LIVDELZI- seladelpar lysine capsule
Livdelzi by
Drug Labeling and Warnings
Livdelzi by is a Prescription medication manufactured, distributed, or labeled by Gilead Sciences, Inc. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LIVDELZI safely and effectively. See full prescribing information for LIVDELZI.
LIVDELZI® (seladelpar) capsules, for oral use
Initial U.S. Approval: 2024INDICATIONS AND USAGE
LIVDELZI is a peroxisome proliferator-activated receptor (PPAR)-delta agonist indicated for the treatment of primary biliary cholangitis (PBC) in combination with ursodeoxycholic acid (UDCA) in adults who have an inadequate response to UDCA, or as monotherapy in patients unable to tolerate UDCA.
This indication is approved under accelerated approval based on a reduction of alkaline phosphatase (ALP). Improvement in survival or prevention of liver decompensation events have not been demonstrated. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s). (1)
Limitations of Use
Use of LIVDELZI is not recommended in patients who have or develop decompensated cirrhosis (e.g., ascites, variceal bleeding, hepatic encephalopathy). (8.7)
DOSAGE AND ADMINISTRATION
The recommended dosage of LIVDELZI is 10 mg orally once daily. Administer LIVDELZI with or without food. (2.1)
DOSAGE FORMS AND STRENGTHS
Capsules: 10 mg (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Fractures: Consider the risk of fracture in patients treated with LIVDELZI. Monitor bone health according to current standards of care. (5.1)
- Liver Test Abnormalities: Obtain baseline clinical and laboratory liver assessments prior to starting LIVDELZI and monitor during treatment. Interrupt or discontinue LIVDELZI if the liver tests worsen. (5.2)
- Biliary Obstruction: Avoid use in patients with complete biliary obstruction. If biliary obstruction is suspected, interrupt LIVDELZI and treat as clinically indicated. (5.3)
ADVERSE REACTIONS
Most common adverse reactions (reported in ≥5% and higher compared to placebo) are headache, abdominal pain, nausea, abdominal distension, and dizziness. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at 1-800-GILEAD-5 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Probenecid: Avoid concomitant use. (7.1)
- Strong CYP2C9 Inhibitors: Monitor for adverse effects. (7.1)
- Dual Moderate CYP2C9 and Moderate to Strong CYP3A4 Inhibitors: Monitor for adverse effects. (7.1)
- CYP2C9 Poor Metabolizers using Moderate to Strong CYP3A4 Inhibitors: Monitor for adverse effects. (7.1)
- Dual or Multiple Clinical Inhibitors of Drug Transporters OATP1B1, OATP1B3, and BCRP: Monitor for adverse effects. (7.1)
- Rifampin: Monitor biochemical response (e.g., ALP and bilirubin) when patients initiate rifampin. (7.1)
- Bile Acid Sequestrants: Administer at least 4 hours before or 4 hours after taking a bile acid sequestrant, or at as great an interval as possible. (7.1)
USE IN SPECIFIC POPULATIONS
Hepatic Impairment: Monitor patients with cirrhosis for evidence of decompensation. Consider discontinuation if patient progresses to moderate or severe hepatic impairment (Child-Pugh B or C). (8.7)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 1/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage and Administration
2.2 Administration Modification for Bile Acid Sequestrants
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Fractures
5.2 Liver Test Abnormalities
5.3 Biliary Obstruction
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on LIVDELZI
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
8.8 CYP2C9 Poor Metabolizers
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
LIVDELZI is indicated for the treatment of primary biliary cholangitis (PBC) in combination with ursodeoxycholic acid (UDCA) in adults who have had an inadequate response to UDCA, or as monotherapy in patients unable to tolerate UDCA.
This indication is approved under accelerated approval based on a reduction of alkaline phosphatase (ALP) [see Clinical Studies (14)]. Improvement in survival or prevention of liver decompensation events have not been demonstrated. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s).
Limitations of Use
Use of LIVDELZI is not recommended in patients who have or develop decompensated cirrhosis (e.g., ascites, variceal bleeding, hepatic encephalopathy) [see Use in Specific Populations (8.7)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage and Administration
The recommended dosage of LIVDELZI is 10 mg orally once daily. Administer LIVDELZI with or without food [see Clinical Pharmacology (12.3)].
2.2 Administration Modification for Bile Acid Sequestrants
Administer LIVDELZI at least 4 hours before or 4 hours after taking bile acid sequestrants, or at as great an interval as possible [see Drug Interactions (7.1)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Fractures
Fractures occurred in 4% of LIVDELZI-treated patients compared to no placebo-treated patients [see Adverse Reactions (6.1)].
Consider the risk of fracture in the care of patients treated with LIVDELZI and monitor bone health according to current standards of care.
5.2 Liver Test Abnormalities
LIVDELZI has been associated with dose-related increases in serum transaminase (aspartate aminotransferase [AST] and alanine aminotransferase [ALT]) levels greater than 3-times upper limit of normal (ULN) in PBC patients receiving 50 mg once daily (5-times higher than the recommended dosage) and 200 mg (20-times higher than the recommended dosage) once daily. Transaminase levels returned to pretreatment levels upon LIVDELZI discontinuation. LIVDELZI 10 mg once daily did not show a similar pattern for increases in transaminase levels [see Overdosage (10)].
Obtain baseline clinical and laboratory assessments at treatment initiation with LIVDELZI and monitor thereafter according to routine patient management. Interrupt LIVDELZI treatment if the liver tests (ALT, AST, total bilirubin [TB], and/or alkaline phosphatase [ALP]) worsen, or the patient develops signs and symptoms consistent with clinical hepatitis (e.g., jaundice, right upper quadrant pain, eosinophilia). Consider permanent discontinuation if liver tests worsen after restarting LIVDELZI.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in labeling:
- Fractures [see Warnings and Precautions (5.1)]
- Liver Test Abnormalities [see Warnings and Precautions (5.2)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In Trial 1, 193 patients were randomized to receive either LIVDELZI 10 mg (N=128) or placebo (N=65) once daily for 12 months [see Clinical Studies (14)]. LIVDELZI or placebo was administered in combination with UDCA in 94% of patients and as monotherapy in 6% of patients who were unable to tolerate UDCA.
Common Adverse Reactions
Table 1 presents common adverse reactions that occurred in Trial 1.
Table 1: Common Adverse Reactions Occurring Through Week 52 in Adult Patients with PBC (Trial 1)* Adverse Reaction† LIVDELZI 10 mg Once Daily
(N=128)
% (n)PLACEBO
(N=65)
% (n)- * Included 12 patients (6%) who were intolerant to UDCA and initiated treatment as monotherapy: 8 patients (6%) in the LIVDELZI 10 mg arm and 4 patients (6%) in the placebo arm.
- † Occurring in greater than or equal to 5% of patients in the LIVDELZI treatment arm and at an incidence greater than or equal to 1% higher than in the placebo arm.
- ‡ The gastrointestinal adverse reactions were mild to moderate without the need for discontinuation of LIVDELZI.
Headache 8% (10) 3% (2) Abdominal pain‡ 7% (9) 2% (1) Nausea‡ 6% (8) 5% (3) Abdominal distension‡ 6% (8) 3% (2) Dizziness 5% (6) 2% (1) Less Common Adverse Reactions
Additional adverse reactions that occurred more frequently in the LIVDELZI-treated patients compared to placebo, but in less than 5% of patients, included dyspepsia, rash, alopecia, anemia, and cough.
Laboratory Abnormalities
Estimated Glomerular Filtration Rate
In Trial 1, LIVDELZI-treated patients developed decreased estimated glomerular filtration rate (eGFR) (serum creatinine elevations) more frequently compared to placebo-treated patients. Ten percent (n=12) of LIVDELZI-treated patients had a decline in eGFR of at least 25%, compared to 2% (n=1) of placebo-treated patients. None of the patients experienced an eGFR decline of 50% or more. The decline in eGFR stabilized or returned towards baseline with ongoing LIVDELZI treatment. None of the patients required discontinuation of LIVDELZI and there were no clinical findings associated with the observed changes in eGFR.
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on LIVDELZI
Table 2 includes clinically significant drug interactions affecting LIVDELZI.
Table 2: Clinically Significant Interactions Affecting LIVDELZI Concomitant Drug or Class Potential Effect on Seladelpar Exposure* Clinical Intervention - * ↑ = Increase, ↓ = Decrease.
Probenecid ↑ seladelpar Avoid concomitant administration of LIVDELZI with probenecid. Strong CYP2C9 Inhibitors ↑ seladelpar Monitor patients for adverse effects during concomitant use of LIVDELZI with strong CYP2C9 inhibitors. Dual Moderate CYP2C9 and Moderate or Strong CYP3A4 Inhibitors (e.g., fluconazole) ↑ seladelpar Monitor patients for adverse effects during concomitant use of LIVDELZI with drugs that are dual moderate CYP2C9 and moderate or strong CYP3A4 inhibitors. CYP2C9 Poor Metabolizers Using Moderate or Strong CYP3A4 Inhibitors ↑ seladelpar Monitor patients who are CYP2C9 poor metabolizers for adverse effects during concomitant use of LIVDELZI with moderate or strong CYP3A4 inhibitor. Dual or Multiple Clinical Inhibitors of Drug Transporters OATP1B1, OATP1B3, and BCRP (e.g, cyclosporine) ↑ seladelpar Monitor patients for adverse effects during concomitant use of LIVDELZI with dual or multiple clinical inhibitors of drug transporters OATP1B1, OATP1B3, and BCRP. Rifampin ↓ seladelpar Concomitant use of LIVDELZI with rifampin, an inducer of metabolizing enzymes, may result in delayed or suboptimal LIVDELZI biochemical response.
Monitor the biochemical response (e.g., ALP and bilirubin) when patients initiate rifampin during LIVDELZI treatment.Bile Acid Sequestrants ↓ seladelpar Bile acid sequestrants may interfere with the action of LIVDELZI by reducing its absorption and systemic exposure, which may reduce LIVDELZI efficacy.
Administer LIVDELZI at least 4 hours before or 4 hours after taking a bile acid sequestrant, or at as great an interval as possible [see Dosage and Administration (2.2)]. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are insufficient data from human pregnancies exposed to LIVDELZI to allow an assessment of a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. In animal reproduction studies, no malformations or effects on embryo-fetal survival occurred in pregnant rats or rabbits after seladelpar treatment at exposures of up to 176-times and 49-times the recommended dose based on AUC (area under the plasma concentration-time curve), respectively. Reduction of fetal growth associated with maternal toxicity occurred in pregnant rabbits at 49-times the recommended dose based on AUC, but not at 3-times the recommended dose. In a pre- and postnatal development study in rats with maternal dosing of seladelpar during organogenesis through lactation, postnatal growth and pre-weaning survival of offspring was reduced at 115-times the recommended dose based on AUC, but not at the lower exposure of 16-times the recommended dose (see Data).
The background risks of major birth defects and miscarriage for the indicated population are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Report pregnancies to Gilead Sciences, Inc. at 1-800-445-3235.
Data
Animal Data
No effects on embryo-fetal development were observed in pregnant rats treated orally with up to 100 mg/kg/day seladelpar (176-times the recommended dose based on AUC) during the period of organogenesis.
Oral administration of 40 mg/kg/day seladelpar in pregnant rabbits (49-times the recommended dose based on AUC) during organogenesis resulted in reduced fetal body weight, which was likely due to maternal toxicity (i.e., decreases in food consumption, body weight, and gravid uterine weight) and distended stomach. No treatment-related fetal malformations or effects on embryo-fetal survival occurred in rabbits at 49-times the recommended dose. No adverse effects on embryo-fetal development were observed at 10 mg/kg/day (3-times the recommended dose based on AUC).
A pre- and postnatal development study was performed using oral administration of seladelpar at doses of 0 (vehicle), 5, 20, or 100 mg/kg/day in pregnant rats during organogenesis through lactation. Treatment with 5 mg/kg/day or higher (4-times the recommended dose based on AUC) resulted in a dose-dependent reduction in pup body weight during the pre-weaning period. The weight reduction in offspring was associated with delays in developmental milestones (i.e., eye opening and pinna unfolding at 5 mg/kg/day and higher; hair growth and sexual maturity at 100 mg/kg/day). Reduction in pup body weight at 100 mg/kg/day (115-times the recommended dose based on AUC), which continued into the post-weaning maturation period, was associated with a slight decrease in pre-weaning survival and was considered adverse. No adverse effects were found in clinical observations, neurobehavioral assessment, or reproductive performance testing in the offspring of females treated with seladelpar. At 20 mg/kg/day (16-times the recommended dose based on AUC), none of the observed effects in offspring were considered to be adverse.
8.2 Lactation
Risk Summary
There are no data on the presence of seladelpar or its metabolite in either human or animal milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for LIVDELZI and any potential adverse effects on the breastfed infant from LIVDELZI or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of LIVDELZI in pediatric patients have not been established.
8.5 Geriatric Use
Of the 128 LIVDELZI-treated patients in Trial 1, 29 (23%) patients were 65 years of age and older and 2 (2%) were 75 years of age and older. No overall differences in safety or effectiveness were observed between patients 65 to 75 years of age and younger adult patients. No dosage adjustment for patients 65 years of age and older is necessary.
Clinical studies of LIVDELZI did not include sufficient numbers of patients 75 years of age and older to determine whether they respond differently from younger adult patients. Because of limited clinical experience with LIVDELZI in patients older than 75 years old, closer monitoring of adverse events in patients older than 75 years is recommended [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
The recommended dosage in patients with mild, moderate, or severe renal impairment is the same as in patients with normal renal function. Patients with end-stage renal disease on dialysis have not been studied [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dosage adjustment is recommended for PBC patients with mild hepatic impairment (Child-Pugh A) [see Clinical Pharmacology (12.3)].
The safety and efficacy of LIVDELZI in patients with decompensated cirrhosis have not been established. Use of LIVDELZI is not recommended in patients who have or develop decompensated cirrhosis (e.g., ascites, variceal bleeding, hepatic encephalopathy).
Monitor patients with cirrhosis for evidence of decompensation. Consider discontinuing LIVDELZI if the patient progresses to moderate or severe hepatic impairment (Child-Pugh B or C).
8.8 CYP2C9 Poor Metabolizers
Monitor CYP2C9 poor metabolizers who receive a concomitant moderate to strong CYP3A4 inhibitor more frequently for adverse reactions.
Seladelpar is a CYP2C9 and CYP3A4 substrate. Increased seladelpar AUC is expected in patients who are CYP2C9 poor metabolizers with concomitant use of a moderate to strong CYP3A4 inhibitor [see Drug Interactions (7.1), Clinical Pharmacology (12.5)].
-
10 OVERDOSAGE
PBC patients who received 5-times the recommended dosage or 20-times the recommended dosage of LIVDELZI experienced an increase in liver transaminases, muscle pain, and/or elevations in creatine phosphokinase, which resolved upon LIVDELZI discontinuation [see Warnings and Precautions (5.2)].
There is no specific treatment for overdose with LIVDELZI. General supportive care of the patient is indicated, as appropriate. If indicated, elimination of unabsorbed drug should be achieved by emesis or gastric lavage; usual precautions should be observed to maintain the airway. Because seladelpar is highly bound to plasma proteins, hemodialysis should not be considered.
-
11 DESCRIPTION
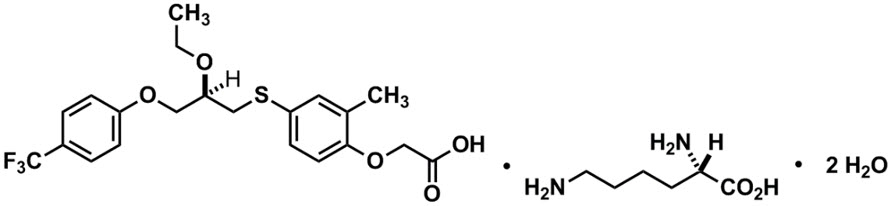
LIVDELZI capsules contain seladelpar lysine, a peroxisome proliferator-activated receptor (PPAR)-delta (δ) agonist. Seladelpar is a single enantiomer of the R-configuration and is present as a lysine dihydrate salt. Seladelpar lysine dihydrate is a white to off-white powder with a molecular formula of C21H23F3O5S ∙C6H14N2O2 ∙2H2O and a molecular weight of 626.7 g/mol. Its solubility in water is pH dependent. It is slightly soluble at low pH and very soluble at high pH. The chemical name for seladelpar lysine dihydrate is 2-[4-[[(2R)-2-ethoxy-3-[4-(trifluoromethyl)phenoxy]propyl]thio]-2-methylphenoxy]acetic acid, lysine dihydrate, and the chemical structure is:
LIVDELZI (seladelpar) capsules are supplied in a 10 mg strength for oral administration. Each capsule contains 14.1 mg of seladelpar lysine and the following inactive ingredients: butylated hydroxytoluene, colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, mannitol, microcrystalline cellulose, and hard gelatin shells.
The light gray opaque (body)/dark blue opaque (cap) capsule shells contain gelatin, titanium dioxide, black iron oxide, yellow iron oxide, red iron oxide and the colorant FD&C Blue #2.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Seladelpar is a peroxisome proliferator-activated receptor (PPAR)-delta (δ) agonist. However, the mechanism by which seladelpar exerts its therapeutic effects in patients with PBC is not well understood. Pharmacological activity that is potentially relevant to therapeutic effects includes inhibition of bile acid synthesis through activation of PPARδ, which is a nuclear receptor expressed in most tissues, including the liver. Published studies show that PPARδ activation by seladelpar reduces bile acid synthesis through Fibroblast Growth Factor 21 (FGF21)-dependent downregulation of CYP7A1, the key enzyme for the synthesis of bile acids from cholesterol.
12.2 Pharmacodynamics
Pharmacodynamic Markers
In patients with PBC treated with 10 mg once daily of LIVDELZI (Trial 1), a greater reduction in mean ALP from baseline was observed as early as 1 month after treatment compared to the placebo group and lower ALP was generally maintained through month 12 [see Clinical Studies (14)].
In another study in which patients with PBC were treated with 2, 5, or 10 mg once daily of seladelpar, a dose dependent reduction in mean ALP was observed.
12.3 Pharmacokinetics
Following a single dose administration, seladelpar systemic exposure increased dose-proportionally from 2 mg (0.2 times the recommended dosage) to 15 mg (1.5 times the recommended dosage) and greater than dose proportionally at higher doses. For a dose increase from 10 mg to 200 mg (20 times the recommended dosage), mean Cmax and mean AUC for seladelpar increased 70-fold and 27-fold, respectively.
Following once daily dosing, seladelpar steady-state was achieved by day 4 and AUC increase was less than 30%. In PBC patients, median (CV) Cmax and AUC for seladelpar was 90.5 (42.5%) ng/mL and 817 (44%) ng*h/mL, respectively at steady state following once daily dosing of 10 mg.
Distribution
Seladelpar steady state apparent volume of distribution was approximately 110.3 L. Seladelpar plasma protein binding is greater than 99%.
Elimination
The apparent oral clearance of seladelpar is 12.6 L/h. Following administration of a single dose of 10 mg seladelpar in healthy subjects, mean elimination half-life was 6 hours for seladelpar. In PBC patients, the half-life range was 3.8 to 6.7 hours for seladelpar.
Metabolism
Seladelpar is primarily metabolized in vitro by CYP2C9 and to a lesser extent by CYP2C8 and CYP3A4, resulting in the three major metabolites: seladelpar sulfoxide (M1), desethyl-seladelpar (M2), and desethyl-seladelpar sulfoxide (M3). The metabolite-to-parent AUC ratios were 0.36, 2.32 and 0.63 for M1, M2 and M3, respectively. Median Tmax for metabolites were 10 hours for M1 and 4 hours for M2 and M3. None of the major metabolites have pharmacological activity.
Excretion
Seladelpar is primarily eliminated in urine as metabolites. Following a single oral dose of 10 mg radiolabeled seladelpar in humans, approximately 73.4% of the dose was recovered in urine (less than 0.01% unchanged) and 19.5% in feces (2.02% unchanged) within 216 hours. Biliary excretion of seladelpar was suggested by an animal study.
Specific Populations
No clinically significant differences in the pharmacokinetics of seladelpar were observed based on age (19 to 79 years old), body mass index (BMI) (17.6 to 45.0 kg/m2), weight (45.8 to 127.5 kg), sex, and race (White, Black, or other).
Patients with Renal Impairment
In subjects with mild (eGFR ≥60 to <90 mL/min/1.73 m2, MDRD), moderate (eGFR ≥30 to <60 mL/min/1.73 m2), and severe (<30 mL/min/1.73 m2 and not on dialysis) renal impairment, the AUCinf of seladelpar was 10% higher, 54% higher, and similar to that in subjects with normal renal function, respectively, after administration of a single 10 mg dose of seladelpar. The difference in Cmax of seladelpar was less than 18% in subjects with renal impairment compared to subjects with normal renal function [see Use in Specific Populations (8.6)]. The pharmacokinetics of seladelpar have not been studied in patients requiring hemodialysis.
Patients with Hepatic Impairment
Hepatic Impairment of various etiologies: Following a single oral dose of 10 mg seladelpar, seladelpar AUC increased 1.1-fold in subjects with mild (Child-Pugh A), 2.5-fold in moderate (Child-Pugh B), and 2.1-fold in severe (Child-Pugh C) hepatic impairment. Seladelpar Cmax increased 1.3-fold in subjects with mild (Child-Pugh A), 5.2-fold in moderate (Child-Pugh B), and 5-fold in severe (Child-Pugh C) hepatic impairment.
Hepatic Impairment in patients with PBC: Compared to PBC patients with mild hepatic impairment (Child-Pugh A) without portal hypertension, seladelpar exposures (Cmax, AUC) were 1.7 to 1.8-fold higher in PBC patients with mild hepatic impairment with portal hypertension, 1.6 to 1.9-fold higher in PBC patients with moderate hepatic impairment (Child-Pugh B), and 2.1 to 2.5-fold higher in PBC patients with severe hepatic impairment (Child-Pugh C) after a single oral dose of 10 mg seladelpar.
Accumulation ratios were less than 1.2-fold in PBC patients with mild hepatic impairment with portal hypertension and PBC patients with moderate hepatic impairment following 10 mg seladelpar once daily dosing for 28 days.
Drug Interaction Studies
Effect of Other Drugs on Seladelpar
Carbamazepine
Seladelpar AUC0–inf decreased by approximately 44% and Cmax by 24% following administration of a single 10 mg seladelpar dose after carbamazepine 300 mg twice daily for 8 days in healthy subjects. The carbamazepine (CYP3A and CYP2C9 inducer) dose was escalated from 100 mg twice daily for 3 days followed by 200 mg twice daily for 4 days to 300 mg twice daily.
Fluconazole
Seladelpar AUC0–inf increased by 2.4-fold and Cmax by 1.4-fold following concomitant use of a single 10 mg seladelpar dose with 400 mg fluconazole (moderate CYP2C9 and CYP3A4 inhibitor) in healthy subjects.
Cyclosporine
Seladelpar AUC0–inf increased by 2.1-fold and Cmax by 2.9-fold following concomitant use of a single 10 mg seladelpar dose with 600 mg cyclosporine (OATP1B1, OATP1B3, and BCRP inhibitor) in healthy subjects.
Probenecid
Seladelpar AUC0–inf increased by 2-fold and Cmax by 4.69-fold following concomitant use of a single 10 mg seladelpar dose with 500 mg probenecid in healthy subjects.
In Vitro Studies
Seladelpar is a substrate of CYP2C9, CYP2C8, CYP3A4, and the transporters BCRP, P-gp, OATP1B1, OATP1B3, and OAT3.
Seladelpar is not a substrate of MATE1, MATE2-K, OAT1, OCT1, or OCT2 transporters.
Effects of Seladelpar on other drugs
In clinical studies, no clinically significant differences in the pharmacokinetics of the following drugs were observed when used concomitantly with seladelpar: tolbutamide (CYP2C9 substrate), midazolam (CYP3A4 substrate), simvastatin (CYP3A4 and OATP substrate), atorvastatin (CYP3A4 and OATP substrate), or rosuvastatin (BCRP and OATP substrate).
12.5 Pharmacogenomics
CYP2C9 activity is decreased in individuals with genetic variants such as CYP2C9*2 and CYP2C9*3. Compared to CYP2C9 normal metabolizers (*1/*1, n=84) after a single dose of seladelpar 1 mg to 15 mg, dose-normalized AUC0–inf was 48% higher in CYP2C9 poor metabolizers (*2/*3, n=2) and 24% higher in CYP2C9 intermediate metabolizers (*1/*2, *1/*8, *1/*3, *2/*2, n=28). Dose-normalized Cmax was similar for CYP2C9 normal, intermediate, and poor metabolizers. Seladelpar pharmacokinetics was not evaluated in patients who are CYP2C9 poor metabolizers with two no function alleles (e.g., *3/*3). CYP2C9 poor metabolizers may have increased AUC when seladelpar is used concomitantly with a moderate to strong CYP3A4 inhibitor [see Drug Interactions (7.1), Use in Specific Populations (8.8)].
The prevalence of CYP2C9 poor metabolizers is approximately 2 to 3% in White populations, 0.5 to 4% in Asian populations, and <1% in African American populations. Additional decreased or nonfunctional alleles (e.g., *5, *6, *11) are more prevalent in African American populations.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 2-year study in CD-1 mice, oral administration of seladelpar produced hepatocellular adenoma or carcinoma at a dose of 5 mg/kg/day in males (6-times the recommended dose based on AUC) and 20 mg/kg/day in females (140-times the recommended dose based on AUC). No tumorigenic effects were observed in female mice at doses of up to 10 mg/kg/day (49-times the recommended dose based on AUC).
In a 2-year study in Sprague-Dawley rats, oral administration of seladelpar produced benign interstitial cell tumors in testes and squamous cell carcinoma of the nonglandular stomach in males at a dose of 30 mg/kg/day (79-times the recommended dose based on AUC). No tumorigenic effects were observed in males at doses of up to 10 mg/kg/day (14-times the recommended dose based on AUC) or in females at doses of up to 30 mg/kg/day (26-times the recommended dose based on AUC).
13.2 Animal Toxicology and/or Pharmacology
In a 2-year study in CD-1 mice, seladelpar produced an increased incidence of lens cataracts at 5 mg/kg/day in both sexes (6-times and 19-times the recommended dose in male and female mice, respectively, based on AUC). In a 2-year study in Sprague-Dawley rats, seladelpar produced an increased incidence of cornea inflammation at 10 mg/kg/day (14-times the recommended dose based on AUC) and cornea mineralization at 30 mg/kg/day (79-times the recommended dose based on AUC), with both effects observed in males only. The incidence of cornea inflammation was not increased in male rats at 3 mg/kg/day (5-times the recommended dose based on AUC).
-
14 CLINICAL STUDIES
The efficacy of LIVDELZI was evaluated in Trial 1 (NCT04620733), a 12-month, randomized, double-blind, placebo-controlled trial. The trial included 193 adult patients with PBC with an inadequate response or intolerance to UDCA. Patients were included in the trial if their ALP was greater than or equal to 1.67-times the ULN and total bilirubin (TB) was less than or equal to 2-times the ULN. Patients were excluded from the trial if they had other chronic liver diseases, clinically important hepatic decompensation including portal hypertension with complications, or cirrhosis with complications (e.g., Model for End Stage Liver Disease [MELD] score of 12 or greater, known esophageal varices or history of variceal bleeds, history of hepatorenal syndrome).
Patients were randomized to receive LIVDELZI 10 mg (N=128) or placebo (N=65) once daily for 12 months. LIVDELZI or placebo was administered in combination with UDCA in 181 (94%) patients during the trial, or as a monotherapy in 12 (6%) patients who were unable to tolerate UDCA.
Baseline Demographics and Characteristics
The mean age of patients was 57 (Range: 28 to 75) years; 95% were female; 88% were White, 6% Asian, 2% Black or African American, and 3% American Indian or Alaska Native. Twenty-nine percent of the patients, 23% in the LIVDELZI 10 mg arm and 42% in the placebo arm, identified as Hispanic/Latino. Thirty-two percent of the patients, 38% in the LIVDELZI 10 mg arm and 20% in the placebo arm, were enrolled in the US.
At baseline, 18 (14%) of the LIVDELZI-treated patients and 9 (14%) of the placebo-treated patients met at least one of the following criteria: Fibroscan >16.9kPa; historical biopsy or radiological evidence suggestive of cirrhosis; platelet count <140,000/µL with at least one additional laboratory finding including serum albumin <3.5 g/dL, INR >1.3, or TB > 1-time ULN; or clinical determination of cirrhosis by the investigator.
The mean baseline ALP concentration was 314 (Range: 161 to 786) units per liter (U/L), corresponding to 2.7-times ULN. The mean baseline TB concentration was 0.8 (Range: 0.3 to 1.9) mg/dL and was less than or equal to the ULN in 87% of the patients. Other mean baseline liver biochemistries were 48 (Range: 9 to 115) U/L for ALT and 40 (Range: 16 to 94) U/L for AST.
Biochemical Results
The primary endpoint was biochemical response at Month 12, where biochemical response was defined as achieving ALP less than 1.67-times ULN, an ALP decrease of greater than or equal to 15% from baseline, and TB less than or equal to ULN. ALP normalization (i.e., ALP less than or equal to ULN) at Month 12 was a key secondary endpoint. The ULN for ALP was defined as 116 U/L. The ULN for TB was defined as 1.1 mg/dL.
Table 3 presents results at Month 12 for the percentage of patients who achieved biochemical response, achieved each component of biochemical response, and achieved ALP normalization. LIVDELZI demonstrated greater improvement on biochemical response and ALP normalization at Month 12 compared to placebo. Overall, 87% of patients had a baseline of TB concentration less than or equal to ULN. Therefore, improvement in ALP was the main contributor to the biochemical response rate results at Month 12.
Table 3: Percentage of Adult Patients with PBC Achieving Biochemical Response and ALP Normalization at Month 12 in Trial 1* LIVDELZI 10 mg Once Daily
(N=128)Placebo
(N=65)Treatment Difference
% (95% CI)†Patients who discontinued treatment prior to Month 12 or who had missing data were considered as non-responders. - * Biochemical response is defined as ALP less than 1.67-times ULN, an ALP decrease of greater than or equal to 15%, and TB less than or equal to ULN.
- † 95% unstratified Miettinen and Nurminen confidence intervals (CIs) are provided.
- ‡ p<0.0001 for LIVDELZI 10 mg versus placebo. P-values were obtained using the Cochran–Mantel–Haenszel test stratified by baseline ALP level (<350 U/L versus ≥350 U/L) and baseline pruritus NRS (<4 versus ≥4).
- § ALP normalization is defined as ALP less than or equal to ULN.
Biochemical Response Rate, n (%)*, ‡ 79 (62) 13 (20) 42 (28, 53) Components of Biochemical Response ALP less than 1.67-times ULN, n (%) 84 (66) 17 (26) 39 (25, 52) Decrease in ALP of at least 15%, n (%) 107 (84) 21 (32) 51 (37, 63) TB less than or equal to ULN, n (%) 104 (81) 50 (77) 4 (-7, 17) ALP Normalization, n (%)‡, § 32 (25) 0 (0) 25 (18, 33) Figure 1 shows the mean (95% CI) levels of ALP over 12 months. There was a trend of lower ALP in LIVDELZI arm compared to placebo arm starting at Month 1 through Month 12.
- * Figure 1 presents means and 95% Wald CIs for baseline, and least squares means and corresponding 95% CIs based on a mixed-effect model for repeated measures (MMRM) for Months 1, 3, 6, 9 and 12. The MMRM adjusts for baseline ALP, baseline ALP level (<350 U/L versus ≥350 U/L), baseline pruritus NRS (<4 versus ≥4), time (in months), treatment arm, treatment-by-baseline ALP interaction, and treatment-by-time interaction. The least squares mean change from baseline in ALP at Month 12 was -134 (-151, -117) U/L and -17 (-40, 6) U/L in the LIVDELZI 10 mg and placebo arms, respectively.
Figure 1: Mean* ALP in Adult Patients with PBC over 12 Months in Trial 1 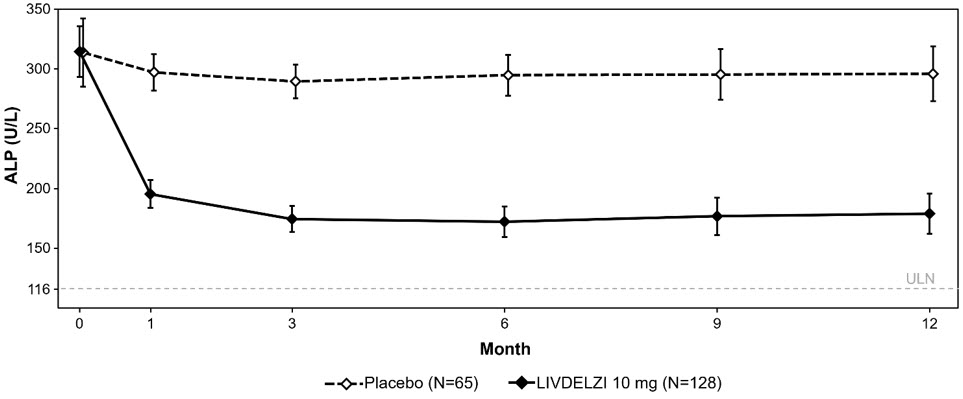
Biochemical response at Month 3 comparing LIVDELZI as a monotherapy to placebo was evaluated in a pooled analysis of a subset of patients from Trial 1 and another randomized, double-blind, placebo-controlled trial in a similar patient population. There was a trend of improvement on biochemical response at Month 3 in the LIVDELZI monotherapy group compared to the placebo group.
Pruritus
A single-item patient-reported outcome (PRO), the pruritus Numerical Rating Scale (NRS), evaluated patients' daily worst itching intensity on an 11-point rating scale with scores ranging from 0 ("no itching") to 10 ("worst itching imaginable") in Trial 1. The pruritus NRS was administered daily in a 14-day run-in period prior to randomization through Month 6.
Table 4 presents the results of the comparison between LIVDELZI and placebo on the key secondary endpoint evaluating the change from baseline in pruritus score at Month 6 in patients with baseline average pruritus scores greater than or equal to 4. The baseline average pruritus score for each patient was calculated by averaging the pruritus NRS scores administered in the run-in period and on Day 1 before treatment initiation. The pruritus scores at Month 6 for each patient were calculated by averaging the pruritus NRS scores within the last week in the month. Patients treated with LIVDELZI demonstrated greater improvement in pruritus compared with placebo.
Table 4: Change from Baseline in Pruritus Score at Month 6 in PBC Patients with Baseline Average Pruritus Score ≥4 in Trial 1* LIVDELZI 10 mg Once Daily
(N=49)Placebo
(N=23)- * Based on least square means from a mixed-effect model for repeated measures (MMRM) for change from baseline at Months 1 (Week 4), 3 (Week 12), and 6 (Week 26) accounting for baseline average pruritus score, baseline ALP level (<350 U/L versus ALP level ≥350 U/L), treatment arm, time (in months), and treatment-by-time interaction.
Baseline Average Pruritus Score, Mean (SD) 6.1 (1.4) 6.6 (1.4) Change from Baseline in Pruritus Score at Month 6* Mean (SE) -3.2 (0.3) -1.7 (0.4) Mean difference vs. placebo (95% CI) -1.5 (-2.5, -0.5)
p=0.0051 -
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
LIVDELZI (seladelpar) capsules are available as 10 mg, light gray opaque body, and a dark blue opaque cap with "CBAY" imprinted on the cap and "10" on the body.
LIVDELZI is packaged in a high density polyethylene bottle, closed with a polypropylene child resistant cap containing an induction seal.
- 10 mg capsules in a 75 cc bottle (30 count) (NDC: 61958-3301-1).
- 10 mg capsules in a 60 cc bottle (30 count) (NDC: 61958-3301-2).
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Fractures
Inform patients that LIVDELZI may increase the risk of bone fractures. Advise patients to call their healthcare provider to report any fractures [see Warnings and Precautions (5.1)].
Liver Test Abnormalities
Instruct patients to report any signs or symptoms of liver-related adverse reactions (e.g., loss of appetite, nausea, increased fatigue, lower extremity edema, abdominal swelling, or jaundice/icterus) to their healthcare provider [see Warnings and Precautions (5.2)].
Biliary Obstruction
Instruct patients to immediately report any signs or symptoms of biliary obstruction (e.g., right upper quadrant pain, jaundice) to their healthcare provider so that LIVDELZI treatment can be interrupted while the patient is being evaluated [see Warnings and Precautions (5.3)].
Pregnancy
Advise patients that there is a pregnancy safety study that captures pregnancy outcomes in women exposed to LIVDELZI during pregnancy, and to report pregnancies and pregnancy outcomes by calling 1-800-445-3235 [see Use in Specific Populations (8.1)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
LIVDELZI (liv del' zee)
(seladelpar)
capsules, for oral use
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 01/2026 What is LIVDELZI? LIVDELZI is a prescription medicine used to treat primary biliary cholangitis (PBC) in combination with ursodeoxycholic acid (UDCA) in adults who have not responded well to UDCA or used alone in patients unable to tolerate UDCA. LIVDELZI is not recommended for use in people who have advanced liver disease (decompensated cirrhosis). Symptoms of advanced liver disease may include confusion, having fluid in the stomach-area (abdomen), black, tarry, or bloody stools, coughing up or vomiting blood, or having vomit that looks like "coffee grounds". It is not known if taking LIVDELZI will improve your chance of survival or prevent liver decompensation. It is not known if LIVDELZI is safe and effective in children. Before taking LIVDELZI, tell your healthcare provider about all your medical conditions, including if you: - have advanced liver disease.
- think you may have a blockage of the bile ducts in your liver (biliary obstruction).
- are pregnant or plan to become pregnant. It is not known if LIVDELZI will harm your unborn baby.
- Pregnancy safety study. If you become pregnant while taking LIVDELZI, tell your healthcare provider right away. There is a pregnancy safety study for women who take LIVDELZI during pregnancy. Talk to your healthcare provider about providing information to the LIVDELZI pregnancy safety study. The purpose of this pregnancy safety study is to capture information about your health and your baby's health. You or your healthcare provider can report your pregnancy by calling 1-800-445-3235.
- are breastfeeding or plan to breastfeed. It is not known if LIVDELZI passes into your breast milk. Talk with your healthcare provider about the best way to feed your baby if you take LIVDELZI.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Certain other medicines may affect the way LIVDELZI works. How should I take LIVDELZI? - Take LIVDELZI exactly as your healthcare provider tells you to.
- Do not take more LIVDELZI than your healthcare provider tells you to.
- Take LIVDELZI by mouth 1 time each day.
- Take LIVDELZI with or without food.
- If you also take medicines to help lower your cholesterol (bile acid binding resins), take LIVDELZI at least 4 hours before or 4 hours after you take the bile acid binding resin. If this is not possible, space the time between taking LIVDELZI and your bile acid binding resin as far apart as possible.
- If you take too much LIVDELZI, call your healthcare provider or get emergency medical help right away.
What are the possible side effects of LIVDELZI? LIVDELZI can cause serious side effects, including: - bone fractures. Taking LIVDELZI may increase your risk of bone fractures. Tell your healthcare provider about any bone fractures, or if you develop pain, or have changes in your ability to move around.
-
changes in liver tests. Increased liver enzymes in the blood have happened when taking more LIVDELZI than prescribed. Your healthcare provider will do tests to check your liver before you start and during treatment with LIVDELZI.
Tell your healthcare provider right away if you have any of the following signs or symptoms of worsening liver problems during treatment with LIVDELZI:
- swelling of your stomach-area (abdomen) from a build-up of fluid
- yellowing of your skin or the whites of your eyes
- pain on the right side of your stomach (abdomen)
- black, tarry, or bloody stools
- coughing up or vomiting blood, or your vomit looks like "coffee grounds"
- mental changes such as confusion, being sleepier than usual or harder to wake up, slurred speech, mood swings, or changes in personality
The most common side effects of LIVDELZI include: - headache
- stomach (abdominal) pain
- nausea
- abdominal swelling (distension)
- dizziness
Tell your healthcare provider if you have any side effect that bothers you or does not go away. These are not all the possible side effects of LIVDELZI. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store LIVDELZI? - Store LIVDELZI at room temperature between 68°F to 77°F (20°C to 25°C)
- Store in the original container to protect LIVDELZI from light.
General information about the safe and effective use of LIVDELZI. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use LIVDELZI for a condition for which it was not prescribed. Do not give LIVDELZI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about LIVDELZI that is written for health professionals. What are the ingredients in LIVDELZI? Active ingredient: seladelpar lysine Inactive ingredients: butylated hydroxytoluene, colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, mannitol, microcrystalline cellulose, and hard gelatin shells. The light gray opaque (body) and dark blue opaque (cap) capsule shells contain gelatin, titanium dioxide, black iron oxide, yellow iron oxide, red iron oxide and the colorant FD&C Blue #2. Distributed by: Gilead Sciences, Inc., Foster City, CA 94404. LIVDELZI is a trademark of Gilead Sciences, Inc., or its related companies. © 2026 Gilead Sciences, Inc. All rights reserved. 217899-GS-002 For more information, call 1-800-445-3235 or go to www.LIVDELZI.com. - PRINCIPAL DISPLAY PANEL - 10 mg Capsule Bottle Label
- PRINCIPAL DISPLAY PANEL - 10 mg Capsule Bottle Label
-
INGREDIENTS AND APPEARANCE
LIVDELZI
seladelpar lysine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61958-3301 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SELADELPAR LYSINE (UNII: N1429130KR) (Seladelpar - UNII:7C00L34NB9) Seladelpar 10 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE 302 (UNII: 91B875MM4H) MICROCRYSTALLINE CELLULOSE 101 (UNII: 7T9FYH5QMK) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) Butylated hydroxytoluene (UNII: 1P9D0Z171K) Product Characteristics Color GRAY, BLUE Score no score Shape CAPSULE Size 19mm Flavor Imprint Code CBAY;10 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-3301-1 30 in 1 BOTTLE; Type 0: Not a Combination Product 08/14/2024 2 NDC: 61958-3301-2 30 in 1 BOTTLE; Type 0: Not a Combination Product 10/31/2025 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA217899 08/14/2024 Labeler - Gilead Sciences, Inc (185049848)

Trademark Results [Livdelzi]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 LIVDELZI 98517068 not registered Live/Pending |
CymaBay Therapeutics, Inc. 2024-04-24 |
 LIVDELZI 98495981 not registered Live/Pending |
CymaBay Therapeutics, Inc. 2024-04-11 |
 LIVDELZI 98146514 not registered Live/Pending |
CymaBay Therapeutics, Inc. 2023-08-23 |
 LIVDELZI 98146510 not registered Live/Pending |
CymaBay Therapeutics, Inc. 2023-08-23 |
 LIVDELZI 88654039 not registered Live/Pending |
CymaBay Therapeutics, Inc. 2019-10-14 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.