ngpt at Week 1 and Week 49 was 259 ± 72 µg/mL and 242 ± 81 µg/mL, respectively; the mean ± SD plasma AUC of avalglucosidase alfa-ngpt at Week 1 and Week 49 was 1,290 ± 420 µg∙h/mL and 1,250 ± 433 µg∙h/mL, respectively. Patients weighing less than 30 kg are expected to have similar AUC following intravenous infusion of 40 mg/kg of NEXVIAZYME every two weeks.
Elimination
The mean total body clearance of avalglucosidase alfa-ngpt was 0.9 L/hour and the mean plasma elimination half-life of avalglucosidase alfa-ngpt was 1.6 hours in LOPD patients.
Metabolism
The protein portion of avalglucosidase alfa-ngpt is expected to be metabolized into small peptides by catabolic pathways.
Specific Populations
Population pharmacokinetic analyses indicated that age, which ranged from 1 to 78 years in the clinical trials, and sex did not significantly influence the pharmacokinetics of avalglucosidase alfa-ngpt in patients with Pompe disease.
Pediatric patients
In 16 patients aged 1 to 12 years with Pompe disease, following a 4-hour intravenous infusion of NEXVIAZYME 20 mg/kg every two weeks and 7-hour intravenous infusion of NEXVIAZYME 40 mg/kg every two weeks, the mean Cmax ranged from 175 to 189 µg/mL and 250 to 403 µg/mL, respectively. The mean AUClast ranged from 805 to 923 µg∙hr/mL for 20 mg/kg every two weeks and 1,720 to 2,630 µg∙hr/mL for 40 mg/kg every two weeks.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADAs in the studies described below with the incidence of ADAs in other studies, including those of NEXVIAZYME or of other avalglucosidase alfa products.
Table 5 presents the incidence of anti-avalglucosidase alfa-ngpt antibodies (referred to as ADA) in NEXVIAZYME-treated patients with Pompe disease [see Clinical Studies (14.1)]. In ERT-naïve LOPD patients who received NEXVIAZYME 20 mg/kg every two weeks for up to 436.2 weeks (with mean of 204.7 weeks), 95% (59/62) of patients developed ADA. The median time to seroconversion was 8 weeks.
ADA cross-reactivity studies showed that antibodies to avalglucosidase alfa-ngpt were cross-reactive to alglucosidase alfa.
Anti-Drug Antibody Effects on Pharmacokinetics
In Study 1, the avalglucosidase alfa-ngpt exposure (e.g., AUC) in the two ADA-negative NEXVIAZYME-treated patients was within the range of patients who developed ADA. Among the patients who developed ADA, the median AUC was similar between Week 1 and Week 49 irrespective of titer values and neutralizing activities of the ADA. There was no identified clinically significant effect of ADA on pharmacokinetics. [see Clinical Pharmacology (12.3)].
Anti-Drug Antibody Effects on Pharmacodynamics
In Study 1, a trend toward decreased pharmacodynamic response as measured by percent change of urinary glucose tetrasaccharides from baseline was observed in NEXVIAZYME-treated patients with an ADA peak titer ≥12,800 compared to those with a lower ADA peak titer.
Anti-Drug Antibody Effects on Safety and Efficacy
Increased incidence of IARs was observed in NEXVIAZYME-treated patients with higher ADA peak titers (>12,800) compared to those with lower ADA peaks. The incidence of hypersensitivity reactions was higher in ERT-experienced adult patients who developed ADA compared to those who were ADA-negative [see Adverse Reactions (6.1)]. In ERT-naïve patients, hypersensitivity reactions occurred irrespective of ADA development and peak ADA titer. In Study 1, there was no identified clinically significant effect of ADA on efficacy.
Table 5: Incidence of Anti-Avalglucosidase Alfa-ngpt Antibodies in NEXVIAZYME-treated Patients with Pompe Disease up to 100 months ERT-Naïve Patients ERT-Experienced Patients Adult and Pediatric Patients*
20 mg/kg every two weeks
(N=62)†Adults
20 mg/kg every two weeks
(N=58)Pediatric Patients
20 mg/kg every two weeks
(N=6)Pediatric Patients
40 mg/kg every two weeks
(N=16)n (%) n (%) n (%) n (%) ‡ ERT-experienced: patients previously treated with alglucosidase alfa within a range of 0.9-9.9 years for adult patients and 0.6-11.8 years for pediatric patients before they received NEXVIAZYME. - * Includes two pediatric patients
- † ERT-naïve: patients only treated with NEXVIAZYME
ADA at baseline 2 (3%) 43 (74%) 1 (17%) 2 (13%) ADA after NEXVIAZYME treatment 59 (95%) 36 (62%) 1 (17%) 9 (56%) Neutralizing Antibody (NAb) After NEXVIAZYME Treatment Both NAb types 14 (23%) 5 (9%) 0 0 Inhibition of enzyme activity 19 (31%) 11 (19%) 0 0 Inhibition of enzyme cellular uptake 26 (42%) 20 (34%) 0 2 (13%) -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals to evaluate the carcinogenic potential or studies to evaluate mutagenic potential have not been performed with avalglucosidase alfa-ngpt.
Intravenous administration of avalglucosidase alfa-ngpt every other day at doses up to 50 mg/kg (exposure not evaluated) had no adverse effects on fertility in male or female mice.
-
14 CLINICAL STUDIES
14.1 Clinical Trial in Patients with Late-Onset Pompe Disease
Study 1 (NCT02782741) was a randomized, double-blinded, multinational, multicenter trial comparing the efficacy and safety of NEXVIAZYME to alglucosidase alfa in treatment-naïve patients with LOPD. One hundred patients (51 in NEXVIAZYME and 49 in alglucosidase alfa) were randomized in a 1:1 ratio based on baseline forced vital capacity ([FVC] % predicted; <55% or ≥55%), sex, age (<18 years or ≥18 years), and country (Japan or not-Japan) to receive 20 mg/kg of NEXVIAZYME or alglucosidase alfa administered intravenously once every two weeks for 49 weeks. After 49 weeks, all randomized patients in Study 1 had the option to enter an open-label extension treatment period to receive NEXVIAZYME and continue treatment up to at least Week 145.
Demographic and Disease Characteristics
Of the 100 randomized patients, 52 were males, the baseline median age was 49 years old (range from 16 to 78), median baseline weight was 76.4 kg (range from 38 to 139 kg), median length of time since diagnosis was 6.9 months (range from 0.3 to 328.4 months), mean age at diagnosis was 46.4 years old (range from 11 to 78), mean FVC (% predicted) at baseline was 62.1% (range from 32 to 85%), and mean 6MWT at baseline was 388.9 meters (range from 118 to 630 meters). The racial groups for the patients consisted of 94 White (94%), 3 Black or African American (3%), and 3 Asian (3%). Fifteen patients were Hispanic/Latino (15%), 76 non-Hispanic/Latino (76%) and 9 were not reported. Five patients (all in the alglucosidase alfa arm) discontinued the study prior to Week 49: four due to adverse events (acute myocardial infarction, arthritis, dyspnea, and urticaria), and one due to withdrawal of consent. All 95 patients who did not discontinue the study prior to Week 49 entered the open-label period (51 from the NEXVIAZYME arm and 44 from the alglucosidase alfa arm). Of the 95 patients, 77 patients had follow-up data on FVC (% predicted) at Week 145 (44 from the NEXVIAZYME arm and 33 from the alglucosidase alfa arm) and 80 patients had follow-up data on 6MWT at Week 145 (45 from the NEXVIAZYME arm and 35 from the alglucosidase alfa arm). Fourteen (14%) patients (7 in each group) discontinued during the Extension Treatment period (5 for adverse event, 1 for poor compliance to protocol and 8 for other reason).
Primary Efficacy Results from the 49-Week Active-Controlled Period and Open-Label Period up to Week 145 in Study 1
The primary endpoint of Study 1 was the change in FVC (% predicted) in the upright position from baseline to Week 49. At Week 49, the least squares (LS) mean change in FVC (% predicted) for patients treated with NEXVIAZYME and alglucosidase alfa was 2.9% and 0.5%, respectively. The estimated treatment difference was 2.4% (95% CI: -0.1, 5.0) favoring NEXVIAZYME (see Table 6). Figure 1 presents the LS mean change from baseline in FVC (% predicted) over time by treatment group up to Week 49.
Table 6: Summary Results of FVC (% predicted) in Upright Position in ERT-Naïve Patients with LOPD (Study 1)* NEXVIAZYME
(n=51)Alglucosidase Alfa
(n=49)- * All randomized patients
- † Estimated using a mixed model for repeated measures (MMRM) including baseline FVC (% predicted, as continuous), sex, baseline age (years), treatment group, visit, and treatment-by-visit interaction term as fixed effects.
- ‡ Noninferiority margin of 1.1% (p=0.0074). Statistical superiority of NEXVIAZYME over alglucosidase alfa was not achieved (p=0.06).
Pretreatment baseline Mean (SD) 62.5 (14.4) 61.6 (12.4) Week 49 Mean (SD) 65.5 (17.4) 61.2 (13.5) Estimated change from baseline to week 49 LS mean (SE) 2.9† (0.9) 0.5† (0.9) Estimated difference between groups in change from baseline to week 49 LS mean (95% CI) 2.4†‡ (-0.1, 5.0) Figure 1: Plot of LS Mean (SE) Change from Baseline of FVC (% predicted) in Upright Position over Time in ERT-Naïve Patients with LOPD (Study 1)* - * All randomized patients
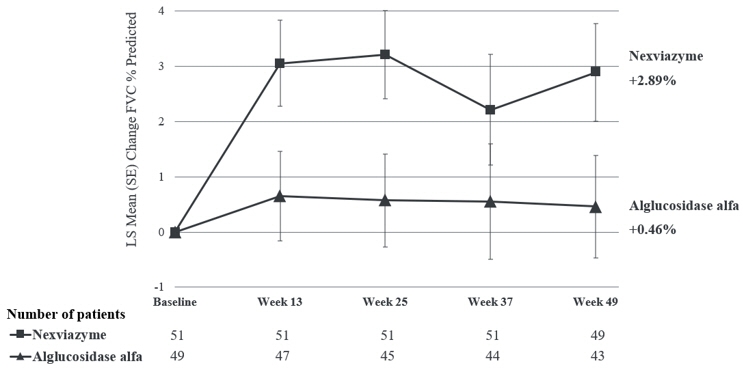
For patients who continued to receive NEXVIAZYME after Week 49 the mean change from baseline to Week 145 in FVC (% predicted) was 1.7 (SD=8.6, 95% CI: -0.9, 4.2), and the mean change from Week 49 to Week 145 was -0.8 (SD=6.2, 95% CI: -2.7, 1.0). For patients who switched from alglucosidase alfa to NEXVIAZYME after Week 49 the mean change from baseline to Week 145 was 0.5 (SD=8.3, 95% CI: -2.3, 3.4), and the mean change in FVC (% predicted) from Week 49 to Week 145 was 0.8 (SD=6.9) (95% CI: -1.6, 3.1).
Efficacy Results for Total Distance Walked in 6 Minutes from the 49-Week Active-Controlled Period and Open-Label Period up to Week 145 in Study 1
The key secondary endpoint of Study 1 was change in total distance walked in 6 minutes (6-Minute Walk Test, 6MWT) from baseline to Week 49. At Week 49, the LS mean change from baseline in 6MWT for patients treated with NEXVIAZYME and alglucosidase alfa was 32.2 meters and 2.2 meters, respectively. The estimated treatment difference was 30 meters (95% CI: 1.3, 58.7) favoring NEXVIAZYME (Table 7). Figure 2 presents the LS mean change from baseline in 6MWT distance over time by treatment group.
Table 7: Summary Results of 6-Minute Walk Test in ERT-Naïve Patients with LOPD (Study 1)* NEXVIAZYME
(n=51)Alglucosidase Alfa
(n=49)- * All randomized patients
- † The MMRM model for 6MWT distance adjusts for baseline FVC (% predicted), baseline 6MWT (distance walked in meters), baseline age (years), sex, treatment group, visit, and treatment-by-visit interaction as fixed effects.
- ‡ p-value at nominal level, without multiplicity adjustment (p=0.04).
Pretreatment baseline Mean (SD) 399.3 (110.9) 378.1 (116.2) Week 49 Mean (SD) 441.3 (109.8) 383.6 (141.1) Estimated change from baseline to week 49 LS mean (SE) 32.2† (9.9) 2.2† (10.4) Estimated difference between groups in change from baseline to week 49 LS mean (95% CI) 30.0†‡ (1.3, 58.7) Figure 2: Plot of LS Mean (SE) Change from Baseline of 6MWT (distance walked, in meters) over Time in ERT-Naïve Patients with LOPD (Study 1)* - * All randomized patients
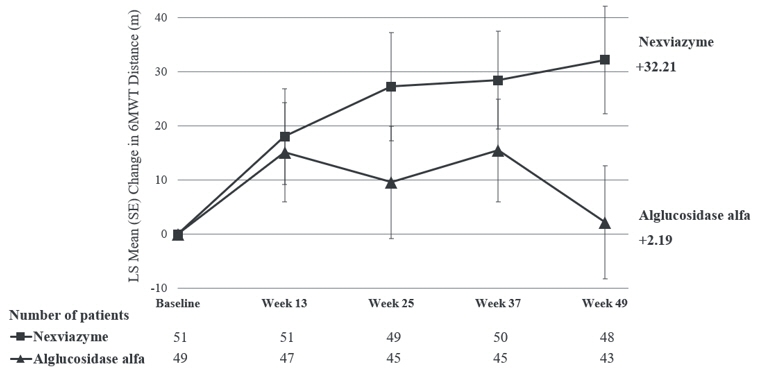
For patients who continued to receive NEXVIAZYME after Week 49 the mean change from baseline to Week 145 in 6MWT was 24.9 (SD=68.6, 95% CI: 4.8, 44.9), and the mean change from Week 49 to Week 145 was -11.0 (SD=33.5, 95% CI: -20.8, -1.2). For patients who switched from alglucosidase alfa to NEXVIAZYME after Week 49 the mean change from baseline to Week 145 in 6MWT was -4.1 (SD=90.4, 95% CI: -34.1, 25.8), and the mean change from Week 49 to Week 145 was -7.7 (SD=50.3, 95% CI: -24.4, 9.0).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
NEXVIAZYME (avalglucosidase alfa-ngpt) for injection is supplied as a sterile, white to pale-yellow lyophilized powder in a single-dose vial. Each vial contains 100 mg of avalglucosidase alfa-ngpt. NEXVIAZYME is available as:
One single-dose vial in a carton (NDC: 58468-0426-1)
-
17 PATIENT COUNSELING INFORMATION
Hypersensitivity Reactions Including Anaphylaxis and Infusion-Associated Reactions (IARs)
Advise the patient and caregiver that reactions related to the infusion may occur during and after NEXVIAZYME treatment, including life-threatening hypersensitivity reactions, including anaphylaxis, and IARs. Inform the patient and caregiver of the signs and symptoms of hypersensitivity reactions and IARs and to seek medical care should signs and symptoms occur [see Warnings and Precautions (5.1, 5.2)].
Risk of Acute Cardiorespiratory Failure
Advise the patient and caregiver that patients with underlying respiratory illness or compromised cardiac or respiratory function may be at risk of acute cardiorespiratory failure from volume overload during NEXVIAZYME infusion and should seek medical care should signs and symptoms occur [see Warnings and Precautions (5.3)].
NEXVIAZYME Exposure During Pregnancy or Lactation
Advise a pregnant or lactating woman exposed to NEXVIAZYME to report NEXVIAZYME exposure to her healthcare provider and by calling 1-800-633-1610, option 1 [see Use in Specific Populations (8.1., 8.2)].
Pompe Registry
Inform the patient and/or caregiver that there is a Pompe Registry to better understand the variability and progression of Pompe disease and to monitor and evaluate long-term effects of NEXVIAZYME. Encourage the patient and/or caregiver to enroll in the Pompe Registry and state that participation is voluntary and may involve long-term follow-up. For more information regarding the registry program, direct the patient and caregiver to visit www.registrynxt.com or call 1-800-745-4447, extension 15500.
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 100 mg Vial Carton
NDC: 58468-0426-1
Rx onlyNexviazyme®
(avalglucosidase alfa-ngpt)
for Injection100 mg per vial
For Intravenous Infusion
after Reconstitution
and DilutionOne single-dose vial
Discard unused portion
-
INGREDIENTS AND APPEARANCE
NEXVIAZYME
avalglucosidase alfa-ngpt injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 58468-0426 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength AVALGLUCOSIDASE ALFA (UNII: EO144CP0X9) (AVALGLUCOSIDASE ALFA - UNII:EO144CP0X9) AVALGLUCOSIDASE ALFA 100 mg in 10 mL Inactive Ingredients Ingredient Name Strength GLYCINE (UNII: TE7660XO1C) 200 mg in 10 mL HISTIDINE (UNII: 4QD397987E) 10.7 mg in 10 mL HISTIDINE MONOHYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) 6.5 mg in 10 mL MANNITOL (UNII: 3OWL53L36A) 200 mg in 10 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 1 mg in 10 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 58468-0426-1 1 in 1 CARTON 08/06/2021 1 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761194 08/06/2021 Labeler - Genzyme Corporation (025322157) Establishment Name Address ID/FEI Business Operations Genzyme Flanders 372153895 ANALYSIS(58468-0426) , API MANUFACTURE(58468-0426) Establishment Name Address ID/FEI Business Operations Genzyme Ireland Limited 985127419 ANALYSIS(58468-0426) , MANUFACTURE(58468-0426) , PACK(58468-0426) , LABEL(58468-0426) Establishment Name Address ID/FEI Business Operations Genzyme Corporation 050424395 PACK(58468-0426) , LABEL(58468-0426) Establishment Name Address ID/FEI Business Operations Eurofins Biopharma Product Testing Ireland Limited 238239933 ANALYSIS(58468-0426) Establishment Name Address ID/FEI Business Operations Genzyme Corporation 968302658 ANALYSIS(58468-0426) Establishment Name Address ID/FEI Business Operations Genzyme Corporation 117450412 ANALYSIS(58468-0426) , API MANUFACTURE(58468-0426) Establishment Name Address ID/FEI Business Operations Genzyme Corporation 034378252 ANALYSIS(58468-0426) , API MANUFACTURE(58468-0426)
Trademark Results [Nexviazyme]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 NEXVIAZYME 88318614 not registered Live/Pending |
Genzyme Corporation 2019-02-27 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.