INLYTA- axitinib tablet, film coated
INLYTA by
Drug Labeling and Warnings
INLYTA by is a Prescription medication manufactured, distributed, or labeled by Pfizer Laboratories Div Pfizer Inc, Upjohn Manufacturing Ireland Unlimited Company, Pfizer Manufacturing Deutschland GmbH, Pfizer Ireland Pharmaceuticals. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use INLYTA safely and effectively. See full prescribing information for INLYTA.
INLYTA® (axitinib) tablets for oral administration
Initial U.S. Approval: 2012RECENT MAJOR CHANGES
Warnings and Precautions, Risk of Impaired Wound Healing (5.8) 01/2020 INDICATIONS AND USAGE
INLYTA is a kinase inhibitor indicated for the treatment of advanced renal cell carcinoma after failure of one prior systemic therapy. (1)
DOSAGE AND ADMINISTRATION
- The starting dose is 5 mg orally twice daily. Dose adjustments can be made based on individual safety and tolerability. (2.1, 2.2)
- Administer INLYTA dose approximately 12 hours apart with or without food. (2.1)
- INLYTA should be swallowed whole with a glass of water. (2.1)
- If a strong CYP3A4/5 inhibitor is required, decrease the INLYTA dose by approximately half. (2.2)
- For patients with moderate hepatic impairment, decrease the starting dose by approximately half. (2.2)
DOSAGE FORMS AND STRENGTHS
1 mg and 5 mg tablets (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Hypertension and Hypertensive Crisis: Hypertension including hypertensive crisis has been observed. Blood pressure should be well-controlled prior to initiating INLYTA. Monitor for hypertension and treat as needed. For persistent hypertension despite use of anti-hypertensive medications, reduce the INLYTA dose. (5.1)
- Arterial and Venous Thromboembolic Events: Arterial and venous thrombotic events have been observed and can be fatal. Use with caution in patients who are at increased risk for these events. (5.2, 5.3)
- Hemorrhage: Hemorrhagic events, including fatal events, have been reported. INLYTA has not been studied in patients with evidence of untreated brain metastasis or recent active gastrointestinal bleeding and should not be used in those patients. (5.4)
- Cardiac Failure: Cardiac failure has been observed and can be fatal. Monitor for signs or symptoms of cardiac failure throughout treatment with INLYTA. (5.5)
- Gastrointestinal Perforation and Fistula Formation: Gastrointestinal perforation and fistula, including death, have occurred. Use with caution in patients at risk for gastrointestinal perforation or fistula. (5.6)
- Hypothyroidism: Hypothyroidism requiring thyroid hormone replacement has been reported. Monitor thyroid function before initiation of, and periodically throughout, treatment with INLYTA. (5.7)
- Risk of Impaired Wound Healing: Withhold INLYTA for at least 2 days prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of INLYTA after resolution of wound healing complications has not been established. (5.8)
- Reversible Posterior Leukoencephalopathy Syndrome (RPLS): RPLS has been observed. Permanently discontinue INLYTA if signs or symptoms of RPLS occur. (5.9)
- Proteinuria: Monitor for proteinuria before initiation of, and periodically throughout, treatment with INLYTA. For moderate to severe proteinuria, reduce the dose or temporarily interrupt treatment with INLYTA. (5.10)
- Elevation of Liver Enzymes: Liver enzyme elevation has been observed during treatment with INLYTA. Monitor ALT, AST and bilirubin before initiation of, and periodically throughout, treatment with INLYTA. (5.11)
- Hepatic Impairment: The starting dose of INLYTA should be decreased if used in patients with moderate hepatic impairment. INLYTA has not been studied in patients with severe hepatic impairment. (2.2, 5.12)
- Embryo-Fetal Toxicity: INLYTA can cause fetal harm. Advise patients of the potential risk to the fetus and to use effective contraception. (5.13, 8.1, 8.3)
ADVERSE REACTIONS
The most common (≥20%) adverse reactions are diarrhea, hypertension, fatigue, decreased appetite, nausea, dysphonia, palmar-plantar erythrodysesthesia (hand-foot) syndrome, weight decreased, vomiting, asthenia, and constipation. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer, Inc at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 1/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
2.2 Dose Modification Guidelines
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypertension and Hypertensive Crisis
5.2 Arterial Thromboembolic Events
5.3 Venous Thromboembolic Events
5.4 Hemorrhage
5.5 Cardiac Failure
5.6 Gastrointestinal Perforation and Fistula Formation
5.7 Thyroid Dysfunction
5.8 Risk of Impaired Wound Healing
5.9 Reversible Posterior Leukoencephalopathy Syndrome
5.10 Proteinuria
5.11 Elevation of Liver Enzymes
5.12 Hepatic Impairment
5.13 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 CYP3A4/5 Inhibitors
7.2 CYP3A4/5 Inducers
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
The recommended starting oral dose of INLYTA is 5 mg twice daily. Administer INLYTA doses approximately 12 hours apart with or without food [see Clinical Pharmacology (12.3)]. INLYTA should be swallowed whole with a glass of water.
If the patient vomits or misses a dose, an additional dose should not be taken. The next prescribed dose should be taken at the usual time.
2.2 Dose Modification Guidelines
Dose increase or reduction is recommended based on individual safety and tolerability.
Over the course of treatment, patients who tolerate INLYTA for at least two consecutive weeks with no adverse reactions >Grade 2 (according to the Common Toxicity Criteria for Adverse Events [CTCAE]), are normotensive, and are not receiving anti-hypertension medication, may have their dose increased. When a dose increase from 5 mg twice daily is recommended, the INLYTA dose may be increased to 7 mg twice daily, and further to 10 mg twice daily using the same criteria.
Over the course of treatment, management of some adverse drug reactions may require temporary interruption or permanent discontinuation and/or dose reduction of INLYTA therapy [see Warnings and Precautions (5)]. If dose reduction from 5 mg twice daily is required, the recommended dose is 3 mg twice daily. If additional dose reduction is required, the recommended dose is 2 mg twice daily.
Strong CYP3A4/5 Inhibitors
The concomitant use of strong CYP3A4/5 inhibitors should be avoided (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, and voriconazole). Selection of an alternate concomitant medication with no or minimal CYP3A4/5 inhibition potential is recommended. Although INLYTA dose adjustment has not been studied in patients receiving strong CYP3A4/5 inhibitors, if a strong CYP3A4/5 inhibitor must be co-administered, a dose decrease of INLYTA by approximately half is recommended, as this dose reduction is predicted to adjust the axitinib area under the plasma concentration vs time curve (AUC) to the range observed without inhibitors. The subsequent doses can be increased or decreased based on individual safety and tolerability. If co-administration of the strong inhibitor is discontinued, the INLYTA dose should be returned (after 3 – 5 half-lives of the inhibitor) to that used prior to initiation of the strong CYP3A4/5 inhibitor [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
Hepatic Impairment
No starting dose adjustment is required when administering INLYTA to patients with mild hepatic impairment (Child-Pugh class A). Based on the pharmacokinetic data, the INLYTA starting dose should be reduced by approximately half in patients with baseline moderate hepatic impairment (Child-Pugh class B). The subsequent doses can be increased or decreased based on individual safety and tolerability. INLYTA has not been studied in patients with severe hepatic impairment (Child-Pugh class C) [see Warnings and Precautions (5.12), Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypertension and Hypertensive Crisis
In a controlled clinical study with INLYTA for the treatment of patients with RCC, hypertension was reported in 145/359 patients (40%) receiving INLYTA and 103/355 patients (29%) receiving sorafenib. Grade 3/4 hypertension was observed in 56/359 patients (16%) receiving INLYTA and 39/355 patients (11%) receiving sorafenib. Hypertensive crisis was reported in 2/359 patients (<1%) receiving INLYTA and none of the patients receiving sorafenib. The median onset time for hypertension (systolic blood pressure >150 mmHg or diastolic blood pressure >100 mmHg) was within the first month of the start of INLYTA treatment and blood pressure increases have been observed as early as 4 days after starting INLYTA. Hypertension was managed with standard antihypertensive therapy. Discontinuation of INLYTA treatment due to hypertension occurred in 1/359 patients (<1%) receiving INLYTA and none of the patients receiving sorafenib [see Adverse Reactions (6.1)].
Blood pressure should be well-controlled prior to initiating INLYTA. Patients should be monitored for hypertension and treated as needed with standard anti-hypertensive therapy. In the case of persistent hypertension despite use of anti-hypertensive medications, reduce the INLYTA dose. Discontinue INLYTA if hypertension is severe and persistent despite anti-hypertensive therapy and dose reduction of INLYTA, and discontinuation should be considered if there is evidence of hypertensive crisis. If INLYTA is interrupted, patients receiving antihypertensive medications should be monitored for hypotension [see Dosage and Administration (2.2)].
5.2 Arterial Thromboembolic Events
In clinical trials, arterial thromboembolic events have been reported, including deaths. In a controlled clinical study with INLYTA for the treatment of patients with RCC, Grade 3/4 arterial thromboembolic events were reported in 4/359 patients (1%) receiving INLYTA and 4/355 patients (1%) receiving sorafenib. Fatal cerebrovascular accident was reported in 1/359 patients (<1%) receiving INLYTA and none of the patients receiving sorafenib [see Adverse Reactions (6.1)].
In clinical trials with INLYTA, arterial thromboembolic events (including transient ischemic attack, cerebrovascular accident, myocardial infarction, and retinal artery occlusion) were reported in 17/715 patients (2%), with two deaths secondary to cerebrovascular accident.
Use INLYTA with caution in patients who are at risk for, or who have a history of, these events. INLYTA has not been studied in patients who had an arterial thromboembolic event within the previous 12 months.
5.3 Venous Thromboembolic Events
In clinical trials, venous thromboembolic events have been reported, including deaths. In a controlled clinical study with INLYTA for the treatment of patients with RCC, venous thromboembolic events were reported in 11/359 patients (3%) receiving INLYTA and 2/355 patients (1%) receiving sorafenib. Grade 3/4 venous thromboembolic events were reported in 9/359 patients (3%) receiving INLYTA (including pulmonary embolism, deep vein thrombosis, retinal vein occlusion and retinal vein thrombosis) and 2/355 patients (1%) receiving sorafenib. Fatal pulmonary embolism was reported in 1/359 patients (<1%) receiving INLYTA and none of the patients receiving sorafenib. In clinical trials with INLYTA, venous thromboembolic events were reported in 22/715 patients (3%), with two deaths secondary to pulmonary embolism.
Use INLYTA with caution in patients who are at risk for, or who have a history of, these events. INLYTA has not been studied in patients who had a venous thromboembolic event within the previous 6 months.
5.4 Hemorrhage
In a controlled clinical study with INLYTA for the treatment of patients with RCC, hemorrhagic events were reported in 58/359 patients (16%) receiving INLYTA and 64/355 patients (18%) receiving sorafenib. Grade 3/4 hemorrhagic events were reported in 5/359 (1%) patients receiving INLYTA (including cerebral hemorrhage, hematuria, hemoptysis, lower gastrointestinal hemorrhage, and melena) and 11/355 (3%) patients receiving sorafenib. Fatal hemorrhage was reported in 1/359 patients (<1%) receiving INLYTA (gastric hemorrhage) and 3/355 patients (1%) receiving sorafenib.
INLYTA has not been studied in patients who have evidence of untreated brain metastasis or recent active gastrointestinal bleeding and should not be used in those patients. If any bleeding requires medical intervention, temporarily interrupt the INLYTA dose.
5.5 Cardiac Failure
In a controlled clinical study with INLYTA for the treatment of patients with RCC, cardiac failure was reported in 6/359 patients (2%) receiving INLYTA and 3/355 patients (1%) receiving sorafenib. Grade 3/4 cardiac failure was observed in 2/359 patients (1%) receiving INLYTA and 1/355 patients (<1%) receiving sorafenib. Fatal cardiac failure was reported in 2/359 patients (1%) receiving INLYTA and 1/355 patients (<1%) receiving sorafenib. Monitor for signs or symptoms of cardiac failure throughout treatment with INLYTA. Management of cardiac failure may require permanent discontinuation of INLYTA.
5.6 Gastrointestinal Perforation and Fistula Formation
In a controlled clinical study with INLYTA for the treatment of patients with RCC, gastrointestinal perforation was reported in 1/359 patients (<1%) receiving INLYTA and none of the patients receiving sorafenib. In clinical trials with INLYTA, gastrointestinal perforation was reported in 5/715 patients (1%), including one death. In addition to cases of gastrointestinal perforation, fistulas were reported in 4/715 patients (1%).
Monitor for symptoms of gastrointestinal perforation or fistula periodically throughout treatment with INLYTA.
5.7 Thyroid Dysfunction
In a controlled clinical study with INLYTA for the treatment of patients with RCC, hypothyroidism was reported in 69/359 patients (19%) receiving INLYTA and 29/355 patients (8%) receiving sorafenib. Hyperthyroidism was reported in 4/359 patients (1%) receiving INLYTA and 4/355 patients (1%) receiving sorafenib. In patients who had thyroid stimulating hormone (TSH) <5 μU/mL before treatment, elevations of TSH to ≥10 μU/mL occurred in 79/245 patients (32%) receiving INLYTA and 25/232 patients (11%) receiving sorafenib [see Adverse Reactions (6.1)].
Monitor thyroid function before initiation of, and periodically throughout, treatment with INLYTA. Treat hypothyroidism and hyperthyroidism according to standard medical practice to maintain euthyroid state.
5.8 Risk of Impaired Wound Healing
Impaired wound healing can occur in patients who receive drugs that inhibit the vascular endothelial growth factor (VEGF) signaling pathway. Therefore, INLYTA has the potential to adversely affect wound healing.
Withhold INLYTA for at least 2 days prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of INLYTA after resolution of wound healing complications has not been established.
5.9 Reversible Posterior Leukoencephalopathy Syndrome
In a controlled clinical study with INLYTA for the treatment of patients with RCC, reversible posterior leukoencephalopathy syndrome (RPLS) was reported in 1/359 patients (<1%) receiving INLYTA and none of the patients receiving sorafenib [see Adverse Reactions (6.1)]. There were two additional reports of RPLS in other clinical trials with INLYTA.
RPLS is a neurological disorder which can present with headache, seizure, lethargy, confusion, blindness and other visual and neurologic disturbances. Mild to severe hypertension may be present. Magnetic resonance imaging is necessary to confirm the diagnosis of RPLS. Discontinue INLYTA in patients developing RPLS. The safety of reinitiating INLYTA therapy in patients previously experiencing RPLS is not known.
5.10 Proteinuria
In a controlled clinical study with INLYTA for the treatment of patients with RCC, proteinuria was reported in 39/359 patients (11%) receiving INLYTA and 26/355 patients (7%) receiving sorafenib. Grade 3 proteinuria was reported in 11/359 patients (3%) receiving INLYTA and 6/355 patients (2%) receiving sorafenib [see Adverse Reactions (6.1)].
Monitoring for proteinuria before initiation of, and periodically throughout, treatment with INLYTA is recommended. For patients who develop moderate to severe proteinuria, reduce the dose or temporarily interrupt INLYTA treatment.
5.11 Elevation of Liver Enzymes
In a controlled clinical study with INLYTA for the treatment of patients with RCC, alanine aminotransferase (ALT) elevations of all grades occurred in 22% of patients on both arms, with Grade 3/4 events in <1% of patients on the INLYTA arm and 2% of patients on the sorafenib arm.
Monitor ALT, aspartate aminotransferase (AST) and bilirubin before initiation of and periodically throughout treatment with INLYTA.
5.12 Hepatic Impairment
The systemic exposure to axitinib was higher in subjects with moderate hepatic impairment (Child-Pugh class B) compared to subjects with normal hepatic function. A dose decrease is recommended when administering INLYTA to patients with moderate hepatic impairment (Child-Pugh class B). INLYTA has not been studied in patients with severe hepatic impairment (Child-Pugh class C) [see Dosage and Administration (2.2), Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)].
5.13 Embryo-Fetal Toxicity
Based on its mechanism of action and findings from animal studies, INLYTA can cause fetal harm when administered to a pregnant woman. There are no available human data to inform the drug-associated risk. In developmental toxicity studies in mice, axitinib was teratogenic, embryotoxic and fetotoxic at maternal exposures that were lower than human exposures at the recommended clinical dose.
Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception during treatment with INLYTA and for 1 week after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with INLYTA and for 1 week after the last dose [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed elsewhere in the labeling [see Warnings and Precautions (5)]:
- Hypertension and hypertensive crisis [see Warnings and Precautions (5.1)]
- Arterial thromboembolic events [see Warnings and Precautions (5.2)]
- Venous thromboembolic events [see Warnings and Precautions (5.3)]
- Hemorrhage [see Warnings and Precautions (5.4)]
- Cardiac failure [see Warnings and Precautions (5.5)]
- Gastrointestinal perforation and fistula formation [see Warnings and Precautions (5.6)]
- Thyroid dysfunction [see Warnings and Precautions (5.7)]
- Reversible posterior leukoencephalopathy syndrome [see Warnings and Precautions (5.9)]
- Proteinuria [see Warnings and Precautions (5.10)]
- Elevation of liver enzymes [see Warnings and Precautions (5.11)]
- Hepatic impairment [see Warnings and Precautions (5.12)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of INLYTA has been evaluated in 715 patients in monotherapy studies, which included 537 patients with advanced RCC. The data described [see Adverse Reactions (6.1)] reflect exposure to INLYTA in 359 patients with advanced RCC who participated in a randomized clinical study versus sorafenib [see Clinical Studies (14)].
The median duration of treatment was 6.4 months (range 0.03 to 22.0) for patients who received INLYTA and 5.0 months (range 0.03 to 20.1) for patients who received sorafenib. Dose modifications or temporary delay of treatment due to an adverse reaction occurred in 199/359 patients (55%) receiving INLYTA and 220/355 patients (62%) receiving sorafenib. Permanent discontinuation due to an adverse reaction occurred in 34/359 patients (9%) receiving INLYTA and 46/355 patients (13%) receiving sorafenib.
The most common (≥20%) adverse reactions observed following treatment with INLYTA were diarrhea, hypertension, fatigue, decreased appetite, nausea, dysphonia, palmar-plantar erythrodysesthesia (hand-foot) syndrome, weight decreased, vomiting, asthenia, and constipation. Table 1 presents adverse reactions reported in ≥10% patients who received INLYTA or sorafenib.
Table 1. Adverse Reactions Occurring in ≥10% of Patients Who Received INLYTA or Sorafenib Adverse Reaction* INLYTA Sorafenib (N=359) (N=355) All Grades† Grade 3/4 All Grades† Grade 3/4 % % % % - * Percentages are treatment-emergent, all-causality events
- † National Cancer Institute Common Terminology Criteria for Adverse Events, Version 3.0
Diarrhea 55 11 53 7 Hypertension 40 16 29 11 Fatigue 39 11 32 5 Decreased appetite 34 5 29 4 Nausea 32 3 22 1 Dysphonia 31 0 14 0 Palmar-plantar erythrodysesthesia syndrome 27 5 51 16 Weight decreased 25 2 21 1 Vomiting 24 3 17 1 Asthenia 21 5 14 3 Constipation 20 1 20 1 Hypothyroidism 19 <1 8 0 Cough 15 1 17 1 Mucosal inflammation 15 1 12 1 Arthralgia 15 2 11 1 Stomatitis 15 1 12 <1 Dyspnea 15 3 12 3 Abdominal pain 14 2 11 1 Headache 14 1 11 0 Pain in extremity 13 1 14 1 Rash 13 <1 32 4 Proteinuria 11 3 7 2 Dysgeusia 11 0 8 0 Dry skin 10 0 11 0 Dyspepsia 10 0 2 0 Pruritus 7 0 12 0 Alopecia 4 0 32 0 Erythema 2 0 10 <1 Selected adverse reactions (all grades) that were reported in <10% of patients treated with INLYTA included dizziness (9%), upper abdominal pain (8%), myalgia (7%), dehydration (6%), epistaxis (6%), anemia (4%), hemorrhoids (4%), hematuria (3%), tinnitus (3%), lipase increased (3%), glossodynia (3%), pulmonary embolism (2%), rectal hemorrhage (2%), hemoptysis (2%), deep vein thrombosis (1%), retinal-vein occlusion/thrombosis (1%), polycythemia (1%), and transient ischemic attack (1%).
Table 2 presents the most common laboratory abnormalities reported in ≥10% patients who received INLYTA or sorafenib.
Table 2. Laboratory Abnormalities Occurring in ≥10% of Patients Who Received INLYTA or Sorafenib Laboratory Abnormality N INLYTA N Sorafenib All Grades* Grade 3/4 All Grades* Grade 3/4 % % % % ALP: alkaline phosphatase; ALT: alanine aminotransferase; AST: aspartate aminotransferase - * National Cancer Institute Common Terminology Criteria for Adverse Events, Version 3.0
Hematology Hemoglobin decreased 320 35 <1 316 52 4 Lymphocytes (absolute) decreased 317 33 3 309 36 4 Platelets decreased 312 15 <1 310 14 0 White blood cells decreased 320 11 0 315 16 <1 Chemistry Creatinine increased 336 55 0 318 41 <1 Bicarbonate decreased 314 44 <1 291 43 0 Hypocalcemia 336 39 1 319 59 2 ALP increased 336 30 1 319 34 1 Hyperglycemia 336 28 2 319 23 2 Lipase increased 338 27 5 319 46 15 Amylase increased 338 25 2 319 33 2 ALT increased 331 22 <1 313 22 2 AST increased 331 20 <1 311 25 1 Hypernatremia 338 17 1 319 13 1 Hypoalbuminemia 337 15 <1 319 18 1 Hyperkalemia 333 15 3 314 10 3 Hypoglycemia 336 11 <1 319 8 <1 Hyponatremia 338 13 4 319 11 2 Hypophosphatemia 336 13 2 318 49 16 Selected laboratory abnormalities (all grades) that were reported in <10% of patients treated with INLYTA included hemoglobin increased (above the upper limit of normal) (9% for INLYTA versus 1% for sorafenib) and hypercalcemia (6% for INLYTA versus 2% for sorafenib).
-
7 DRUG INTERACTIONS
7.1 CYP3A4/5 Inhibitors
Co-administration of ketoconazole, a strong inhibitor of CYP3A4/5, increased the plasma exposure of axitinib in healthy volunteers. Co-administration of INLYTA with strong CYP3A4/5 inhibitors should be avoided. Grapefruit or grapefruit juice may also increase axitinib plasma concentrations and should be avoided. Selection of concomitant medication with no or minimal CYP3A4/5 inhibition potential is recommended. If a strong CYP3A4/5 inhibitor must be co-administered, the INLYTA dose should be reduced [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
7.2 CYP3A4/5 Inducers
Co-administration of rifampin, a strong inducer of CYP3A4/5, reduced the plasma exposure of axitinib in healthy volunteers. Co-administration of INLYTA with strong CYP3A4/5 inducers (e.g., rifampin, dexamethasone, phenytoin, carbamazepine, rifabutin, rifapentin, phenobarbital, and St. John's wort) should be avoided. Selection of concomitant medication with no or minimal CYP3A4/5 induction potential is recommended [see Dosage and Administration (2.2, Clinical Pharmacology (12.3)]. Moderate CYP3A4/5 inducers (e.g., bosentan, efavirenz, etravirine, modafinil, and nafcillin) may also reduce the plasma exposure of axitinib and should be avoided if possible.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animal studies and its mechanism of action, INLYTA can cause fetal harm when administered to a pregnant woman. There are no available human data to inform the drug-associated risk. In developmental toxicity studies, axitinib was teratogenic, embryotoxic and fetotoxic in mice at exposures lower than human exposures at the recommended starting dose (see Data). Advise females of reproductive potential of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated populations are unknown. However, the background risk in the United States (U.S.) general population of major birth defects is 2%–4% and of miscarriage is 15%–20% of clinically recognized pregnancies.
Data
Animal Data
Oral axitinib administered twice daily to female mice prior to mating and through the first week of pregnancy caused an increase in post-implantation loss at all doses tested (≥15 mg/kg/dose, approximately 10 times the systemic exposure (AUC) in patients at the recommended starting dose). In an embryo-fetal developmental toxicity study, pregnant mice received oral doses of 0.15, 0.5 and 1.5 mg/kg/dose axitinib twice daily during the period of organogenesis. Embryo-fetal toxicities observed in the absence of maternal toxicity included malformation (cleft palate) at 1.5 mg/kg/dose (approximately 0.5 times the AUC in patients at the recommended starting dose) and variation in skeletal ossification at ≥0.5 mg/kg/dose (approximately 0.15 times the AUC in patients at the recommended starting dose).
8.2 Lactation
Risk Summary
There are no data on the presence of axitinib in human milk, or its effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions in a breastfed child from INLYTA, advise lactating women not to breastfeed during treatment and for 2 weeks after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Based on findings in animal studies, INLYTA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Females of reproductive potential should have a pregnancy test prior to starting treatment with INLYTA.
Contraception
Females
INLYTA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with INLYTA and for 1 week after the last dose.
Infertility
Females and Males
Based on findings in animals, INLYTA may impair fertility in females and males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and efficacy of INLYTA in pediatric patients have not been studied.
Juvenile Animal Toxicity Data
Toxicities in bone and teeth were observed in immature mice and dogs administered oral axitinib twice daily for 1 month or longer. Effects in bone consisted of thickened growth plates in mice and dogs at ≥15 mg/kg/dose (approximately 6 and 15 times, respectively, the systemic exposure (AUC) in patients at the recommended starting dose). Abnormalities in growing incisor teeth (including dental caries, malocclusions and broken and/or missing teeth) were observed in mice administered oral axitinib twice daily at ≥5 mg/kg/dose (approximately 1.5 times the AUC in patients at the recommended starting dose). Other toxicities of potential concern to pediatric patients have not been evaluated in juvenile animals.
8.5 Geriatric Use
In a controlled clinical study with INLYTA for the treatment of patients with RCC, 123/359 patients (34%) treated with INLYTA were ≥65 years of age. Although greater sensitivity in some older individuals cannot be ruled out, no overall differences were observed in the safety and effectiveness of INLYTA between patients who were ≥65 years of age and younger.
No dosage adjustment is required in elderly patients [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
In a dedicated hepatic impairment trial, compared to subjects with normal hepatic function, systemic exposure following a single dose of INLYTA was similar in subjects with baseline mild hepatic impairment (Child-Pugh class A) and higher in subjects with baseline moderate hepatic impairment (Child-Pugh class B).
No starting dose adjustment is required when administering INLYTA to patients with mild hepatic impairment (Child-Pugh class A). A starting dose decrease is recommended when administering INLYTA to patients with moderate hepatic impairment (Child-Pugh class B) [see Dosage and Administration (2.2), Warnings and Precautions (5.12), Clinical Pharmacology (12.3)].
INLYTA has not been studied in subjects with severe hepatic impairment (Child-Pugh class C).
8.7 Renal Impairment
No dedicated renal impairment trial for axitinib has been conducted. Based on the population pharmacokinetic analyses, no significant difference in axitinib clearance was observed in patients with pre-existing mild to severe renal impairment (15 mL/min ≤creatinine clearance [CLcr] <89 mL/min) [see Clinical Pharmacology (12.3)]. No starting dose adjustment is needed for patients with pre-existing mild to severe renal impairment. Caution should be used in patients with end-stage renal disease (CLcr <15 mL/min).
-
10 OVERDOSAGE
There is no specific treatment for INLYTA overdose.
In a controlled clinical study with INLYTA for the treatment of patients with RCC, 1 patient inadvertently received a dose of 20 mg twice daily for 4 days and experienced dizziness (Grade 1).
In a clinical dose finding study with INLYTA, subjects who received starting doses of 10 mg twice daily or 20 mg twice daily experienced adverse reactions which included hypertension, seizures associated with hypertension, and fatal hemoptysis.
In cases of suspected overdose, INLYTA should be withheld and supportive care instituted.
-
11 DESCRIPTION
INLYTA (axitinib) is a kinase inhibitor. Axitinib has the chemical name N-methyl-2-[3-((E)-2-pyridin-2-yl-vinyl)-1H-indazol-6-ylsulfanyl]-benzamide. The molecular formula is C22H18N4OS and the molecular weight is 386.47 Daltons. The chemical structure is:
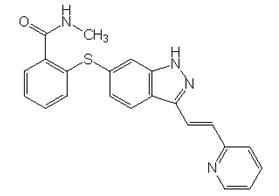
Axitinib is a white to light-yellow powder with a pKa of 4.8. The solubility of axitinib in aqueous media over the range pH 1.1 to pH 7.8 is in excess of 0.2 µg/mL. The partition coefficient (n-octanol/water) is 3.5.
INLYTA is supplied as red, film-coated tablets containing either 1 mg or 5 mg of axitinib together with microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, magnesium stearate, and Opadry® II red 32K15441 as inactive ingredients. The Opadry II red 32K15441 film coating contains lactose monohydrate, HPMC 2910/Hypromellose 15cP, titanium dioxide, triacetin (glycerol triacetate), and red iron oxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Axitinib has been shown to inhibit receptor tyrosine kinases including vascular endothelial growth factor receptors (VEGFR)-1, VEGFR-2, and VEGFR-3 at therapeutic plasma concentrations. These receptors are implicated in pathologic angiogenesis, tumor growth, and cancer progression. VEGF-mediated endothelial cell proliferation and survival were inhibited by axitinib in vitro and in mouse models. Axitinib was shown to inhibit tumor growth and phosphorylation of VEGFR-2 in tumor xenograft mouse models.
12.2 Pharmacodynamics
The effect of a single oral dose of INLYTA (5 mg) in the absence and presence of 400 mg ketoconazole on the QTc interval was evaluated in a randomized, single-blinded, two-way crossover study in 35 healthy subjects. No large changes in mean QTc interval (i.e., >20 ms) from placebo were detected up to 3 hours post-dose. However, small increases in mean QTc interval (i.e., <10 ms) cannot be ruled out.
12.3 Pharmacokinetics
The population pharmacokinetic analysis pooled data from 17 trials in healthy subjects and patients with cancer. A two-compartment disposition model with first-order absorption and lag-time adequately describes the axitinib concentration-time profile.
Absorption and Distribution
Following single oral 5-mg dose administration, the median Tmax ranged from 2.5 to 4.1 hours. Based on the plasma half-life, steady state is expected within 2 to 3 days of dosing. Dosing of axitinib at 5 mg twice daily resulted in approximately 1.4-fold accumulation compared to administration of a single dose. At steady state, axitinib exhibits approximately linear pharmacokinetics within the 1-mg to 20-mg dose range. The mean absolute bioavailability of axitinib after an oral 5 mg dose is 58%.
Compared to overnight fasting, administration of INLYTA with a moderate fat meal resulted in 10% lower AUC and a high fat, high-calorie meal resulted in 19% higher AUC. INLYTA can be administered with or without food [see Dosage and Administration (2.1)].
Axitinib is highly bound (>99%) to human plasma proteins with preferential binding to albumin and moderate binding to α1-acid glycoprotein. In patients with advanced RCC (n=20), at the 5 mg twice daily dose in the fed state, the geometric mean (CV%) Cmax and AUC0–24 were 27.8 (79%) ng/mL and 265 (77%) ng.h/mL, respectively. The geometric mean (CV%) clearance and apparent volume of distribution were 38 (80%) L/h and 160 (105%) L, respectively.
Metabolism and Elimination
The plasma half-life of INLYTA ranges from 2.5 to 6.1 hours. Axitinib is metabolized primarily in the liver by CYP3A4/5 and to a lesser extent by CYP1A2, CYP2C19, and UGT1A1. Following oral administration of a 5-mg radioactive dose of axitinib, approximately 41% of the radioactivity was recovered in feces and approximately 23% was recovered in urine. Unchanged axitinib, accounting for 12% of the dose, was the major component identified in feces. Unchanged axitinib was not detected in urine; the carboxylic acid and sulfoxide metabolites accounted for the majority of radioactivity in urine. In plasma, the N-glucuronide metabolite represented the predominant radioactive component (50% of circulating radioactivity) and unchanged axitinib and the sulfoxide metabolite each accounted for approximately 20% of the circulating radioactivity.
The sulfoxide and N-glucuronide metabolites show approximately ≥400-fold less in vitro potency against VEGFR-2 compared to axitinib.
Drug-Drug Interactions
Effects of Other Drugs on INLYTA
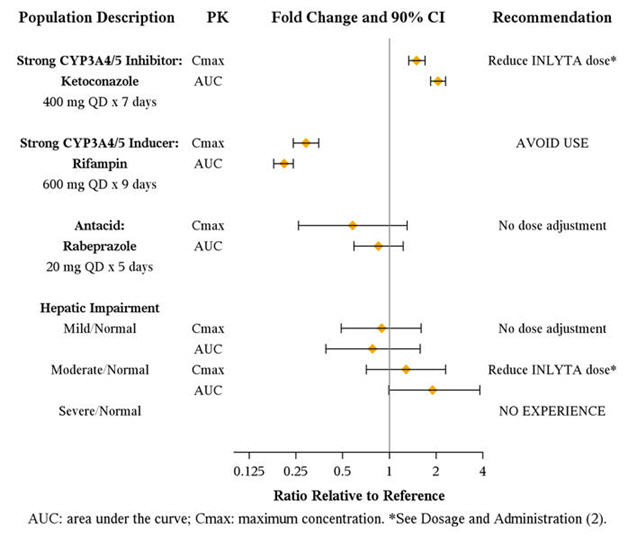
Axitinib is metabolized primarily in the liver by CYP3A4/5. Additionally, the aqueous solubility of axitinib is pH dependent, with higher pH resulting in lower solubility. The effects of a strong CYP3A4/5 inhibitor, a strong CYP3A4/5 inducer, and an antacid on the pharmacokinetics of axitinib are presented in Figure 1 [see Dosage and Administration (2.2) and Drug Interactions (7.1, 7.2)].
Figure 1. Impact of Co-administered Drugs and Hepatic Impairment on Axitinib Pharmacokinetics
Effects of INLYTA on Other Drugs
In vitro studies demonstrated that axitinib has the potential to inhibit CYP1A2 and CYP2C8. However, co-administration of axitinib with paclitaxel, a CYP2C8 substrate, did not increase plasma concentrations of paclitaxel in patients.
In vitro studies indicated that axitinib does not inhibit CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, or UGT1A1 at therapeutic plasma concentrations. In vitro studies in human hepatocytes indicated that axitinib does not induce CYP1A1, CYP1A2, or CYP3A4/5.
Axitinib is an inhibitor of the efflux transporter P-glycoprotein (P-gp) in vitro. However, INLYTA is not expected to inhibit P-gp at therapeutic plasma concentrations.
Specific Populations
Hepatic Impairment
The effects of hepatic impairment on the pharmacokinetics of axitinib are presented in Figure 1 [see Dosage and Administration (2.2), Warnings and Precautions (5.12), Use in Specific Populations (8.6)].
Renal Impairment
Population pharmacokinetic analysis (based on pre-existing renal function) was carried out in 590 healthy volunteers and patients, including five with severe renal impairment (15 mL/min ≤CLcr <29 mL/min), 64 with moderate renal impairment (30 mL/min ≤CLcr <59 mL/min), and 139 with mild renal impairment (60 mL/min ≤CLcr <89 mL/min). Mild to severe renal impairment did not have meaningful effects on the pharmacokinetics of axitinib. Data from only one patient with end-stage renal disease are available [see Use in Specific Populations (8.7)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with axitinib.
Axitinib was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay and was not clastogenic in the in vitro human lymphocyte chromosome aberration assay. Axitinib was genotoxic in the in vivo mouse bone marrow micronucleus assay.
INLYTA has the potential to impair reproductive function and fertility in humans. In repeat-dose toxicology studies, findings in the male reproductive tract were observed in the testes/epididymis (decreased organ weight, atrophy or degeneration, decreased numbers of germinal cells, hypospermia or abnormal sperm forms, reduced sperm density and count) at ≥15 mg/kg/dose administered orally twice daily in mice (approximately 7 times the systemic exposure (AUC) in patients at the recommended starting dose) and ≥1.5 mg/kg/dose administered orally twice daily in dogs (approximately 0.1 times the AUC in patients at the recommended starting dose). Findings in the female reproductive tract in mice and dogs included signs of delayed sexual maturity, reduced or absent corpora lutea, decreased uterine weights and uterine atrophy at ≥5 mg/kg/dose (approximately 1.5 or 0.3 times the AUC in patients at the recommended starting dose compared to mice and dogs, respectively).
In a fertility study in mice, axitinib did not affect mating or fertility rate when administered orally twice daily to males at any dose tested up to 50 mg/kg/dose following at least 70 days of administration (approximately 57 times the AUC in patients at the recommended starting dose). In female mice, reduced fertility and embryonic viability were observed at all doses tested (≥15 mg/kg/dose administered orally twice daily) following at least 15 days of treatment with axitinib (approximately 10 times the AUC in patients at the recommended starting dose).
-
14 CLINICAL STUDIES
The safety and efficacy of INLYTA were evaluated in a randomized, open-label, multicenter Phase 3 study. Patients (N=723) with advanced RCC whose disease had progressed on or after treatment with 1 prior systemic therapy, including sunitinib-, bevacizumab-, temsirolimus-, or cytokine-containing regimens were randomized (1:1) to receive INLYTA (N=361) or sorafenib (N=362). Progression-free survival (PFS) was assessed by a blinded independent central review committee. Other endpoints included objective response rate (ORR) and overall survival (OS).
Of the patients enrolled in this study, 389 patients (54%) had received 1 prior sunitinib-based therapy, 251 patients (35%) had received 1 prior cytokine-based therapy (interleukin-2 or interferon-alfa), 59 patients (8%) had received 1 prior bevacizumab-based therapy, and 24 patients (3%) had received 1 prior temsirolimus-based therapy. The baseline demographic and disease characteristics were similar between the INLYTA and sorafenib groups with regard to age (median 61 years), gender (72% male), race (75% white, 21% Asian), Eastern Cooperative Oncology Group (ECOG) performance status (55% 0, 45% 1), and histology (99% clear cell).
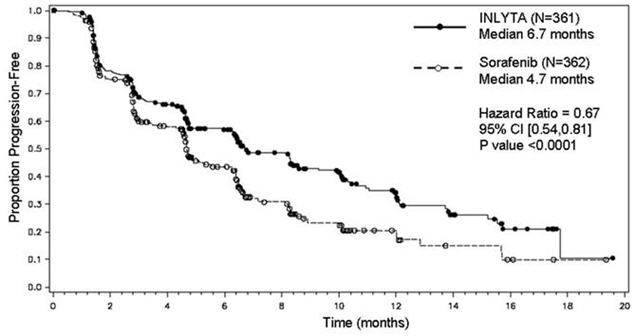
There was a statistically significant advantage for INLYTA over sorafenib for the endpoint of PFS (see Table 3 and Figure 2). There was no statistically significant difference between the arms in OS.
Table 3. Efficacy Results Endpoint/Study Population INLYTA Sorafenib HR (95% CI) P-value CI: Confidence interval; HR: Hazard ratio (INLYTA/sorafenib); ITT: Intent to treat; ORR: Objective response rate; NS: Not significant; OS: Overall survival; PFS: Progression-free survival - * Time from randomization to progression or death due to any cause, whichever occurs first.
- † Assessed by independent radiology review according to RECIST.
- ‡ One-sided p-value from a log-rank test of treatment stratified by ECOG performance status and prior therapy (comparison is considered statistically significant if the one-sided p-value is <0.023).
- § Risk ratio is used for ORR. A risk ratio >1 indicated a higher likelihood of responding in the axitinib arm; a risk ratio <1 indicated a higher likelihood of responding in the sorafenib arm.
- ¶ P-value not included since it was not adjusted for multiple testing.
Overall ITT N= 361 N = 362 Median PFS*,† in months (95% CI) 6.7 (6.3, 8.6) 4.7 (4.6, 5.6) 0.67 (0.54, 0.81) <0.0001‡ Median OS in months (95% CI) 20.1 (16.7, 23.4) 19.2 (17.5, 22.3) 0.97 (0.80, 1.17) NS ORR % (95% CI) 19.4 (15.4, 23.9) 9.4 (6.6, 12.9) 2.06§ (1.41, 3.00) -¶ PFS by prior treatment Sunitinib-refractory subgroup N=194 N=195 Median, months (95% CI) 4.8 (4.5, 6.4) 3.4 (2.8, 4.7) 0.74 (0.57, 0.96) -¶ Cytokine-refractory subgroup N=126 N=125 Median, months (95% CI) 12.1 (10.1, 13.9) 6.5 (6.3, 8.3) 0.46 (0.32, 0.68) -¶ Figure 2. Kaplan-Meier Curve for Progression Free Survival by Independent Assessment (Intent-to-Treat Population)
-
16 HOW SUPPLIED/STORAGE AND HANDLING
INLYTA tablets are supplied as follows:
- 1 mg tablets are red film-coated, oval tablets debossed with "Pfizer" on one side and "1 XNB" on the other; available in bottles of 180: NDC: 0069-0145-01.
- 5 mg tablets are red film-coated, triangular tablets debossed with "Pfizer" on one side and "5 XNB" on the other; available in bottles of 60: NDC: 0069-0151-11.
- Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Hypertension
Advise patients that hypertension may develop during INLYTA treatment and that blood pressure should be monitored regularly during treatment [see Warnings and Precautions (5.1)].
Arterial/Venous Thromboembolic Events
Advise patients that arterial and venous thromboembolic events have been observed during INLYTA treatment and to inform their doctor if they experience symptoms suggestive of thromboembolic events [see Warnings and Precautions (5.2, 5.3)].
Hemorrhage
Advise patients that INLYTA may increase the risk of bleeding and to promptly inform their doctor of any bleeding episodes [see Warnings and Precautions (5.4)].
Cardiac Failure
Advise patients that cardiac failure may develop during INLYTA treatment and that signs or symptoms of cardiac failure should be regularly monitored for during treatment [see Warnings and Precautions (5.5)].
Gastrointestinal Disorders
Advise patients that gastrointestinal disorders such as diarrhea, nausea, vomiting, and constipation may develop during INLYTA treatment and to seek immediate medical attention if they experience persistent or severe abdominal pain because cases of gastrointestinal perforation and fistula have been reported in patients taking INLYTA [see Warnings and Precautions (5.6) and Adverse Reactions (6.1)].
Abnormal Thyroid Function
Advise patients that abnormal thyroid function may develop during INLYTA treatment and to inform their doctor if symptoms of abnormal thyroid function occur [see Warnings and Precautions (5.7)].
Risk of Impaired Wound Healing
Advise patients that INLYTA may impair wound healing. Advise patients to inform their healthcare provider of any planned surgical procedure [see Warnings and Precautions (5.8)].
Reversible Posterior Leukoencephalopathy Syndrome
Advise patients to inform their doctor if they have worsening of neurological function consistent with RPLS (headache, seizure, lethargy, confusion, blindness and other visual and neurologic disturbances) [see Warnings and Precautions (5.9)].
Embryo-Fetal Toxicity
Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of the pregnancy [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with INLYTA and for 1 week after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 1 week following the last dose [see Warnings and Precautions (5.13) and Use in Specific Populations (8.3)].
Lactation
Advise patients not to breastfeed while taking INLYTA and for 2 weeks after receiving the last dose [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: January 2020 PATIENT INFORMATION
INLYTA® (in-ly-ta)
(axitinib)
tabletsWhat is INLYTA?
INLYTA is a prescription medicine used to treat advanced kidney cancer (advanced renal cell carcinoma or RCC) when 1 prior drug treatment regimen for your RCC has not worked.
It is not known if INLYTA is safe and effective in children.Before taking INLYTA, tell your healthcare provider about all of your medical conditions, including if you: - have high blood pressure
- have thyroid problems
- have liver problems
- have a history of blood clots in your veins or arteries (types of blood vessels), including stroke, heart attack, or change in vision
- have any bleeding problems
- have a history of heart failure
- have an unhealed wound
- plan to have surgery or have had a recent surgery. You should stop taking INLYTA for at least 2 days before planned surgery. See "What are the possible side effects of INLYTA?"
- have any other medical conditions
- are pregnant or plan to become pregnant. Taking INLYTA during pregnancy can harm your unborn baby. You should not become pregnant while taking INLYTA. Talk to your healthcare provider if you are pregnant or plan to become pregnant.
- are able to become pregnant. You should have a pregnancy test before you start treatment with INLYTA. Use effective birth control during treatment and for at least 1 week after your last dose of INLYTA. Talk to your healthcare provider about birth control methods that you can use to prevent pregnancy during this time.
- are breastfeeding or plan to breastfeed. It is not known if INLYTA passes into your breast milk. Do not breastfeed during treatment and for at least 2 weeks after your last dose of INLYTA.
- Use effective birth control during treatment and for at least 1 week after your last dose of INLYTA.
- If your female partner becomes pregnant during your treatment with INLYTA, tell your healthcare provider right away.
Talk with your healthcare provider before you start taking any new medicine. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.How should I take INLYTA? - Take INLYTA exactly as prescribed by your healthcare provider.
- Your healthcare provider may change your dose if needed.
- INLYTA can be taken with or without food.
- Take INLYTA 2 times a day about 12 hours apart.
- Swallow INLYTA tablets whole with a glass of water.
- Your healthcare provider should check your blood pressure regularly during treatment with INLYTA.
- If you vomit or miss a dose of INLYTA, take your next dose at your regular time. Do not take two doses at the same time.
- If you take too much INLYTA, call your healthcare provider or go to the nearest hospital emergency room right away.
What should I avoid while taking INLYTA? - Do not drink grapefruit juice or eat grapefruit. Grapefruit may increase the amount of INLYTA in your blood.
What are the possible side effects of INLYTA?
INLYTA may cause serious side effects, including:- High blood pressure (hypertension). High blood pressure is common with INLYTA, and may sometimes be severe. Your healthcare provider should check your blood pressure regularly during treatment with INLYTA. If you develop blood pressure problems, your healthcare provider may prescribe medicine to treat your high blood pressure, lower your dose, or stop your treatment with INLYTA.
- Problem with blood clots in your veins or arteries. INLYTA can cause blood clots which can be serious, and sometimes lead to death. Get emergency help and call your healthcare provider if you get any of the following symptoms:
- chest pain or pressure
- pain in your arms, back, neck or jaw
- shortness of breath
- numbness or weakness on one side of your body
- trouble talking
- headache
- vision changes
-
Bleeding. INLYTA can cause bleeding which can be serious, and sometimes lead to death. Call your healthcare provider right away or get medical help if you develop any of the following signs or symptoms:
- unexpected bleeding or bleeding that lasts a long time, such as:
- unusual bleeding from the gums
- menstrual bleeding or vaginal bleeding that is heavier than normal
- bleeding that is severe or you cannot control
- pink or brown urine
- red or black stools (looks like tar)
- bruises that happen without a known cause or get larger
- cough up blood or blood clots
- vomit blood or your vomit looks like "coffee grounds"
-
- unexpected pain, swelling, or joint pain
- headaches, feeling dizzy or weak
- Heart failure. Your healthcare provider should check you for signs or symptoms of heart failure regularly during treatment with INLYTA. Heart failure can be serious and can sometimes lead to death. Tell your healthcare provider if you have any of the following symptoms during your treatment with INLYTA:
- tiredness
- swelling of your stomach-area (abdomen), legs or ankles
- shortness of breath
- protruding neck veins
-
Tear in your stomach or intestinal wall (perforation). A tear in your stomach or intestinal wall can be serious and can sometimes lead to death. Get medical help right away if you get the following symptoms:
- severe stomach-area (abdominal) pain or stomach-area pain that does not go away
- vomit blood
- red or black stools
- Thyroid gland problems. Your healthcare provider should do blood tests to check your thyroid gland function before and during your treatment with INLYTA. Tell your healthcare provider if you have any of the following symptoms during your treatment with INLYTA:
- tiredness that worsens or that does not go away
- feeling hot or cold
- your voice deepens
- weight gain or weight loss
- hair loss
- muscle cramps and aches
-
Risk of wound healing problems. Wounds may not heal properly during INLYTA treatment. Tell your healthcare provider if you plan to have any surgery before starting or during treatment with INLYTA.
- You should stop taking INLYTA at least 2 days before planned surgery.
- Your healthcare provider should tell you when you may start taking INLYTA again after surgery.
- Reversible Posterior Leukoencephalopathy Syndrome (RPLS). A condition called reversible posterior leukoencephalopathy syndrome (RPLS) can happen while taking INLYTA. Call your healthcare provider right away if you get:
- headache
- seizures
- weakness
- confusion
- high blood pressure
- blindness or change in vision
- problems thinking
- Protein in your urine. Your healthcare provider should check your urine for protein before and during your treatment with INLYTA. If you develop protein in your urine, your healthcare provider may decrease your dose of INLYTA or stop your treatment.
- Change in liver function. Your healthcare provider should do blood tests before and during your treatment with INLYTA to check your liver function.
- diarrhea (frequent or loose bowel movements)
- tiredness or feeling weak
- decreased appetite
- nausea
- hoarseness
- rash, redness, itching or peeling of your skin on your hands and feet
- decreased weight
- vomiting
- constipation
INLYTA may cause fertility problems in males and females, which may affect your ability to have a child. Talk to your healthcare provider if this is a concern for you.
These are not all of the possible side effects of INLYTA
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store INLYTA? - Store INLYTA at room temperature between 68°F to 77°F (20°C to 25°C).
General information about the safe and effective use of INLYTA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use INLYTA for a condition for which it was not prescribed. Do not give INLYTA to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about INLYTA that is written for health professionals.What are the ingredients in INLYTA?
Active ingredient: axitinib
Inactive ingredients: microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, magnesium stearate, and Opadry® II red 32K15441. The Opadry II red 32K15441 film coating contains: lactose monohydrate, HPMC 2910/Hypromellose 15cP, titanium dioxide, triacetin (glycerol triacetate), and red iron oxide.
LAB-0439-5.0 For more information, go to www.inlyta.com or call 8770744-5675
This product's label may have been updated. For current full prescribing information, please visit www.pfizer.com. - PRINCIPAL DISPLAY PANEL - 1 mg Tablet Bottle Label
- PRINCIPAL DISPLAY PANEL - 5 mg Tablet Bottle Label
-
INGREDIENTS AND APPEARANCE
INLYTA
axitinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0069-0145 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength AXITINIB (UNII: C9LVQ0YUXG) (AXITINIB - UNII:C9LVQ0YUXG) AXITINIB 1 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIACETIN (UNII: XHX3C3X673) FERRIC OXIDE RED (UNII: 1K09F3G675) HYPROMELLOSE 2910 (15000 MPA.S) (UNII: 288VBX44JC) Product Characteristics Color RED Score no score Shape OVAL Size 9mm Flavor Imprint Code Pfizer;1;XNB Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0069-0145-01 180 in 1 BOTTLE; Type 0: Not a Combination Product 01/27/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA202324 01/27/2012 INLYTA
axitinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0069-0151 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength AXITINIB (UNII: C9LVQ0YUXG) (AXITINIB - UNII:C9LVQ0YUXG) AXITINIB 5 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIACETIN (UNII: XHX3C3X673) FERRIC OXIDE RED (UNII: 1K09F3G675) HYPROMELLOSE 2910 (15000 MPA.S) (UNII: 288VBX44JC) Product Characteristics Color RED Score no score Shape TRIANGLE Size 8mm Flavor Imprint Code Pfizer;5;XNB Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0069-0151-11 60 in 1 BOTTLE; Type 0: Not a Combination Product 01/27/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA202324 01/27/2012 Labeler - Pfizer Laboratories Div Pfizer Inc (134489525) Establishment Name Address ID/FEI Business Operations Pfizer Pharmaceuticals LLC 829084552 ANALYSIS(0069-0151, 0069-0145) , PACK(0069-0151, 0069-0145) Establishment Name Address ID/FEI Business Operations Upjohn Manufacturing Ireland Unlimited Company 986030667 ANALYSIS(0069-0145, 0069-0151) , API MANUFACTURE(0069-0145, 0069-0151) Establishment Name Address ID/FEI Business Operations Pfizer Manufacturing Deutschland GmbH 341970073 ANALYSIS(0069-0145, 0069-0151) , LABEL(0069-0145, 0069-0151) , MANUFACTURE(0069-0145, 0069-0151) , PACK(0069-0145, 0069-0151)


Trademark Results [INLYTA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 INLYTA 85226554 not registered Dead/Abandoned |
Pfizer Inc. 2011-01-26 |
 INLYTA 78554237 3349442 Live/Registered |
Pfizer Inc. 2005-01-26 |
 INLYTA 76294810 not registered Dead/Abandoned |
Pfizer Inc. 2001-08-02 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.