BELEODAQ- belinostat injection, powder, lyophilized, for solution
Beleodaq by
Drug Labeling and Warnings
Beleodaq by is a Prescription medication manufactured, distributed, or labeled by Spectrum Pharmaceuticals, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BELEODAQ safely and effectively. See full prescribing information for BELEODAQ.
BELEODAQ® (belinostat) for injection, for intravenous use
Initial U.S. Approval: 2014INDICATIONS AND USAGE
Beleodaq is a histone deacetylase inhibitor indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL). This indication is approved under accelerated approval based on tumor response rate and duration of response. An improvement in survival or disease-related symptoms has not been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trial. (1)
DOSAGE AND ADMINISTRATION
- Recommended dosage of Beleodaq is 1,000 mg/m2 administered over 30 minutes by intravenous infusion once daily on days 1-5 of a 21-day cycle. Cycles can be repeated until disease progression or unacceptable toxicity. (2.1)
- Treatment discontinuation or interruption with or without dosage reductions by 25% may be needed to manage adverse reactions (2.2)
DOSAGE FORMS AND STRENGTHS
For injection: 500 mg, lyophilized powder in single-dose vial for reconstitution (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Hematologic Toxicity: Thrombocytopenia, leukopenia (neutropenia and lymphopenia), and anemia: Monitor blood counts and modify dosage for hematologic toxicities. (2.2, 5.1)
- Infection: Serious and fatal infections (e.g., pneumonia and sepsis) (5.2)
- Hepatotoxicity: Beleodaq may cause hepatic toxicity and liver function test abnormalities. Monitor liver function tests and omit or modify dosage for hepatic toxicities. (2.2, 5.3)
- Tumor lysis syndrome: Monitor patients with advanced stage disease and/or high tumor burden and take appropriate precautions (5.4)
- Embryo-fetal toxicity: Beleodaq may cause fetal harm when administered to a pregnant woman. Advise women of potential harm to the fetus and to avoid pregnancy while receiving Beleodaq (5.6)
ADVERSE REACTIONS
The most common adverse reactions (>25%) are nausea, fatigue, pyrexia, anemia, and vomiting. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Spectrum Pharmaceuticals, Inc. at 1-888-292-9617 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
USE IN SPECIFIC POPULATIONS
Nursing Mothers: Women should be advised against breastfeeding while being treated with Beleodaq. (8.3)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2017
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
2.2 Dosage Modification for Hematologic and Non-Hematologic Toxicities
2.3 Patients with Reduced UGT1A1 Activity
2.4 Preparation and Administration Precautions
2.5 Reconstitution and Infusion Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hematologic Toxicity
5.2 Infections
5.3 Hepatotoxicity
5.4 Tumor Lysis Syndrome
5.5 Gastrointestinal Toxicity
5.6 Embryo-fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 UGT1A1 Inhibitors
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Use in Patients with Hepatic Impairment
8.7 Use in Patients with Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Beleodaq is indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL).
This indication is approved under accelerated approval based on tumor response rate and duration of response [see Clinical Studies (14)]. An improvement in survival or disease-related symptoms has not been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trial.
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
The recommended dosage of Beleodaq is 1,000 mg/m2 administered over 30 minutes by intravenous infusion once daily on Days 1-5 of a 21-day cycle. Cycles can be repeated every 21 days until disease progression or unacceptable toxicity.
2.2 Dosage Modification for Hematologic and Non-Hematologic Toxicities
Table 1 displays the recommended Beleodaq dosage modifications for hematologic and non-hematologic toxicities. Base dosage adjustments for thrombocytopenia and neutropenia on platelet and absolute neutrophil nadir (lowest value) counts in the preceding cycle of therapy.
- Absolute neutrophil count (ANC) should be greater than or equal to 1.0 x 109/L and the platelet count should be greater than or equal to 50 x 109/L prior to the start of each cycle and prior to resuming treatment following toxicity. Resume subsequent treatment with Beleodaq according to the guidelines described in Table 1 below. Discontinue Beleodaq in patients who have recurrent ANC nadirs less than 0.5 x 109/L and/or recurrent platelet count nadirs less than 25 x 109/L after two dosage reductions.
- Other toxicities must be NCI-CTCAE Grade 2 or less prior to re-treatment.
Monitor complete blood counts at baseline and weekly. Perform serum chemistry tests, including renal and hepatic functions prior to the start of the first dose of each cycle.
Table 1: Dosage Modifications for Hematologic and Non-Hematologic Toxicities - * For nausea, vomiting, and diarrhea, only dose modify if the duration is greater than 7 days with supportive management
Dosage Modification
Dosage Modifications due to Hematologic Toxicities
Platelet count ≥ 25 x 109/L and nadir ANC ≥ 0.5 x 109/L
No Change
Nadir ANC < 0.5 x 109/L (any platelet count)
Decrease dosage by 25% (750 mg/m2)
Platelet count < 25 x 109/L (any nadir ANC)
Dosage Modifications due to Non-Hematologic Toxicities
Any CTCAE Grade 3 or 4 adverse reaction*
Decrease dosage by 25% (750 mg/m2)
Recurrence of CTCAE Grade 3 or 4 adverse reaction after two dosage reductions
Discontinue Beleodaq
2.3 Patients with Reduced UGT1A1 Activity
Reduce the starting dose of Beleodaq to 750 mg/m2 in patients known to be homozygous for the UGT1A1*28 allele [see Clinical Pharmacology (12.5)].
2.4 Preparation and Administration Precautions
As with other potentially cytotoxic anticancer agents, exercise care in the handling and preparation of solutions prepared with Beleodaq.
2.5 Reconstitution and Infusion Instructions
- Aseptically reconstitute each vial of Beleodaq by adding 9 mL of Sterile Water for injection, USP, into the Beleodaq vial with a suitable syringe to achieve a concentration of 50 mg of belinostat per mL. Swirl the contents of the vial until there are no visible particles in the resulting solution. The reconstituted product may be stored for up to 12 hours at ambient temperature (15-25°C; 59-77°F).
- Aseptically withdraw the volume needed for the required dosage (based on the 50 mg/mL concentration and the patient’s BSA [m2]) and transfer to an infusion bag containing 250 mL of 0.9 % Sodium Chloride injection. The infusion bag with drug solution may be stored at ambient room temperature (15-25°C; 59-77°F) for up to 36 hours including infusion time.
- Visually inspect the solution for particulate matter. Do not use if cloudiness or particulates are observed.
- Connect the infusion bag containing drug solution to an infusion set with a 0.22 µm in-line filter for administration.
- Infuse intravenously over 30 minutes. If infusion site pain or other symptoms potentially attributable to the infusion occur, the infusion time may be extended to 45 minutes.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hematologic Toxicity
Beleodaq can cause thrombocytopenia, leukopenia (neutropenia and lymphopenia), and/or anemia; monitor blood counts weekly during treatment, and modify dosage as necessary [see Dosage and Administration (2.2) and Adverse Reactions(6.1)].
5.2 Infections
Serious and sometimes fatal infections, including pneumonia and sepsis, have occurred with Beleodaq. Do not administer Beleodaq to patients with an active infection. Patients with a history of extensive or intensive chemotherapy may be at higher risk of life threatening infections [see Adverse Reactions (6.1)].
5.3 Hepatotoxicity
Beleodaq can cause fatal hepatotoxicity and liver function test abnormalities [see Adverse Reactions (6.1)]. Monitor liver function tests before treatment and before the start of each cycle. Interrupt or adjust dosage until recovery, or permanently discontinue Beleodaq based on the severity of the hepatic toxicity[see Dosage and Administration (2.2)].
5.4 Tumor Lysis Syndrome
Tumor lysis syndrome has occurred in Beleodaq-treated patients in the clinical trial of patients with relapsed or refractory PTCL [see Clinical Studies (14)]. Monitor patients with advanced stage disease and/or high tumor burden and take appropriate precautions [see Adverse Reactions (6.1)].
5.5 Gastrointestinal Toxicity
Nausea, vomiting and diarrhea occur with Beleodaq [see Adverse Reactions (6.1)] and may require the use of antiemetic and antidiarrheal medications.
5.6 Embryo-fetal Toxicity
Beleodaq can cause fetal harm when administered to a pregnant woman. Beleodaq may cause teratogenicity and/or embryo-fetal lethality because it is genotoxic and targets actively dividing cells [see Nonclinical Toxicology (13.1)]. Women of childbearing potential should be advised to avoid pregnancy while receiving Beleodaq. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of potential hazard to the fetus [see Use in Specific Populations (8.1)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described in more detail in other sections of the prescribing information.
- Hematologic Toxicity [see Warnings and Precautions (5.1)]
- Infection [see Warnings and Precautions (5.2)]
- Hepatotoxicity [see Warnings and Precautions (5.3)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.4)]
- Gastrointestinal Toxicity [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of Beleodaq may not reflect the rates observed in practice.
Adverse Reactions in Patients with Peripheral T-Cell Lymphoma
The safety of Beleodaq was evaluated in 129 patients with relapsed or refractory PTCL in the single arm clinical trial in which patients were administered Beleodaq at a dosage of 1,000 mg/m2 administered over 30 minutes by IV infusion once daily on Days 1-5 of a 21-day cycle [see Clinical Studies (14)]. The median duration of treatment was 2 cycles (range 1 – 33 cycles).
The most common adverse reactions observed in the trial of patients with relapsed or refractory PTCL treated with Beleodaq were nausea, fatigue, pyrexia, anemia, and vomiting [see Clinical Studies (14)]. Table 2 summarizes the adverse reactions regardless of causality from the trial in patients with relapsed or refractory PTCL.
Table 2: Adverse Reactions Occurring in ≥ 10% of Patients with Relapsed or Refractory PTCL (NCI-CTC Grade 1-4) Adverse Reactions
Percentage of Patients (%)
(N=129)
All Grades
Grade 3 or 4
All Adverse Reactions
97
61
Nausea
42
1
Fatigue
37
5
Pyrexia
35
2
Anemia
32
11
Vomiting
29
1
Constipation
23
1
Diarrhea
23
2
Dyspnea
22
6
Rash
20
1
Peripheral Edema
20
0
Cough
19
0
Thrombocytopenia
16
7
Pruritus
16
3
Chills
16
1
Increased Blood Lactate Dehydrogenase
16
2
Decreased Appetite
15
2
Headache
15
0
Infusion Site Pain
14
0
Hypokalemia
12
4
Prolonged QT
11
4
Abdominal pain
11
1
Hypotension
10
3
Phlebitis
10
1
Dizziness
10
0
Note: Adverse reactions are listed by order of incidence in the “All Grades” category first, then by incidence in “the Grade 3 or 4” category; Measured by National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 3.0
Serious Adverse Reactions
Sixty-one patients (47.3%) experienced serious adverse reactions while taking Beleodaq or within 30 days after their last dose of Beleodaq. The most common serious adverse reactions (> 2%) were pneumonia, pyrexia, infection, anemia, increased creatinine, thrombocytopenia, and multi-organ failure. One treatment-related death associated with hepatic failure was reported in the trial.
One patient with baseline hyperuricemia and bulky disease experienced Grade 4 tumor lysis syndrome during the first cycle of treatment and died due to multi-organ failure. A treatment-related death from ventricular fibrillation was reported in another monotherapy clinical trial with Beleodaq. ECG analysis did not identify QTc prolongation.
Discontinuations due to Adverse Reactions
Twenty-five patients (19.4%) discontinued treatment with Beleodaq due to adverse reactions. The adverse reactions reported most frequently as the reason for discontinuation of treatment included anemia, febrile neutropenia, fatigue, and multi-organ failure.
Dosage Modifications due to Adverse Reactions
In the trial, dosage adjustments due to adverse reactions occurred in 12% of Beleodaq-treated patients.
-
7 DRUG INTERACTIONS
7.1 UGT1A1 Inhibitors
Belinostat is primarily metabolized by UGT1A1. Avoid concomitant administration of Beleodaq with strong inhibitors of UGT1A1 [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category D [see Warnings and Precautions (5.6)].
Risk Summary
Beleodaq may cause teratogenicity and/or embryo-fetal lethality because it is a genotoxic drug and targets actively dividing cells [see Nonclinical Toxicology (13.1)]. Women should avoid pregnancy while receiving Beleodaq. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of potential hazard to the fetus.
Animal Data
No reproductive and developmental animal toxicology studies have been conducted with belinostat.
8.3 Nursing Mothers
It is not known whether belinostat is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from Beleodaq, a decision should be made whether to discontinue nursing or discontinue drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
The safety and effectiveness of Beleodaq in pediatric patients have not been established.
8.5 Geriatric Use
In the single-arm trial, 48% of patients (n = 62) were ≥ 65 years of age and 10% of patients (n=13) were ≥ 75 years of age [see Clinical Studies (14)]. The median age of the trial population was 63 years. Patients ≥ 65 years of age had a higher response rate to Beleodaq treatment than patients < 65 years of age (36% versus 16%) while no meaningful differences in response rate were observed between patients ≥ 75 years of age and those < 75 years of age. No clinically meaningful differences in serious adverse reactions were observed in patients based on age (< 65 years compared with ≥ 65 years or < 75 years of age compared with ≥ 75 years of age).
8.6 Use in Patients with Hepatic Impairment
Belinostat is metabolized in the liver and hepatic impairment is expected to increase exposure to belinostat. Patients with moderate and severe hepatic impairment (total bilirubin >1.5 x upper limit of normal (ULN)) were excluded from clinical trials.
There is insufficient data to recommend a dose of Beleodaq in patients with moderate and severe hepatic impairment [see Clinical Pharmacology (12.3)].
8.7 Use in Patients with Renal Impairment
Belinostat exposure is not altered in patients with Creatinine Clearance (CLcr) > 39 mL/min. There is insufficient data to recommend a dose of Beleodaq in patients with CLcr ≤ 39 mL/min. [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
No specific information is available on the treatment of overdosage of Beleodaq. There is no antidote for Beleodaq and it is not known if Beleodaq is dialyzable. If an overdose occurs, general supportive measures should be instituted as deemed necessary by the treating physician. The elimination half-life of belinostat is 1.1 hours [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
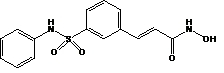
Beleodaq is a histone deacetylase inhibitor with a sulfonamide-hydroxamide structure. The chemical name of belinostat is (2E)-N-hydroxy-3-[3-(phenylsulfamoyl)phenyl]prop-2-enamide. The structural formula is as follows:
The molecular formula is C15H14N2O4S and the molecular weight is 318.35 g/mol.
Belinostat is a white to off-white powder. It is slightly soluble in distilled water (0.14 mg/mL) and polyethylene glycol 400 (about 1.5 mg/mL), and is freely soluble in ethanol (> 200 mg/mL). The pKa values are 7.87 and 8.71 by potentiometry and 7.86 and 8.59 by UV.
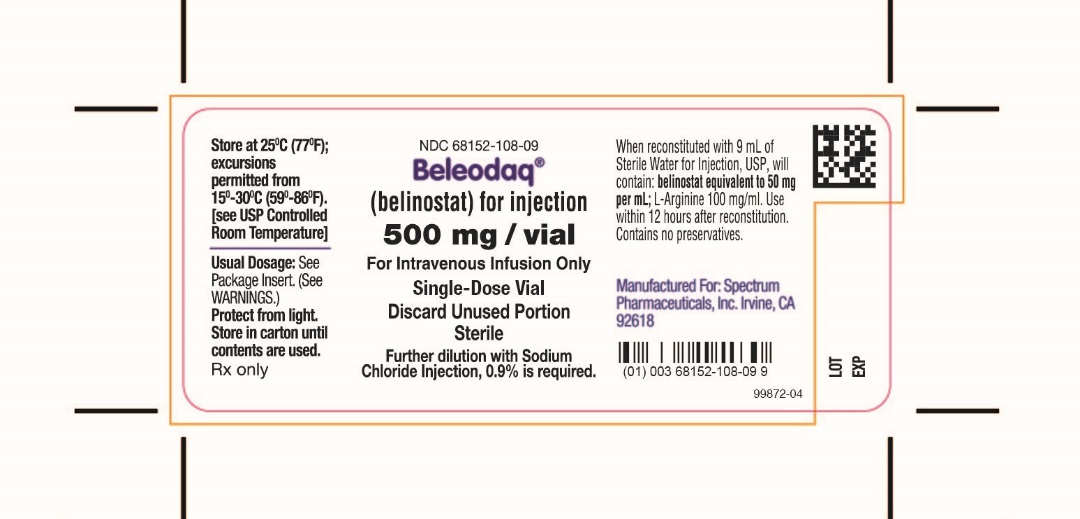
Beleodaq (belinostat) for injection is supplied as a sterile lyophilized yellow powder containing 500 mg belinostat as the active ingredient. Each vial also contains 1000 mg L-Arginine, USP as an inactive ingredient. The drug product is supplied in a single-dose 30 mL clear glass vial with a coated stopper and aluminum crimp seal with “flip-off” cap. Beleodaq is intended for intravenous administration after reconstitution with 9 mL Sterile Water for injection, and the reconstituted solution is further diluted with 250 mL of sterile 0.9% Sodium Chloride injection prior to infusion [see Dosage and Administration (2)].
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Beleodaq is a histone deacetylase (HDAC) inhibitor. HDACs catalyze the removal of acetyl groups from the lysine residues of histones and some non-histone proteins. In vitro, belinostat caused the accumulation of acetylated histones and other proteins, inducing cell cycle arrest and/or apoptosis of some transformed cells. Belinostat shows preferential cytotoxicity towards tumor cells compared to normal cells. Belinostat inhibited the enzymatic activity of histone deacetylases at nanomolar concentrations (<250 nM).
12.2 Pharmacodynamics
Cardiac Electrophysiology
Multiple clinical trials have been conducted with Beleodaq, in many of which ECG data were collected and analyzed by a central laboratory. Analysis of clinical ECG and belinostat plasma concentration data demonstrated no meaningful effect of Beleodaq on cardiac repolarization. None of the trials showed any clinically relevant changes caused by Beleodaq on heart rate, PR duration or QRS duration as measures of autonomic state, atrio-ventricular conduction or depolarization; there were no cases of Torsades de Pointes.
12.3 Pharmacokinetics
The pharmacokinetic characteristics of belinostat were analyzed from pooled data from phase 1/2 clinical studies that used doses of belinostat ranging from 150 to 1200 mg/m2. The total mean plasma clearance and elimination half-life were 1240 mL/min and 1.1 hours, respectively. The total clearance approximates average hepatic blood flow (1500 mL/min), suggesting high hepatic extraction (clearance being flow dependent).
Distribution
The mean belinostat volume of distribution approaches total body water, indicating that belinostat has limited body tissue distribution. In vitro plasma studies have shown that between 92.9% and 95.8% of belinostat is bound to protein in an equilibrium dialysis assay, and was independent of belinostat plasma concentrations from 500 to 25,000 ng/mL.
Elimination
Metabolism
Belinostat is primarily metabolized by hepatic UGT1A1. Strong UGT1A1 inhibitors are expected to increase exposure to belinostat. Belinostat also undergoes hepatic metabolism by CYP2A6, CYP2C9, and CYP3A4 enzymes to form belinostat amide and belinostat acid. The enzymes responsible for the formation of methyl belinostat and 3-(anilinosulfonyl)-benzenecarboxylic acid, (3-ASBA) are not known.
Excretion
Following a single dose of [14C]-labelled belinostat (100 μCi, 1500 mg) administered as a 30-minute intravenous infusion in patients with recurrent or progressive malignancy (N=6), fecal excretion accounted for a mean (± SD) of 9.7% (± 6.5%) of the administered radioactive belinostat dose over 168 hours. The mean (± SD) of the administered radioactive belinostat dose that was excreted in urine over 168 hours was 84.8% (± 9.8%), of which unchanged belinostat accounted for only 1.7%.
Drug Interaction Studies
In vitro studies showed belinostat and its metabolites (including belinostat glucuronide, belinostat amide, methyl belinostat) inhibited metabolic activities of CYP2C8 and CYP2C9. Other metabolites (3-ASBA and belinostat acid) inhibited CYP2C8.
In cancer patients, co-administration of Beleodaq (1,000 mg/m2) and warfarin (5 mg), a known CYP2C9 substrate, did not increase the AUC or Cmax of either R- or S-warfarin.
Belinostat is likely a glycoprotein (P-gp) substrate but is unlikely to inhibit P-gp.
12.5 Pharmacogenomics
UGT1A1 activity is reduced in individuals with genetic polymorphisms that lead to reduced enzyme activity such as the UGT1A1*28 polymorphism. Approximately 20% of the black population, 10% of the white population, and 2% of the Asian population are homozygous for the UGT1A1*28 allele. Additional reduced function alleles may be more prevalent in specific populations.
Because belinostat is primarily (80 -90%) metabolized by UGT1A1, the clearance of belinostat could be decreased in patients with reduced UGT1A1 activity (e.g., patients with UGT1A1*28 allele). Reduce the starting dose of Beleodaq to 750 mg/m2 in patients known to be homozygous for the UGT1A1*28 allele to minimize dose limiting toxicities.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with belinostat.
Belinostat was genotoxic in a bacterial reverse mutation test (Ames assay), an in vitro mouse lymphoma cell mutagenesis assay, and an in vivo rat micronucleus assay.
Beleodaq may impair male fertility. Fertility studies using belinostat were not conducted. However, belinostat effects on male reproductive organs observed during the 24-week repeat-dose dog toxicology study included reduced organ weights of the testes/epididymides that correlated with a delay in testicular maturation.
-
14 CLINICAL STUDIES
Relapsed or Refractory Peripheral T-cell Lymphoma (PTCL)
In an open-label, single-arm, non-randomized international trial conducted at 62 centers, 129 patients with relapsed or refractory PTCL were treated with Beleodaq 1,000 mg/m2 administered over 30 minutes via IV infusion once daily on Days 1-5 of a 21-day cycle. There were 120 patients who had histologically confirmed PTCL by central review evaluable for efficacy. Patients were treated with repeat cycles every three weeks until disease progression or unacceptable toxicity.
Efficacy was evaluated using response rate (complete response and partial response) as assessed by an independent review committee (IRC) using the International Workshop Criteria (IWC) (Cheson 2007). Response assessments were evaluated every 6 weeks for the first 12 months and then every 12 weeks until 2 years from the start of study treatment. Duration of response was measured from the first day of documented response to disease progression or death. Response and progression of disease were evaluated by the IRC using the IWC.
Table 3 summarizes the baseline demographic and disease characteristics of the study population, who were evaluable for efficacy.
Table 3 Baseline Patient Characteristics (PTCL Population) Characteristics
Evaluable Patients
(N=120)
Age (years)
Median (range)
64 (29-81)
Sex, %
Male
Female
52
48
Race, %
White
Black
Asian
Latin
Other
88
6
3
3
2
PTCL Subtype Based on Central Diagnosis, %
PTCL Unspecified (NOS)
Angioimmunoblastic T-cell lymphoma (AITL)
ALK-1 negative anaplastic large cell lymphoma (ALCL)
Other
64
18
11
7
Baseline Platelet Count, %
≥100,000/μL
<100,000/μL
83
17
ECOG Performance Status, %
0
1
2
3
34
43
22
1
Median time (months) from Initial PTCL Diagnosis (Range)
12 (2.6 – 266.4)
Median Number of Prior Systemic Therapies (Range)
2 (1-8)
In all evaluable patients (N = 120) treated with Beleodaq, the overall response rate per central review using IWC was 25.8% (n = 31) (Table 4) with rates of 23.4% for PTCL, NOS and 45.5% for AITL, the two largest subtypes enrolled.
Table 4: Response Analysis per Central Assessment Using IWC in Patients with Relapsed or Refractory PTCL Evaluable Patients
(N=120)Response Rate
n (%)
(95% CI)
- CR+PR
31 (25.8)
18.3-34.6
- CR
13 (10.8)
5.9-17.8
- PR
18 (15.0)
9.1 – 22.7
CI=confidence interval, CR=complete response, PR=partial response
The median duration of response based on the first date of response to disease progression or death was 8.4 months (95% CI: 4.5 – 29.4). Of the responders, the median time to response was 5.6 weeks (range 4.3 - 50.4 weeks). Nine patients (7.5%) were able to proceed to a stem cell transplant after treatment with Beleodaq.
-
15 REFERENCES
- 1. OSHA Hazardous Drugs. OSHA. [Accessed on 14 May 2014, from http://www.osha.gov/SLTC/hazardousdrugs/index.html].
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
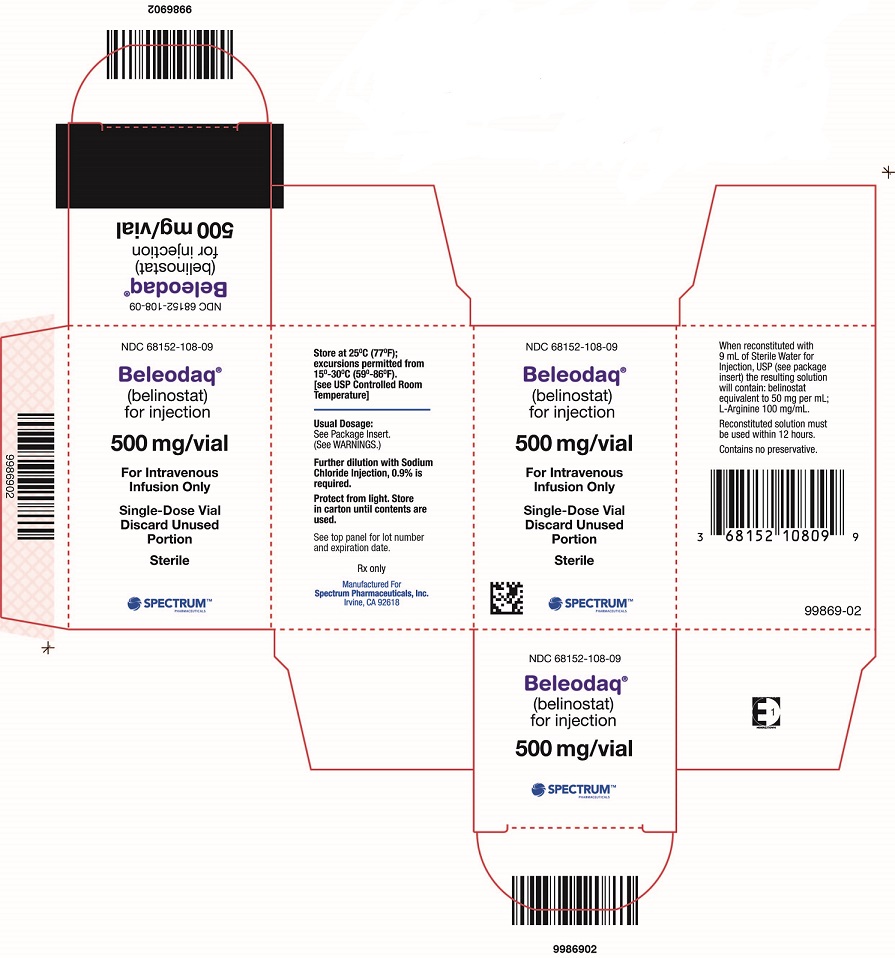
Beleodaq (belinostat) for injection is supplied in single vial cartons; each 30 mL clear vial contains sterile, lyophilized powder equivalent to 500 mg belinostat.
NDC: 68152-108-09: Individual carton of Beleodaq 30 mL single-dose vial containing 500 mg belinostat.
16.2 Storage and Handling
Store Beleodaq (belinostat) for injection at room temperature 20°C to 25°C (68°F to 77°F). Excursions are permitted between 15°C and 30°C (59°F and 86°F). Retain in original package until use. [see USP Controlled Room Temperature].
Beleodaq is a cytotoxic drug. Follow special handling and disposal procedures [see References (15)1].
-
17 PATIENT COUNSELING INFORMATION
Physicians should discuss the FDA approved Patient Information Leaflet with patients prior to treatment with Beleodaq. Instruct patients to read the Patient Information Leaflet carefully.
Advise the patient or the caregiver to read the FDA-approved patient labeling (Patient Information).
Advise patients or their caregivers:
- To report symptoms of nausea, vomiting and diarrhea so that appropriate antiemetic and antidiarrheal medications can be administered [see Warnings and Precautions (5.5)].
- To report any symptoms of thrombocytopenia, leukopenia (neutropenia and lymphopenia), and anemia [see Warnings and Precautions (5.1)].
- To immediately report symptoms of infection (e.g., pyrexia) [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
- Of the potential risk to the fetus and for women to avoid pregnancy while receiving Beleodaq [see Warnings and Precautions (5.6)].
- To understand the importance of monitoring liver function test abnormalities and to immediately report potential symptoms of liver injury [see Dosage and Administration (2.2) and Warnings and Precautions (5.3)].
Manufactured for:
Spectrum Pharmaceuticals, Inc.
Irvine, CA 92618
Beleodaq is a registered trademark of Spectrum Pharmaceuticals, Inc. All rights are reserved.
U.S. Patent: 6,888,027
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
BELEODAQ®(Bē-lēo-dak)
(belinostat) for injection, for intravenous use
Read this Patient Information before you receive treatment with Beleodaq and each time you receive Beleodaq. There may be new information. This leaflet does not take the place of talking to your doctor about your medical condition or your treatment.
What is Beleodaq?
Beleodaq is a prescription medicine used to treat people with a type of cancer called peripheral T-cell Lymphoma (PTCL) that comes back or does not respond to cancer treatment.
It is not known if Beleodaq is safe and effective in children.
What should I tell my doctor before receiving Beleodaq?
Before receiving Beleodaq, tell your doctor about all of your medical conditions, including if you:
- have an infection
- have had chemotherapy treatment
- have liver or kidney problems
- have nausea, vomiting, or diarrhea
- are pregnant or plan to become pregnant. Beleodaq can harm your unborn baby. You should not become pregnant while receiving Beleodaq. Tell your doctor right away if you become pregnant while receiving Beleodaq.
- are breastfeeding or plan to breastfeed. It is not known if Beleodaq passes into your breast milk. You and your doctor should decide if you will receive Beleodaq or breastfeed. You should not do both.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How will I receive Beleodaq?
- Beleodaq will be given to you by intravenous (IV) injection into your vein, usually over 30 minutes.
- Beleodaq is given one time a day on Days 1 through 5 of a 21-day cycle of treatment.
- You should have regular blood tests before and during your treatment with Beleodaq.
- Your doctor may change your dose of Beleodaq, change when you receive your treatment, or stop treatment if you have certain side effects while receiving Beleodaq.
What are the possible side effects of Beleodaq?
Beleodaq may cause serious side effects, including:
-
Low blood cell counts. Your doctor will do blood tests to check your blood counts during your treatment with Beleodaq.
- Low platelet counts can cause unusual bleeding or bruising under your skin.
- Low red blood cell counts may make you feel weak, tired, or you get tired easily, you look pale, or you feel short of breath.
- Low white blood cell counts can cause you to get infections, which may be serious.
- Serious infections. People receiving Beleodaq may develop serious infections that can sometimes lead to death. You may have a greater risk of life-threatening infections if you have had chemotherapy in the past. Tell your doctor right away if you have any of the following signs or symptoms of an infection: fever, flu-like symptoms, cough, shortness of breath, burning with urination, muscle aches, or worsening skin problems.
- Liver problems. Beleodaq may cause liver problems which can lead to death. Your doctor will do blood tests during your treatment with Beleodaq to check for liver problems. Tell your doctor right away if you have any of the following signs or symptoms of liver problems: yellowing of the skin or the white part of your eyes (jaundice), dark urine, itching, or pain in the right upper stomach area.
- Tumor Lysis Syndrome (TLS). TLS is caused by a fast breakdown of cancer cells. Your doctor will check you for TLS during treatment with Beleodaq.
- Nausea, vomiting, and diarrhea are common with Beleodaq and can sometimes be serious. Tell your doctor if you develop nausea, vomiting or diarrhea. Your doctor may prescribe medicines to help prevent or treat these side effects.
Common side effects of Beleodaq include fatigue, fever, and low red blood cell count.
- Tell your doctor if you have any side effect that bothers you or that does not go away.
- These are not all the possible side effects of Beleodaq. Call your doctor for medical advice about side effects. You can report side effects to FDA at 1-800-FDA-1088.
General information about Beleodaq
- Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or doctor for information about Beleodaq that is written for health professionals.
What are the ingredients in Beleodaq?
Active ingredient: belinostat
Inactive ingredients: L-ArginineManufactured for: Spectrum Pharmaceuticals, Inc. Irvine, CA 92618
Beleodaq is a registered trademark of Spectrum Pharmaceuticals, Inc. All rights reserved.
For more information, go to www.Beleodaq.com or call 1-888-292-9617.
- This Patient Information has been approved by the U.S. Food and Drug Administration. Issued: April 2017
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
Carton Label
NDC: 68152-108-09
Beleodaq® (belinostat) for injection
500 mg/vial
For Intravenous Infusion Only
Single-Dose Vial
Vial Label
NDC: 68152-108-09
Beleodaq® (belinostat) for injection
500 mg/vial
For Intravenous Infusion Only
Single-Dose Vial
-
INGREDIENTS AND APPEARANCE
BELEODAQ
belinostat injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68152-108 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BELINOSTAT (UNII: F4H96P17NZ) (BELINOSTAT - UNII:F4H96P17NZ) BELINOSTAT 500 mg in 10 mL Inactive Ingredients Ingredient Name Strength ARGININE (UNII: 94ZLA3W45F) 1000 mg in 10 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68152-108-09 1 in 1 CARTON 07/21/2014 1 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA206256 07/21/2014 Labeler - Spectrum Pharmaceuticals, Inc. (790888002)
Trademark Results [Beleodaq]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 BELEODAQ 86140026 4626277 Live/Registered |
ACROTECH BIOPHARMA LLC 2013-12-10 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.