KENGREAL- cangrelor injection, powder, lyophilized, for solution
KENGREAL by
Drug Labeling and Warnings
KENGREAL by is a Prescription medication manufactured, distributed, or labeled by Chiesi USA, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use KENGREAL safely and effectively. See full prescribing information for KENGREAL.
KENGREAL® (cangrelor) for injection, for intravenous use
Initial U.S. Approval: 2015
INDICATIONS AND USAGE
KENGREAL is a P2Y12 platelet inhibitor indicated as an adjunct to percutaneous coronary intervention (PCI) for reducing the risk of periprocedural myocardial infarction (MI), repeat coronary revascularization, and stent thrombosis (ST) in patients in who have not been treated with a P2Y12 platelet inhibitor and are not being given a glycoprotein IIb/IIIa inhibitor. (1)
DOSAGE AND ADMINISTRATION
- KENGREAL is intended for administration via a dedicated IV line, only after reconstitution and dilution. (2.3)
- Administer 30 mcg/kg intravenous (IV) bolus prior to PCI followed immediately by a 4 mcg/kg/min IV infusion for at least 2 hours or duration of procedure, whichever is longer. (2.1)
- To maintain platelet inhibition after discontinuation of KENGREAL infusion, administer an oral P2Y12 platelet inhibitor. (2.2)
DOSAGE FORMS AND STRENGTHS
Single-use 10 mL vial containing 50 mg KENGREAL as a lyophilized powder for reconstitution (3.0)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Bleeding: Like other drugs that inhibit platelet P2Y12 function, KENGREAL can increase the risk of bleeding (5.1)
ADVERSE REACTIONS
The most common adverse reaction is bleeding. (5.1, 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Chiesi USA, Inc. at 1-888-661-9260 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
Revised: 10/2019
- KENGREAL is intended for administration via a dedicated IV line, only after reconstitution and dilution. (2.3)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
2.2 Transitioning Patients to Oral P2Y12 Therapy
2.3 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Significant Active Bleeding
4.2 Hypersensitivity
5 WARNINGS AND PRECAUTIONS
5.1 Bleeding
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Thienopyridines
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 CHAMPION PHOENIX Trial
14.2 CHAMPION PCI and PLATFORM Trials
16 HOW SUPPLIED/STORAGE AND HANDLING
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
KENGREAL is indicated as an adjunct to percutaneous coronary intervention (PCI) to reduce the risk of periprocedural myocardial infarction (MI), repeat coronary revascularization, and stent thrombosis (ST) in patients who have not been treated with a P2Y12 platelet inhibitor and are not being given a glycoprotein IIb/IIIa inhibitor [see Clinical Studies (14.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
The recommended dosage of KENGREAL is a 30 mcg/kg IV bolus followed immediately by a 4 mcg/kg/min IV infusion. Initiate the bolus infusion prior to PCI. The maintenance infusion should ordinarily be continued for at least 2 hours or for the duration of PCI, whichever is longer.
2.2 Transitioning Patients to Oral P2Y12 Therapy
To maintain platelet inhibition after discontinuation of KENGREAL infusion, administer an oral P2Y12 platelet inhibitor, as described below:
- Ticagrelor: 180 mg at any time during KENGREAL infusion or immediately after discontinuation [see Clinical Pharmacology (12.2)].
- Prasugrel: 60 mg immediately after discontinuation of KENGREAL [see Drug Interactions (7.1) and Clinical Pharmacology (12.2)].
- Clopidogrel: 600 mg immediately after discontinuation of KENGREAL [see Drug Interactions (7.1) and Clinical Pharmacology (12.2)].
2.3 Preparation and Administration
KENGREAL is intended for IV administration, after reconstitution and dilution.
Preparation
Reconstitute the vial prior to dilution in a bag. For each 50 mg/vial, reconstitute by adding 5 mL of Sterile Water for Injection. Swirl gently until all material is dissolved. Avoid vigorous mixing. Allow any foam to settle. Ensure that the contents of the vial are fully dissolved and the reconstituted material is a clear, colorless to pale yellow solution. Parenteral drug products should be inspected visually for particulate matter after reconstitution.
Before administration, each reconstituted vial must be diluted further with Normal Saline (Sodium Chloride Injection 0.9% USP) or 5% Dextrose Injection USP.
Withdraw the contents from one reconstituted vial and add to one 250 mL saline bag. Mix the bag thoroughly. This dilution will result in a concentration of 200 mcg/mL and should be sufficient for at least 2 hours of dosing. Patients 100 kg and over will require a minimum of 2 bags.
Reconstituted KENGREAL should be diluted immediately. Diluted KENGREAL is stable for up to 12 hours in 5% Dextrose Injection and 24 hours in Normal Saline at Room Temperature. Discard any unused portion of reconstituted solution remaining in the vial.
Administration
Administer KENGREAL via a dedicated IV line.
Administer the bolus volume rapidly (<1 minute), from the diluted bag via manual IV push or pump. Ensure the bolus is completely administered before the start of PCI. Start the infusion immediately after administration of the bolus [see Dosage and Administration (2.1)].
- Ticagrelor: 180 mg at any time during KENGREAL infusion or immediately after discontinuation [see Clinical Pharmacology (12.2)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
4.1 Significant Active Bleeding
KENGREAL is contraindicated in patients with significant active bleeding [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
4.2 Hypersensitivity
KENGREAL is contraindicated in patients with known hypersensitivity (e.g., anaphylaxis) to KENGREAL or any component of the product [see Adverse Reactions (6.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Bleeding
Drugs that inhibit platelet P2Y12 function, including KENGREAL, increase the risk of bleeding.
In CHAMPION PHOENIX bleeding events of all severities were more common with KENGREAL than with clopidogrel [see Adverse Reactions (6.1)]. Bleeding complications with KENGREAL were consistent across a variety of clinically important subgroups (see Figure 1).
Once KENGREAL is discontinued, there is no antiplatelet effect after an hour [see Clinical Pharmacology (12.3)].
-
6 ADVERSE REACTIONS
The following adverse reactions are also discussed elsewhere in the labeling:
- Bleeding [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of KENGREAL has been evaluated in 13,301 subjects in controlled trials, in whom, 5,529 were in the CHAMPION PHOENIX trial.
Bleeding
There was a greater incidence of bleeding with KENGREAL than with clopidogrel. No baseline demographic factor altered the relative risk of bleeding with KENGREAL (see Table 1 and Figure 1).
Table 1: Major Bleeding Results in the CHAMPION PHOENIX Study (Non-CABG related Bleeding)a CHAMPION PHOENIX KENGREAL
(N=5529)Clopidogrel
(N=5527)Any GUSTO bleeding, n (%) 857 (15.5) 602 (10.9) Severe/life-threatening b 11 (0.2) 6 (0.1) Moderate c 21 (0.4) 14 (0.3) Mild d 825 (14.9) 582 (10.5) Any TIMI bleeding, n (%) 45 (0.8) 17 (0.3) Major e 12 (0.2) 6 (0.1) Minor f 33 (0.6) 11 (0.2) Abbreviations: GUSTO: Global Utilization of Streptokinase and Tissue Plasminogen Activator for Occluded Arteries; TIMI: Thrombolysis in Myocardial Infarction
a Safety population is all randomized subjects who received at least one dose of study drug
b intracranial hemorrhage or bleeding resulting in substantial hemodynamic compromise requiring treatment
c requiring blood transfusion but not resulting in hemodynamic compromise
d all other bleeding not included in severe or moderate
e any intracranial hemorrhage, or any overt bleeding associated with a reduction in hemoglobin of ≥5 g/dL (or, when hemoglobin is not available, an absolute reduction in hematocrit ≥15%)
f any overt sign of bleeding (including observation by imaging techniques) that is associated with a reduction in hemoglobin of ≥3 g/dL and <5 g/dL (or, when hemoglobin is not available, an absolute reduction in hematocrit of ≥9% and <15%)
Figure 1: Bleeding Results in the CHAMPION PHOENIX Studya (All Non-CABG related)
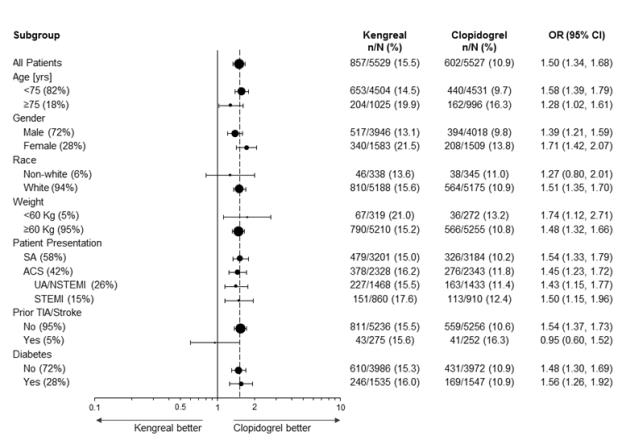
a Safety population is all randomized subjects who received at least one dose of study drug
Note: The figure above presents effects in various subgroups most of which are baseline characteristics and most of which were pre-specified (patient presentation and weight were not pre-specified subgroups). The 95% confidence limits that are shown do not take into account how many comparisons were made, nor do they reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
Drug Discontinuation
In CHAMPION PHOENIX the rate of discontinuation for bleeding events was 0.3% for KENGREAL and 0.1% for clopidogrel. Discontinuation for non-bleeding adverse events was low and similar for KENGREAL (0.6%) and for clopidogrel (0.4%). Coronary artery dissection, coronary artery perforation, and dyspnea were the most frequent events leading to discontinuation in patients treated with KENGREAL.
Non-Bleeding Adverse Reactions
Hypersensitivity
Serious cases of hypersensitivity were more frequent with KENGREAL (7/13301) than with control (2/12861). These included anaphylactic reactions, anaphylactic shock, bronchospasm, angioedema, and stridor.
Decreased renal function
Worsening renal function was reported in 3.2% of KENGREAL patients with severe renal impairment (creatinine clearance <30 mL/min) compared to 1.4% of clopidogrel patients with severe renal impairment.
Dyspnea
Dyspnea was reported more frequently in patients treated with KENGREAL (1.3%) than with control (0.4%).
-
7 DRUG INTERACTIONS
7.1 Thienopyridines
Clopidogrel or prasugrel administered during KENGREAL infusion will have no antiplatelet effect until the next dose is administered. Therefore, administer clopidogrel or prasugrel after KENGREAL infusion is discontinued [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on cangrelor use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. Untreated myocardial infarction can be fatal to the pregnant women and fetus (see Clinical Considerations).
In animal reproduction studies, continuous infusion of cangrelor in pregnant rats and rabbits throughout organogenesis at dose approximately 2-times the maximum recommended human dose (MRHD) did not result in fetal malformations (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Myocardial infarction is a medical emergency in pregnancy which can be fatal to the pregnant woman and fetus if left untreated. Life-sustaining therapy for the pregnant woman should not be withheld due to potential concerns regarding the effects of cangrelor on the fetus.
Labor or delivery
Cangrelor use during labor and delivery may increase the risk for maternal bleeding and hemorrhage. Performance of neuraxial blockade procedures is not advised during cangrelor use due to potential risk of spinal hematoma. When possible, discontinue cangrelor 1 hour prior to labor, delivery, or neuraxial blockade [see Clinical Pharmacology (12.2)].
Data
Animal Data
A prenatal and postnatal development study in female rats demonstrated a slight increase in the incidence of maternal mortality in dams treated at doses up to 30 mcg/kg/min (approximately 7.5 times the MRHD) cangrelor continuous infusion from Day 6 of gestation up to Day 23 post-partum. Pregnancy rates, gestation index, length of gestation, numbers of live, dead and malformed pups, sex ratio, live birth index, and lactation of the maternal animals were unaffected.
Cangrelor administered at dose levels of ≥ 3 mcg/kg/min in pregnant rats from Day 6 to 17 post-coitum resulted in dose-related fetal growth retardation characterized by increased incidences of incomplete ossification and unossified hind limb metatarsals.
An embryo-fetal development study in rabbits administered 4, 12, or 36 mcg/kg/min cangrelor continuous IV infusion from Day 6 to Day 19 post-coitum resulted in increased incidences of abortion and intrauterine losses at ≥12 mcg/kg/min (3 times the MRHD). Fetal growth retardation occurred at 36 mcg/kg/min (9 times the MRHD) and was characterized by decreased fetal weights, slight reduction in ossification, and a slight increase in skeletal variants.
Cangrelor did not produce malformations in either the rat or rabbit embryo-fetal development studies and is not considered to be a teratogen.
8.2 Lactation
Risk Summary
There are no data on the presence of cangrelor in human milk or animal milk, the effects on the breastfed infant, or the effects on milk production. However, due to its short-half life, cangrelor exposure is expected to be very low in the breastfed infant.
8.5 Geriatric Use
In CHAMPION PHOENIX, 18% of patients were ≥75 years. No overall differences in safety or effectiveness were observed between these patients and those patients <75 years [see Clinical Studies (14.1)].
8.6 Renal Impairment
No dosage adjustment is required for patients with mild, moderate, or severe renal impairment [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
KENGREAL has not been studied in patients with hepatic impairment. However, the metabolism of KENGREAL is not dependent of hepatic function, so dosage adjustment is not required for patients with hepatic impairment [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There is no specific treatment to reverse the antiplatelet effect of KENGREAL but the effect is gone within one hour after the drug is discontinued.
In clinical trials, 36 patients received an overdose of KENGREAL, ranging from 36 to 300 mcg/kg (bolus dose) or 4.8 to 13.7 mcg/kg/min (infusion dose). The maximum overdose received was 10 times the PCI bolus dose or 3.5 times the PCI infusion dose in 4 patients. No clinical sequela were noted as a result of overdose following completion of KENGREAL therapy.
-
11 DESCRIPTION
KENGREAL is a direct-acting P2Y12 platelet receptor inhibitor that blocks adenosine diphosphate (ADP)-induced platelet activation and aggregation. The chemical structure is similar to adenosine triphosphate (ATP).
The chemical name of KENGREAL is tetrasodium salt of N6-[2-(methylthio)ethyl]-2-[(3,3,3,-trifluoropropyl)-5’-adenylic acid, monanhydride with (dichloromethylene) bisphosphonic acid.
The empirical formula of KENGREAL is C17H21N5Cl2F3Na4O12P3S2 and the molecular weight is 864.3 g/mol.
The chemical structure is represented below:
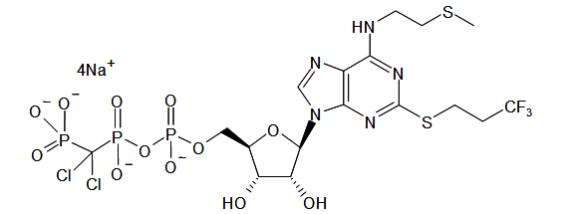
Cangrelor for Injection is a sterile white to off-white lyophilized powder for IV infusion. In addition to the active ingredient, cangrelor, each single use vial contains mannitol, sorbitol, and sodium hydroxide to adjust the pH.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Cangrelor is a direct P2Y12 platelet receptor inhibitor that blocks ADP-induced platelet activation and aggregation. Cangrelor binds selectively and reversibly to the P2Y12 receptor to prevent further signaling and platelet activation.
12.2 Pharmacodynamics
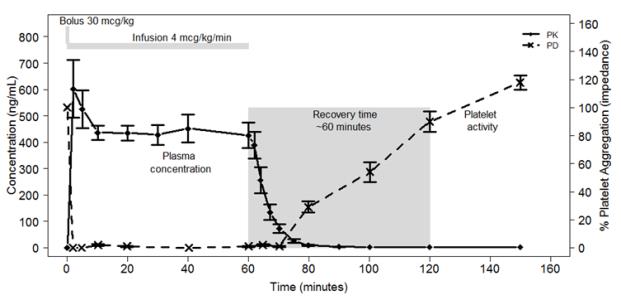
Cangrelor inhibits activation and aggregation of platelets. After administration of a 30 mcg/kg IV bolus followed by a 4 mcg/kg/min IV infusion, platelet inhibition occurs within 2 minutes.
Figure 2 shows the effect on platelet activity, and its relation to cangrelor plasma concentration, of administering a 30 mcg/kg IV bolus, followed by a 1-hour 4 mcg/kg/min IV infusion, of cangrelor. The anti-platelet effect is maintained for the duration of the infusion. After discontinuation of the infusion, the anti-platelet effect decreases rapidly and platelet function returns to normal within 1 hour.
Figure 2: Cangrelor PD Characteristics
12.3 Pharmacokinetics
KENGREAL administered intravenously has linear pharmacokinetics in both healthy volunteers and patients. KENGREAL is rapidly distributed and metabolized, reaching Cmax within 2 minutes after administration of an intravenous bolus followed by infusion.
Distribution
In a study in healthy volunteers, KENGREAL administration at a dose of 30 mcg/kg bolus plus 4 mcg/kg/min showed a volume of distribution of 3.9 L. Plasma protein binding of KENGREAL is about 97-98%.
Metabolism
KENGREAL is deactivated rapidly in the circulation by dephosphorylation to its primary metabolite, a nucleoside, which has negligible anti-platelet activity. KENGREAL’s metabolism is independent of hepatic function and it does not interfere with other drugs metabolized by hepatic enzymes.
Elimination
Following IV administration of [3H] KENGREAL 58% of radioactivity was recovered in urine. The remaining 35% of radioactivity was in feces, presumably following biliary excretion. The average elimination half-life of KENGREAL is about 3-6 minutes.
Specific Populations
KENGREAL pharmacokinetics are not affected by sex, age, renal status or hepatic function. No dose adjustment is needed for these factors [see Use in Specific Populations (8)].
Weight
Although weight was a significant covariate for PK with higher clearance in heavier patients, the impact of weight on drug exposure is accounted by the use of weight-based dosing.
Drug-Drug Interactions
Co-administration of cangrelor with unfractionated heparin, aspirin, and nitroglycerin was formally studied in healthy subjects, with no evidence of an effect on the PK/PD of cangrelor.
In clinical trials cangrelor has been co-administered with bivalirudin, low molecular weight heparin, clopidogrel, prasugrel, and ticagrelor without clinically detectable interactions.
The expected antiplatelet effect of a 600 mg loading dose of clopidogrel or a 60 mg loading dose of prasugrel was blocked when clopidogrel or prasugrel was administered during a cangrelor infusion [see Drug Interactions (7.1)].
In contrast, the antiplatelet effect of a 180 mg ticagrelor loading dose was not altered significantly when ticagrelor was administered during cangrelor infusion [see Drug Interactions (7.1)].
Figure 3: Inhibition (Mean) of 20 µM ADP-induced Platelet Aggregation (IPA) Measured by Light Transmission Aggregometry after Cangrelor 30 mcg/kg Bolus and 120-minute 4 mcg/kg Infusion with Transition to Other Oral P2Y12 Platelet Inhibitors.
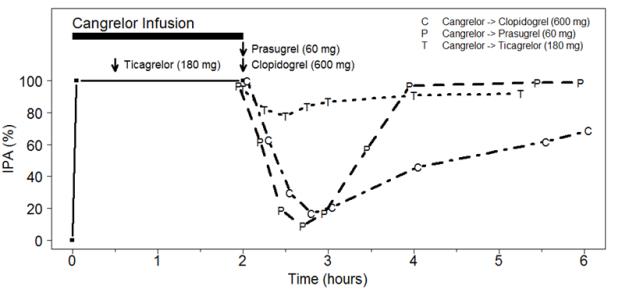
As shown in Figure 3, discontinuation of cangrelor infusion, followed by administration of the irreversible P2Y12 platelet inhibitors clopidogrel or prasugrel led to a 1-hour decrease in IPA followed by an increase in inhibition of platelet aggregation beginning at about one hour. This time course of platelet inhibition reflects the pharmacokinetics of cangrelor (offset) followed by the absorption and metabolism of clopidogrel and prasugrel to active metabolites (onset). Administration of ticagrelor, a reversible P2Y12 platelet inhibitor, during the cangrelor infusion led to minimal decrease in platelet inhibition for approximately 0.5 hour following discontinuation of the cangrelor infusion. Administering ticagrelor during cangrelor infusion does not attenuate the anti-platelet effect of ticagrelor.
In vitro studies suggest that neither cangrelor nor its major metabolites inhibit the activity of the hepatic CYP isoenzymes at therapeutic concentrations. Therefore, cangrelor administration is not expected to interfere with the hepatic metabolism of other concomitantly administered therapeutic agents.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
No carcinogenicity studies were conducted.
Mutagenesis
Cangrelor was non-mutagenic and non-clastogenic in genetic toxicology studies, including in vitro bacterial gene mutation assay, mouse lymphoma thymidine kinase assay, chromosome aberration assay in human peripheral lymphocytes, and in vivo bone marrow micronucleus assay in mice.
Impairment of Fertility
Cangrelor had no significant effect on male or female rats fertility treated by continuous infusion for 28 days, or on early embryonic development at steady state plasma concentration (Css) of approximately the same as that achieved in the PCI setting at the MRHD.
-
14 CLINICAL STUDIES
14.1 CHAMPION PHOENIX Trial
The CHAMPION PHOENIX trial was intended to test whether faster platelet inhibition with cangrelor at the time of PCI would reduce the rate of periprocedural thrombotic events compared to a drug with a slower antiplatelet effect, clopidogrel, given at about the time of PCI. It was a randomized, double-blind study in which patients with coronary artery disease (stable angina, UA/NSTEMI, STEMI) requiring PCI and receiving standard therapy including aspirin and heparin or bivalirudin were randomized 1:1 to KENGREAL (n=5472) or to clopidogrel 300 or 600 mg (n=5470). Patients who had already taken an oral P2Y12 platelet inhibitor were not eligible to enroll. Patients administered glycoprotein IIb/IIIa inhibitors (GPI) or for whom GPI use was planned were also not eligible to enroll. PHOENIX was thus a study of people undergoing PCI who had not been previously treated with anti-platelet therapy other than aspirin.
The primary outcome measure was the first occurrence of any one of the composite endpoint of all-cause mortality, myocardial infarction (MI), ischemia-driven revascularization (IDR), and stent thrombosis (ST) within 48 hours after randomization.
KENGREAL was administered as 30 mcg/kg bolus followed by 4 mcg/kg/min infusion for 2 to 4 hours. Clopidogrel 600 mg was administered immediately at the end of the KENGREAL infusion in patients randomized to KENGREAL. Clopidogrel 300 mg or 600 mg was administered shortly before PCI or shortly afterward, in patients randomized to clopidogrel.
KENGREAL significantly reduced the occurrence of primary composite endpoint events compared to clopidogrel (relative risk reduction [RRR] 22%). Most of the effect was a reduction in post-procedural MIs detected solely by elevations in CK-MB (type 4a MI). KENGREAL did not reduce the risk of death. Table 2 shows the study results for the primary composite endpoint and the contribution of each component to the primary endpoint.
Table 2: Primary Endpoint and Its Component Events at 48 Hours in CHAMPION PHOENIX (mITT populationa) KENGREAL
(N=5470)
n (%)Clopidogrel
(N=5469)
n (%)KENGREAL vs. clopidogrel OR (95% CI) p-value Primary Endpoint
Death/MI/IDR/ST257 (4.7) 322 (5.9) 0.78 (0.66,0.93) b 0.005 Death 18 (0.3) 18 (0.3) MI 202 (3.7) 254 (4.6) IDR 10 (0.2) 14 (0.3) ST 27 (0.5) 36 (0.7) Note: if a subject had more than one event at 48 hours, then worst outcome counted (death >MI >IDR >ST)
aThe mITT population is all randomized subjects who received at least one dose of study drug and underwent the index PCI procedure
b Based on logistic model adjusted for loading dose and baseline patient status for primary endpoint
A supplementary analysis was also performed omitting two subcomponent events of the primary endpoint that were of lesser clinical significance: intraprocedural stent thrombosis (defined as a new or increasing thrombus within or adjacent to a deployed stent occurring during the index PCI procedure), and myocardial infarction with less than a 10-fold increase in CK-MB, or with less than a 5-fold increase in CK-MB in the presence of new Q waves or new left bundle branch block (LBBB). These results are shown in Table 3.
Table 3: Supplementary Endpoint and Its Component Events at 48 Hours in CHAMPION PHOENIX (mITT population) n (%) KENGREAL
(N=5470)Clopidogrel
(N=5469)KENGREAL vs. clopidogrel
OR (95% CI)Supplementary Endpoint
Death/SCAI-MI/IDR/ARC-ST79 (1.4) 114 (2.1) 0.69 (0.52,0.92) Death 18 (0.3) 18 (0.3) SCAI-MIa 48 (0.9) 80 (1.5) IDR 13 (0.2) 16 (0.3) ARC-STb 0 (0.0) 0 (0.0) Note: if a subject had more than one event at 48 hours, then worst outcome counted (death >SCAI-MI >IDR >ARC-ST)
aSCAI MI: CK-MB ≥10X ULN, or CK-MB ≥5X ULN with new Q waves or new LBBB
bARC-ST defined according to the ARC definition [Cutlip et al. 2007]
The effect of KENGREAL appeared to be consistent in a variety of pre-specified and other clinically important subgroups (see Figure 4).
Figure 4: CHAMPION PHOENIX Study: Primary Efficacy Endpoint by Subgroup (mITT Populationa)
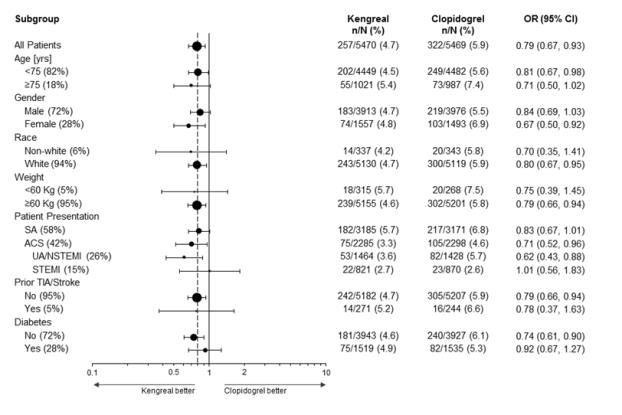
a The mITT population is all randomized subjects who received at least one dose of study drug and underwent the index PCI procedure.
Note: The figure above presents effects in various subgroups most of which are baseline characteristics and most of which were pre-specified (patient presentation and weight were not pre-specified subgroups). The 95% confidence limits that are shown do not take into account how many comparisons were made, nor do they reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
14.2 CHAMPION PCI and PLATFORM Trials
Two additional concurrent clinical trials of the effect of KENGREAL in patients undergoing percutaneous coronary intervention, CHAMPION PCI and CHAMPION PLATFORM were conducted and terminated early for futility. They were completed prior to the design and conduct of CHAMPION PHOENIX. The comparative characteristics and outcomes of each trial are shown in Table 4.
Table 4: Summary of the CHAMPION Trials PCI PLATFORM PHOENIX Subjects Randomized (% of planned enrollment) 8,846 (99%) 5,346 (84%) 11,145 (100%) Primary Endpoint at 48 hours Death, MI, or IDR Death, MI, or IDR Death, MI, IDR, or ST Outcome of primary analysis, OR (95% CI) 1.05 (0.88, 1.24) 0.87 (0.71, 1.07) 0.78 (0.66, 0.93) Clopidogrel dose and time in clopidogrel arm 600 mg immediately before PCI 600 mg immediately after PCI 300 or 600 mg shortly before or shortly after PCI Population enrolled
(%)Stable angina 15% 5% 58% UA/NSTEMI 74% 95% 26% STEMI 11% Excluded 16% -
16 HOW SUPPLIED/STORAGE AND HANDLING
KENGREAL is supplied as a sterile lyophilized powder in single-use 10 mL vials.
- NDC # 10122-620-01: 10 mL vial containing 50 mg cangrelor
- NDC # 10122-620-10: 10 count of 10 mL vials containing 50 mg cangrelor
Vials of KENGREAL should be stored at USP Controlled Room Temperature, [20°C to 25°C (68°F to 77°F) with excursions between 15°C and 30°C (59°F and 86°F) permitted].
KENGREAL® is a registered trademeark of Chiesi Farmaceutici S.p.A.
Distributed by:
Chiesi USA, Inc.
Cary, NC 27518CTK-001-0119-01-SPL
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel
NDC: 10122-620-10
Kengreal®
(cangrelor) for injection
50 mg per vial
Rx Only
10 Single Use Vials
-
INGREDIENTS AND APPEARANCE
KENGREAL
cangrelor injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 10122-620 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CANGRELOR (UNII: 6AQ1Y404U7) (CANGRELOR - UNII:6AQ1Y404U7) CANGRELOR 50 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) 158 mg SORBITOL (UNII: 506T60A25R) 52 mg SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 10122-620-10 10 in 1 CARTON 07/08/2015 1 NDC: 10122-620-01 1 in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA204958 07/08/2015 Labeler - Chiesi USA, Inc. (088084228) Registrant - Chiesi USA, Inc. (088084228)
Trademark Results [KENGREAL]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 KENGREAL 87442665 not registered Live/Pending |
Chiesi Farmaceutici S.P.A. 2017-05-09 |
 KENGREAL 86166110 4867615 Live/Registered |
CHIESI FARMACEUTICI S.P.A. 2014-01-15 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.