LUMOXITI- moxetumomab pasudotox injection, powder, lyophilized, for solution IV STABILIZER- polysorbate 80 solution
IV STABILIZER by
Drug Labeling and Warnings
IV STABILIZER by is a Prescription medication manufactured, distributed, or labeled by Innate Pharma, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LUMOXITI safely and effectively. See full prescribing information for LUMOXITI.
LUMOXITI™ (moxetumomab pasudotox-tdfk) for injection, for intravenous use
Initial U.S. Approval: 2018WARNING: CAPILLARY LEAK SYNDROME and HEMOLYTIC UREMIC SYNDROME
See full prescribing information for complete boxed warning.
- Capillary Leak Syndrome (CLS), including life-threatening cases, occurred in patients receiving LUMOXITI. Delay dosing or discontinue LUMOXITI as recommended. (2.3, 5.1)
- Hemolytic Uremic Syndrome (HUS), including life-threatening cases, occurred in patients receiving LUMOXITI. Discontinue LUMOXITI in patients with HUS. (2.3, 5.2)
INDICATIONS AND USAGE
LUMOXITI is a CD22-directed cytotoxin indicated for the treatment of adult patients with relapsed or refractory hairy cell leukemia (HCL) who received at least two prior systemic therapies, including treatment with a purine nucleoside analog (PNA). (1)
Limitations of Use
Not recommended in patients with severe renal impairment (CrCl ≤ 29 mL/min). (1)
DOSAGE AND ADMINISTRATION
- Recommended dosage: 0.04 mg/kg as an intravenous infusion over 30 minutes on Days 1, 3, and 5 of each 28-day cycle. (2.1)
- Maintain adequate hydration throughout treatment. (2.2)
- Consider low-dose aspirin on Days 1 to 8 of each 28-day cycle. (2.2)
- Premedicate with an acetaminophen antipyretic, antihistamine, and H2-receptor antagonist prior to all infusions. (2.2)
- See full prescribing information for instructions on reconstitution of lyophilized cake or powder, and preparation and administration of reconstituted drug. (2.4)
DOSAGE FORMS AND STRENGTHS
For injection: 1 mg lyophilized cake or powder in a single-dose vial for reconstitution and further dilution. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Renal Toxicity: Monitor for changes in renal function prior to each infusion and as clinically indicated. Delay dosing until recovery. (5.3)
- Infusion Related Reactions: Pre-medicate and if a severe infusion related reaction occurs, interrupt the LUMOXITI infusion and institute appropriate medical management. (5.4)
- Electrolyte Abnormalities: Monitor serum electrolytes prior to each dose and on Day 8 of each treatment cycle. Monitoring mid-cycle is also recommended. (5.5)
ADVERSE REACTIONS
Most common (≥ 20%) adverse reactions are infusion related reactions, edema, nausea, fatigue, headache, pyrexia, constipation, anemia, and diarrhea. Most common (≥ 50%) laboratory abnormalities are creatinine increased, ALT increased, hypoalbuminemia, AST increased, hypocalcemia, and hypophosphatemia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Innate Pharma at 1-888-501-0998 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 4/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: CAPILLARY LEAK SYNDROME and HEMOLYTIC UREMIC SYNDROME
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Recommended Concomitant Treatment
2.3 Monitoring to Assess Safety
2.4 Instructions for Reconstitution, Dilution, and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Capillary Leak Syndrome (CLS)
5.2 Hemolytic Uremic Syndrome (HUS)
5.3 Renal Toxicity
5.4 Infusion Related Reactions
5.5 Electrolyte Abnormalities
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: CAPILLARY LEAK SYNDROME and HEMOLYTIC UREMIC SYNDROME
- Capillary Leak Syndrome (CLS), including life-threatening cases, occurred in patients receiving LUMOXITI. Monitor weight and blood pressure; check labs, including albumin, if CLS is suspected. Delay dosing or discontinue LUMOXITI as recommended [see Dosage and Administration (2.3) and Warnings and Precautions (5.1)].
- Hemolytic Uremic Syndrome (HUS), including life-threatening cases, occurred in patients receiving LUMOXITI. Monitor hemoglobin, platelet count, serum creatinine, and ensure adequate hydration. Discontinue LUMOXITI in patients with HUS [see Dosage and Administration (2.3) and Warnings and Precautions (5.2)].
-
1 INDICATIONS AND USAGE
LUMOXITI is indicated for the treatment of adult patients with relapsed or refractory hairy cell leukemia (HCL) who received at least two prior systemic therapies, including treatment with a purine nucleoside analog (PNA).
Limitations of Use
LUMOXITI is not recommended in patients with severe renal impairment (CrCl ≤ 29 mL/min) [see Dosage and Administration (2.3), Warnings and Precautions (5.3), and Use in Specific Populations (8.5)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dose of LUMOXITI is 0.04 mg/kg administered as a 30-minute intravenous infusion on Days 1, 3, and 5 of each 28-day cycle. Continue LUMOXITI treatment for a maximum of 6 cycles, disease progression, or unacceptable toxicity.
2.2 Recommended Concomitant Treatment
Hydration
Intravenously administer 1 L of isotonic solution (e.g., 5% Dextrose Injection, USP and 0.45% or 0.9% Sodium Chloride Injection, USP) over 2-4 hours before and after each LUMOXITI infusion. Administer 0.5 L to patients under 50 kg.
Advise all patients to adequately hydrate with up to 3 L (twelve 8-oz glasses) of oral fluids (e.g., water, milk, or juice) per 24 hours on Days 1 through 8 of each 28-day cycle. In patients under 50 kg, up to 2 L (eight 8-oz glasses) per 24 hours is recommended.
Monitor fluid balance and serum electrolytes to avoid fluid overload and/or electrolyte abnormalities [see Warnings and Precautions (5.2, 5.5)].
Thromboprophylaxis
Consider low-dose aspirin on Days 1 through 8 of each 28-day cycle.
Monitor for signs and symptoms of thrombosis [see Warnings and Precautions (5.2)].
Premedication
Premedicate 30-90 minutes prior to each LUMOXITI infusion with:
- An antihistamine (e.g., hydroxyzine or diphenhydramine)
- Acetaminophen antipyretic
- A histamine-2 receptor antagonist (e.g., ranitidine, famotidine, or cimetidine)
If a severe infusion related reaction occurs, interrupt the LUMOXITI infusion and institute appropriate medical management. Administer an oral or intravenous corticosteroid approximately 30 minutes before resuming, and before each LUMOXITI infusion thereafter [see Warnings and Precautions (5.4)].
2.3 Monitoring to Assess Safety
Manage adverse reactions by withholding and/or discontinuing LUMOXITI as described below.
Identify Capillary Leak Syndrome (CLS) and Hemolytic Uremic Syndrome (HUS) based on clinical presentation (see Table 1).
Table 1: Monitoring for CLS and HUS CLS HUS Adverse reactions graded by the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03. Monitoring Parameter Before every infusion, check: - Weight
- Blood pressure
Before every infusion, check: - Hemoglobin levels
- Platelet count
- Serum creatinine
Assessment - If weight has increased by 5.5 pounds (2.5 kg) or 5% or greater from Day 1 of the cycle and the patient is hypotensive, promptly check for peripheral edema, hypoalbuminemia, and respiratory symptoms, including shortness of breath and cough.
- If CLS is suspected, check for a decrease in oxygen saturation and evidence of pulmonary edema and/or serosal effusions.
If HUS is suspected, promptly check blood LDH, indirect bilirubin, and blood smear schistocytes for evidence of hemolysis. Capillary Leak Syndrome (CLS)
Patients who experience Grade 2 or higher CLS should receive appropriate supportive measures, including treatment with oral or intravenous corticosteroids, with monitoring of weight, albumin levels, and blood pressure until resolution [see Warnings and Precautions (5.1)].
Table 2: CLS Grading and Management Guidance CLS Grade LUMOXITI Dosing Per National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03. Grade 2
Symptomatic; medical intervention indicatedDelay dosing until recovery of symptoms. Grade 3
Severe symptoms; medical intervention indicatedDiscontinue LUMOXITI. Grade 4
Life-threatening consequences; urgent intervention indicatedHemolytic Uremic Syndrome (HUS)
Discontinue LUMOXITI in patients with HUS. Treat with appropriate supportive measures and fluid replacement, with monitoring of blood chemistry, complete blood counts, and renal function until resolution [see Warnings and Precautions (5.2)].
Increased Creatinine
For patients with baseline serum creatinine within normal limits, delay dosing for Grade 2 or higher creatinine increases (greater than 1.5-times baseline or the upper limit of normal). Resume LUMOXITI upon recovery to Grade 1 (1- to 1.5-times baseline, or between the upper limit of normal and 1.5-times the upper limit of normal).
For patients with baseline serum creatinine of Grades 1 or 2, delay dosing for creatinine increases to Grade 3 or higher (greater than 3-times baseline or the upper limit of normal). Resume LUMOXITI upon recovery to baseline grade or lower [see Warnings and Precautions (5.3)].
2.4 Instructions for Reconstitution, Dilution, and Administration
LUMOXITI must be reconstituted and diluted by a healthcare provider using aseptic technique. Refer to the Healthcare Provider Instructions for Use for LUMOXITI for full reconstitution, dilution, and administration information.
Step 1: Calculate Dose
- Calculate the dose (mg) and the number of LUMOXITI vials (1 mg/vial) to be reconstituted. The final concentration of the reconstituted LUMOXITI solution is 1 mg/mL.
- DO NOT round down for partial vials.
- Individualize dosing based on the patient's actual body weight prior to the first dose of the first treatment cycle.
- A change in dose should only be made between cycles when a change in weight of greater than 10% is observed from the weight used to calculate the first dose of the first treatment cycle. No change in dose should be made during a particular cycle.
Step 2: Reconstitution
Reconstitute LUMOXITI vials with Sterile Water for Injection, USP only.
- Reconstitute each LUMOXITI (1 mg/vial) with 1.1 mL Sterile Water for Injection, USP. The resulting 1 mg/mL solution allows a withdrawal volume of 1 mL.
- Direct the Sterile Water for Injection, USP along the walls of the vial and not directly at the lyophilized cake or powder.
- DO NOT reconstitute LUMOXITI vials with the IV Solution Stabilizer.
- Gently swirl the vial until completely dissolved. Invert the vial to ensure all cake or powder in the vial is dissolved. Do not shake.
- Visually inspect that the reconstituted solution is clear to slightly opalescent, colorless to slightly yellow, and free from visible particles. Do not use if solution is cloudy, discolored, or contains any particles.
- Use reconstituted solution immediately. Do not store reconstituted LUMOXITI vials. See Table 3 for storage times and conditions for the reconstituted solution.
Step 3: Dilution
Add the IV Solution Stabilizer to the infusion bag prior to adding LUMOXITI solution to the infusion bag. Vial of IV Solution Stabilizer is packaged separately.
- Obtain a 50 mL 0.9% Sodium Chloride Injection, USP infusion bag.
- Add 1 mL IV Solution Stabilizer to the infusion bag containing 50 mL 0.9% Sodium Chloride Injection, USP.
- Only one vial of IV Solution Stabilizer should be used per administration of LUMOXITI.
- Gently invert the bag to mix the solution. Do not shake.
- Withdraw the required volume (calculated from Step 1) of LUMOXITI solution from the reconstituted vial(s).
- Inject LUMOXITI into the infusion bag containing 50 mL 0.9% Sodium Chloride Injection, USP and 1 mL IV Solution Stabilizer.
- Gently invert the bag to mix the solution. Do not shake.
- Discard any partially used or empty vials of LUMOXITI and IV Solution Stabilizer.
- See Table 3 for storage times and conditions for the diluted solution.
Step 4: Administration Instructions
For intravenous infusion only.
- Administer the diluted solution intravenously over 30 minutes.
- Do not mix LUMOXITI, or administer as an infusion with other medicinal products.
- After the infusion, flush the intravenous administration line with of 0.9% Sodium Chloride Injection, USP at the same rate as the infusion. This ensures that the full LUMOXITI dose is delivered.
Table 3: Storage Times and Conditions for Reconstituted and Diluted LUMOXITI Solution Reconstituted Solution Diluted LUMOXITI Solution in Infusion Bag After Dilution Administration LUMOXITI does not contain bacteriostatic preservatives. Use reconstituted solution immediately.
DO NOT STORE reconstituted LUMOXITI vials.Use diluted solution immediately or after storage at room temperature (20°C to 25°C; 68°F to 77°F) for up to 4 hours or store refrigerated at 2°C to 8°C (36°F to 46°F) for up to 24 hours.
PROTECT FROM LIGHT.
DO NOT FREEZE.
DO NOT SHAKE.If the diluted solution is refrigerated (2°C to 8°C; 36°F to 46°F), allow it to equilibrate at room temperature (20°C to 25°C; 68°F to 77°F) for no more than 4 hours prior to administration.
Administer diluted solution within 24 hours of reconstitution as a 30-minute infusion.
PROTECT FROM LIGHT. - 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Capillary Leak Syndrome (CLS)
Capillary leak syndrome (CLS), including life-threatening cases, has been reported among patients treated with LUMOXITI and is characterized by hypoalbuminemia, hypotension, symptoms of fluid overload, and hemoconcentration. In the combined safety database of HCL patients treated with LUMOXITI, CLS occurred in 34% (44/129) of patients, including Grade 2 in 23% (30/129), Grade 3 in 1.6% (2/129), and Grade 4 in 2% (3/129).
Most cases of CLS occurred in the first 8 days (range: 1 to 19) of a treatment cycle, however, cases have also been reported on other days throughout the cycle. The median time to resolution of CLS was 12 days (range: 1 to 53).
Monitor patient weight and blood pressure prior to each LUMOXITI infusion and as clinically indicated during treatment. Assess patients for signs and symptoms of CLS, including weight gain (increase in 5.5 pounds (2.5 kg) or ≥ 5% from Day 1 of current cycle), hypotension, peripheral edema, shortness of breath or cough, and pulmonary edema and/or serosal effusions. In addition, the following changes in laboratory parameters may help identify CLS: hypoalbuminemia, elevated hematocrit, leukocytosis, and thrombocytosis [see Dosage and Administration (2.3)].
CLS may be life-threatening or fatal if treatment is delayed. Counsel patients to seek immediate medical attention should signs or symptoms of CLS occur at any time. Patients who develop CLS should receive appropriate supportive measures, including concomitant oral or intravenous corticosteroids, and hospitalization as clinically indicated. Withhold LUMOXITI for Grade 2 CLS until resolution, and permanently discontinue for Grade ≥ 3 CLS [see Dosage and Administration (2.3)].
5.2 Hemolytic Uremic Syndrome (HUS)
Hemolytic Uremic Syndrome (HUS), including life threatening cases, has been reported in patients treated with LUMOXITI and is characterized by the triad of microangiopathic hemolytic anemia, thrombocytopenia, and progressive renal failure. In the combined safety database of HCL patients treated with LUMOXITI, HUS occurred in 7% (9/129) of patients, including Grade 3 in 3% (4/129) and Grade 4 in 0.8% (1/129).
Most cases of HUS occurred in the first 9 days (range: 1 to 16) of a treatment cycle, however, cases have also been reported on other days throughout the cycle. The median time to resolution of HUS was 11.5 days (range: 2 to 44). All cases resolved, including those who discontinued LUMOXITI.
Avoid LUMOXITI in patients with prior history of severe thrombotic microangiopathy (TMA) or HUS. Administer prophylactic intravenous fluids before and after LUMOXITI infusions [see Dosage and Administration (2.2)]. In Study 1053, patients with a platelet count ≥ 100,000/mm3 received low-dose aspirin on Days 1 through 8 of each 28-day cycle for prophylaxis of thrombosis.
Monitor blood chemistry and complete blood counts prior to each dose and on Day 8 of each treatment cycle. Monitoring mid-cycle is also recommended. Consider the diagnosis of HUS in patients who develop hemolytic anemia, worsening or sudden onset of thrombocytopenia, increase in creatinine levels, elevation of bilirubin and/or LDH, and have evidence of hemolysis based on peripheral blood smear schistocytes [see Dosage and Administration (2.3)].
The events of HUS may be life-threatening if treatment is delayed with increased risk of progressive renal failure requiring dialysis. If HUS is suspected initiate appropriate supportive measures, including fluid repletion, hemodynamic monitoring, and consider hospitalization as clinically indicated. Discontinue LUMOXITI in patients with HUS [see Dosage and Administration (2.3)].
5.3 Renal Toxicity
Renal toxicity has been reported in patients treated with LUMOXITI therapy. In the combined safety database of HCL patients treated with LUMOXITI, 26% (34/129) reported adverse events of renal toxicity, including acute kidney injury (2.3%), renal failure (2.3%), renal impairment (1.6%), serum creatinine increased (17%), and proteinuria (8%). Grade 3 acute kidney injury occurred in 1.6% (2/129) of patients. All other events were mild to moderate in severity.
Based on laboratory findings, during treatment, serum creatinine increased by two or more grades from baseline in 22% (29/129) of patients, including increases of Grade 3 in 1.6% (2/129) of patients. At the end of treatment, serum creatinine levels remained elevated at 1.5- to 3-times the upper limit of normal in 5% of patients. Patients who experience HUS, those ≥ 65 years of age, or those with baseline renal impairment may be at increased risk for worsening of renal function following treatment with LUMOXITI [see Use in Specific Populations (8.5)].
Monitor renal function prior to each infusion of LUMOXITI, and as clinically indicated throughout treatment. Delay LUMOXITI dosing in patients with Grade ≥ 3 elevations in creatinine, or upon worsening from baseline by ≥ 2 grades [see Dosage and Administration (2.3)].
5.4 Infusion Related Reactions
Infusion related reactions occurred in patients treated with LUMOXITI, and were defined as the occurrence of any one of the following events on the day of study drug infusion: chills, cough, dizziness, dyspnea, feeling hot, flushing, headache, hypertension, hypotension, infusion related reaction, myalgia nausea, pyrexia, sinus tachycardia, tachycardia, vomiting, or wheezing. In Study 1053, infusion related reactions occurred in 50% (40/80) of patients. Grade 3 infusion related events as defined, occurred in 11% (9/80) of LUMOXITI-treated patients. The most frequently reported infusion related events were nausea (15%), pyrexia (14%), chills (14%), vomiting (11%), headache (9%), and infusion related reaction (9%).
Infusion related reactions may occur during any cycle of treatment with LUMOXITI. Prior to each dose of LUMOXITI, premedicate with antihistamines and antipyretics. If a severe infusion related reaction occurs, interrupt the LUMOXITI infusion and institute appropriate medical management. Administer an oral or intravenous corticosteroid approximately 30 minutes before resuming, or before the next LUMOXITI infusion [see Dosage and Administration (2.2)].
5.5 Electrolyte Abnormalities
In the combined safety database of HCL patients treated with LUMOXITI, electrolyte abnormalities occurred in 57% (73/129) of patients with the most common electrolyte abnormality being hypocalcemia occurring in 25% of patients. Grade 3 electrolyte abnormalities occurred in 14% (18/129) of patients and Grade 4 electrolyte abnormalities occurred in 0.8% (1/129) of patients. Electrolyte abnormalities co-occurred in the same treatment cycle with CLS, HUS, fluid retention, or renal toxicity in 37% (48/129) of patients.
Monitor serum electrolytes prior to each dose and on Day 8 of each treatment cycle. Monitoring mid-cycle is also recommended.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling.
- Capillary Leak Syndrome [see Warnings and Precautions (5.1)]
- Hemolytic Uremic Syndrome [see Warnings and Precautions (5.2)]
- Renal Toxicity [see Warnings and Precautions (5.3)]
- Infusion Related Reactions [see Warnings and Precautions (5.4)]
- Electrolyte Abnormalities [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
As clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data described in this section reflect exposure to LUMOXITI in 80 patients with previously treated HCL in Study 1053 [see Clinical Studies (14)]. Patients received LUMOXITI 0.04 mg/kg as an intravenous infusion over 30 minutes on Days 1, 3, and 5 of each 28-day cycle for a maximum of 6 cycles or until disease progression or unacceptable toxicity.
The median duration of treatment with LUMOXITI was 5.7 months (range: 0.9 to 6.7), with a median of 6 treatment cycles started in each patient.
The most common non-laboratory adverse reactions (≥ 20%) of any grade were infusion related reactions, edema, nausea, fatigue, headache, pyrexia, constipation, anemia, and diarrhea. The most common Grade 3 or 4 adverse reactions (reported in at least ≥ 5% of patients) were hypertension, febrile neutropenia, and HUS.
The most common laboratory abnormalities (≥ 20%) of any grade were creatinine increased, ALT increased, hypoalbuminemia, AST increased, hypocalcemia, hypophosphatemia, hemoglobin decreased, neutrophil count decreased, hyponatremia, blood bilirubin increased, hypokalemia, GGT increased, hypomagnesemia, platelet count decreased, hyperuricemia, and alkaline phosphate increased.
Adverse reactions resulting in permanent discontinuation of LUMOXITI occurred in 15% (12/80) of patients. The most common adverse reaction leading to LUMOXITI discontinuation was HUS (5%). The most common adverse reaction resulting in dose delays, omissions, or interruptions was pyrexia (3.8%).
Tables 4 and 5 present the frequency category of adverse reactions and key laboratory abnormalities observed in patients with relapsed or refractory HCL treated with LUMOXITI.
Table 4: Adverse Reactions* in ≥ 20% (All Grades) of Patients with HCL in Study 1053 LUMOXITI
N=80All Grades (%) Grade 3 (%) - * Per National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03.
- † Infusion related reactions: includes patients who were reported to have one or more infusion event that may be infusion-related on the day of study drug infusion.
General Disorders and Administration Site Conditions Edema peripheral 39 - Fatigue 34 - Pyrexia 31 1.3 Gastrointestinal Disorders Nausea 35 2.5 Constipation 23 - Diarrhea 21 - Injury, Poisoning, and Procedural Complications Infusion related reactions† 50 3.8 Nervous System Disorders Headache 33 - Blood and Lymphatic System Disorders Anemia 21 10 Fluid retention occurred in 63% (50/80) of patients treated with LUMOXITI in Study 1053, including Grade 3 in 1.3% (1/80) of patients. Fluid retention included all preferred terms of edema peripheral (39%), face edema (14%), abdominal distension (13%), weight increased (8%), pleural effusion (6%), edema (5%), peripheral swelling (5%), localized edema (3.8%), ascites (1.3%), fluid overload (1.3%), fluid retention (1.3%), and pericardial effusion (1.3%). Of the fifty patients with fluid retention, 29% of patients required diuretics.
Ocular adverse events occurred, including: blurred vision (9%), dry eye (8%), cataracts (5%), ocular discomfort and/or pain (4%), ocular swelling/periorbital edema (4%), conjunctivitis (1.3%), conjunctival hemorrhage (1.3%), and ocular discharge (1.3%).
Table 5: Laboratory Abnormalities* in ≥ 20% (All Grades) Reported in Patients with HCL in Study 1053 LUMOXITI
N=80All Grades (%) Grade 3 (%) Grade 4 (%) ALT=alanine aminotransferase; AST=aspartate aminotransferase; GGT=gamma glutamyl transferase - * Per National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03 and based on laboratory measurements worsening from baseline
Hematology Hemoglobin decreased 43 15 - Neutrophil count decreased 41 11 20 Platelet count decreased 21 11 3.8 Chemistry Creatinine increased 96 2.5 - ALT increased 65 3.8 - Hypoalbuminemia 64 1.3 - AST increased 55 1.3 - Hypocalcemia 54 - - Hypophosphatemia 53 14 - Hyponatremia 41 8.8 - Blood Bilirubin increased 30 1.3 - Hypokalemia 25 1.3 1.3 GGT increased 25 - - Hypomagnesemia 23 1.3 - Hyperuricemia 21 - 2.5 Alkaline phosphatase increased 20 - - 6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to moxetumomab pasudotox-tdfk in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
The immunogenicity of LUMOXITI was evaluated using electrochemiluminescent (ECL)-based immunoassay to test for anti-moxetumomab pasudotox-tdfk antibodies (ADA). For patients whose serum tested positive for ADA, a cell-based assay was performed to detect neutralizing antibodies (nAb). In Study 1053, 59% (45/76) of patients tested positive for ADA prior to any treatment with moxetumomab pasudotox-tdfk. Seventy out of 80 subjects tested ADA positive at any point during the study and were subsequently tested for nAb. The results showed that 67 of 70 subjects were nAb-positive. Among these 67 patients who tested nAb-positive, 99% (66/67) had ADA specific to the PE38 binding domain, and 54% (36/67) also had ADA specific to the CD22 binding domain. In 41 out of 73 patients who had baseline and post-baseline ADA results, the median fold increase from baseline (Cycle 1, Day 1) in ADA titer was 3.75- (range: 0 to 240), 54- (range: 0 to 2560), 120- (range: 0 to 1920), and 128- (range: 0 to 2560) fold at Cycles 2, 3, 5, and end-of-treatment, respectively. Patients who tested positive for ADA had decreased systemic moxetumomab pasudotox-tdfk concentrations [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action and findings in non-pregnant female animals, LUMOXITI is expected to cause maternal and embryo-fetal toxicity when administered to a pregnant woman [see Clinical Pharmacology (12.1) and Nonclinical Toxicology (13.2)]. There are no available data on LUMOXITI use in pregnant women to inform a drug-associated risk of major birth defects and miscarriage. Animal reproduction or developmental toxicity studies have not been conducted with LUMOXITI. Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.2 Lactation
Risk Summary
No data are available regarding the presence of moxetumomab pasudotox-tdfk in human milk, the effects on the breastfed child, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for LUMOXITI and any potential adverse effects on the breastfed child from LUMOXITI or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Contraception
Females
To avoid potential exposure to the fetus, women of reproductive potential should use effective contraception during treatment with LUMOXITI and for at least 30 days after the last dose is received. Verify the pregnancy status of females of reproductive potential prior to initiating LUMOXITI.
8.5 Geriatric Use
In the combined safety database of HCL patients treated with LUMOXITI, 31% (40/129) of patients treated with LUMOXITI were 65 years of age or older and 8% (10/129) were 75 years of age or older. Exploratory analyses across this population suggest a higher incidence of adverse reactions leading to drug discontinuation (23% versus 7%) and renal toxicity (40% versus 20%) for patients 65 years of age or older as compared to those younger than 65 years. Clinical studies of LUMOXITI did not include sufficient numbers of subjects aged 65 and over to determine whether there were differences in efficacy between younger and older patients.
-
11 DESCRIPTION
Moxetumomab pasudotox-tdfk is a CD22-directed cytotoxin. Moxetumomab pasudotox-tdfk is composed of a recombinant, murine immunoglobulin variable domain genetically fused to a truncated form of Pseudomonas exotoxin, PE38, that inhibits protein synthesis. Moxetumomab pasudotox-tdfk has an approximate molecular weight of 63 kDa and is produced in E. coli cells by recombinant DNA technology. During the moxetumomab pasudotox-tdfk manufacturing process, fermentation is carried out in nutrient medium containing the antibiotic kanamycin. However, kanamycin is cleared in the manufacturing process and is not detectable in the final product.
LUMOXITI (moxetumomab pasudotox-tdfk) for injection is supplied as a sterile, preservative-free, white to off-white lyophilized cake or powder in a single-dose vial for reconstitution and dilution prior to intravenous infusion. Each single-dose vial contains 1 mg moxetumomab pasudotox-tdfk, glycine (80 mg), polysorbate 80 (0.2 mg), sodium phosphate monobasic monohydrate (3.4 mg), sucrose (40 mg), and sodium hydroxide to adjust pH to 7.4. After reconstitution with 1.1 mL Sterile Water for Injection, USP, the resulting 1 mg/mL solution allows a withdrawal volume of 1 mL. Prior to intravenous infusion, the reconstituted vial(s) of solution are added to an infusion bag containing 50 mL of 0.9% Sodium Chloride Injection, USP and 1 mL of IV Solution Stabilizer.
IV Solution Stabilizer is a sterile, preservative-free, colorless to slightly yellow, clear solution free from visible particles and supplied in a single-dose vial. Each vial contains 1 mL solution. Each vial contains citric acid monohydrate (0.7 mg), polysorbate 80 (6.5 mg), sodium citrate dihydrate (6.4 mg), and Water for Injection, USP. The pH is 6.0.
The LUMOXITI and IV Solution Stabilizer vial stoppers are not made with natural rubber latex.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Moxetumomab pasudotox-tdfk is a CD22-directed cytotoxin. Moxetumomab pasudotox-tdfk binds CD22 on the cell surface of B-cells and is internalized. Moxetumomab pasudotox-tdfk internalization results in ADP-ribosylation of elongation factor 2, inhibition of protein synthesis, and apoptotic cell death.
12.2 Pharmacodynamics
The presence of moxetumomab pasudotox-tdfk may interfere with detection of cellular CD22, therefore, total peripheral blood B-cell counts (including normal B cells and HCL cells) were quantified using a standard assay for CD19+ B cells as a surrogate. In patients with HCL, treatment with LUMOXITI at the approved recommended dosage resulted in a reduction of circulating CD19+ B cells. The circulating CD19+ B cells on Day 8 were reduced by 89% from baseline following the first three infusions of LUMOXITI. B cell reduction was sustained for at least 1-month post-treatment.
Total counts of CD3+ T cells, CD4+ T cells, CD8+ T cells, and CD16+/CD56 Natural Killer cells and quantitative immunoglobulin (Ig) A, G, and M levels were evaluated throughout the course of treatment. On Day 8, median cell counts were reduced from baseline for the following populations: CD3+ T cells (-21%), CD4+ T cells (-20%), CD8+ T cells (-23%), and CD16+/CD56 Natural Killer cells (-47%). All monitored cell counts returned to, or were elevated above baseline levels on Day 29 and thereafter. At baseline, the median IgA, IgG, and IgM levels were 107 mg/dL (11-260), 834 mg/dL (387-3003), and 42 mg/dL (6-380), respectively, and remained generally unchanged at the end of treatment.
12.3 Pharmacokinetics
The pharmacokinetics (PK) of moxetumomab pasudotox-tdfk was studied in patients with HCL at doses ranging from 0.005 to 0.05 mg/kg (about 0.1 to 1.3 times the approved recommended dosage) administered intravenously over 30 minutes on Days 1, 3, and 5 of a 28-day cycle. Moxetumomab pasudotox-tdfk concentrations increased dose-proportionally over the studied dose range. The mean steady state moxetumomab pasudotox-tdfk exposures at the approved recommended dosages were 379 ng/mL (range: 20 to 862; SD: 262) for Cmax and 626 ng∙hour/mL (range: 5 to 1960; SD: 610) for AUC0-last. No systemic accumulation of moxetumomab pasudotox-tdfk was observed. Baseline CD19+ B cells were evaluated for association with the PK exposure and higher PK exposures were significantly associated with low baseline CD19+ counts (p < 0.001).
Distribution
The population PK model estimated mean volume of distribution of moxetumomab pasudotox-tdfk was 6.5 L (SD 2.4).
Elimination
The mean elimination half-life of moxetumomab pasudotox-tdfk was 1.4 hours (range: 0.8 to 1.8; SD: 0.35). The population PK model estimated mean systemic clearance of moxetumomab pasudotox-tdfk after the first dose of the first cycle was 25 L/hour (SD: 29.0) and after subsequent dosing was 4 L/hour (SD: 4.4).
Specific Populations
No clinically significant differences in the pharmacokinetics of moxetumomab pasudotox-tdfk were observed for age (36 to 84 years), sex, race (White and non-White), body weight (42 to 152 kg), mild hepatic impairment (total bilirubin ≤ upper limit of normal [ULN] and AST > ULN, or total bilirubin > 1 to 1.5 times ULN and any AST), mild renal impairment (CLcr 60-89 mL/min; n=40), or moderate renal impairment (CLcr 30-59 mL/min; n=4) based on population PK analysis.
The pharmacokinetics of moxetumomab pasudotox-tdfk in patients with moderate to severe hepatic impairment (total bilirubin > 1.5 ULN) or severe renal impairment (CrCl ≤ 29 mL/min) is unknown.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies have been conducted to assess the carcinogenic or genotoxic potential of moxetumomab pasudotox-tdfk. Animal fertility studies have not been conducted with moxetumomab pasudotox-tdfk.
13.2 Animal Toxicology and/or Pharmacology
At a human equivalent dose > 3 times the recommended dose, degeneration of heart tissue was observed in cynomolgus monkeys. At a human equivalent dose > 10 times the recommended dose, gliosis in the brain, axonal degeneration in the spinal cord, and body tremors were observed in cynomolgus monkeys.
-
14 CLINICAL STUDIES
The efficacy of LUMOXITI was based upon Study 1053 titled "A Pivotal Multicenter Trial of Moxetumomab Pasudotox in Relapsed/Refractory Hairy Cell Leukemia" (NCT01829711). Study 1053 was conducted in patients with histologically confirmed HCL or HCL variant with a need for therapy based on presence of cytopenias or splenomegaly and who had received prior treatment with at least 2 systemic therapies, including 1 purine nucleoside analog (PNA). Eligible patients had serum creatinine ≤ 1.5 mg/dL or creatinine clearance ≥ 60 mL/min as estimated by the Cockcroft Gault equation.
A total of 80 patients were enrolled; 77 with classic HCL and 3 with HCL variant. The median age was 60 years (range: 34 to 84) years, 79% were male, and 94% were Caucasian. At baseline, 98% of patients had an ECOG performance status of 0 or 1. The median number of prior treatments was 3 (range: 2 to 11); all patients received prior PNA therapy, including 29% in combination with rituximab. The most common other prior treatment regimens were rituximab monotherapy (51%), interferon-alpha (25%), and a BRAF inhibitor (18%). At baseline, 33% (26/80) of patients had low hemoglobin (< 10 g/dL), 68% (54/80) of patients had neutropenia (< 1000/mm3), and 84% (67/80) patients had baseline platelet counts < 100,000/mm3. About 35% of patients had enlarged spleens (≥ 14 cm, assessed by BICR) at baseline.
Patients received LUMOXITI 0.04 mg/kg as an intravenous infusion over 30 minutes on Days 1, 3, and 5 of each 28-day cycle for a maximum of 6 cycles or until documentation of complete response (CR), disease progression, or unacceptable toxicity. The median duration of follow-up was 16.7 months (range: 2 to 49). An independent review committee (IRC) performed efficacy evaluations using blood, bone marrow, and imaging criteria adapted from previous HCL studies and consensus guidelines.
Efficacy of LUMOXITI in HCL was evaluated by the IRC-assessed rate of durable CR, as confirmed by maintenance of hematologic remission (hemoglobin ≥ 11 g/dL, neutrophils ≥ 1500/mm3, and platelets ≥ 100,000/mm3 without transfusions or growth factor for at least 4 weeks) more than 180 days after IRC-assessed CR. The IRC-assessed durable CR rate was 30% (24/80 patients; 95% CI: 20, 41).
Additional efficacy outcome measures included overall response rate (ORR), CR, and duration of response (see Table 6).
Table 6: Additional Efficacy Results in Patients with HCL in Study 1053 Independent Review Committee (IRC) Assessed
N=80CI=Confidence Interval; NR=Not Reached; + indicates censored observations - * ORR defined as best overall response of CR or PR.
- † CR defined as clearing of the bone marrow of hairy cells by routine Hematoxylin & Eosin stain, radiologic resolution of pre-existing lymphadenopathy and/or organomegaly, and hematologic remission.
- ‡ PR defined as ≥ 50% decrease or normalization (< 500/mm3) in peripheral blood lymphocyte count, reduction of pre-existing lymphadenopathy and/or organomegaly, and hematologic remission.
Overall Response Rate Overall Response Rate* (%) [95% CI] 75 [64, 84] Complete Response† (%) [95% CI] 41 [30, 53] Partial Response‡ (%) [95% CI] 34 [24, 45] Duration of Response Median in months [range] NR [0+ to 43+] Duration of CR Median in months [range] NR [0+ to 40+] The median time to ORR and CR was 5.7 months (range: 1.8 to 12.9) and 5.9 months (range 1.8 to 13.2), respectively. Sixty-four patients (80%) had normalization of hematologic parameters and achieved hematologic remission, with a median time to hematologic remission of 1.1 months (range: 0.2 to 13) and with a median duration of hematologic remission not reached (range: 0.3 to 48.2+).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
- LUMOXITI (moxetumomab pasudotox-tdfk) for injection is supplied as a sterile, preservative-free, white to off-white lyophilized cake or powder in a 1 mg single-dose vial. Each carton (NDC: 73380-4700-1) contains one single-dose vial.
- IV Solution Stabilizer is supplied as a sterile, preservative-free, colorless to slightly yellow, clear solution free from visible particles in a 1 mL single-dose vial. The IV Solution Stabilizer is packaged separately from LUMOXITI. Each carton (NDC: 73380-4715-9) contains one single-dose vial. Do not use the IV Solution Stabilizer to reconstitute LUMOXITI.
Only one vial of IV Solution Stabilizer should be used per administration of LUMOXITI.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Capillary Leak Syndrome
Advise patients on the risk of developing capillary leak syndrome. Advise patients to immediately report any symptoms suggestive of capillary leak syndrome, such as difficulty breathing, rapid weight gain, hypotension, or swelling of their arms, legs, and/or face to their healthcare provider for further evaluation [see Warnings and Precautions (5.1)].
Hemolytic Uremic Syndrome
Advise patients on the risk of developing hemolytic uremic syndrome. Advise patients on the importance of maintaining high fluid intake, and the need for frequent monitoring of blood chemistry values [see Warnings and Precautions (5.2)].
Renal Toxicity
Inform patients that taking LUMOXITI may cause decreased renal function. Advise patients to report any changes to urine output to their healthcare provider for further evaluation [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
Infusion Related Reactions
Advise patients to contact their healthcare provider immediately for signs or symptoms of infusion related reactions [see Warnings and Precautions (5.4)].
Electrolyte Abnormalities
Advise patients to report symptoms of electrolyte abnormalities (e.g., muscle cramping, paresthesias, irregular or fast heartbeat, nausea, seizures) to their healthcare provider immediately [see Warnings and Precautions (5.5)].
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Issued: September 2018 MEDICATION GUIDE
LUMOXITI™ (loo-MOCKS-eh-tee)
(moxetumomab pasudotox-tdfk)
for injectionWhat is the most important information I should know about LUMOXITI?
LUMOXITI can cause serious side effects, including:- Capillary Leak Syndrome (CLS). LUMOXITI can cause fluid to leak from small blood vessels into your body's tissues. This condition is called "Capillary Leak Syndrome" (CLS). CLS can quickly cause you to have symptoms that may become life-threatening if not treated right away. Get emergency medical help right away if you develop any of the following symptoms of CLS:
- swelling of your face, arms, or legs
- fast weight gain (increase in 5.5 pounds from Day 1 of your current cycle)
- weakness or dizziness
- shortness of breath or trouble breathing
- cough
- low blood pressure
Your healthcare provider will check your weight and blood pressure before you receive each dose of LUMOXITI and as needed during treatment. - Hemolytic Uremic Syndrome (HUS). Hemolytic uremic syndrome is a condition that affects your blood cells and kidneys and may be life-threatening if not treated right away. Get emergency medical help right away if you develop any of the following symptoms of HUS:
- decrease in the amount of urine or dark urine (tea-colored)
- unusual bleeding or bruising of your skin
- stomach pain
- vomiting
- fever
- feeling tired
- changes in mood or behavior
- confusion
- seizures
- shortness of breath
- fast heartbeat
Your healthcare provider will do blood tests to check your blood cells and kidneys before you receive each dose of LUMOXITI and during treatment as recommended by your healthcare provider. If you develop any of these symptoms of CLS or HUS, your healthcare provider may monitor you in the hospital.
Getting medical treatment right away may help keep these problems from becoming more serious.
Your healthcare provider will check you for these problems during your treatment with LUMOXITI. Your healthcare provider may delay or completely stop treatment with LUMOXITI if you have severe side effects.
See "What are the possible side effects of LUMOXITI?" below for other side effects of LUMOXITI.What is LUMOXITI?
LUMOXITI is a prescription medicine used to treat adults with hairy cell leukemia (HCL)- that has come back or has not responded to previous treatment, and
- have received at least 2 other treatments, including a type of medicine called purine nucleoside analog (PNA).
Before you receive LUMOXITI, tell your healthcare provider about all your medical conditions, including if you: - have had conditions that affect your blood and blood vessels called HUS or severe thrombotic microangiopathy (TMA)
- have kidney problems
- are pregnant or plan to become pregnant. LUMOXITI may harm your unborn baby.
- If you are a female who can become pregnant, you should use effective birth control during treatment with LUMOXITI and for at least 30 days after your last dose of LUMOXITI.
- If you are a female who can become pregnant, your healthcare provider will perform a pregnancy test before you start treatment with LUMOXITI.
- Tell your healthcare provider right away if you become pregnant during treatment with LUMOXITI.
- are breastfeeding or plan to breastfeed. It is not known if LUMOXITI passes into your breast milk. You and your healthcare provider should decide if you will receive LUMOXITI or breastfeed. You should not do both.
Know the medicines you take. Keep a list of your medicines with you and show it to your healthcare provider when you get a new medicine.How will I receive LUMOXITI? - Your healthcare provider will give you LUMOXITI into your vein through an intravenous (IV) line over 30 minutes.
- LUMOXITI is usually given on Day 1, Day 3, and Day 5 of a 28-day treatment cycle. This is 1 treatment cycle. You may receive up to 6 treatment cycles.
- Your healthcare provider will give you medicines and IV fluids before and after your infusions.
- It is important for you to drink the additional prescribed amount of fluids (water, milk, or juice) of up to twelve 8-oz glasses every 24 hours on Days 1 through 8 of each 28-day treatment cycle when you receive LUMOXITI infusions.
- Your healthcare provider will decide how many treatment cycles you need.
- If you miss any appointments, call your healthcare provider as soon as possible to reschedule your appointment.
What are the possible side effects of LUMOXITI?
LUMOXITI can cause serious side effects, including:- See "What is the most important information I should know about LUMOXITI?"
- Kidney problems. LUMOXITI may cause kidney problems. People who have HUS, are 65 years of age or older, or those who have kidney problems before starting treatment with LUMOXITI may have an increased risk of worse kidney problems after treatment with LUMOXITI. Tell your healthcare provider right away if you have any changes in the amount you urinate. Your healthcare provider will do tests to check your kidneys before you receive each dose of LUMOXITI and as needed during treatment. Your healthcare provider may delay your treatment with LUMOXITI if you have severe kidney problems.
- Infusion reactions. LUMOXITI can cause infusion reactions that are common but can also be serious. Infusion reactions may happen on the day you receive your LUMOXITI infusion. Signs and symptoms of infusion reactions may include:
- chills
- cough
- dizziness
- shortness of breath or wheezing
- feeling hot or flushing
- fast heartbeat
- headache
- changes in blood pressure
- muscle pain
- nausea
- fever
- vomiting
Your healthcare provider may give you medicines to take before and after each LUMOXITI infusion. - Electrolyte problems. Tell your healthcare provider if you get any of the following symptoms of electrolyte problems:
- muscle cramps
- numbness or tingling
- abnormal or fast heartbeat
- nausea
- seizures
Your healthcare provider will do blood tests to check your electrolytes before you receive each dose of LUMOXITI and during treatment as recommended by your healthcare provider. The most common side effects of LUMOXITI include: - swelling in your face, arms, or legs
- nausea
- feeling tired
- headache
- fever
- constipation
- low red blood cells (anemia)
- diarrhea
These are not all the possible side effects of LUMOXITI.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of LUMOXITI.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about LUMOXITI that is written for health professionals.What are the ingredients in LUMOXITI?
Active ingredient: moxetumomab pasudotox-tdfk
Inactive ingredients: glycine, polysorbate 80, sodium phosphate monobasic monohydrate, sucrose, and sodium hydroxide
Inactive ingredients of IV Solution Stabilizer: citric acid monohydrate, polysorbate 80, sodium citrate dihydrate, Water for Injection, USP
LUMOXITI is a trademark of Innate Pharma.© Innate Pharma 2020
Distributed by: Innate Pharma Inc., Rockville, MD 20850
Manufactured by: Innate Pharma Inc., Rockville, MD 20850; U.S. License No. 2206
For more information, go to www.LUMOXITI.com or call 1-888-501-0998. -
LUMOXITI™ (moxetumomab pasudotox-tdfk)for injection
Healthcare Provider Instructions for Use
Important Information
Read the following instructions before reconstitution, dilution, and administration of LUMOXITI.
- LUMOXITI must be prepared by a healthcare professional using proper aseptic technique.
- Do not freeze or shake LUMOXITI or IV Solution Stabilizer.
- Provide each patient the Medication Guide packaged with LUMOXITI prior to each treatment cycle to inform them of the risks and benefits of LUMOXITI.
- See Full Prescribing Information for more information on LUMOXITI.
For questions, call Innate Pharma at 1-888-501-0998.
How Supplied
- LUMOXITI and IV Solution Stabilizer are packaged separately.
- Prior to preparation, LUMOXITI and IV Solution Stabilizer should be stored at 2°C to 8°C (36°F to 46°F) in original cartons to protect from light.
LUMOXITI (moxetumomab pasudotox-tdfk) - Each single-dose vial supplies LUMOXITI 1 mg/vial (moxetumomab pasudotox-tdfk) for injection as a lyophilized cake or powder for reconstitution and dilution prior to intravenous infusion.
- Multiple vials of LUMOXITI may be required to administer a single dose (See Step 1: Calculate Dose).
- Reconstitute LUMOXITI vials with Sterile Water for Injection, USP only (not supplied).
IV Solution Stabilizer - Each single-dose vial contains 1 mL IV Solution Stabilizer.
- Only one vial of IV Solution Stabilizer is needed per administration of LUMOXITI, regardless of the number of vials of LUMOXITI used to prepare the infusion.
- Do not use IV Solution Stabilizer to reconstitute LUMOXITI.
- Do not flush IV lines with IV Solution Stabilizer.

Storage and Handling of Reconstituted and Diluted LUMOXITI
Table 1. Storage Times and Conditions for Reconstituted and Diluted LUMOXITI Solution Reconstituted Solution Diluted LUMOXITI Solution in Infusion Bag After Dilution Administration LUMOXITI does not contain bacteriostatic preservatives.
Use reconstituted solution immediately.
DO NOT STORE reconstituted LUMOXITI vials.Use diluted solution immediately or after storage at room temperature (20°C to 25°C; 68°F to 77°F) for up to 4 hours or store refrigerated at 2°C to 8°C (36°F to 46°F) for up to 24 hours.
PROTECT FROM LIGHT.
DO NOT FREEZE.
DO NOT SHAKE.If the diluted solution is refrigerated (2°C to 8°C; 36°F to 46°F), allow it to equilibrate at room temperature (20°C to 25°C; 68°F to 77°F) for no more than 4 hours prior to administration.
Administer diluted solution within 24 hours of reconstitution as a 30-minute infusion.
PROTECT FROM LIGHT.Calculate the dose (mg) and the number of LUMOXITI vials (1 mg/vial) to be reconstituted. The final concentration of the reconstituted LUMOXITI solution is 1 mg/mL.
- Individualize dosing based on the patient's actual body weight prior to the first dose of the first treatment cycle.
- A change in dose should only be made between cycles when a change in weight of greater than 10% is observed from the weight used to calculate the first dose of the first treatment cycle. No change in dose should be made during a particular cycle.
- Do not round down the dose for partial vials.
Step 2: Gather Supplies
- LUMOXITI 1 mg/vial (number of vials to be reconstituted are based on Step 1)
- 1 vial of IV Solution Stabilizer (packaged separately)
- alcohol swabs
- 1 infusion bag containing 50 mL of 0.9% Sodium Chloride Injection, USP
- Sterile Water for Injection, USP
- syringes and needles
Step 3: Reconstitution
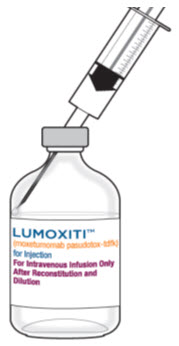
Reconstitute each LUMOXITI vial with 1.1 mL Sterile Water for Injection, USP using aseptic technique.

- Direct the Sterile Water for Injection, USP slowly along the walls of the LUMOXITI vial and not directly at the lyophilized cake or powder (see figure below).
- Do not reconstitute LUMOXITI vials with the IV Solution Stabilizer.
- Gently swirl the vial until completely dissolved. Invert the vial to ensure all cake or powder in the vial is dissolved. Do not shake.
Visually inspect that the reconstituted solution is clear to slightly opalescent, colorless to slightly yellow, and free from visible particles.
- Do not use if solution is cloudy, discolored, or contains any particles.
The resulting 1 mg/mL solution allows a withdrawal volume of 1 mL.
Use reconstituted solution immediately. Do not store reconstituted LUMOXITI vials. See Table 1 for storage times and conditions for the reconstituted solution.
Step 4: Preparation of Infusion Bag with IV Solution Stabilizer


Obtain a 50 mL 0.9% Sodium Chloride Injection, USP infusion bag.
Only one vial of IV Solution Stabilizer is needed per administration of LUMOXITI, regardless of the number of vials of LUMOXITI used to prepare the infusion.
- Add 1 mL IV Solution Stabilizer to the infusion bag containing 50 mL 0.9% Sodium Chloride Injection, USP.
- Gently invert the bag to mix the solution. Do not shake.
Step 5: Dilution
Slowly withdraw the required volume of reconstituted LUMOXITI solution needed from each vial, per the calculated dose based on the patient's actual body weight (kg).
- Inject LUMOXITI into the infusion bag containing 50 mL 0.9% Sodium Chloride Injection, USP and 1 mL IV Solution Stabilizer.
- Gently invert the bag to mix the solution. Do not shake.
- Discard any partially used or empty vials of LUMOXITI and IV Solution Stabilizer.
- See Table 1 for storage times and conditions for the diluted solution.
Step 6: Intravenous Hydration and Pre-infusion Medications
Administer intravenous hydration and premedication to the patient.
- Intravenously administer 1 L of isotonic solution (e.g. 5% Dextrose Injection, USP and 0.45% or 0.9% Sodium Chloride Injection, USP) over 2 to 4 hours before each LUMOXITI infusion. Administer 0.5 L to patients under 50 kg.
- Premedicate 30 to 90 minutes prior to each LUMOXITI infusion with an antihistamine (e.g. hydroxyzine or diphenhydramine), acetaminophen, and a histamine-2 receptor antagonist (e.g. ranitidine, famotidine, or cimetidine).
Step 7: Administration
Infuse the diluted LUMOXITI solution intravenously over 30 minutes.
- Do not mix LUMOXITI, or administer as an infusion with other medicinal products.
- After the infusion, flush the intravenous administration line with of 0.9% Sodium Chloride Injection, USP at the same rate as the infusion. This ensures that the full LUMOXITI dose is delivered.
Step 8: Post-infusion Medications
Administer post-infusion medications.
- Intravenously administer 1 L of isotonic solution (e.g. 5% Dextrose Injection, USP and 0.45% or 0.9% Sodium Chloride Injection, USP) over 2 to 4 hours after each LUMOXITI infusion. Administer 0.5 L to patients under 50 kg.
- Consider oral antihistamines and acetaminophen for up to 24 hours following LUMOXITI infusions.
- Consider oral corticosteroid (e.g. 4 mg dexamethasone) to manage nausea and vomiting.
Maintain adequate oral fluid intake.
- Advise all patients to adequately hydrate with up to 3 L (twelve 8-oz glasses) of oral fluids (e.g. water, milk, or juice) per 24 hours on Days 1 through 8 of each 28-day treatment cycle. In patients under 50 kg, up to 2 L (eight 8-oz glasses) per 24-hour period is recommended.
Consider low-dose aspirin on Days 1 through 8 of each 28-day treatment cycle.
This Healthcare Provider Instructions for Use has been approved by the U.S. Food and Drug Administration.
LUMOXITI is a trademark of Innate Pharma. © Innate Pharma 2020
Distributed by: Innate Pharma Inc., Rockville, MD 20850
Manufactured by: Innate Pharma Inc., Rockville, MD 20850; U.S. License No. 2206For more information, go to www.LUMOXITI.com or call 1-888-501-0998.
Revised: 4/2020 -
PRINCIPAL DISPLAY PANEL - 1 mg Vial Carton
NDC: 73380-4700-1
Rx onlyLUMOXITI™
(moxetumomab pasudotox-tdfk)
for Injection1 mg/vial
For Intravenous Infusion Only After Reconstitution and Dilution
ATTENTION PHARMACIST:
Use ONLY with IV Solution Stabilizer for LUMOXITI™;
packaged separately, to prepare the infusion
bag prior to adding reconstituted LUMOXITI™Keep vial in original carton to protect from light.
No preservatives.Dispense the accompanying
Medication Guide to each patient.One single-dose vial
Discard unused portioninnate pharma

-
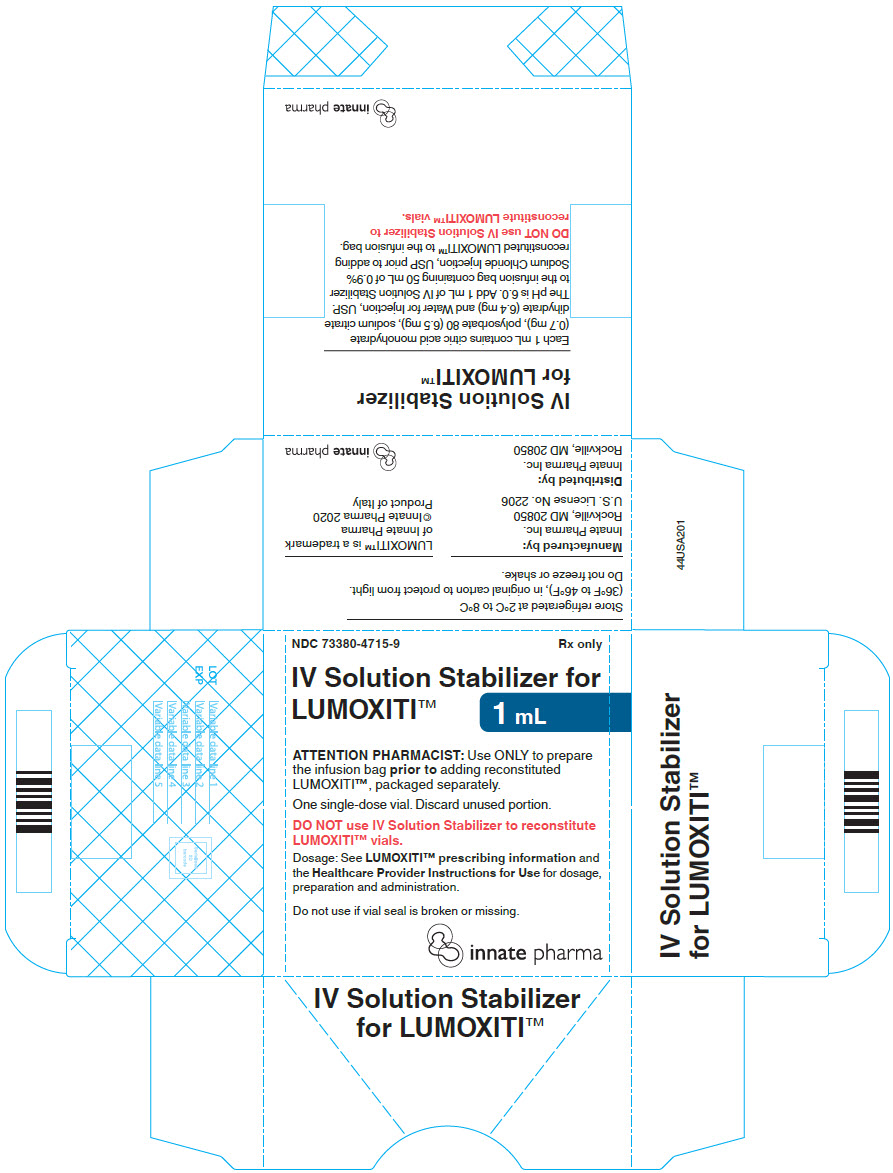
PRINCIPAL DISPLAY PANEL - 1 mL Vial Carton
NDC: 73380-4715-9
Rx onlyIV Solution Stabilizer for
LUMOXITI™1 mL
ATTENTION PHARMACIST: Use ONLY to prepare
the infusion bag prior to adding reconstituted
LUMOXITI™, packaged separately.One single-dose vial. Discard unused portion.
DO NOT use IV Solution Stabilizer to reconstitute
LUMOXITI™ vials.Dosage: See LUMOXITI™ prescribing information and
the Healthcare Provider Instructions for Use for dosage,
preparation and administration.Do not use if vial seal is broken or missing.
innate pharma
-
INGREDIENTS AND APPEARANCE
LUMOXITI
moxetumomab pasudotox injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 73380-4700 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MOXETUMOMAB PASUDOTOX (UNII: 2NDX4B6N8F) (MOXETUMOMAB PASUDOTOX - UNII:2NDX4B6N8F) MOXETUMOMAB PASUDOTOX 1 mg in 1 mL Inactive Ingredients Ingredient Name Strength GLYCINE (UNII: TE7660XO1C) 80 mg in 1 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.2 mg in 1 mL SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) 3.4 mg in 1 mL SUCROSE (UNII: C151H8M554) 40 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 73380-4700-1 1 in 1 CARTON 05/15/2020 1 1 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761104 05/15/2020 IV STABILIZER
polysorbate 80 solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 73380-4715 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength POLYSORBATE 80 (UNII: 6OZP39ZG8H) (POLYSORBATE 80 - UNII:6OZP39ZG8H) POLYSORBATE 80 6.5 mg in 1 mL Inactive Ingredients Ingredient Name Strength CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) 0.7 mg in 1 mL TRISODIUM CITRATE DIHYDRATE (UNII: B22547B95K) 6.4 mg in 1 mL WATER (UNII: 059QF0KO0R) 1 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 73380-4715-9 1 in 1 CARTON 05/15/2020 1 1 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761104 05/15/2020 Labeler - Innate Pharma, Inc. (117202909)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.