REBLOZYL- luspatercept injection, powder, lyophilized, for solution
Reblozyl by
Drug Labeling and Warnings
Reblozyl by is a Prescription medication manufactured, distributed, or labeled by Celgene, Celgene Corporation. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use REBLOZYL safely and effectively. See full prescribing information for REBLOZYL.
REBLOZYL® (luspatercept-aamt) for injection, for subcutaneous use
Initial U.S. Approval: 2019RECENT MAJOR CHANGES
INDICATIONS AND USAGE
REBLOZYL is an erythroid maturation agent indicated for the treatment of:
- Anemia in adult patients with beta thalassemia who require regular red blood cell (RBC) transfusions (1.1).
- Anemia failing an erythropoiesis stimulating agent and requiring 2 or more RBC units over 8 weeks in adult patients with very low- to intermediate-risk myelodysplastic syndromes with ring sideroblasts (MDS-RS) or with myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T) (1.2).
- Limitations of Use: REBLOZYL is not indicated for use as a substitute for RBC transfusions in patients who require immediate correction of anemia (1.3).
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
None (4).
WARNINGS AND PRECAUTIONS
- Thrombosis/Thromboembolism: Increased risk in patients with beta thalassemia. Monitor patients for signs and symptoms of thromboembolic events and institute treatment promptly (5.1).
- Hypertension: Monitor blood pressure (BP) during treatment. Initiate anti-hypertensive treatment if necessary (5.2).
- Embryo-Fetal Toxicity: May cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and use of effective contraception (5.3, 8.1, 8.3).
ADVERSE REACTIONS
The most common (>10%) adverse reactions were fatigue, headache, musculoskeletal pain, arthralgia, dizziness/vertigo, nausea, diarrhea, cough, abdominal pain, dyspnea, and hypersensitivity (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Celgene Corporation at 1-888-423-5436 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Beta Thalassemia
1.2 Myelodysplastic Syndromes with Ring Sideroblasts or Myelodysplastic/ Myeloproliferative Neoplasm with Ring Sideroblasts and Thrombocytosis Associated Anemia
1.3 Limitations Of Use
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Beta Thalassemia
2.2 Recommended Dosage for Myelodysplastic Syndromes with Ring Sideroblasts (MDS-RS) or Myelodysplastic/ Myeloproliferative Neoplasm with Ring Sideroblasts and Thrombocytosis (MDS/MPN-RS-T) Associated Anemia
2.3 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Thrombosis/Thromboembolism
5.2 Hypertension
5.3 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Beta Thalassemia
14.2 Myelodysplastic Syndromes with Ring Sideroblasts or Myelodysplastic / Myeloproliferative Neoplasm with Ring Sideroblasts and Thrombocytosis Associated Anemia
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Beta Thalassemia
REBLOZYL is indicated for the treatment of anemia in adult patients with beta thalassemia who require regular red blood cell (RBC) transfusions.
1.2 Myelodysplastic Syndromes with Ring Sideroblasts or Myelodysplastic/ Myeloproliferative Neoplasm with Ring Sideroblasts and Thrombocytosis Associated Anemia
REBLOZYL is indicated for the treatment of anemia failing an erythropoiesis stimulating agent and requiring 2 or more red blood cell units over 8 weeks in adult patients with very low- to intermediate-risk myelodysplastic syndromes with ring sideroblasts (MDS-RS) or with myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T).
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Beta Thalassemia
The recommended starting dose of REBLOZYL is 1 mg/kg once every 3 weeks by subcutaneous injection for patients with beta thalassemia. Prior to each REBLOZYL dose, review the patient's hemoglobin and transfusion record. Titrate the dose based on responses according to Table 1. Interrupt treatment for adverse reactions as described in Table 2. Discontinue REBLOZYL if a patient does not experience a decrease in transfusion burden after 9 weeks of treatment (administration of 3 doses) at the maximum dose level or if unacceptable toxicity occurs at any time.
If a planned administration of REBLOZYL is delayed or missed, administer REBLOZYL as soon as possible and continue dosing as prescribed, with at least 3 weeks between doses.
Dose Modifications for Response
Assess and review hemoglobin results prior to each administration of REBLOZYL. If an RBC transfusion occurred prior to dosing, use the pretransfusion hemoglobin for dose evaluation.
If a patient does not achieve a reduction in RBC transfusion burden after at least 2 consecutive doses (6 weeks) at the 1 mg/kg starting dose, increase the REBLOZYL dose to 1.25 mg/kg. Do not increase the dose beyond the maximum dose of 1.25 mg/kg. In the absence of transfusions, if hemoglobin increase is greater than 2 g/dL within 3 weeks or the predose hemoglobin is greater than or equal to 11.5 g/dL, reduce the dose or interrupt treatment with REBLOZYL as described in Table 1.
Dose level modifications for response are provided in Table 1.
Table 1: Beta Thalassemia - REBLOZYL Dose Titration for Response REBLOZYL
Dosing Recommendation*- * Do not increase the dose if the patient is experiencing an adverse reaction as described in Table 2.
Starting Dose - 1 mg/kg every 3 weeks
Dose Increases for Insufficient Response at Initiation of Treatment No reduction in RBC transfusion burden after at least 2 consecutive doses (6 weeks) at the 1 mg/kg starting dose - Increase the dose to 1.25 mg/kg every 3 weeks
No reduction in RBC transfusion burden after 3 consecutive doses (9 weeks) at 1.25 mg/kg - Discontinue treatment
Dose Modifications for Predose Hemoglobin Levels or Rapid Hemoglobin Rise Predose hemoglobin is greater than or equal to 11.5 g/dL in the absence of transfusions - Interrupt treatment
- Restart when the hemoglobin is no more than 11 g/dL
Increase in hemoglobin greater than 2 g/dL within 3 weeks in the absence of transfusions and - current dose is 1.25 mg/kg
- current dose is 1 mg/kg
- current dose is 0.8 mg/kg
- current dose is 0.6 mg/kg
- Reduce dose to 1 mg/kg
- Reduce dose to 0.8 mg/kg
- Reduce dose to 0.6 mg/kg
- Discontinue treatment
Dose Modifications for Toxicity
For patients experiencing Grade 3 or higher adverse reactions, modify treatment as described in Table 2.
Table 2: Beta Thalassemia - REBLOZYL Dosing Modifications for Adverse Reactions REBLOZYL
Dosing Recommendation*- * Grade 1 is mild, Grade 2 is moderate, Grade 3 is severe, and Grade 4 is life-threatening.
Grade 3 or 4 hypersensitivity reactions - Discontinue treatment
Other Grade 3 or 4 adverse reactions - Interrupt treatment
- Restart when the adverse reaction resolves to no more than Grade 1
2.2 Recommended Dosage for Myelodysplastic Syndromes with Ring Sideroblasts (MDS-RS) or Myelodysplastic/ Myeloproliferative Neoplasm with Ring Sideroblasts and Thrombocytosis (MDS/MPN-RS-T) Associated Anemia
The recommended starting dose of REBLOZYL is 1 mg/kg once every 3 weeks by subcutaneous injection for patients with anemia of MDS-RS or MDS/MPN-RS-T. Prior to each REBLOZYL dose, review the patient's hemoglobin and transfusion record. Titrate the dose based on responses according to Table 3. Interrupt treatment for adverse reactions as described in Table 4. Discontinue REBLOZYL if a patient does not experience a decrease in transfusion burden after 9 weeks of treatment (administration of 3 doses) at the maximum dose level or if unacceptable toxicity occurs at any time.
If a planned administration of REBLOZYL is delayed or missed, administer REBLOZYL as soon as possible and continue dosing as prescribed, with at least 3 weeks between doses.
Dose Modifications for Response
Assess and review hemoglobin results prior to each administration of REBLOZYL. If an RBC transfusion occurred prior to dosing, use the pretransfusion hemoglobin for dose evaluation.
If a patient is not RBC transfusion-free after at least 2 consecutive doses (6 weeks) at the 1 mg/kg starting dose, increase the REBLOZYL dose to 1.33 mg/kg (Table 3). If a patient is not RBC transfusion-free after at least 2 consecutive doses (6 weeks) at the 1.33 mg /kg dose level, increase the REBLOZYL dose to 1.75 mg/kg. Do not increase the dose more frequently than every 6 weeks (2 doses) or beyond the maximum dose of 1.75 mg/kg.
In the absence of transfusions, if hemoglobin increase is greater than 2 g/dL within 3 weeks or if the predose hemoglobin is greater than or equal to 11.5 g/dL, reduce the dose or interrupt treatment with REBLOZYL as described in Table 3. If, upon dose reduction, the patient loses response (i.e., requires a transfusion) or hemoglobin concentration drops by 1 g/dL or more in 3 weeks in the absence of transfusion, increase the dose by one dose level. Wait a minimum of 6 weeks between dose increases.
Dose modifications for response are provided in Table 3.
Table 3: MDS-RS and MDS/MPN-RS-T Associated Anemia - REBLOZYL Dose Titration for Response REBLOZYL
Dosing Recommendation*- * Do not increase the dose if the patient is experiencing an adverse reaction as described in Table 4.
Starting Dose - 1 mg/kg every 3 weeks
Dose Increases for Insufficient Response at Initiation of Treatment Not RBC transfusion-free after at least 2 consecutive doses (6 weeks) at the 1 mg/kg starting dose - Increase the dose to 1.33 mg/kg every 3 weeks
Not RBC transfusion-free after at least 2 consecutive doses (6 weeks) at 1.33 mg/kg - Increase the dose to 1.75 mg/kg every 3 weeks
No reduction in RBC transfusion burden after at least 3 consecutive doses (9 weeks) at 1.75 mg/kg - Discontinue treatment
Dose Modifications for Predose Hemoglobin Levels or Rapid Hemoglobin Rise Predose hemoglobin is greater than or equal to 11.5 g/dL in the absence of transfusions - Interrupt treatment
- Restart when the hemoglobin is no more than 11 g/dL
Increase in hemoglobin greater than 2 g/dL within 3 weeks in the absence of transfusions and - current dose is 1.75 mg/kg
- current dose is 1.33 mg/kg
- current dose is 1 mg/kg
- current dose is 0.8 mg/kg
- current dose is 0.6 mg/kg
- Reduce dose to 1.33 mg/kg
- Reduce dose to 1 mg/kg
- Reduce dose to 0.8 mg/kg
- Reduce dose to 0.6 mg/kg
- Discontinue treatment
Dose Modifications for Toxicity
For patients experiencing Grade 3 or higher adverse reactions, modify treatment as described in Table 4.
Table 4: MDS-RS and MDS/MPN-RS-T Associated Anemia - REBLOZYL Dosing Modifications for Adverse Reactions REBLOZYL
Dosing Recommendation*- * Grade 1 is mild, Grade 2 is moderate, Grade 3 is severe, and Grade 4 is life-threatening.
- † Per Table 3 dose reductions above.
Grade 3 or 4 hypersensitivity reactions - Discontinue treatment
Other Grade 3 or 4 adverse reactions - Interrupt treatment
- When the adverse reaction resolves to no more than Grade 1, restart treatment at the next lower dose level†
- If the dose delay is > 12 consecutive weeks, discontinue treatment
2.3 Preparation and Administration
REBLOZYL should be reconstituted and administered by a healthcare professional.
Reconstitute REBLOZYL with Sterile Water for Injection, USP only.
Table 5: Reconstitution Volumes Vial Size Amount of Sterile Water for Injection, USP required for reconstitution Final Concentration Deliverable Volume 25 mg vial 0.68 mL 25 mg/0.5 mL 0.5 mL 75 mg vial 1.6 mL 75 mg/1.5 mL 1.5 mL (50 mg/mL) Reconstitute the number of REBLOZYL vials to achieve the appropriate dose based on the patient's weight. Use a syringe with suitable graduations for reconstitution to ensure accurate dosage.
Reconstitution Instructions
- Reconstitute with Sterile Water for Injection, USP using volumes described in Table 5 (Reconstitution volumes) with the stream directed onto the lyophilized powder. Allow to stand for one minute.
- Discard the needle and syringe used for reconstitution. The needle and syringe used for reconstitution should not be used for subcutaneous injections.
- Gently swirl the vial in a circular motion for 30 seconds. Stop swirling and let the vial sit in an upright position for 30 seconds.
- Inspect the vial for undissolved particles in the solution. If undissolved powder is observed, repeat step 3 until the powder is completely dissolved.
- Invert the vial and gently swirl in an inverted position for 30 seconds. Bring the vial back to the upright position, and let it sit for 30 seconds.
- Repeat step 5 seven more times to ensure complete reconstitution of material on the sides of the vial.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit. REBLOZYL is a colorless to slightly yellow, clear to slightly opalescent solution which is free of foreign particulate matter. Do not use if undissolved product or foreign particulate matter are observed.
- If the reconstituted solution is not used immediately:
- Store at room temperature at 20°C to 25°C (68°F to 77°F) in the original vial for up to 8 hours. Discard if not used within 8 hours of reconstitution.
- Alternatively, store refrigerated at 2°C to 8°C (36°F to 46°F) for up to 24 hours in the original vial. Remove from refrigerated condition 15-30 minutes prior to injection to allow solution to reach room temperature for a more comfortable injection. Discard if not used within 24 hours of reconstitution.
- Do not freeze the reconstituted solution.
Discard any unused portion. Do not pool unused portions from the vials. Do not administer more than 1 dose from a vial. Do not mix with other medications.
Instructions for Subcutaneous Administration
Calculate the exact total dosing volume of 50 mg/mL solution required for the patient.
Slowly withdraw the dosing volume of the reconstituted REBLOZYL solution from the single-dose vial(s) into a syringe. Divide doses requiring larger reconstituted volumes (i.e., greater than 1.2 mL) into separate similar volume injections and inject into separate sites. If multiple injections are required, use a new syringe and needle for each subcutaneous injection.
Administer the injection subcutaneously into the upper arm, thigh, and/or abdomen.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Thrombosis/Thromboembolism
In adult patients with beta thalassemia, thromboembolic events (TEE) were reported in 8/223 (3.6%) REBLOZYL-treated patients. Reported TEEs included deep vein thromboses, pulmonary embolus, portal vein thrombosis, and ischemic strokes. Patients with known risk factors for thromboembolism, e.g. splenectomy or concomitant use of hormone replacement therapy, may be at further increased risk of thromboembolic conditions. Consider thromboprophylaxis in patients with beta thalassemia at increased risk of TEE. Monitor patients receiving REBLOZYL for signs and symptoms of thromboembolic events and institute treatment promptly.
5.2 Hypertension
Hypertension was reported in 10.7% (61/571) of REBLOZYL-treated patients. Across clinical studies, the incidence of grade 3-4 hypertension ranged from 1.8% to 8.6%. In adult patients with beta thalassemia with normal baseline blood pressure, 13 (6.2%) patients developed systolic blood pressure (SBP) ≥130 mm Hg and 33 (16.6%) patients developed diastolic blood pressure (DBP) ≥80 mm Hg. In adult patients with MDS with normal baseline blood pressure, 26 (29.9%) patients developed SBP ≥130 mm Hg and 23 (16.4%) patients developed DBP ≥80 mm Hg. Monitor blood pressure prior to each administration. Manage new-onset hypertension or exacerbations of preexisting hypertension using anti-hypertensive agents.
5.3 Embryo-Fetal Toxicity
Based on findings from animal reproductive studies, REBLOZYL may cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of luspatercept-aamt to pregnant rats and rabbits during organogenesis resulted in adverse developmental outcomes including increased embryo-fetal mortality, alterations to growth, and structural abnormalities at exposures (based on area under the curve [AUC]) above those occurring at the maximum recommended human dose (MRHD) of 1.75 mg/kg.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use an effective method of contraception during treatment with REBLOZYL and for at least 3 months after the final dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Thrombosis/Thromboembolism [see Warnings and Precautions (5.1)]
- Hypertension [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in the WARNINGS AND PRECAUTIONS reflect exposure to REBLOZYL as a single agent administered across a range of doses (0.125 mg/kg to 1.75 mg/kg) in 571 patients in 4 trials.
Beta Thalassemia
The safety of REBLOZYL in patients with beta thalassemia was evaluated in the BELIEVE trial [see Clinical Studies (14.1)]. Key eligibility criteria included adult patients with beta thalassemia (with the exception of patients with hemoglobin S or alpha-thalassemia disease) without major organ damage or recent DVT stroke and platelet counts less than or equal to 1000 × 109/L.
Patients received a starting dose of REBLOZYL 1 mg/kg subcutaneous injection every 3 weeks. Overall, 53% of patients had their dose increased to 1.25 mg/kg (46% REBLOZYL, n = 223) or placebo (66%, n = 109). The median duration of treatment was similar between the REBLOZYL and placebo arms (63.3 weeks vs. 62.1 weeks, respectively). Per protocol, patients in the REBLOZYL and placebo arms were to remain on therapy for at least 48 weeks in the double-blind phase of the trial.
Among patients receiving REBLOZYL, 94% were exposed for 6 months or longer and 72% were exposed for greater than one year.
The median age of patients who received REBLOZYL was 30 years (range: 18, 66); 59% female; 54% White and 36% Asian.
Serious adverse reactions occurred in 3.6% of patients on REBLOZYL. Serious adverse reactions reported in 1% of patients were cerebrovascular accident and deep vein thrombosis. A fatal adverse reaction occurred in one patient treated with REBLOZYL who died due to an unconfirmed case of AML (M6).
Permanent discontinuation due to an adverse reaction (Grades 1-4) occurred in 5.4% of patients who received REBLOZYL. Most frequent adverse reactions requiring permanent discontinuation in patients who received REBLOZYL included arthralgia (1%), back pain (1%), bone pain (<1%), and headache (<1%).
Dosage reductions due to an adverse reaction occurred in 2.7% of patients who received REBLOZYL. Most frequent adverse reactions requiring dosage reduction in >0.5% of patients who received REBLOZYL included hypertension and headache.
Dosage interruptions due to an adverse reaction occurred in 15.2% of patients who received REBLOZYL. Most frequent adverse reactions requiring dosage interruption in >1% of patients who received REBLOZYL included upper respiratory tract infection, ALT increase, and cough.
The most common adverse reactions (at least 10% for REBLOZYL and 1% more than placebo) were headache (26%), bone pain (20%), arthralgia (19%), fatigue (14%), cough (14%), abdominal pain (14%), diarrhea (12%), and dizziness (11%).
Table 6 summarizes the adverse reactions in BELIEVE.
Table 6: Adverse Drug Reactions (>5%) in Patients with Beta Thalassemia Receiving REBLOZYL with a Difference Between Arms of 1% in BELIEVE Trial Body System REBLOZYL
(N=223)Placebo
(N=109)Adverse Reaction All Grades
n (%)Grades ≥3*
n (%)All Grades
n (%)Grades ≥3
n (%)- * Limited to Grade 3 reactions with the exception of 4 events of Grade 4 hyperuricemia.
- † Grouped term includes abdominal pain and abdominal pain upper.
- ‡ Grouped term includes essential hypertension, hypertension, and hypertensive crisis.
Musculoskeletal and connective tissue disorders Bone Pain 44 (20) 3 (1) 9 (8) 0 (0) Arthralgia 43 (19) 0 (0) 13 (12) 0 (0) Infections and infestation Influenza 19 (9) 0 (0) 6 (6) 0 (0) Viral Upper Respiratory Infection 14 (6) 1 (0.4) 2 (2) 0 (0) Nervous system disorders Headache 58 (26) 1 (<1) 26 (24) 1 (1) Dizziness 25 (11) 0 (0) 5 (5) 0 (0) General disorders and administration site conditions Fatigue 30 (14) 0 (0) 14 (13) 0 (0) Gastrointestinal disorders Abdominal Pain † 31 (14) 0 (0) 13 (12) 0 (0) Diarrhea 27 (12) 1 (<1) 11 (10) 0 (0) Nausea 20 (9) 0 (0) 6 (6) 0 (0) Vascular disorders Hypertension ‡ 18 (8) 4 (2) 3 (3) 0 (0) Metabolism and nutrition disorders Hyperuricemia 16 (7) 6 (3) 0 (0) 0 (0) Respiratory, thoracic and mediastinal disorders Cough 32 (14) 0 (0) 12 (11) 0 (0) Clinically relevant adverse reactions in <5% of patients include vertigo/vertigo positional, syncope/presyncope, injection site reactions and hypersensitivity.
Liver function abnormalities in the BELIEVE trial are shown in Table 7.
Table 7: Liver Function Laboratory Abnormalities in Patients with Beta Thalassemia in the BELIEVE Trial REBLOZYL
N = 223
n (%)Placebo
N = 109
n (%)ALP = alkaline phosphatase; ALT = alanine aminotransferase; AST = aspartate aminotransferase; ULN = upper limit of normal. ALT ≥ 3 × ULN 26 (12) 13 (12) AST ≥ 3 × ULN 25 (11) 5 (5) ALP ≥ 2 × ULN 17 (8) 1 (<1) Total bilirubin ≥ 2 × ULN 143 (64) 51 (47) Direct bilirubin ≥ 2 × ULN 13 (6) 4 (4) Myelodysplastic Syndromes with Ring Sideroblasts or Myelodysplastic / Myeloproliferative Neoplasm with Ring Sideroblasts and Thrombocytosis Associated Anemia
The safety of REBLOZYL at the recommended dose and schedule was evaluated in 242 patients with MDS with ring sideroblasts (n=192) or other myeloid neoplasms (n=50). The safety population included 63% males and 37% females of median age 72 years (range, 30 – 95 years); of these patients, 81% were White, 0.4% Black, 0.4% Other, and race was not reported in 18.2% of patients. The median time on treatment with REBLOZYL was 50.4 weeks (range, 3 – 221 weeks); 67% of patients were exposed for 6 months or longer and 49% were exposed for greater than one year.
Among the 242 patients treated with REBLOZYL, 5 (2.1%) had a fatal adverse reaction, 11 (4.5%) discontinued due to an adverse reaction, and 7 (2.9%) had a dose reduction due to an adverse reaction. The most common (≥10%) all-grade adverse reactions included fatigue, musculoskeletal pain, dizziness, diarrhea, nausea, hypersensitivity reactions, hypertension, headache, upper respiratory tract infection, bronchitis, and urinary tract infection. The most common (≥2%) Grade ≥ 3 adverse reactions included fatigue, hypertension, syncope and musculoskeletal pain. Selected laboratory abnormalities that changed from Grade 0-1 at baseline to Grade ≥ 2 at any time during the studies in at least 10% of patients included creatinine clearance decreased, total bilirubin increased, and alanine aminotransferase increased.
Table 8 shows the most common adverse reactions for patients treated with REBLOZYL or placebo through the first 8 cycles in the MEDALIST trial [see Clinical Studies (14.2)].
Table 8: Adverse Reactions (≥5%) in Patients Receiving REBLOZYL with a Difference Between Arms of >2% in MEDALIST Trial Through Cycle 8 Body System /Adverse Reaction REBLOZYL
(N=153)Placebo
(N=76)All Grades
n (%)Grade 3
n (%)All Grades
n (%)Grade 3
n (%)- * Includes asthenic conditions.
- † Reaction includes similar/grouped terms.
General disorders and administration site conditions Fatigue *, † 63 (41) 11 (7) 17 (22) 2 (3) Musculoskeletal and connective tissue disorders Musculoskeletal pain † 30 (20) 3 (2) 11 (14) 0 (0) Nervous system disorders Dizziness/vertigo 28 (18) 1 (<1) 5 (7) 1 (1) Headache † 21 (14) 0 (0) 5 (7) 0 (0) Syncope / presyncope 8 (5) 5 (3) 0 (0) 0 (0) Gastrointestinal disorders Nausea † 25 (16) 1 (<1) 8 (11) 0 (0) Diarrhea † 25 (16) 0 (0) 7 (9) 0 (0) Respiratory, thoracic and mediastinal disorders Dyspnea † 20 (13) 2 (1) 4 (5) 1 (1) Immune system disorders Hypersensitivity reactions † 15 (10) 1 (<1) 5 (7) 0 (0) Renal and urinary disorders Renal impairment † 12 (8) 3 (2) 3 (4) 0 (0) Cardiac disorders Tachycardia † 12 (8) 0 (0) 1 (1) 0 (0) Injury poisoning and procedural complications Injection site reactions 10 (7) 0 (0) 3 (4) 0 (0) Infections and infestations Upper respiratory tract infection 10 (7) 1 (<1) 2 (3) 0 (0) Influenza / influenza like illness 9 (6) 0 (0) 2 (3) 0 (0) Other clinically relevant adverse reactions reported in <5% of patients include bronchitis, urinary tract infection, and hypertension [see Warnings and Precautions (5.2)].
Shifts from Grades 0-1 to Grades 2-4 abnormalities for selected laboratory tests during the first 8 cycles in the MEDALIST trial are shown in Table 9.
Table 9: Selected Grades 2-4 Treatment-Emergent Laboratory Abnormalities Through Cycle 8 in the MEDALIST Trial Parameter REBLOZYL Placebo N* n (%) N* n (%) ALT = alanine aminotransferase; AST = aspartate aminotransferase. - * Number of patients at Grades 0-1 at baseline.
ALT elevated 151 13 (9) 74 5 (7) AST elevated 152 6 (4) 76 0 (0) Total bilirubin elevated 140 17 (12) 66 3 (5) Creatinine clearance reduced 113 30 (27) 62 13 (21) 6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to luspatercept in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
Of 284 patients with beta thalassemia who were treated with REBLOZYL and evaluable for the presence of anti-luspatercept-aamt antibodies, 4 patients (1.4%) tested positive for treatment-emergent anti-luspatercept-aamt antibodies, including 2 patients (0.7%) who had neutralizing antibodies.
Of 260 patients with MDS who were treated with REBLOZYL and evaluable for the presence of anti-luspatercept-aamt antibodies, 23 patients (8.9%) tested positive for treatment-emergent anti-luspatercept-aamt antibodies, including 9 patients (3.5%) who had neutralizing antibodies.
Luspatercept-aamt serum concentration tended to decrease in the presence of neutralizing antibodies. There were no severe acute systemic hypersensitivity reactions reported for patients with anti-luspatercept-aamt antibodies in REBLOZYL clinical trials, and there was no association between hypersensitivity type reaction or injection site reaction and presence of anti-luspatercept-aamt antibodies.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animal reproduction studies, REBLOZYL may cause fetal harm when administered to a pregnant woman. There are no available data on REBLOZYL use in pregnant women to inform a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal reproduction studies, administration of luspatercept-aamt to pregnant rats and rabbits during the period of organogenesis resulted in adverse developmental outcomes including embryo-fetal mortality, alterations to growth, and structural abnormalities at exposures (based on area under the curve [AUC]) above those occurring at the maximum recommended human dose (MRHD) (see Data). Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In embryo-fetal development studies, luspatercept-aamt was administered subcutaneously at 5, 15, or 30 mg/kg on gestation days 3 and 10 (rats) or 5, 20, or 40 mg/kg on gestation days 4 and 11 (rabbits). Effects in both species included reductions in numbers of live fetuses and fetal body weights, and increases in resorptions, post-implantation losses, and skeletal variations (such as asymmetric sternal centra in rats and angulated hyoid in rabbits). Effects were observed at exposures (based on AUC) approximately 7-times (rats) and 16-times (rabbits) the MRHD of 1.75 mg/kg.
In a pre- and postnatal development study, pregnant rats were administered luspatercept-aamt subcutaneously at 3, 10, or 30 mg/kg once every 2 weeks during organogenesis and through weaning, gestation day 6 through postnatal day 20. At all dose levels lower F1 pup body weights and adverse kidney findings (such as membranoproliferative glomerulonephritis, tubular atrophy/hypoplasia, and vessel ectasia occasionally associated with hemorrhage) were observed. These effects were observed at exposures (based on AUC) approximately 1.6-times the MRHD of 1.75 mg/kg.
8.2 Lactation
Risk Summary
Luspatercept-aamt was detected in milk of lactating rats. When a drug is present in animal milk, it is likely that the drug will be present in human milk. There are no data on the presence of REBLOZYL in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in the breastfed child, advise patients that breastfeeding is not recommended during treatment with REBLOZYL, and for 3 months after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential before starting REBLOZYL treatment.
Contraception
Females
REBLOZYL may cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)]. Advise female patients of reproductive potential to use effective contraception during treatment with REBLOZYL and for at least 3 months after the last dose.
Infertility
Females
Based on findings in animals, REBLOZYL may impair female fertility [see Nonclinical Toxicology (13.1)]. Adverse effects on fertility in female rats were reversible after a 14-week recovery period.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established. Based on findings in juvenile animals, REBLOZYL is not recommended for use in pediatric patients [see Non-Clinical Toxicology (13.1)].
8.5 Geriatric Use
Clinical studies of REBLOZYL in beta thalassemia did not include sufficient numbers of patients age 65 years and older to determine whether they respond differently from younger patients.
Clinical studies of REBLOZYL for treatment of anemia in MDS-RS and MDS/MPN-RS-T included 206 (79%) patients ≥ 65 years of age and 93 (36%) patients ≥ 75 years of age. No differences in safety or effectiveness were observed between older (≥ 65 years) and younger patients.
-
11 DESCRIPTION
Luspatercept-aamt is an erythroid maturation agent. Luspatercept-aamt is a receptor fusion protein consisting of a modified extracellular domain of the human activin receptor type IIB linked to a human IgG1 Fc domain with a calculated molecular mass of approximately 76 kD. Luspatercept is produced in Chinese hamster ovary cells by recombinant DNA technology.
REBLOZYL (luspatercept-aamt) for injection is a sterile, preservative-free, white to off-white, lyophilized powder in single-dose vials for subcutaneous use after reconstitution.
Each 25 mg single-dose vial provides nominal 25 mg of luspatercept-aamt and citric acid monohydrate (0.085 mg), polysorbate 80 (0.10 mg), sucrose (45.0 mg), and tri-sodium citrate dihydrate (1.35 mg) at pH 6.5. After reconstitution with 0.68 mL Sterile Water for Injection USP, the resulting concentration is 25 mg/0.5 mL of luspatercept-aamt and the nominal deliverable volume is 0.5 mL.
Each 75 mg single-dose vial provides nominal 75 mg of luspatercept-aamt and citric acid monohydrate (0.254 mg), polysorbate 80 (0.30 mg), sucrose (135 mg), and tri-sodium citrate dihydrate (4.06 mg) at pH 6.5. After reconstitution with 1.6 mL Sterile Water for Injection USP, the resulting concentration is 75 mg/1.5 mL (50 mg/mL) of luspatercept-aamt and the nominal deliverable volume is 1.5 mL.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Luspatercept-aamt is a recombinant fusion protein that binds several endogenous TGF-β superfamily ligands, thereby diminishing Smad2/3 signaling. Luspatercept-aamt promoted erythroid maturation through differentiation of late-stage erythroid precursors (normoblasts) in mice. In models of β-thalassemia and MDS, luspatercept-aamt decreased abnormally elevated Smad2/3 signaling and improved hematology parameters associated with ineffective erythropoiesis in mice.
12.2 Pharmacodynamics
Increases in Hemoglobin in Patients with Low RBC Transfusion Burden
In patients having received < 4 units of RBC transfusion within 8 weeks prior to study, hemoglobin increased within 7 days of initiating REBLOZYL and correlated with the time to luspatercept-aamt maximum serum concentration (Cmax). The greatest Hgb increase occurred after the first dose; approximately 0.75 g/dL at a dose of 0.6 to 1.25 times the recommended starting dose for beta thalassemia, or approximately 1 g/dL at a dose of 0.75 to 1.75 times the recommended starting dose for MDS. Additional smaller increases were observed after subsequent doses. Hemoglobin levels returned to baseline approximately 6 to 8 weeks from the last dose following administration of luspatercept-aamt (0.6 to 1.75 mg/kg).
Increasing luspatercept-aamt serum exposure (AUC) was associated with greater Hgb increase in patients with beta thalassemia or MDS who had a baseline transfusion burden < 4 units/8 weeks. Increasing luspatercept-aamt serum exposure (time-averaged AUC) was associated with greater probability of achieving transfusion independence for at least 8 consecutive weeks in patients with MDS requiring transfusions (≥ 2 units of RBC transfusion within 8 weeks).
12.3 Pharmacokinetics
Luspatercept-aamt exhibited linear pharmacokinetics (PK) over the dose range of 0.2 to 1.25 mg/kg (0.2 to 1.25 times the recommended starting dosage) in patients with beta thalassemia, and from 0.125 mg/kg to 1.75 mg/kg in patients with MDS. The mean (% coefficient of variation [%CV]) steady-state AUC at the starting dose of 1 mg/kg was 126 (35.9%) day∙µg/mL for patients with beta thalassemia and 145 (38.3%) day∙µg/mL for patients with MDS. Luspatercept-aamt serum concentration reached steady state after 3 doses when administered every 3 weeks. The accumulation ratio of luspatercept-aamt was approximately 1.5.
Absorption
The median (range) time to maximum concentration (Tmax) of luspatercept-aamt was observed at approximately 7 [6 to 10] days post-dose in adult patients with beta thalassemia or 7 [5 to 21] days post-dose in adult patients with MDS. The absorption of luspatercept-aamt was not significantly affected by the subcutaneous injection sites (upper arm, thigh, or abdomen).
Distribution
The mean (%CV) apparent volume of distribution (Vd/F) of luspatercept-aamt was 7.1 (26.7%) L for patients with beta thalassemia, and 9.7 (26.5%) for patients with MDS.
Elimination
The mean (%CV) half-life (t1/2) of luspatercept-aamt was approximately 11 (25.7%) days and the mean (%CV) apparent total clearance (CL/F) was 0.44 (38.5%) L/day in patients with beta thalassemia. The mean (%CV) t1/2 of luspatercept-aamt was approximately 13 (31.6%) days and the mean (%CV) CL/F was 0.52 (41.2%) L/day in patients with MDS.
Specific Populations
No clinically significant differences in the luspatercept-aamt PK was observed based on age (18 to 95 years), sex, race/ethnicity (Asian, White), mild to severe hepatic impairment (total bilirubin ≤ upper limit of normal [ULN] and aspartate aminotransaminase [AST] or alanine transaminase [ALT] > ULN, or total bilirubin > ULN and any AST or ALT), mild to moderate renal impairment (estimated glomerular filtration rate [eGFR] 30 to 89 mL/min/1.73 m2), baseline albumin (30 to 56 g/L), baseline serum erythropoietin (2.4 to 2450 U/L), red blood cell (RBC) transfusion burden (0 to 43 units/24 weeks), beta thalassemia genotype (β0/β0 vs. non-β0/β0), splenectomy, and ring sideroblasts status in MDS (negative vs. positive). The effect of AST or ALT >3 × ULN and the effect of severe renal impairment (eGFR <30 mL/min/1.73 m2) on luspatercept-aamt PK is unknown.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity or mutagenicity studies have been conducted with luspatercept-aamt.
In a repeat-dose toxicity study, juvenile rats were administered luspatercept-aamt subcutaneously at 1, 3, or 10 mg/kg once every 2 weeks from postnatal day 7 to 91. Hematologic malignancies (granulocytic leukemia, lymphocytic leukemia, malignant lymphoma) were observed at 10 mg/kg resulting in exposures (based on area under the curve [AUC]) approximately 4.4 times the maximum recommended human dose (MRHD) of 1.75 mg/kg.
In a combined male and female fertility and early embryonic development study in rats, luspatercept-aamt was administered subcutaneously to animals at doses of 1 to 15 mg/kg. There were significant reductions in the average numbers of corpora lutea, implantations, and viable embryos in luspatercept-aamt-treated females. Effects on female fertility were observed at the highest dose with exposures (based on AUC) approximately 7-times the MRHD of 1.75 mg/kg. Adverse effects on fertility in female rats were reversible after a 14-week recovery period. No adverse effects were noted in male rats.
-
14 CLINICAL STUDIES
14.1 Beta Thalassemia
The efficacy of REBLOZYL was evaluated in adult patients with beta thalassemia in the BELIEVE trial (NCT02604433). BELIEVE was a multicenter, randomized, double-blind, placebo-controlled trial in which (n=336) patients with beta thalassemia requiring regular red blood cell transfusions (6-20 RBC units per 24 weeks) with no transfusion-free period greater than 35 days during that period were randomized 2:1 to REBLOZYL (n=224) or placebo (n=112). In BELIEVE, REBLOZYL was administered subcutaneously once every 3 weeks as long as a reduction in transfusion requirement was observed or until unacceptable toxicity. All patients were eligible to receive best supportive care, which included RBC transfusions; iron-chelating agents; use of antibiotic, antiviral, and antifungal therapy; and/or nutritional support, as needed.
The BELIEVE trial excluded patients with a diagnosis of Hemoglobin S/β-thalassemia or isolated alpha (α)-thalassemia (e.g., Hemoglobin H) or who had major organ damage (liver disease, heart disease, lung disease, renal insufficiency). Patients with recent deep vein thrombosis or stroke or recent use of ESA, immunosuppressant, or hydroxyurea therapy were also excluded. The median age was 30 years (range: 18-66). The trial was comprised of patients who were 42% male, 54.2% White, 34.8% Asian, and 0.3% Black or African American. The percent of patients reporting their race as "other" was 7.7%, and race was not collected or reported for 3% of patients.
Table 10 summarizes the baseline disease-related characteristics in the BELIEVE study.
Table 10: Baseline Disease Characteristics of Patients with Beta Thalassemia in BELIEVE Disease Characteristic REBLOZYL
(N=224)Placebo
(N=112)HbE=hemoglobin E. - * "Missing" category includes patients in the population who had no result for the parameter listed.
Beta thalassemia diagnosis, n (%) Beta-thalassemia 174 (77.7) 83 (74.1) HbE/beta thalassemia 31 (13.8) 21 (18.8) Beta thalassemia combined with alpha-thalassemia 18 (8) 8 (7.1) Missing * 1 (0.4) 0 Baseline transfusion burden 12 weeks prior to randomization Median (min, max) (Units/12 weeks) 6.12 (3, 14) 6.27 (3, 12) Beta thalassemia gene mutation grouping, n (%) β0/β0 68 (30.4) 35 (31.3) Non-β0/β0 155 (69.2) 77 (68.8) Missing * 1 (0.4) 0 Baseline serum ferritin level (μg/L) N 220 111 Median (min, max) 1441.25 (88, 6400) 1301.50 (136, 6400) Splenectomy, n (%) Yes 129 (57.6) 65 (58) No 95 (42.4) 47 (42) Age patient started regular transfusions (years) N 169 85 Median (min, max) 2 (0, 52) 2 (0, 51) The efficacy of REBLOZYL in adult patients with beta thalassemia was established based upon the proportion of patients achieving RBC transfusion burden reduction (≥33% reduction from baseline) with a reduction of at least 2 units from Week 13 to Week 24.
Efficacy results are shown in Table 11.
Table 11: Efficacy Results in Beta Thalassemia - BELIEVE Endpoint REBLOZYL
(N=224)Placebo
(N=112)Risk Difference
(95% CI)p-value ≥33% Reduction from baseline in RBC transfusion burden with a reduction of at least 2 units for 12 consecutive weeks Primary endpoint – Week 13 to Week 24 48 (21.4) 5 (4.5) 17.0
(10.4, 23.6)<0.0001 Week 37 to Week 48 44 (19.6) 4 (3.6) 16.1
(9.8, 22.4)<0.0001 ≥50% Reduction from baseline in RBC transfusion burden with a reduction of at least 2 units for 12 consecutive weeks Week 13 to Week 24 17 (7.6) 2 (1.8) 5.8
(1.6, 10.1)0.0303 Week 37 to Week 48 23 (10.3) 1 (0.9) 9.4
(5, 13.7)0.0017 14.2 Myelodysplastic Syndromes with Ring Sideroblasts or Myelodysplastic / Myeloproliferative Neoplasm with Ring Sideroblasts and Thrombocytosis Associated Anemia
The efficacy of REBLOZYL was evaluated in the MEDALIST trial (NCT02631070), a multi-center, randomized, double-blind, placebo-controlled trial in patients with IPSS-R very low, low, or intermediate-risk myelodysplastic syndromes who have ring sideroblasts and require red blood cell transfusions (2 or more RBC units over 8 weeks). For eligibility, patients were required to have had an inadequate response to prior treatment with an erythropoiesis-stimulating agent (ESA), be intolerant of ESAs, or have a serum erythropoietin > 200 U/L. The MEDALIST trial excluded patients with deletion 5q (del 5q), white blood cell count > 13 Gi/L, neutrophils < 0.5 Gi/L, platelets < 50 Gi/L, or with prior use of a disease modifying agent for treatment of MDS.
The MEDALIST trial included 229 patients randomized 2:1 to REBLOZYL (n=153) or placebo (n=76). Randomization was stratified by baseline RBC transfusion burden and baseline IPSS-R. Treatment was started at 1 mg/kg subcutaneously every 3 weeks; the dose could be increased after completion of the first 2 cycles if the patient had at least one RBC transfusion in the prior 6 weeks. Two dose level increases were allowed (to 1.33 mg/kg and to 1.75 mg/kg). Doses were held and subsequently reduced for adverse reactions, reduced if the hemoglobin increased by ≥ 2 g/dL from the prior cycle, and held if the predose hemoglobin was ≥ 11.5 g/dL.
All patients received best supportive care, which included RBC transfusions as needed. The primary efficacy assessment was conducted after completion of 24 weeks on study drug. Patients with a decrease in transfusion requirement or increase in hemoglobin could continue on blinded study drug thereafter until unacceptable toxicity, loss of efficacy, or disease progression. The median age of the 229 study participants was 71 years (range: 26, 95 years). The trial population was 63% male and 69% White. Table 12 summarizes the baseline disease-related characteristics in the MEDALIST study.
Table 12: Baseline Disease Characteristics of Patients in MEDALIST Disease Characteristic REBLOZYL
(N=153)Placebo
(N=76)EPO=erythropoietin; IPSS R=International Prognostic Scoring System-Revised; ITT=intent-to-treat; MDS=myelodysplastic syndromes; RARS=refractory anemia with ring sideroblasts; RBC=red blood cell; RCMD=refractory cytopenia with multilineage dysplasia; SD=standard deviation; WHO=World Health Organization. - * Time since original MDS diagnosis was defined as the number of years from the date of original diagnosis to the date of informed consent.
- † Baseline EPO was defined as the highest EPO value within 35 days of the first dose of study drug.
- ‡ Includes MDS-RS-MLD and MDS-RS-SLD.
- § Includes MDS-EB-1, MDS-EB-2, and MDS-U.
Time Since Original MDS Diagnosis * (months) Median (range) 44.0 (3, 421) 36.1 (4, 193) Serum EPO (U/L) Categories †, n (%) < 200 88 (57.5) 50 (65.8) 200 to 500 43 (28.1) 15 (19.7) > 500 21 (13.7) 11 (14.5) Missing 1 (0.7) 0 Diagnosis per WHO Criteria, n (%) MDS-RS ‡ 135 (88.2) 65 (85.5) MDS/MPN-RS-T 14 (9.2) 9 (11.8) Other § 4 (2.6) 2 (2.6) IPSS-R Classification Risk Category, n (%) Very low 18 (11.8) 6 (7.9) Low 109 (71.2) 57 (75) Intermediate 25 (16.3) 13 (17.1) High 1 (0.7) 0 RBC Transfusions/8 Weeks Over 16 Weeks Categories, n (%) < 4 units 46 (30.1) 20 (26.3) ≥ 4 and < 6 units 41 (26.8) 23 (30.3) ≥ 6 units 66 (43.1) 33 (43.4) The efficacy of REBLOZYL in adult patients with MDS-RS and MDS-RS-T was established based upon the proportion of patients who were red blood cell transfusion independent (RBC-TI), defined as the absence of any RBC transfusion during any consecutive 8-week period occurring entirely within Weeks 1 through 24.
The efficacy results are shown in Tables 13 and 14.
Table 13: Efficacy Results in MEDALIST Endpoint REBLOZYL
(N=153)
n, %
(95% CI)Placebo
(N=76)
n, %
(95% CI)Common Risk Difference
(95% CI)p-value - * The median (range) duration of treatment was 49 weeks (6 to 114 weeks) on the REBLOZYL arm and 24 weeks (7 to 89 weeks) on the placebo arm.
RBC-TI ≥ 8 weeks during Weeks 1-24 58 (37.9)
(30.2, 46.1)10 (13.2)
(6.5, 22.9)24.6
(14.5, 34.6)<0.0001 RBC-TI ≥ 12 weeks during Weeks 1-24 43 (28.1)
(21.1, 35.9)6 (7.9)
(3.0, 16.4)20.0
(10.9, 29.1)0.0002 RBC-TI ≥ 12 weeks during Weeks 1-48* 51 (33.3)
(25.9, 41.4)9 (11.8)
(5.6, 21.3)21.4
(11.2, 31.5)0.0003 Table 14 shows the proportion of patients who achieved RBC-TI ≥ 8 weeks during Weeks 1-24 by diagnosis and baseline transfusion requirement.
Table 14: RBC-TI ≥ 8 weeks during Weeks 1-24 By Diagnosis and Baseline Transfusion Burden in MEDALIST Responders / N % Response (95% CI) REBLOZYL Placebo REBLOZYL Placebo - * Includes MDS-EB-1, MDS-EB-2, and MDS-U.
- † Includes patients who received 3.5 units.
- ‡ Includes patients who received 5.5 units.
WHO 2016 Diagnosis MDS-RS 46 / 135 8 / 65 34.1 (26.1, 42.7) 12.3 (5.5, 22.8) MDS/MPN-RS-T 9 / 14 2 / 9 64.3 (35.1, 87.2) 22.2 (2.8, 60.0) Other * 3 / 4 0 / 2 75.0 (19.4, 99.4) 0.0 (0.0, 84.2) Baseline RBC Transfusion Burden 2 - 3 units/8 weeks † 37 / 46 8 / 20 80.4 (66.1, 90.6) 40.0 (19.1, 63.9) 4 - 5 units/8 weeks ‡ 15 / 41 1 / 23 36.6 (22.1, 53.1) 4.3 (0.1, 21.9) ≥ 6 units/8 weeks 6 / 66 1 / 33 9.1 (3.4, 18.7) 3.0 (0.1, 15.8) -
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
REBLOZYL (luspatercept-aamt) for injection is a white to off-white lyophilized powder supplied in a single-dose vial. Each carton contains one vial.
REBLOZYL 25 mg/vial (NDC: 59572-711-01)
REBLOZYL 75 mg/vial (NDC: 59572-775-01) -
17 PATIENT COUNSELING INFORMATION
Discuss the following with patients prior to and during treatment with REBLOZYL.
Thromboembolic Events
Advise beta thalassemia patients of the potential risk of thromboembolic events. Review known risk factors for developing thromboembolic events and advise patients to reduce modifiable risk factors (e.g., smoking, use of oral contraceptives) [see Warnings and Precautions (5.1)].
Effects on Blood Pressure
Caution patients that REBLOZYL may cause an increase in blood pressure [see Warnings and Precautions (5.2)].
Embryo-Fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception while receiving REBLOZYL and for at least 3 months after the final dose. Advise females to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, during treatment with REBLOZYL [see Warnings and Precautions (5.3) and Use in Specific Populations (8.1)].
Lactation
Advise females not to breastfeed during treatment with REBLOZYL and for 3 months after the final dose [see Use in Specific Populations (8.2)].
-
SPL UNCLASSIFIED SECTION
Manufactured by:
Celgene Corporation
86 Morris Avenue
Summit, NJ 07901
U.S. License No. 2114Jointly Marketed by:
Acceleron Pharma, Inc.
Cambridge, MA 02139REBLOZYL® is a registered trademark of Celgene Corporation.
Patent: www.celgene.com/therapies
© 2016-2020 Celgene Corporation.
All Rights Reserved.
REBPI.002
-
PATIENT PACKAGE INSERT
-
PRINCIPAL DISPLAY PANEL - 25 mg Vial Label
NDC: 59572-711-01
Reblozyl®
(luspatercept-aamt)
for Injection25 mg/vial
For Subcutaneous Use Only
Reconstitute prior
to administrationLOT
EXP
-
PRINCIPAL DISPLAY PANEL - 25 mg Vial Carton
NDC: 59572-711-01
Rx onlyReblozyl®
(luspatercept-aamt)
for Injection25 mg/vial
For Subcutaneous Use Only
Reconstitute with Sterile Water
for Injection USP, prior to
administration.One Single-Dose Vial
Discard Unused Portion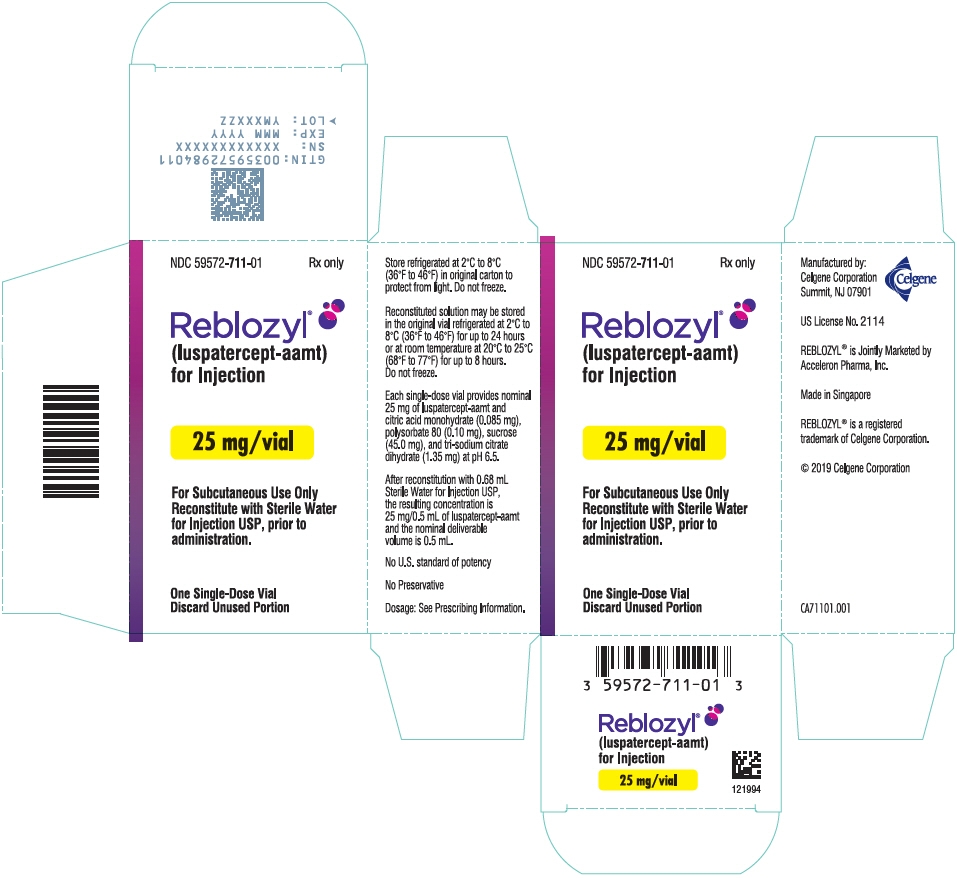
-
PRINCIPAL DISPLAY PANEL - 75 mg Vial Label
NDC: 59572-775-01
Reblozyl®
(luspatercept-aamt)
for Injection75 mg/vial
For Subcutaneous Use Only
Reconstitute prior
to administrationLOT
EXP
-
PRINCIPAL DISPLAY PANEL - 75 mg Vial Carton
NDC: 59572-775-01
Rx onlyReblozyl®
(luspatercept-aamt)
for Injection75 mg/vial
For Subcutaneous Use Only
Reconstitute with Sterile Water
for Injection USP, prior to
administration.One Single-Dose Vial
Discard Unused Portion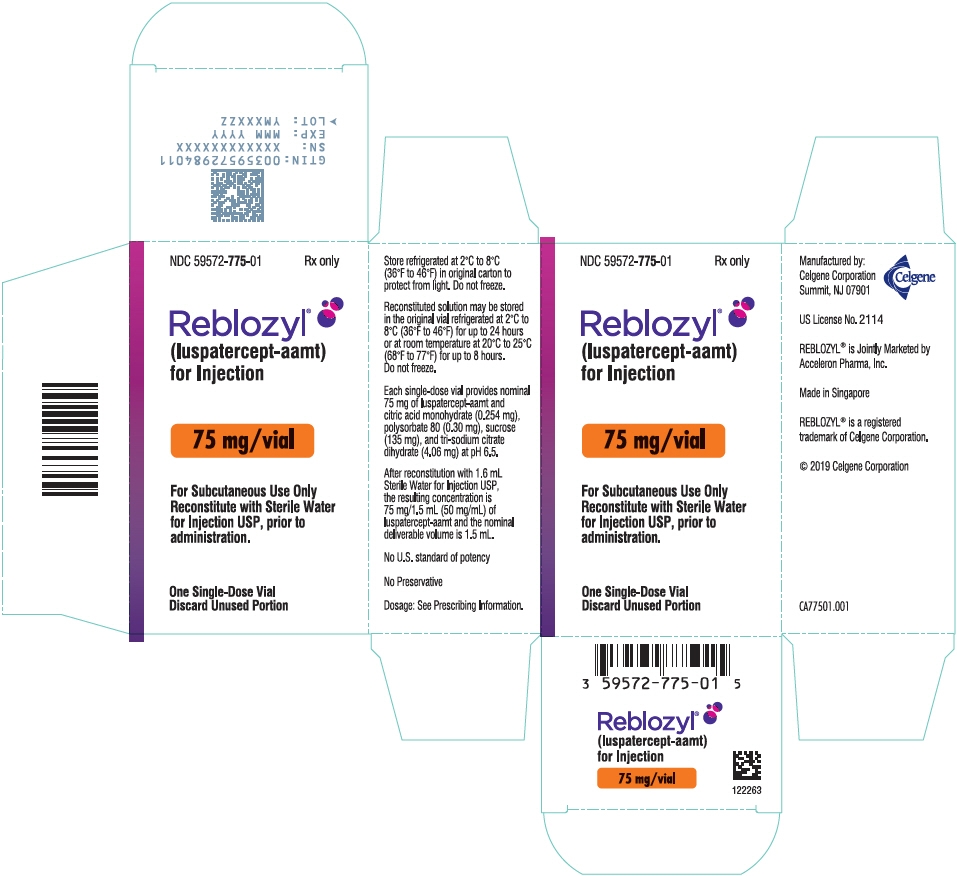
-
INGREDIENTS AND APPEARANCE
REBLOZYL
luspatercept injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59572-711 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LUSPATERCEPT (UNII: AQK7UBA1LS) (LUSPATERCEPT - UNII:AQK7UBA1LS) LUSPATERCEPT 25 mg Inactive Ingredients Ingredient Name Strength CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) TRISODIUM CITRATE DIHYDRATE (UNII: B22547B95K) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SUCROSE (UNII: C151H8M554) HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R) Product Characteristics Color WHITE (white to off-white) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59572-711-01 1 in 1 CARTON 11/08/2019 1 1 in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761136 11/08/2019 REBLOZYL
luspatercept injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59572-775 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LUSPATERCEPT (UNII: AQK7UBA1LS) (LUSPATERCEPT - UNII:AQK7UBA1LS) LUSPATERCEPT 75 mg Inactive Ingredients Ingredient Name Strength CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) TRISODIUM CITRATE DIHYDRATE (UNII: B22547B95K) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SUCROSE (UNII: C151H8M554) HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R) Product Characteristics Color WHITE (white to off-white) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59572-775-01 1 in 1 CARTON 11/08/2019 1 1 in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761136 11/08/2019 Labeler - Celgene (174201137) Registrant - Celgene Corporation (174201137)
Trademark Results [Reblozyl]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 REBLOZYL 90033523 not registered Live/Pending |
Celgene Corporation 2020-07-02 |
 REBLOZYL 88822744 not registered Live/Pending |
Celgene Corporation 2020-03-05 |
 REBLOZYL 87329959 5321484 Live/Registered |
Celgene Corporation 2017-02-09 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.