DOXERCALCIFEROL injection, solution
Doxercalciferol by
Drug Labeling and Warnings
Doxercalciferol by is a Prescription medication manufactured, distributed, or labeled by Meitheal Pharmaceuticals Inc., Nanjing King-Friend Biochemical Pharmaceutical Co., Ltd.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DOXERCALCIFEROL INJECTION safely and effectively. See full prescribing information for DOXERCALCIFEROL INJECTION.
DOXERCALCIFEROL injection, for intravenous use
Initial U.S. Approval: 1999INDICATIONS AND USAGE
Doxercalciferol Injection is a synthetic vitamin D2 analog:
- Doxercalciferol Injection is indicated for the treatment of secondary hyperparathyroidism in adult patients with CKD on dialysis. (1)
DOSAGE AND ADMINISTRATION
- Before initiating treatment, ensure serum calcium is not above the upper limit of normal. (2.1)
- Dosage for doxercalciferol injection in patients with CKD on dialysis: Initiate dosing at 4 mcg by bolus intravenous administration three times weekly at the end of dialysis (no more frequently than every other day). Maximum dose is 18 mcg weekly. (2.4)
- Target the maintenance dose of doxercalciferol injection to intact parathyroid hormone (PTH) levels within the desired therapeutic range and serum calcium within normal limits. (2)
- See Full Prescribing Information for dose titration, laboratory monitoring, and important administration instructions. (2)
DOSAGE FORMS AND STRENGTHS
- Injection: (3)
- 4 mcg per 2 mL (2 mcg per mL) multi-dose vial
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Hypercalcemia: Can occur during treatment with doxercalciferol and can lead to cardiac arrhythmias and seizures. Severe hypercalcemia may require emergency attention. Risk may be increased when used concomitantly with high dose calcium preparations, thiazide diuretics, or vitamin D compounds. Monitor serum calcium prior to initiation and during treatment and adjust dose accordingly. (2, 5.1)
- Digitalis Toxicity: Hypercalcemia increases the risk of digitalis toxicity. In patients using digitalis compounds, monitor serum calcium and patients for signs and symptoms of digitalis toxicity. Increase frequency of monitoring when initiating or adjusting the dose of doxercalciferol. (5.2)
- Serious Hypersensitivity Reactions: Anaphylaxis, with symptoms of angioedema, hypotension, unresponsiveness, chest discomfort, shortness of breath, and cardiopulmonary arrest, has been reported in hemodialysis patients after administration of doxercalciferol. Monitor patients upon treatment initiation for hypersensitivity reactions. Should a reaction occur, discontinue and treat. (5.3)
- Adynamic Bone Disease: May develop and increase risk of fractures if intact PTH levels are suppressed to abnormally low levels. Monitor intact PTH levels to avoid oversuppression and adjust dose if needed. (5.4)
ADVERSE REACTIONS
The most common adverse reactions in patients with Stage 3 or 4 CKD (incidence >5%) were infection, urinary tract infection, chest pain, angina pectoris, constipation, dyspepsia, anemia, leucopenia, dehydration, edema, depression, hypertonia, insomnia, asthenia, paresthesia, cough increased, dyspnea, pruritus, sinusitis, and rhinitis. (6.1)
The most common adverse reactions in patients with CKD on dialysis (incidence >5%) were headache, malaise, edema, nausea/vomiting, dyspnea, dizziness, pruritus, and bradycardia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Meitheal Pharmaceuticals Inc. at 1-844-824-8426 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Cytochrome P450 inhibitors: Formation of the active doxercalciferol moiety may be hindered and may necessitate dosage adjustment. Monitor intact PTH and serum calcium concentrations closely. (7)
- Enzyme inducers: Formation of the active doxercalciferol moiety may be affected and may necessitate dosage adjustment. Monitor intact PTH and serum calcium concentrations closely. (7)
- Magnesium-containing products: Combined use may cause hypermagnesemia. Monitor serum magnesium concentrations more frequently and adjust dose as needed. (7)
- Cholestyramine: May impair absorption of doxercalciferol capsules. Administer doxercalciferol capsules at least 1 hour before or 4 to 6 hours after taking cholestyramine. (7)
- Mineral oil or other substances that may affect absorption of fat: May impair absorption of doxercalciferol capsules. Administer doxercalciferol capsules at least 1 hour before or 4 to 6 hours after taking substances that may affect absorption.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 4/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Prior to Initiation of Doxercalciferol Injection
2.4 Important Administration Instructions for Doxercalciferol Injection
2.5 Dosage Recommendations for Doxercalciferol Injection in Patients with CKD on Dialysis
2.6 Drug Interactions that May Require Dosage Adjustments of Doxercalciferol Injection
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypercalcemia
5.2 Digitalis Toxicity
5.3 Serious Hypersensitivity Reactions
5.4 Adynamic Bone Disease
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Clinical Studies of Doxercalciferol Capsules in Patients with Stage 3 or 4 CKD
14.2 Clinical Studies of Doxercalciferol Capsules in Patients with CKD on Dialysis
14.3 Clinical Studies of Doxercalciferol Injection in Patients with CKD on Dialysis
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Prior to Initiation of Doxercalciferol Injection
- Ensure serum calcium is not above the upper limit of normal before initiating treatment with doxercalciferol injection [see Warnings and Precautions (5.1)].
2.4 Important Administration Instructions for Doxercalciferol Injection
- Administer doxercalciferol injection intravenously as a bolus dose at the end of dialysis.
- Inspect doxercalciferol injection visually prior to administration; the solution should appear clear and colorless. Do not use if the solution is not clear or particles are present.
- After initial vial use, store opened multi-dose vial for up to 3 days at 2°C to 8°C (36°F to 46°F). Discard unused portion of multi-dose vial after 3 days [see How Supplied/Storage and Handling (16)].
2.5 Dosage Recommendations for Doxercalciferol Injection in Patients with CKD on Dialysis
- Initiate doxercalciferol injection at a dose of 4 mcg given by bolus intravenous administration three times weekly at the end of dialysis (no more frequently than every other day).
- Target the maintenance dose of doxercalciferol to intact parathyroid hormone (PTH) levels within the desired therapeutic range and serum calcium within normal limits.
- Monitor serum calcium, phosphorus, and intact PTH levels weekly after initiation of therapy or dose adjustment.
- Titrate the dose of doxercalciferol injection based on intact PTH. The dose may be increased at 8-week intervals by 1 mcg to 2 mcg if intact PTH is not lowered by 50% and fails to reach the target range. The maximum dose is 18 mcg weekly. Prior to raising the dose, ensure serum calcium is within normal limits.
- Suspend or decrease the dose if intact PTH is persistently and abnormally low to reduce the risk of adynamic bone disease [see Warnings and Precautions (5.4)] or if serum calcium is consistently above the normal range to reduce the risk of hypercalcemia [see Warnings and Precautions (5.1)]. If suspended, the drug should be restarted one week later at a dose that is at least 1 mcg lower.
2.6 Drug Interactions that May Require Dosage Adjustments of Doxercalciferol Injection
- Increased monitoring of serum calcium and dose adjustment of doxercalciferol may be necessary when given concomitantly with drugs that may increase the risk of hypercalcemia [see Drug Interactions (7)].
- Increased monitoring of both serum calcium and intact PTH as well as dose adjustment of doxercalciferol may be necessary when given concomitantly with cytochrome P450 inhibitors or enzyme inducers [see Drug Interactions (7)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Doxercalciferol is contraindicated in patients with:
- Hypercalcemia [see Warnings and Precautions (5.1)]
- Vitamin D toxicity [see Warnings and Precautions (5.1)]
- Known hypersensitivity to doxercalciferol or any of the inactive ingredients of doxercalciferol injection; serious hypersensitivity reactions including anaphylaxis and angioedema have been reported [see Warnings and Precautions (5.3), Adverse Reactions (6.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypercalcemia
Hypercalcemia may occur during doxercalciferol treatment. Acute hypercalcemia may increase the risk of cardiac arrhythmias and seizures and may potentiate the effect of digitalis on the heart [see Warnings and Precautions (5.2)]. Chronic hypercalcemia can lead to generalized vascular calcification and other soft-tissue calcification. Severe hypercalcemia may require emergency attention.
Hypercalcemia may be exacerbated by concomitant administration of high doses of calcium-containing preparations, thiazide diuretics, or other vitamin D compounds [see Drug Interactions (7)]. In addition, high intake of calcium and phosphate concomitantly with vitamin D compounds may lead to hypercalciuria and hyperphosphatemia. Patients with a history of hypercalcemia prior to initiating therapy may be at increased risk for development of hypercalcemia with doxercalciferol. In these circumstances, frequent serum calcium monitoring and doxercalciferol dose adjustments may be required.
When initiating doxercalciferol or adjusting doxercalciferol dose, measure serum calcium frequently (weekly in patients with CKD on dialysis or every 2 weeks for patients with stage 3 or 4 CKD). Once a maintenance dose has been established, measure serum calcium monthly for 3 months and then every 3 months. If hypercalcemia occurs, reduce the dose or discontinue doxercalciferol until serum calcium is normal [see Dosage and Administration (2)].
Inform patients about the symptoms of elevated calcium (feeling tired, difficulty thinking clearly, loss of appetite, nausea, vomiting, constipation, increased thirst, increased urination and weight loss) and instruct them to report new or worsening symptoms when they occur.
5.2 Digitalis Toxicity
Doxercalciferol can cause hypercalcemia [see Warnings and Precautions (5.1)] which increases the risk of digitalis toxicity. In patients using doxercalciferol concomitantly with digitalis compounds, monitor both serum calcium and patients for signs and symptoms of digitalis toxicity. Increase the frequency of monitoring when initiating or adjusting the dose of doxercalciferol [see Drug Interactions (7)].
5.3 Serious Hypersensitivity Reactions
Serious hypersensitivity reactions, including fatal outcome, have been reported post marketing in patients on hemodialysis following administration of doxercalciferol injection. Hypersensitivity reactions include anaphylaxis with symptoms of angioedema (involving face, lips, tongue and airways), hypotension, unresponsiveness, chest discomfort, shortness of breath, and cardiopulmonary arrest. These reactions may occur separately or together.
Monitor patients receiving doxercalciferol upon initiation of treatment for hypersensitivity reactions. Should a hypersensitivity reaction occur, discontinue doxercalciferol, monitor and treat if indicated [see Contraindications (4)].
5.4 Adynamic Bone Disease
Adynamic bone disease with subsequent increased risk of fractures may develop if intact PTH levels are suppressed by doxercalciferol to abnormally low levels. Monitor intact PTH levels to avoid oversuppression and adjust the doxercalciferol dose, if needed [see Dosage and Administration (2)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in another section of the label:
- Hypercalcemia [see Warnings and Precautions (5.1)]
- Serious Hypersensitivity Reactions [see Warnings and Precautions (5.3)]
- Adynamic Bone Disease [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse reactions in patients with stage 3 or 4 CKD
Doxercalciferol capsules have been evaluated in two placebo-controlled, double-blind 24 week studies in patients with Stage 3 or 4 CKD. Patients were treated with doxercalciferol capsules (n=27) or placebo (n=28) [see Clinical Studies (14.1)]. Adverse reactions occurring in the doxercalciferol capsules group at a frequency of 5% or greater and more frequently than in the placebo group are presented in Table 1.
Table 1: Adverse Reactions Occurring in ≥5% Doxercalciferol Capsule-Treated Patients with CKD on Predialysis and Greater than Placebo in Two Double-Blind Clinical Studies * Pooled data on adverse reactions from clinical study reports (Studies BCI-CH-115 and BCI-CH-119).
Adverse Reaction* Doxercalciferol (n=27)
%Placebo (n=28)
%Infection/bacterial infection/viral infection 30 25 Constipation 26 11 Rhinitis 22 11 Anemia 19 4 Cough 19 4 Dyspnea 19 11 Paresthesia 15 11 Asthenia 15 11 Insomnia 15 4 Hypertonia 11 4 Angina pectoris 8 0 Dehydration 7 4 Depression 7 0 Dyspepsia 7 4 Edema 7 4 Urinary tract infection 7 4 Leukopenia 7 0 Chest pain 7 4 Pruritus 7 4 Sinusitis 7 4 Adverse reactions in patients with CKD on dialysis
Doxercalciferol capsules have been evaluated in two placebo-controlled, double-blind studies in patients with CKD on hemodialysis. Patients were treated with doxercalciferol capsules (n=61) or placebo (n=61) [see Clinical Studies (14.2)]. After randomization to two groups, eligible patients underwent an 8-week washout period during which no vitamin D derivatives were administered to either group. Subsequently, all patients received doxercalciferol capsules in an open-label fashion for 16 weeks followed by a double-blind period of 8 weeks during which patients received either doxercalciferol capsules or placebo. Adverse reactions occurring in the doxercalciferol capsule groups at a frequency of 2% or greater, and more frequently than in the placebo group are presented in Table 2.
Table 2: Adverse Reactions Occurring in ≥2% Doxercalciferol Capsule-Treated Patients with CKD on Dialysis and Greater than Placebo in Two Double-Blind Clinical Studies * A patient who reported the same medical term more than once was counted only once for that medical term.
Adverse Reaction* Doxercalciferol (n=61)
%Placebo (n=61)
%Edema 34 21 Malaise 28 20 Headache 28 18 Nausea/Vomiting 21 20 Dizziness 12 10 Dyspnea 12 7 Pruritus 8 7 Bradycardia 7 5 Anorexia 5 3 Dyspepsia 5 2 Arthralgia 5 0 Weight increase 5 0 Abscess 3 0 Sleep disorder 3 0 Doxercalciferol Injection
Adverse reactions in patients with CKD on hemodialysis
Doxercalciferol injection has been studied in 70 patients with CKD on hemodialysis in two 12-week, open-label, single-arm, multicenter studies [see Clinical Studies (14.3)]. The incidence of hypercalcemia and hyperphosphatemia increased during therapy with doxercalciferol injection. Patients with higher pretreatment serum levels of calcium (>10.5 mg/dL) or phosphorus (>6.9 mg/dL) were more likely to experience hypercalcemia or hyperphosphatemia.
There was no placebo group included in the studies of doxercalciferol injection. Adverse reactions in patients with CKD on hemodialysis receiving doxercalciferol injection are expected to be similar to those reported in placebo-controlled studies of doxercalciferol capsules presented in Table 2.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of doxercalciferol. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate their frequency or to establish a causal relationship to drug exposure.
Hypersensitivity reactions, including fatal outcome, have been reported in patients on hemodialysis following administration of doxercalciferol injection. Hypersensitivity reactions include anaphylaxis with symptoms of angioedema (involving face, lips, tongue and airways), hypotension, unresponsiveness, chest discomfort, shortness of breath, cardiopulmonary arrest, pruritus, and skin burning sensation.
-
7 DRUG INTERACTIONS
Tables 3 and 4 include clinically significant drug interactions with doxercalciferol.
Table 3: Clinically Significant Drug Interactions with Doxercalciferol Injection and Doxercalciferol Capsules Drugs that May Increase the Risk of Hypercalcemia Clinical Impact Concomitant administration of high doses of calcium-containing preparations or other vitamin D compounds may increase the risk of hypercalcemia. Thiazide diuretics are known to induce hypercalcemia by reducing excretion of calcium in the urine. Examples Calcium-containing products, other vitamin D compounds or thiazide diuretics Intervention Monitor serum calcium concentrations more frequently and adjust doxercalciferol dose as needed [see Warnings and Precautions (5.1)]. Digitalis Compounds Clinical Impact Doxercalciferol can cause hypercalcemia which can potentiate the risk of digitalis toxicity.
InterventionMonitor patients for signs and symptoms of digitalis toxicity and increase frequency of serum calcium monitoring when initiating or adjusting the dose of doxercalciferol in patients receiving digitalis compounds [see Warnings and Precautions (5.2)]. Cytochrome P450 Inhibitors Clinical Impact Doxercalciferol is activated by CYP 27 in the liver. Cytochrome P450 inhibitors may inhibit the 25-hydroxylation of doxercalciferol and thus reduce the formation of active doxercalciferol moiety [see Clinical Pharmacology (12.3)]. Examples Ketoconazole and erythromycin Intervention If a patient initiates or discontinues therapy with a cytochrome P450 inhibitor, dose adjustment of doxercalciferol may be necessary. Monitor intact PTH and serum calcium concentrations closely. Enzyme Inducers Clinical Impact Doxercalciferol is activated by CYP 27 in the liver. Enzyme inducers may affect the 25-hydroxylation of doxercalciferol [see Clinical Pharmacology (12.3)]. Examples Glutethimide and phenobarbital Intervention If a patient initiates or discontinues therapy with an enzyme inducer, dose adjustment of doxercalciferol may be necessary. Monitor intact PTH and serum calcium concentrations closely. Magnesium-containing Products Clinical Impact Concomitant administration of doxercalciferol and high doses of magnesium-containing products may increase the risk of hypermagnesemia. Examples Magnesium-containing products such as antacids Intervention Avoid use of magnesium-containing products and doxercalciferol in patients on chronic renal dialysis. Table 4: Clinically Significant Drug Interactions with Doxercalciferol Capsules Cholestyramine Clinical Impact Cholestyramine has been reported to reduce intestinal absorption of fat-soluble vitamins. Therefore, it may impair intestinal absorption of doxercalciferol capsules. Intervention Administer doxercalciferol capsules at least 1 hour before or 4 to 6 hours after taking cholestyramine. Mineral Oil or other Substances that May Affect Absorption of Fat Clinical Impact The use of mineral oil or other substances that may affect absorption of fat may influence the absorption and availability of doxercalciferol. Intervention Administer doxercalciferol capsules at least 1 hour before or 4 to 6 hours after taking mineral oil or other substances that may affect absorption of fat. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The limited available data with doxercalciferol in pregnant women are insufficient to identify a drug-associated risk for major birth defects, miscarriage or adverse maternal or fetal outcomes. There are risks to the mother and fetus associated with chronic kidney disease in pregnancy [see Clinical Considerations]. In reproduction studies in rats and rabbits administered doxercalciferol during organogenesis at up to 20 mcg/kg/day and 0.1 mcg/kg/day, respectively (approximately 25 times (rats) and less than (rabbits) the maximum recommended human oral dose of 60 mcg/week based on mcg/m2 body surface area), no adverse developmental effects were observed [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively.
8.2 Lactation
Risk Summary
There is no information available on the presence of doxercalciferol in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production. Infants exposed to doxercalciferol through breast milk should be monitored for signs and symptoms of hypercalcemia [see Clinical Considerations].
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for doxercalciferol and any potential adverse effects on the breastfed child from doxercalciferol or from the underlying maternal condition.
8.4 Pediatric Use
Safety and efficacy of doxercalciferol in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies of doxercalciferol did not include sufficient numbers of patients 65 years or over to determine whether they respond differently from younger subjects. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic or cardiac function, and of concomitant disease or other drug therapy.
-
10 OVERDOSAGE
Overdosage of doxercalciferol may lead to hypercalcemia, hypercalciuria, and hyperphosphatemia [see Warnings and Precautions (5.1)]. The treatment of acute overdosage should consist of supportive measures and discontinuation of doxercalciferol administration. Serum calcium levels should be measured until normal.
Based on similarities between doxercalciferol and its active metabolite, 1α,25-(OH)2D2, it is expected that doxercalciferol is not removed from the blood by dialysis.
-
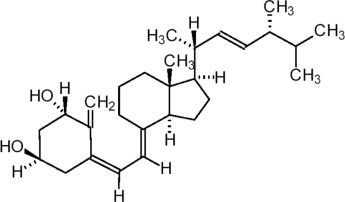
11 DESCRIPTION
Doxercalciferol Injection contains doxercalciferol, which is a synthetic vitamin D2 analog. Doxercalciferol undergoes metabolic activation in vivo to form 1α,25-dihydroxyvitamin D2 (1α,25-(OH)2D2), a naturally occurring, biologically active form of vitamin D2.
Doxercalciferol is a colorless crystalline compound with a calculated molecular weight of 412.65 and a molecular formula of C28H44O2. It is soluble in oils and organic solvents, but is relatively insoluble in water. Chemically, doxercalciferol is (1α,3β,5Z,7E,22E)-9,10-secoergosta-5,7,10(19),22-tetraene-1,3-diol. The structural formula is:
Doxercalciferol Injection 2 mL multi-dose vials contain 4 mcg per 2 mL (2 mcg per mL) of doxercalciferol. Each milliliter (mL) of solution contains 2 mcg doxercalciferol and the following inactive ingredients: butylated hydroxytoluene, 20 mcg; edetate disodium, 1.1 mg; dehydrated alcohol, 59.25 mg; polysorbate 20, 10 mg; sodium chloride, 1.5 mg; disodium hydrogen phosphate, anhydrous, 7.63 mg; and monosodium phosphate monohydrate, 1.8 mg.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Doxercalciferol is a synthetic vitamin D2 analog that requires metabolic activation to form the active 1α,25-(OH)2D2 metabolite, which binds to the vitamin D receptor (VDR) to result in the selective activation of vitamin D responsive pathways. Vitamin D and doxercalciferol have been shown to reduce PTH levels by inhibiting PTH synthesis and secretion.
12.3 Pharmacokinetics
Absorption
In healthy volunteers, peak blood levels of 1α,25-(OH)2D2, the major metabolite of doxercalciferol, are attained at 8 hours after a single intravenous dose of doxercalciferol and at 11 to 12 hours following capsule doses.
Elimination
The mean elimination half-life of 1α,25-(OH)2D2 after an oral dose is approximately 32 to 37 hours with a range of up to 96 hours.
Specific Populations
Patients with renal impairment
The mean elimination half-life of 1α,25-(OH)2D2 in patients with end-stage renal disease (ESRD) and in healthy volunteers appears to be similar following an oral dose. Hemodialysis causes a temporary increase in 1α,25-(OH)2D2 mean concentrations, presumably due to volume contraction. 1α,25-(OH)2D2 is not removed from blood during hemodialysis.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 104-week carcinogenicity study in rats, there was an increased incidence of benign and malignant adrenal pheochromocytomas in both males and females at oral doses of 0.04, 0.13, and 0.39 mcg/kg/day (less than the maximum recommended human oral dose of 60 mcg/week based on mcg/m2 body surface area). This increased incidence of pheochromocytomas in rats may be due to altered calcium homeostasis by doxercalciferol. No evidence of genetic toxicity was observed in an in vitro bacterial mutagenicity assay (Ames test) or a mouse lymphoma gene mutation assay. Doxercalciferol caused structural chromatid and chromosome aberrations in an in vitro human lymphocyte clastogenicity assay with metabolic activation. However, doxercalciferol was negative in an in vivo mouse micronucleus clastogenicity assay.
Doxercalciferol had no effect on male or female fertility in rats at oral doses up to 2.5 mcg/kg/day (approximately 3 times the maximum recommended human oral dose of 60 mcg/week based on mcg/m2 body surface area).
-
14 CLINICAL STUDIES
14.1 Clinical Studies of Doxercalciferol Capsules in Patients with Stage 3 or 4 CKD
The safety and effectiveness of doxercalciferol capsules were evaluated in two clinical studies in 55 patients with Stage 3 or 4 CKD. Eighty-two percent of the patients were male, the average age was 65 years, 51% were Caucasian, 40% African-American, and the average serum intact PTH level at baseline was 195 pg/mL. While levels of 25-(OH) vitamin D were not evaluated at baseline, retrospective assessments of stored serum revealed that the mean ± SD serum 25-(OH) vitamin D was 19 ± 8 ng/mL (range: <5 to 54 ng/mL) in the study population.
After randomization to two groups, eligible patients underwent an 8-week washout period during which no vitamin D derivatives were administered to either group. Subsequently, one group received doxercalciferol capsules and the other placebo during the double-blind period of 24 weeks. The initial dose of doxercalciferol capsules was 1 mcg per day. The dosage of doxercalciferol capsules was adjusted as necessary by the investigator to reduce intact PTH levels to a target of ≥30% below postwashout baseline. The maximum dosage was limited to 3.5 mcg per day. If at any time during the trial intact PTH fell below 15 pg/mL, doxercalciferol capsules were immediately suspended and restarted at a lower dosage the following week.
Decreases in the mean plasma intact PTH from baseline values were calculated using as baseline the average of the last 2 values obtained during the 8-week washout phase. In analyses of pooled data from the two studies, intact PTH levels decreased from baseline by an average of 101 pg/mL in the doxercalciferol capsules group and by 4 pg/mL in the placebo group (p<0.001). Twenty (74%) of 27 subjects in the doxercalciferol capsules group achieved mean plasma intact PTH suppression of ≥30% from baseline for the last four weeks of treatment, whereas two (7%) of the 28 subjects treated with placebo achieved this level of intact PTH suppression.
14.2 Clinical Studies of Doxercalciferol Capsules in Patients with CKD on Dialysis
The safety and effectiveness of doxercalciferol capsules were evaluated in two double-blind, placebo-controlled, multicenter clinical studies (Study A and Study B) in a total of 138 patients with CKD on hemodialysis. Patients in Study A were an average age of 52 years (range: 22 to 75), were 55% male, and were 58% African-American, 31% Caucasian, and 11% Hispanic, and had been on hemodialysis for an average of 53 months. Patients in Study B were an average of 52 years (range: 27 to 75), were 45% male, and 99% African-American, and 1% Caucasian, and had been on hemodialysis for an average of 56 months. After randomization to two groups, eligible patients underwent an 8-week washout period during which no vitamin D derivatives were administered to either group. Subsequently, all patients received doxercalciferol capsules in an open-label fashion for 16 weeks followed by a double-blind period of 8 weeks during which patients received either doxercalciferol capsules or placebo. The initial dose of doxercalciferol capsules during the open-label phase was 10 mcg after each dialysis session (3 times weekly) for a total of 30 mcg per week. The dosage of doxercalciferol was adjusted as necessary by the investigator to achieve intact PTH levels within 150 pg/mL to 300 pg/mL. The maximum dosage was limited to 20 mcg after each dialysis session (60 mcg/week). If at any time during the trial intact PTH fell below 150 pg/mL, doxercalciferol was immediately suspended and restarted at a lower dosage the following week. Mean weekly doses during the 16-week open-label period ranged from 15 mcg to 29 mcg in Study A and from 19 mcg to 28 mcg in Study B.
One hundred and six (77%) of the 138 patients who were treated with doxercalciferol capsules during the 16-week open-label phase achieved intact PTH levels ≤300 pg/mL. Ninety-four (68%) of these patients exhibited plasma intact PTH levels ≤300 pg/mL on at least 3 occasions. Eighty-seven (63%) patients had plasma intact PTH levels <150 pg/mL on at least one occasion during the open-label phase of study participation.
Decreases in plasma intact PTH from baseline values were calculated using as baseline the average of the last 3 values obtained during the 8-week washout phase and are displayed in Table 5.
Table 5: Intact PTH Summary Data for Patients with CKD on Dialysis Receiving Doxercalciferol Capsules in Studies A and B * All subjects; last value carried to discontinuation.
NA = not applicable
Intact PTH (pg/mL) means ± SD (n)*
p-value vs Baseline p-value vs PlaceboDoxercalciferol Capsules Placebo Study A Baseline 797.2 ± 443.8 (30) 847.1 ± 765.5 (32) NA – 0.97 – Week 16
(open-label)384.3 ± 397.8 (24)
<0.001526.5 ± 872.2 (29)
<0.0010.72 – Week 24
(double-blind)404.4 ± 262.9 (21)
<0.001672.6 ± 356.9 (24)
0.700.008 – Study B Baseline 973.9 ± 567.0 (41) 990.4 ± 488.3 (35) NA – 0.81 – Week 16
(open-label)476.1 ± 444.5 (37)
<0.001485.9 ± 443.4 (32)
<0.0010.91 – Week 24
(double-blind)459.8 ± 443.0 (35)
<0.001871.9 ± 623.6 (30)
<0.065<0.001 – Doxercalciferol capsules treatment resulted in a statistically significant reduction from baseline in mean intact PTH levels during the 16-week open-label treatment period in more than 94% of the 138 treated patients. During the double-blind period (weeks 17 to 24), the reduction in mean intact PTH levels was maintained in the doxercalciferol capsules treatment group compared to a return to near baseline in the placebo group.
14.3 Clinical Studies of Doxercalciferol Injection in Patients with CKD on Dialysis
The safety and effectiveness of doxercalciferol injection were evaluated in two open-label, single-arm, multicenter clinical studies (Study C and Study D) in a total of 70 patients with CKD on hemodialysis. Patients in Study C were an average age of 54 years (range: 23 to 73), were 50% male, and were 61% African-American, 25% Caucasian, and 14% Hispanic, and had been on hemodialysis for an average of 65 months. Patients in Study D were an average age of 51 years (range: 28 to 76), were 48% male, and 100% African-American and had been on hemodialysis for an average of 61 months. This group of 70 of the 138 patients who had been treated with doxercalciferol capsules in prior clinical studies (Study A and Study B) received doxercalciferol injection in an open-label fashion for 12 weeks following an 8-week washout (control) period. Dosing of doxercalciferol injection was initiated at the rate of 4 mcg administered at the end of each dialysis session (3 times weekly) for a total of 12 mcg per week. The dosage of doxercalciferol was adjusted to achieve intact PTH levels (measured weekly) within a targeted range of 150 pg/mL to 300 pg/mL. The dosage was increased by 2 mcg per dialysis session after 8 weeks of treatment if the intact PTH levels remained above 300 pg/mL and were greater than 50% of baseline levels. The maximum dosage was limited to 18 mcg per week. If at any time during the study intact PTH fell below 150 pg/mL, doxercalciferol injection was immediately suspended and restarted at a lower dosage the following week. Mean weekly doses ranged from ranged from 9 mcg to 13 mcg in Study C and ranged from 9 mcg to 12 mcg in Study D.
Fifty-two (74%) of the 70 patients who were treated with doxercalciferol injection achieved intact PTH levels ≤300 pg/mL. Forty-one (59%) of these patients exhibited plasma intact PTH levels ≤300 pg/mL on at least 3 occasions. Thirty-six (51%) patients had plasma intact PTH levels <150 pg/mL on at least one occasion during study participation. Decreases in plasma intact PTH from baseline values were calculated using as baseline the average of the last 3 values obtained during the 8-week washout period and are displayed in Table 6.
Table 6: Intact PTH Summary Data for Patients with CKD on Dialysis Receiving Doxercalciferol Injection in Studies C and D * Values were carried forward for the two patients on study for 10 weeks
† Treatment intact PTH minus baseline intact PTH
‡ Wilcoxon one-sample test
Intact PTH Level Study C (n=28) Study D (n=42) Combined Protocols (n=70) Baseline (Mean of Weeks -2, -1, and 0) Mean (SE) 698 (60) 762 (65) 736 (46) Median 562 648 634 On-treatment (Week 12*) Mean (SE) 406 (63) 426 (60) 418 (43) Median 311 292 292 Change from Baseline† Mean (SE) -292 (55) -336 (41) -318 (33) Median -274 -315 -304 P-value‡ 0.004 0.001 <0.001 Doxercalciferol treatment resulted in at least 30% reduction from baseline in mean intact PTH levels during the 12-week open-label treatment period in more than 92% of the 70 treated patients.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Doxercalciferol Injection is a clear, colorless solution supplied in 2 mL amber glass multi-dose vials. Each vial includes an aluminum seal and an orange plastic flip-off cap, and is supplied as follows:
NDC Doxercalciferol Injection (2 mcg per mL) Package Factor 71288-802-03 4 mcg per 2 mL Multi-Dose Vial 50 vials per carton Storage and Handling
Store at 20° to 25°C (68° to 77°F); excursions permitted between 15° and 30°C (59° and 86°F). [See USP Controlled Room Temperature.] Store unopened vial in original carton.
Opened vials: Store for up to 3 days at 2° to 8°C (36° to 46°F). Discard unused portion after 3 days.
Protect from light.
Sterile, Nonpyrogenic.
The container closure is not made with natural rubber latex. -
17 PATIENT COUNSELING INFORMATION
Hypercalcemia
Advise patients to contact a health care provider if they develop symptoms of elevated calcium (e.g. feeling tired, difficulty thinking clearly, loss of appetite, nausea, vomiting, constipation, increased thirst, increased urination and weight loss) [see Warnings and Precautions (5.1)].
Hypersensitivity
Inform patients that hypersensitivity reactions can occur with doxercalciferol [see Warnings and Precautions (5.3)].
Monitoring
Inform patients that they will need routine monitoring of laboratory parameters such as calcium and intact PTH while receiving doxercalciferol. Inform patients that more frequent monitoring is necessary during the initiation of therapy, following dose changes or when potentially interacting medications are started or discontinued [see Dosage and Administration (2), Drug Interactions (7)].
Drug Interactions
Advise patients to inform their physician of all medications, including prescription and nonprescription drugs, and supplements they are taking. Advise patients to also inform their physician that they are receiving doxercalciferol if a new medication is prescribed [see Drug Interactions (7)].
meitheal®
Mfd. for Meitheal Pharmaceuticals
Chicago, IL 60631 (USA)
©2019 Meitheal Pharmaceuticals Inc.April 2019
810032 - PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
DOXERCALCIFEROL
doxercalciferol injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 71288-802 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Doxercalciferol (UNII: 3DIZ9LF5Y9) (Doxercalciferol - UNII:3DIZ9LF5Y9) Doxercalciferol 4 ug in 2 mL Inactive Ingredients Ingredient Name Strength Alcohol (UNII: 3K9958V90M) Polysorbate 20 (UNII: 7T1F30V5YH) Sodium Chloride (UNII: 451W47IQ8X) BUTYLATED HYDROXYTOLUENE (UNII: 1P9D0Z171K) SODIUM PHOSPHATE, DIBASIC, ANHYDROUS (UNII: 22ADO53M6F) SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) EDETATE DISODIUM (UNII: 7FLD91C86K) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71288-802-03 50 in 1 CARTON 02/11/2020 1 NDC: 71288-802-02 2 mL in 1 VIAL, MULTI-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA211670 02/11/2020 Labeler - Meitheal Pharmaceuticals Inc. (080548348) Establishment Name Address ID/FEI Business Operations Nanjing King-Friend Biochemical Pharmaceutical Co., Ltd. 421297554 MANUFACTURE(71288-802)

© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.