PALSONIFY- paltusotine tablet, film coated
PALSONIFY by
Drug Labeling and Warnings
PALSONIFY by is a Prescription medication manufactured, distributed, or labeled by Crinetics Pharmaceuticals, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PALSONIFY™ safely and effectively. See full prescribing information for PALSONIFY.
PALSONIFY (paltusotine) tablets, for oral use
Initial U.S. Approval: 2025INDICATIONS AND USAGE
PALSONIFY is a somatostatin receptor agonist indicated for the treatment of adults with acromegaly who had an inadequate response to surgery and/or for whom surgery is not an option (1).
DOSAGE AND ADMINISTRATION
- Take orally once daily with water on an empty stomach (at least 6 hours after a meal) and at least 1 hour before the next meal (2.1).
- Recommended initial dosage is 40 mg once daily. During initiation, PALSONIFY may be temporarily reduced to 20 mg once daily if needed, based on tolerability. Once adverse reactions have resolved, resume PALSONIFY 40 mg once daily (2.2).
- After 2 to 4 weeks, based on IGF-1 levels, titrate to 60 mg once daily (2.2).
DOSAGE FORMS AND STRENGTHS
Tablets: 20 mg, 30 mg (3)
CONTRAINDICATIONS
- None (4)
WARNINGS AND PRECAUTIONS
- Cholelithiasis and its Complications: Monitor periodically. If complications of cholelithiasis occur, discontinue PALSONIFY and treat appropriately (5.1).
- Hyperglycemia and Hypoglycemia: Monitor glucose and adjust antidiabetic treatment as needed (5.2).
- Cardiovascular Abnormalities: Bradycardia or conduction abnormalities may occur. Dosage adjustments of concomitantly used drugs with bradycardia effects may be necessary (5.3).
- Thyroid Function Abnormalities: Hypothyroidism may occur. Assess thyroid function periodically (5.4).
- Steatorrhea and Malabsorption of Dietary Fats: New onset steatorrhea, stool discoloration, loose stools, abdominal bloating, and weight loss may occur. If new occurrence or worsening of these symptoms are reported, evaluate for potential pancreatic exocrine insufficiency (5.5).
- Vitamin B12 Deficiency: Monitor vitamin B12 levels during treatment if indicated (5.6).
ADVERSE REACTIONS
Most common adverse reactions (≥5%) are diarrhea, abdominal pain, nausea, decreased appetite, sinus bradycardia, hyperglycemia, palpitations, and gastroenteritis (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Crinetics Pharmaceuticals, Inc. at toll-free phone 1-833-CRN-INFO or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Strong CYP3A4 Inducers: may decrease paltusotine exposure. May require PALSONIFY dosage increase (2.3, 7.1).
- Moderate CYP3A4 Inducers: may decrease paltusotine exposure. May require PALSONIFY dosage increase (2.3, 7.1).
- Proton Pump Inhibitors: may decrease paltusotine exposure. May require PALSONIFY dosage increase (2.3, 7.1)
- Cyclosporine: may decrease cyclosporine exposure. May require cyclosporine dosage adjustment (7.2).
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 9/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
2.2 Recommended Dosage, Titration, and Monitoring
2.3 Dosage Modifications for Drug Interactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Cholelithiasis and its Complications
5.2 Hyperglycemia and Hypoglycemia
5.3 Cardiovascular Abnormalities
5.4 Thyroid Function Abnormalities
5.5 Steatorrhea and Malabsorption of Dietary Fats
5.6 Vitamin B12 Deficiency
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on PALSONIFY
7.2 Effect of PALSONIFY on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Phototoxicity
14 CLINICAL STUDIES
14.1 Study 1: Adults with Acromegaly Naïve or Previously Treated on a Somatostatin Analog
14.2 Study 2: Adults with Acromegaly Previously Controlled on a Somatostatin Analog
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
- Take PALSONIFY orally once daily with water on an empty stomach, at least 6 hours after a meal (e.g., after overnight fasting) and at least 1 hour before the next meal [see Clinical Pharmacology (12.3)].
2.2 Recommended Dosage, Titration, and Monitoring
- The recommended initial dosage of PALSONIFY is 40 mg once daily.
- During initiation period, PALSONIFY may be temporarily reduced to 20 mg once daily if needed, based on tolerability [see Adverse Reactions (6.1)]. Once adverse reactions have resolved, resume PALSONIFY 40 mg once daily.
- After 2 to 4 weeks on PALSONIFY 40 mg once daily, based on IGF-1 levels, titrate to a PALSONIFY dosage of 60 mg once daily.
2.3 Dosage Modifications for Drug Interactions
Concomitant Use with Strong CYP3A4 Inducers
- Patients taking strong CYP3A4 inducers may require an increased dosage of PALSONIFY. Do not exceed three-fold the PALSONIFY dosage prior to concomitant use or 120 mg daily, whichever is less [see Drug Interactions (7.1)].
Concomitant Use with Moderate CYP3A4 Inducers
- Patients taking moderate CYP3A4 inducers may require an increased dosage of PALSONIFY. Do not exceed two-fold the PALSONIFY dosage prior to concomitant use or 120 mg daily, whichever is less [see Drug Interactions (7.1)].
Concomitant Use with Proton Pump Inhibitors
- Patients taking proton pump inhibitors may require an increased dosage of PALSONIFY. Avoid concomitant use of proton pump inhibitors in patients who are already on PALSONIFY 60 mg [see Drug Interactions (7.1)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Cholelithiasis and its Complications
PALSONIFY may inhibit gallbladder contractility and decrease bile secretion, which may lead to gallbladder stones or sludge. Cholelithiasis was reported in participants treated with PALSONIFY in clinical trials. Complications of cholelithiasis, such as acute cholecystitis and pancreatitis, have also been reported with the use of PALSONIFY [see Adverse Reactions (6.1)]. Monitor patients periodically. If complications of cholelithiasis occur, discontinue PALSONIFY and treat appropriately.
5.2 Hyperglycemia and Hypoglycemia
PALSONIFY may alter the balance between the counter-regulatory hormones, insulin, glucagon, and growth hormone, which may result in hypoglycemia, hyperglycemia, or diabetes mellitus. Hyperglycemia was reported in participants treated with PALSONIFY in clinical trials [see Adverse Reactions (6.1)]. Monitor blood glucose levels when PALSONIFY treatment is initiated or when the dose is altered. Adjust antidiabetic treatment accordingly.
5.3 Cardiovascular Abnormalities
Cardiac conduction abnormalities and other ECG changes such as PR interval prolongation have occurred during treatment with PALSONIFY. Bradycardia, sinus arrest, and atrioventricular block were reported in participants treated with PALSONIFY in clinical trials [see Adverse Reactions (6.1)]. These ECG changes may occur in patients with acromegaly. Dosage adjustments of concomitantly used drugs that have bradycardia effects (e.g., beta-blockers) may be necessary.
5.4 Thyroid Function Abnormalities
Somatostatin analogs may suppress the secretion of thyroid-stimulating hormone, which may result in hypothyroidism. Periodic assessment of thyroid function (TSH, total, and/or free T4) is recommended during treatment with PALSONIFY.
5.5 Steatorrhea and Malabsorption of Dietary Fats
New onset steatorrhea, stool discoloration and loose stools have been reported in patients receiving somatostatin analogs. Somatostatin analogs reversibly inhibit secretion of pancreatic enzymes and bile acids, which may result in malabsorption of dietary fats and subsequent symptoms of steatorrhea, loose stools, abdominal bloating, and weight loss. If new occurrence or worsening of these symptoms are reported in patients receiving PALSONIFY, evaluate patients for potential pancreatic exocrine insufficiency and manage accordingly.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Cholelithiasis and Complications of Cholelithiasis [see Warnings and Precautions (5.1)]
- Hyperglycemia and Hypoglycemia [see Warnings and Precautions (5.2)]
- Cardiovascular Abnormalities [see Warnings and Precautions (5.3)]
- Thyroid Function Abnormalities [see Warnings and Precautions (5.4)]
- Steatorrhea and Malabsorption of Dietary Fats [see Warnings and Precautions (5.5)]
- Changes in Vitamin B12 Levels [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of PALSONIFY was evaluated in adults with acromegaly in two randomized, double-blind, placebo-controlled Phase 3 studies. Study 1 was a 24-week, randomized, placebo-controlled study in 111 adults who were naive or previously treated on a somatostatin analog and biochemically uncontrolled at randomization. Participants in Study 1 had a mean age of 47 years (range: 18 to 80 years) and were randomized to PALSONIFY (n=54) or placebo (n=57) [see Clinical Studies (14.1)]. Study 2 was a 36-week, randomized, placebo-controlled study in 58 adults who were biochemically controlled on injectable depot formulations of octreotide or lanreotide. Participants in Study 2 had a mean age of 55 years (range: 29 to 84 years) and were randomized to PALSONIFY (n=30) or placebo (n=28) [see Clinical Studies (14.2)].
Adverse Reactions
Adverse reactions that occurred in ≥5% of PALSONIFY-treated participants and 5% greater incidence than placebo in the randomized-controlled phase of Study 1 and Study 2 are presented in Table 1 and Table 2, respectively. Adverse reactions presented in Table 1 and Table 2 exclude events which occurred after a participant received rescue therapy (Study 1 PALSONIFY n=1, placebo n=13; Study 2 PALSONIFY n=1, placebo n=17).
Table 1. Adverse Reactions Occurring in ≥5% of PALSONIFY-Treated Participants and 5% Greater Incidence than Placebo-Treated Participants During the Randomized Controlled Period of Study 1 a Abdominal pain also includes abdominal discomfort.
b Hyperglycemia also includes impaired fasting glucose and diabetes mellitus.
Adverse Reaction PALSONIFY
N=54
n (%)Placebo
N=57
n (%)Diarrhea 18 (33) 8 (14) Abdominal paina 10 (19) 3 (5) Nausea 5 (9) 1 (2) Sinus bradycardia 4 (7) 0 Hyperglycemiab 4 (7) 1 (2) Table 2. Adverse Reactions Occurring in ≥5% of PALSONIFY-Treated Participants and 5% Greater Incidence than Placebo-Treated Participants During the Randomized Controlled Period of Study 2 a Decreased appetite also includes early satiety.
Adverse Reaction PALSONIFY
N=30
n (%)Placebo
N=28
n (%)Diarrhea 7 (23) 3 (11) Nausea 4 (13) 1 (4) Decreased appetitea 3 (10) 0 Palpitations 2 (7) 0 Gastroenteritis 2 (7) 0 Gastrointestinal
Gastrointestinal adverse reactions, including diarrhea, nausea, and abdominal pain were reported in participants from Studies 1 and 2 (see Table 1 and Table 2). Most gastrointestinal adverse reactions occurred within the first two months of PALSONIFY treatment initiation and had a median duration ranging between 6 to 18 days.
Cholelithiasis and its Complications
Cholelithiasis and its complications were reported in 10/173 (6%) participants during Studies 1 and 2 as follows: cholelithiasis (n=8), acute cholecystitis, biliary colic, and bile duct stone (one participant each). The majority of the events occurred within the first nine months of treatment. One PALSONIFY-treated participant who experienced obstructive pancreatitis required cholecystectomy [see Warnings and Precautions (5.1)].
Hyperglycemia
During Study 1, an increase from baseline to Week 24 in mean fasting plasma glucose (FPG) of 6.9 mg/dL and hemoglobin A1c (HbA1c) of 0.26% was observed in the PALSONIFY arm, and an increase in mean FPG of 1.5 mg/dL and HbA1c of 0.04% was observed in the placebo arm. Of the 34 PALSONIFY-treated participants who had normal baseline FPG, 25 (74%) developed at least one glucose value ≥100 mg/dL. Of the 31 placebo-treated participants who had a normal baseline FPG, 9 (29%) developed at least one elevated glucose value.
All participants in Study 2 were previously treated with other somatostatin analog products known to increase glucose levels. During Study 2, a decrease from baseline to Week 36 in mean FPG of 1.8 mg/dL and HbA1c of 0.05% was observed in the PALSONIFY arm, and a decrease in mean FPG of 11.7 mg/dL and HbA1c of 0.25% was observed in the placebo arm. Of the 5 PALSONIFY-treated participants who had normal baseline FPG, 4 (80%) developed at least one glucose value ≥100 mg/dL. Of the 11 placebo-treated participants who had a normal baseline FPG, 2 (18%) developed at least one elevated glucose value.
Hypoglycemia
During PALSONIFY clinical development program, 5 participants in the open-label extension phase reported hypoglycemia. Most participants had a history of diabetes at baseline and were treated with antidiabetic medications, such as insulin and sulfonylureas.
Cardiac
Bradycardia was reported in 4 (7%) participants in the paltusotine arm versus none in placebo in Study 1, and in 1 (3%) participant in the paltusotine arm versus none in placebo in Study 2. Bradycardia was asymptomatic and occurred within the first three months of treatment.
During the PALSONIFY clinical development program, 3 PALSONIFY-treated participants with preexisting cardiovascular comorbidities experienced serious cardiac adverse events during the open-label extension phase: sinus arrest (2 participants) and complete atrioventricular block (one participant).
Ocular
Findings of ocular phototoxicity were observed in a nonclinical study [see Nonclinical Toxicology (13.2)]. Due to these findings, ocular assessments were conducted during the open-label period of Studies 1 and 2. The following retinal observations were noted: drusen; dry age-related macular degeneration, early stage; retinal pigment epithelium changes; epiretinal membrane; diabetic retinopathy; retinoschisis; and hypertensive retinopathy. Baseline assessments were not available for comparison.
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on PALSONIFY
Table 3. Clinically Significant Interactions Affecting PALSONIFY Strong CYP3A4 Inducers Intervention Concomitant use of PALSONIFY with strong CYP3A4 inducers may require an increased dosage of PALSONIFY, not to exceed three-fold the dose prior to concomitant use or 120 mg daily, whichever is less. Clinical Impact Concomitant use of PALSONIFY with strong CYP3A4 inducers reduced paltusotine exposure and may affect therapeutic response [see Clinical Pharmacology (12.3)]. Moderate CYP3A4 Inducers Intervention Concomitant use of PALSONIFY with moderate CYP3A4 inducers may require an increased dosage of PALSONIFY, not to exceed two-fold the dose prior to concomitant use or 120 mg daily, whichever is less. Clinical Impact Concomitant use of PALSONIFY with moderate CYP3A4 inducers resulted in a decrease in paltusotine exposure [see Clinical Pharmacology (12.3)]. Proton Pump Inhibitors Intervention Concomitant use of PALSONIFY with PPIs may require an increased dosage of PALSONIFY. Patients who are already on PALSONIFY 60 mg should avoid concomitant use with proton pump inhibitors. Clinical Impact Concomitant use of PALSONIFY with proton pump inhibitors demonstrated a dose-dependent decrease in paltusotine exposure [see Clinical Pharmacology (12.3)]. 7.2 Effect of PALSONIFY on Other Drugs
Table 4. Clinically Significant Interactions Affecting Other Drugs Cyclosporine Intervention Adjustment of cyclosporine dose to maintain therapeutic levels may be necessary. Follow recommended therapeutic drug monitoring for cyclosporine. Clinical Impact Concomitant use of PALSONIFY with cyclosporine resulted in a decrease in cyclosporine bioavailability [see Clinical Pharmacology (12.3)]. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The available data with PALSONIFY use in pregnant women are insufficient to identify a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. In animal reproduction studies, no malformations were observed with oral administration of paltusotine to pregnant rats and rabbits during organogenesis at exposures 11 and 3 times the human exposure at the maximum recommended human dose (MRHD) of 60 mg once daily, respectively (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Animal Data
In an embryo-fetal development study in pregnant rats, paltusotine was administered orally at 25, 75, and 500 mg/kg/day during the period of organogenesis from gestation Day 7 to 17. The NOAEL for maternal and embryo-fetal developmental toxicity was 500 mg/kg/day, as no paltusotine-related effects were observed on any ovarian, uterine or litter parameters (approximately 11 times the MRHD based on AUC).
In an embryo-fetal development study in pregnant rabbits, paltusotine was administered orally at 10, 25, and 75 mg/kg/day during the period of organogenesis from gestation Day 7 to 19. Maternal findings included an increased incidence of spontaneous abortions at 75 mg/kg/day, with decreased body weight gain, body weights, and food consumption. No fetal abnormalities were observed at any dose. Lower fetal body weight was noted at 75 mg/kg/day. The NOAEL for maternal and embryo-fetal developmental toxicity was 25 mg/kg/day (approximately 3 times the MRHD based on AUC).
In a pre- and postnatal development study, pregnant rats were given paltusotine orally at the doses of 25, 75, and 500 mg/kg/day from gestation Day 6 to lactation Day 20. Paltusotine had no effect on the growth and development of offspring up to the highest tested dose of 500 mg/kg (5 times MRHD based on AUC). In a single dose pharmacokinetic study, oral administration of paltusotine at 25 mg/kg or 500 mg/kg paltusotine to pregnant rats showed measurable paltusotine concentrations in embryos and fetuses, demonstrating placental transfer. Overall, the embryo and fetal concentrations were generally low when compared to maternal concentrations.
8.2 Lactation
Risk Summary
There is no information available on the presence of paltusotine in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production. Paltusotine is present in animal milk (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for PALSONIFY and any potential adverse effects on the breastfed infant from PALSONIFY or from the underlying maternal condition.
Data
Paltusotine concentrations were elevated 2.4- to 3.8-fold in rat milk compared to plasma during animal studies. Paltusotine concentrations in plasma and maternal milk suggest that rat pups were likely exposed in utero and/or via lactational transfer. The concentration of paltusotine in animal milk does not necessarily predict the concentration of paltusotine in human milk.
8.4 Pediatric Use
The safety and efficacy of PALSONIFY have not been established in pediatric patients.
8.5 Geriatric Use
In the Phase 3 studies 17/84 (20.2%) PALSONIFY-treated participants were ≥65 years of age. No overall differences in safety or effectiveness of PALSONIFY have been observed between participants 65 years of age and older and younger adult participants. No dose adjustments are required based on age [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
No dosage adjustment of PALSONIFY is recommended for patients with hepatic impairment. In a clinical pharmacology study in participants with varying degrees of hepatic impairment, exposures of paltusotine were similar across all hepatic impairment groups when compared with participants with normal hepatic function [see Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
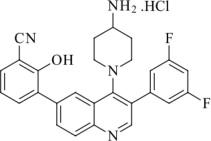
PALSONIFY tablets contain paltusotine hydrochloride, a somatostatin receptor agonist. Paltusotine is known chemically as 3-[4-(4-Amino-1-piperidinyl)-3-(3,5-difluorophenyl)-6-quinolinyl]-2-hydroxybenzonitrile hydrochloride. The molecular weight of paltusotine hydrochloride is 492.95 g/mol (C27H22F2N4O·HCl).
PALSONIFY tablets for oral administration contain 20 mg of paltusotine (equivalent to 21.6 mg of paltusotine hydrochloride) or 30 mg of paltusotine (equivalent to 32.4 mg of paltusotine hydrochloride). Each tablet contains the following inactive ingredients: colloidal silicon dioxide, copovidone, crospovidone, magnesium stearate, mannitol, and microcrystalline cellulose. Additionally, the 20 mg tablets contain Opadry pink coating (hypromellose, iron oxide red, iron oxide yellow, titanium dioxide, and triacetin) and the 30 mg tablets contain Opadry yellow coating (hypromellose, iron oxide yellow, titanium dioxide, and triacetin).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Similar to the natural hormone somatostatin, paltusotine suppresses growth hormone (GH) and insulin-like growth factor-1 (IGF-1) secretion. Paltusotine exerts its pharmacological activity via selective agonism (>4000-fold) at somatostatin receptor 2 (SSTR2) and exhibits little or no affinity for other SST receptor subtypes. Paltusotine inhibited cyclic adenosine monophosphate accumulation via human SSTR2 activation with an average drug (agonist) concentration that results in half-maximal response (EC50) of 0.25 nM.
12.2 Pharmacodynamics
In patients with acromegaly, there was a dose-dependent reduction in IGF-1 levels over the therapeutic dose range of 20 to 60 mg.
Paltusotine may inhibit gallbladder contractility and decrease bile secretion, which may lead to gallbladder stones or sludge [see Warnings and Precautions (5.1)].
12.3 Pharmacokinetics
Paltusotine exhibited dose-proportional increases in exposures for doses ranging from 20 mg (lowest approved recommended dosage) to 120 mg (2 times the highest approved recommended dosage) in healthy participants. Apparent dose proportional increase was observed for mean steady-state trough concentrations up to 60 mg once daily in participants with acromegaly. Following once daily administration, paltusotine reaches steady-state exposure within one week.
Absorption
Following oral administration of paltusotine, the median time to maximum plasma concentration (tmax) is 1 to 4 hours regardless of post dose fasting duration.
Effect of Food
Relative to administration in the fasted state, administration of paltusotine with a high-fat meal (800 to 1000 calories, 50% to 60% fat) reduced the AUC by 85% and the Cmax by 81%. Administration of paltusotine with a low-fat meal (400 to 500 calories, 25% fat) reduced AUC by 72% and the Cmax by 68%.
Distribution
The volume of distribution (Vz) of paltusotine is 220 L. Paltusotine is highly plasma protein bound (99%).
Elimination
After maximal concentrations were attained, paltusotine concentration declined with apparent terminal half-life (t½) of 28 hours.
Metabolism
Paltusotine is metabolized primarily in the liver via glucuronidation and oxidation. In vitro, glucuronidation was the major metabolic pathway and is primarily mediated by UGT1A1 and UGT1A9. Oxidation was a secondary pathway and was primarily catalyzed by CYP3A4/5 with a minor contribution from CYP2D6.
Excretion
Following oral administration of radiolabeled paltusotine, fecal excretion was the predominant route of elimination with observed mean recovery of total administered radioactivity being 90% in feces and 3.9% in urine. Unchanged paltusotine was a major component in excreta.
Specific Populations
Effects of Age, Body Weight, Sex, Race, Renal Function, and UGT1A1 Polymorphism
Based on population pharmacokinetics data, no clinically significant difference in the pharmacokinetics of paltusotine was observed based on age (18 to 84 years), body weight (45 to 138 kg), sex, race (White, Asian, Other), renal function (52 to 148 mL/min/1.73 m2; eGFR), or UGT1A1 polymorphism.
Hepatic Impairment
Paltusotine AUC did not change in participants with mild hepatic impairment (Child-Pugh A) compared to participants with normal hepatic function. Paltusotine AUC decreased by 25% in moderate (Child-Pugh B) and 10% in severe (Child-Pugh C) hepatic impairment.
Strong CYP3A4 Inducers:
Paltusotine Cmax and AUC decreased by approximately 44% and 70%, respectively, following concomitant administration of carbamazepine (a strong inducer of multiple enzymes and transporters [CYP3A4, UGT1A1, and P-gp]).
Moderate CYP3A4 Inducers:
Drugs such as efavirenz are predicted to decrease Cmax and AUC of paltusotine approximately 5% and 30%, respectively.
Proton Pump Inhibitors (PPIs):
Paltusotine exhibited pH-dependent aqueous solubility. Paltusotine AUC was decreased by 21% with 20 mg dose and 42% with 60 mg dose levels.
Other Drugs:
Weak CYP3A4 inducers or inhibitors of CYP3A4/5, P-gp, or UGT1A1 are predicted to not cause clinically significant changes in paltusotine pharmacokinetics.
Cyclosporine:
Concomitant use of PALSONIFY decreased cyclosporine Cmax and AUC in whole blood by 53% and 35%, respectively.
Other Drugs:
Paltusotine did not show clinically significant changes in the pharmacokinetics of other drugs that were CYP3A4/5 or MATE substrates. Paltusotine is predicted to not cause clinically significant changes in the pharmacokinetics of other drugs that are CYP2D6 or P-gp substrates.
In Vitro Studies
In vitro studies suggest paltusotine is an inhibitor of CYP2D6, CYP3A4, and CYP2C19 and a weak inhibitor of UGT1A1, UGT1A3, and UGT1A9 but is unlikely to meaningfully inhibit these enzymes at clinically relevant concentrations. Paltusotine does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, UGT1A6, or UGT2B15. Paltusotine is not an inducer of CYP1A2, CYP2B6, or CYP3A4.
Paltusotine is a substrate of P-gp and BCRP transporters. In vitro studies suggest paltusotine is an inhibitor of P-gp, MATE-1, MATE2-K, and BSEP transporters but is unlikely to meaningfully inhibit these transporters at clinically relevant concentrations. Paltusotine does not inhibit OAT1, OAT3, or OCT1.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis and Mutagenesis
No carcinogenic potential was demonstrated in transgenic (rasH2) mice when administering paltusotine by oral gavage for 26 weeks at doses up to 300 mg/kg/day (2.8-times the clinical dose of 60 mg based on AUC). Additionally, no carcinogenic effects were noted when administering paltusotine by oral gavage for 88 to 90 weeks at doses up to 500 mg/kg/day (34-times the clinical dose of 60 mg based on AUC) in rats. Paltusotine was not genotoxic in the in vitro bacterial reverse mutation test (Ames), in vitro micronucleus assay, or in the in vivo rat bone marrow micronucleus assay.
Impairment of Fertility
Animal studies have been conducted to evaluate the effect of paltusotine on fertility and early embryonic development of rats at doses of up to 500 mg/kg/day (18-times the clinical dose of 60 mg based on AUC). There were no paltusotine-related effects on any mating or fertility parameter in males or females at any dose. In addition, there were no paltusotine-related effects on reproductive organ weights or sperm parameters in males at any dose. Based on the finding of decreased corpora lutea in females at 500 mg/kg/day (which resulted in fewer implantation sites and live embryos/litter), the reproductive no-observed-adverse-effect-level (NOAEL) in females was considered to be 75 mg/kg/day (5-times the clinical dose of 60 mg based on AUC).
13.2 Phototoxicity
Paltusotine absorbs light at 203 to 350 nm wavelength and exhibited a positive phototoxicity signal in vitro as indicated by reduced viability of BALB/c 3T3 mouse fibroblasts. In a phototoxicity study in pigmented Long-Evans rats, oral administration of paltusotine for three consecutive days followed by ultraviolet radiation (UVR) exposure resulted in non-dose dependent ocular toxicities including corneal dystrophy, unilateral inferior focal retinopathy, and unilateral retinal degeneration/necrosis at systemic exposures approximately 2-times the exposure at the MRHD (based on body surface area). The clinical significance of these findings to humans is unknown.
-
14 CLINICAL STUDIES
The effectiveness of PALSONIFY for treatment of adults with acromegaly was evaluated in two randomized, double-blind, parallel group, placebo-controlled clinical studies.
14.1 Study 1: Adults with Acromegaly Naïve or Previously Treated on a Somatostatin Analog
Study 1 (NCT05192382) enrolled 111 adult participants with biochemically uncontrolled acromegaly. Participants were either treatment naïve (n=46/111) or had no treatment within the previous 4 months prior to screening (n=36/111) (‘Not Medically Treated’ group) or were previously treated on a somatostatin receptor analog and then washed out of treatment during screening (n=29/111) (‘Washout’ group). The mean age at enrollment was 47 years (range: 18 to 80 years); 53% were female; and 52% were White, 31% Asian, 3% Black or African American, 9% Other, and 5% Unknown race. The mean duration since diagnosis of acromegaly was 87 months. Prior to study participation, 95% of participants had received pituitary surgery (mean duration 78 months prior to study participation). Of the 111 participants, 86 (78%) had macroadenomas (>10 mm), 9 (8%) had microadenomas (≤10 mm), and tumor size was unknown in 16 (14%) participants. In the ‘Not Medically Treated’ group, IGF-1 levels were required to be ≥1.3×ULN at screening. In the ‘Washout’ group, IGF-1 levels were required to be ≤1.0×ULN at screening and ≥1.1×ULN with at least a 30% rise in IGF-1 after washout. Participants were randomized to receive either PALSONIFY (n=54) or placebo (n=57) for the 24-week treatment period.
Dose
The starting dose was 20 mg daily, followed by dose increase to 40 mg daily after 2 weeks. The dose could be titrated from 40 mg to a maximum dose of 60 mg based on IGF-1 value during the first 12 weeks of treatment. After Week 12, the PALSONIFY dose was maintained until the end of the randomized controlled period of the study (Week 24). The dose could be down titrated at any time during the study based on tolerability. Rescue therapy with standard of care treatment was initiated if a participant had evidence of uncontrolled acromegaly based on IGF-1 levels and symptoms. Fourteen (13%) participants received rescue therapy during the study: one (2%) participant in PALSONIFY arm and 13 (23%) participants in placebo arm.
Efficacy Assessment and Results
The primary endpoint was the proportion of PALSONIFY participants achieving biochemical control (defined as IGF-1 level ≤1.0×ULN) compared to placebo-treated participants. At Week 24, 56% of PALSONIFY participants achieved biochemical control compared to 5% of placebo-treated participants (p-value <0.0001) (Table 5).
Table 5. Proportion of Participants Achieving Biochemical Control (IGF-1 Levels ≤1.0×ULN) at Week 24 in Adults with Acromegaly (Study 1) a The baseline mean IGF-1 was 2.3×ULN in the ‘Not Medically Treated’ group and 1.5×ULN in the ‘Washout’ group.
IGF-1=insulin-like growth factor-1; ULN=upper limit of normal
IGF-1 at Week 24 is based on the average of the last 2 measurements of IGF-1 collected at Weeks 22 and 24. When one of the two last IGF-1 measurements was missing a single value was used. Week 24 is the end of the randomized controlled portion of the study; if a participant received rescue therapy, the last assessment prior to rescue is used.
IGF-1 Normalization PALSONIFY
(N=54)Placebo
(N=57)p-value Proportion of participants who achieved response in IGF-1 at Week 24 (≤1.0×ULN)a 56% 5% <0.0001 The majority of participants who achieved IGF-1 normalization during Study 1 did so within the first 2 to 4 weeks following initiation of treatment, with sustained response through the end of the treatment period.
A posthoc subgroup analysis for the primary efficacy endpoint evaluating the response rate in participants who were naïve to medical treatment, who had not achieved biochemical control on prior medical therapy, or for whom prior biochemical control status was unknown (Group A) and participants who demonstrated prior response to medical therapy who were either washed out from the previous therapy prior to baseline or had documented biochemical control on prior medical therapy (Group B) is provided below (Table 6).
Table 6. Proportion of Participants Achieving Biochemical Control (IGF-1 Levels ≤1.0×ULN) at Week 24 in Adults with Acromegaly Based on Prior Biochemical Control (Study 1) N=total number of participants per treatment arm; n=number of participants with event; Nx=total number of participants in the subgroup; CI=confidence interval
a Continuity-corrected Newcombe-Wilson confidence limits were used for the 95% confidence interval.
b Participants who were not medically treated recently and had not achieved biochemical control on previous medical therapy.
c Participants who were not medically treated recently and for whom biochemical control on previous medical therapy was unknown.
d Participants who were either washed out or not medically treated recently but had achieved biochemical control on previous medical therapy.
IGF-1 Normalization PALSONIFY
(N=54)
(n/Nx)Placebo
(N=57)
(n/Nx)Treatment Difference
(95% CI)aGroup A: Treatment naïve, uncontrolled on prior therapy, or with unknown biochemical control on prior therapy(s) 34%
(11/32)3%
(1/35)32%
(11%, 51%)Treatment naïve 23%
(5/22)4%
(1/23)18%
(-6%, 42%)Absence of biochemical control on prior treatmentb 57%
(4/7)0%
(0/6)57%
(-4%, 88%)Unknown biochemical control on prior treatmentc 67%
(2/3)0%
(0/6)67%
(-6%, 98%)Group B: Responders to prior treatmentd 86%
(19/22)9%
(2/22)77%
(46%, 90%)In Study 1, PALSONIFY-treated participants had numerically lower (versus placebo) severity of symptom scores associated with acromegaly as measured by the patient-reported symptom severity instrument, which assessed headaches, joint pain, sweating, fatigue, weakness, swelling, and/or numbness/tingling.
14.2 Study 2: Adults with Acromegaly Previously Controlled on a Somatostatin Analog
Study 2 (NCT04837040) enrolled 58 participants who were previously biochemically controlled (defined as IGF-1 levels ≤1.0×ULN during screening and at randomization) on injectable depot octreotide or lanreotide somatostatin analog formulations. The mean age at enrollment was 55 years (range: 29 to 84 years); 55% were female; and 72% were White, 3% Asian, 5% Black or African American, 12% Other, and 7% Unknown race. The mean duration since diagnosis of acromegaly was 155 months. Prior to study participation, 86% of participants had received pituitary surgery (mean duration 138 months prior to study participation). Of the 58 participants, 33 (57%) had macroadenomas (>10 mm), 11 (19%) had microadenomas (≤10 mm), and tumor size was unknown in 14 (24%) participants. Participants were randomized to receive either PALSONIFY (n=30) or placebo (n=28) for the 36-week treatment period.
Dose
The starting dose was 40 mg, and the dose could be titrated from 40 mg to a maximum of 60 mg based on IGF-1 value during the first 24 weeks of treatment. After Week 24, the PALSONIFY dose was maintained until the end of the randomized controlled period of the study (Week 36). The dose could be down titrated at any time during the study based on tolerability. Rescue therapy with standard of care treatment was initiated if a participant had evidence of uncontrolled acromegaly based on IGF-1 levels and symptoms. Eighteen (31%) participants received rescue therapy during the study: one (3%) participant in PALSONIFY arm and 17 (61%) participants in placebo arm.
Efficacy Assessment and Results
The primary endpoint was the proportion of PALSONIFY participants with biochemical response maintenance (i.e., IGF-1 ≤1.0×ULN) compared to placebo-treated participants. At Week 36, 83% of PALSONIFY participants maintained biochemical control compared to 4% of placebo-treated participants (p-value <0.0001) (Table 7).
Table 7. Proportion of Participants Maintaining Biochemical Control (IGF-1 Levels ≤1.0×ULN) in Adults with Acromegaly and Previously Maintained on a Somatostatin Analog Injection (Study 2) a The baseline mean IGF-1 was 0.83×ULN. Of enrolled participants, 59% were previously treated with octreotide and 41% previously treated with lanreotide.
IGF-1=insulin-like growth factor-1; ULN=upper limit of normal
Week 36 is the end of the randomized controlled portion of the study; if a participant received rescue therapy, the last assessment prior to rescue is used.
IGF-1 Normalization PALSONIFY
(N=30)Placebo
(N=28)p-value Proportion of participants who maintained response in IGF-1 at Week 36 (≤1.0×ULN)a 83% 4% <0.0001 In Study 2, PALSONIFY-treated participants had numerically lower (versus placebo) severity of symptom scores associated with acromegaly as measured by the patient-reported symptom severity instrument, which assessed headaches, joint pain, sweating, fatigue, weakness, swelling, and/or numbness/tingling.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
PALSONIFY is provided in two strengths: 20 mg and 30 mg.
Table 8. PALSONIFY Configurations and NDC Numbers Strength Tablet Color/Shape Imprint NDC Package Size 20 mg Pink; biconvex oval shape “PAL” on one side; “20” on other side 84015-920-60 Bottle of 60 tablets 30 mg Yellow; biconvex oval shape “PAL” on one side; “30” on other side 84015-930-60 Bottle of 60 tablets Storage and Handling
- PALSONIFY tablets are packaged in a child-resistant container; keep out of reach of children.
- Store tablets at 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature]. Excursions permitted 15°C to 30°C (59°F to 86°F).
- Store and dispense in original container or USP equivalent tight container.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Cholelithiasis and its Complications
Advise patients to contact their healthcare provider if they experience signs or symptoms of gallstones (cholelithiasis) or complications of cholelithiasis (e.g., cholecystitis, cholangitis, and pancreatitis) [see Warnings and Precautions (5.1)].
Hyperglycemia and Hypoglycemia
Advise patients to contact their healthcare provider if they have problems with blood sugar levels, either hyperglycemia or hypoglycemia [see Warnings and Precautions (5.2)].
Cardiovascular Abnormalities
Advise patients to contact their healthcare provider if they notice irregular heartbeat [see Warnings and Precautions (5.3)].
Thyroid Function Abnormalities
Advise patients that their thyroid function may be assessed periodically [see Warnings and Precautions (5.4)].
Steatorrhea and Malabsorption of Dietary Fats
Advise patients to contact their healthcare provider if they experience new or worsening symptoms of steatorrhea, loose stools, abdominal bloating, weight loss [see Warnings and Precautions (5.5)].
Vitamin B12 Deficiency
Advise patients that Vitamin B12 levels may be monitored during treatment [see Warnings and Precautions (5.6)].
PALSONIFY is a trademark of Crinetics Pharmaceuticals, Inc.
©2025 Crinetics Pharmaceuticals, Inc. All rights reserved.
Manufactured for:
Crinetics Pharmaceuticals, Inc.
San Diego, CA 92121 USA -
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration
Issued: 09/2025
Patient Information
PALSONIFY™ [pal-SAHN-ih-fy]
(paltusotine)
tablets, for oral useWhat is PALSONIFY?
- PALSONIFY is a prescription medicine used to treat adults with acromegaly for whom surgery was not effective or surgery is not an option.
- It is not known if PALSONIFY is safe and effective in children.
Before you take PALSONIFY, tell your healthcare provider about all of your medical conditions, including if you:
- have gallbladder problems.
- have blood sugar control problems (low blood sugar or high blood sugar).
- have problems with low heart rate.
- are pregnant or plan to become pregnant. It is not known if PALSONIFY will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if PALSONIFY passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby if you take PALSONIFY.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. PALSONIFY may affect the way other medicines work, and other medicines may affect how PALSONIFY works.
Especially tell your healthcare provider if you take medicine that can slow the heart rate.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.How should I take PALSONIFY?
- Take PALSONIFY exactly as your healthcare provider tells you to take it.
- Take PALSONIFY by mouth 1 time a day with a glass of water on an empty stomach.
- Take PALSONIFY at least 6 hours after a meal (for example, after an overnight fast) and at least 1 hour before the next meal.
What are the possible side effects of PALSONIFY?
PALSONIFY may cause serious side effects including:
- gallbladder problems. PALSONIFY may cause problems with the gallbladder. Tell your healthcare provider if you have sudden pain in the upper right area of your stomach (abdomen), sudden pain in your right shoulder or between your shoulder blades, yellowing of your skin or the whites of your eyes, or pale stools.
- blood sugar problems. PALSONIFY may change hormone levels that can lead to high blood sugar (hyperglycemia), low blood sugar (hypoglycemia), or diabetes. Tell your healthcare provider if you have problems with high or low blood sugar. Your healthcare provider will check your blood sugar when you start taking PALSONIFY or when your dose is changed.
- heart rate problems. Tell your healthcare provider if your heart is not beating normally.
- thyroid problems. PALSONIFY may keep your thyroid from releasing thyroid hormones, leading to hypothyroidism. Your thyroid function may be checked from time to time during your treatment with PALSONIFY.
- fatty stools. PALSONIFY may cause your body to have problems with absorbing dietary fats. Tell your healthcare provider if you have any new or worsening symptoms including fatty stools, changes in the color of your stools, loose stools, stomach (abdominal) bloating, or weight loss.
- low vitamin B12 levels in your blood. Your healthcare provider may check your vitamin B12 levels during treatment with PALSONIFY.
The most common side effects of PALSONIFY include: - diarrhea
- stomach area (abdominal) pain
- nausea and vomiting
- decreased appetite
- slow heart rate (bradycardia)
- high blood sugar levels (hyperglycemia)
- irregular heartbeat (palpitations)
These are not all the possible side effects of PALSONIFY. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects of PALSONIFY. You may report side effects to FDA at 1-800-FDA-1088.How should I store PALSONIFY?
- Store PALSONIFY at room temperature between 68°F to 77°F (20°C to 25°C).
- Store PALSONIFY in the original container.
General information about the safe and effective use of PALSONIFY.
Medicines are sometimes prescribed for purposes other than those listed in the Patient Information leaflet. Do not use PALSONIFY for a condition for which it was not prescribed. Do not give PALSONIFY to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about PALSONIFY that is written for health professionals.What are the ingredients in PALSONIFY?
Active ingredient: paltusotine
Inactive ingredients: colloidal silicon dioxide, copovidone, crospovidone, magnesium stearate, mannitol, and microcrystalline cellulose. Additionally, the 20 mg tablets contain Opadry pink coating (hypromellose, iron oxide red, iron oxide yellow, titanium dioxide, and triacetin) and the 30 mg tablets contain Opadry yellow coating (hypromellose, iron oxide yellow, titanium dioxide, and triacetin).
Manufactured for:
Crinetics Pharmaceuticals, Inc.
San Diego, California 92121
For more information about PALSONIFY call 1-833-CRN-INFO (1-833-276-4636) or go to www.PALSONIFY.com. -
PRINCIPAL DISPLAY PANEL
Principal Display Panel
NDC: 84015-920-60
Rx Only
Once-Daily Oral
Palsonify™
(paltusotine) tablets
20 mg
60 Tablets
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel
NDC: 84015-930-60
Rx Only
Once-Daily Oral
Palsonify™
(paltusotine) tablets
30 mg
60 Tablets
-
INGREDIENTS AND APPEARANCE
PALSONIFY
paltusotine tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 84015-920 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PALTUSOTINE HYDROCHLORIDE (UNII: FZT5Z83JNS) (PALTUSOTINE - UNII:F2IBD1GMD3) PALTUSOTINE 20 mg Inactive Ingredients Ingredient Name Strength COPOVIDONE K25-31 (UNII: D9C330MD8B) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIACETIN (UNII: XHX3C3X673) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color PINK (opadry pink) Score no score Shape OVAL (OVAL) Size 16mm Flavor Imprint Code PAL;20 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 84015-920-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 09/25/2025 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219070 09/25/2025 PALSONIFY
paltusotine tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 84015-930 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PALTUSOTINE HYDROCHLORIDE (UNII: FZT5Z83JNS) (PALTUSOTINE - UNII:F2IBD1GMD3) PALTUSOTINE 30 mg Inactive Ingredients Ingredient Name Strength COPOVIDONE K25-31 (UNII: D9C330MD8B) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIACETIN (UNII: XHX3C3X673) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color YELLOW (opadry yellow) Score no score Shape OVAL (OVAL) Size 18mm Flavor Imprint Code PAL;30 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 84015-930-60 60 in 1 BOTTLE; Type 0: Not a Combination Product 09/25/2025 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219070 09/25/2025 Labeler - Crinetics Pharmaceuticals, Inc. (828902515)


© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.