Mercaptopurine by Quinn Pharmaceuticals / Stason Pharmaceuticals, Inc. MERCAPTOPURINE tablet
Mercaptopurine by
Drug Labeling and Warnings
Mercaptopurine by is a Prescription medication manufactured, distributed, or labeled by Quinn Pharmaceuticals, Stason Pharmaceuticals, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
- CAUTION
-
DESCRIPTION
Mercaptopurine was synthesized and developed by Hitchings, Elion, and associates at the Wellcome Research Laboratories.
Mercaptopurine, known chemically as 6 H-purine-6-thione, 1,7-dihydro-, monohydrate, is an analogue of the purine bases adenine and hypoxanthine. Its structural formula is:
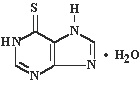
Mercaptopurine is available in tablet form for oral administration. Each scored tablet contains 50 mg mercaptopurine and the inactive ingredients corn and potato starch, lactose, magnesium stearate, and stearic acid.
Each tablet meets the requirements of Test 2 for Dissolution in the USP monograph for Mercaptopurine Tablets, USP.
-
CLINICAL PHARMACOLOGY
Mechanism of Action
Mercaptopurine (6-MP) competes with hypoxanthine and guanine for the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRTase) and is itself converted to thioinosinic acid (TIMP). This intracellular nucleotide inhibits several reactions involving inosinic acid (IMP), including the conversion of IMP to xanthylic acid (XMP) and the conversion of IMP to adenylic acid (AMP) via adenylosuccinate (SAMP). In addition, 6-methylthioinosinate (MTIMP) is formed by the methylation of TIMP. Both TIMP and MTIMP have been reported to inhibit glutamine-5-phosphoribosylpyrophosphate amidotransferase, the first enzyme unique to the de novo pathway for purine ribonucleotide synthesis. Experiments indicate that radiolabeled mercaptopurine may be recovered from the DNA in the form of deoxythioguanosine. Some mercaptopurine is converted to nucleotide derivatives of 6-thioguanine (6-TG) by the sequential actions of inosinate (IMP) dehydrogenase and xanthylate (XMP) aminase, converting TIMP to thioguanylic acid (TGMP).
Animal tumors that are resistant to mercaptopurine often have lost the ability to convert mercaptopurine to TIMP. However, it is clear that resistance to mercaptopurine may be acquired by other means as well, particularly in human leukemias.
It is not known exactly which of any one or more of the biochemical effects of mercaptopurine and its metabolites are directly or predominantly responsible for cell death.
Pharmacokinetics
Clinical studies have shown that the absorption of an oral dose of mercaptopurine in humans is incomplete and variable, averaging approximately 50% of the administered dose. The factors influencing absorption are unknown. Intravenous administration of an investigational preparation of mercaptopurine revealed a plasma half-disappearance time of 21 minutes in pediatric patients and 47 minutes in adults. The volume of distribution usually exceeded that of the total body water.
Following the oral administration of 35S-6-mercaptopurine in one subject, a total of 46% of the dose could be accounted for in the urine (as parent drug and metabolites) in the first 24 hours. There is negligible entry of mercaptopurine into cerebrospinal fluid.
Plasma protein binding averages 19% over the concentration range 10 to 50 mcg/mL (a concentration only achieved by intravenous administration of mercaptopurine at doses exceeding 5 to 10 mg/kg).
A reduction in mercaptopurine dosage is required if patients are receiving both mercaptopurine and allopurinol (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
Metabolism and Genetic Polymorphism
Several published studies indicate that patients with reduced TPMT or NUDT15 activity receiving usual doses of mercaptopurine, accumulate excessive cellular concentrations of active 6-TGNs, and are at higher risk for severe myelosuppression. In a study of 1028 children with ALL, the approximate tolerated mercaptopurine dosage range for patients with TPMT and/or NUDT15 deficiency on mercaptopurine maintenance therapy (as a percentage of the planned dosage) was as follows: heterozygous for either TPMT or NUDT15, 50-90%; heterozygous for both TPMT and NUDT15, 30-50%; homozygous for either TPMT or NUDT15, 5-10%.
Approximately 0.3% (1:300) of patients of European or African ancestry have two loss-of-function alleles of the TPMT gene and have little or no TPMT activity (homozygous deficient or poor metabolizers), and approximately 10% of patients have one loss-of-function TPMT allele leading to intermediate TPMT activity (heterozygous deficient or intermediate metabolizers). The TPMT*2, TPMT*3A, and TPMT*3C alleles account for about 95% of individuals with reduced levels of TPMT activity. NUDT15 deficiency is detected in <1% of patients of European or African ancestry. Among patients of East Asian ancestry (i.e., Chinese, Japanese, Vietnamese), 2% have two loss-of-function alleles of the NUDT15 gene, and approximately 21% have one loss-of-function allele. The p.R139C variant of NUDT15 (present on the *2 and *3 alleles) is the most commonly observed, but other less common loss-of-function NUDT15 alleles have been observed.
Consider all clinical information when interpreting results from phenotypic testing used to determine the level of thiopurine nucleotides or TPMT activity in erythrocytes, since some coadministered drugs can influence measurement of TPMT activity in blood, and blood from recent transfusions will misrepresent a patient’s actual TPMT activity. -
INDICATIONS AND USAGE
Mercaptopurine is indicated for maintenance therapy of acute lymphatic (lymphocytic, lymphoblastic) leukemia as part of a combination regimen. The response to this agent depends upon the particular subclassification of acute lymphatic leukemia and the age of the patient (pediatric or adult).
Mercaptopurine is not effective for prophylaxis or treatment of central nervous system leukemia.
Mercaptopurine is not effective in acute myelogenous leukemia, chronic lymphatic leukemia, the lymphomas (including Hodgkins Disease), or solid tumors.
-
CONTRAINDICATIONS
Mercaptopurine should not be used in patients whose disease has demonstrated prior resistance to this drug. In animals and humans, there is usually complete cross-resistance between mercaptopurine and thioguanine.
Mercaptopurine should not be used in patients who have a hypersensitivity to mercaptopurine or any component of the formulation.
-
WARNINGS
Mercaptopurine is mutagenic in animals and humans, carcinogenic in animals, and may increase the patient's risk of neoplasia. Cases of hepatosplenic T-cell lymphoma have been reported in patients treated with mercaptopurine for inflammatory bowel disease. The safety and efficacy of mercaptopurine in patients with inflammatory bowel disease have not been established.
Bone Marrow Toxicity
The most consistent, dose-related toxicity is bone marrow suppression. This may be manifest by anemia, leukopenia, thrombocytopenia, or any combination of these. Any of these findings may also reflect progression of the underlying disease. In many patients with severe depression of the formed elements of the blood due to mercaptopurine, the bone marrow appears hypoplastic on aspiration or biopsy, whereas in other cases it may appear normocellular. The qualitative changes in the erythroid elements toward the megaloblastic series, characteristically seen with the folic acid antagonists and some other antimetabolites, are not seen with this drug. Life-threatening infections and bleeding have been observed as a consequence of mercaptopurine-induced granulocytopenia and thrombocytopenia. Since mercaptopurine may have a delayed effect, it is important to withdraw the medication temporarily at the first sign of an unexpected abnormally large fall in any of the formed elements of the blood, if not attributable to another drug or disease process.
Evaluate patients with repeated severe myelosuppression for thiopurine S-methyltransferase (TPMT) or nucleotide diphosphatase (NUDT15) deficiency. TPMT genotyping or phenotyping (red blood cell TPMT activity) and NUDT15 genotyping can identify patients who have reduced activity of these enzymes. Patients with homozygous TPMT or NUDT15 deficiency require substantial dosage reductions.
Bone marrow toxicity may be more profound in patients treated with concomitant allopurinol (see PRECAUTIONS: Drug Interactions and DOSAGE AND ADMINISTRATION). This problem could be exacerbated by coadministration with drugs that inhibit TPMT, such as olsalazine, mesalazine, or sulphasalazine
Hepatotoxicity
Mercaptopurine is hepatotoxic in animals and humans. A small number of deaths have been reported that may have been attributed to hepatic necrosis due to administration of mercaptopurine. Hepatic injury can occur with any dosage, but seems to occur with more frequency when doses of 2.5 mg/kg/day are exceeded. The histologic pattern of mercaptopurine hepatotoxicity includes features of both intrahepatic cholestasis and parenchymal cell necrosis, either of which may predominate. It is not clear how much of the hepatic damage is due to direct toxicity from the drug and how much may be due to a hypersensitivity reaction. In some patients jaundice has cleared following withdrawal of mercaptopurine and reappeared with its reintroduction.
Published reports have cited widely varying incidences of overt hepatotoxicity. In a large series of patients with various neoplastic diseases, mercaptopurine was administered orally in doses ranging from 2.5 mg/kg to 5.0 mg/kg without evidence of hepatotoxicity. It was noted by the authors that no definite clinical evidence of liver damage could be ascribed to the drug, although an occasional case of serum hepatitis did occur in patients receiving 6-MP who previously had transfusions. In reports of smaller cohorts of adult and pediatric leukemic patients, the incidence of hepatotoxicity ranged from 0% to 6%. In an isolated report by Einhorn and Davidsohn, jaundice was observed more frequently (40%), especially when doses exceeded 2.5 mg/kg. Usually, clinically detectable jaundice appears early in the course of treatment (1 to 2 months). However, jaundice has been reported as early as 1 week and as late as 8 years after the start of treatment with mercaptopurine. The hepatotoxicity has been associated in some cases with anorexia, diarrhea, jaundice and ascites. Hepatic encephalopathy has occurred.
Monitoring of serum transaminase levels, alkaline phosphatase, and bilirubin levels may allow early detection of hepatotoxicity. It is advisable to monitor these liver function tests at weekly intervals when first beginning therapy and at monthly intervals thereafter. Liver function tests may be advisable more frequently in patients who are receiving mercaptopurine with other hepatotoxic drugs or with known pre-existing liver disease. The onset of clinical jaundice, hepatomegaly, or anorexia with tenderness in the right hypochondrium are immediate indications for withholding mercaptopurine until the exact etiology can be identified. Likewise, any evidence of deterioration in liver function studies, toxic hepatitis, or biliary stasis should prompt discontinuation of the drug and a search for an etiology of the hepatotoxicity.
The concomitant administration of mercaptopurine with other hepatotoxic agents requires especially careful clinical and biochemical monitoring of hepatic function. Combination therapy involving mercaptopurine with other drugs not felt to be hepatotoxic should nevertheless be approached with caution. The combination of mercaptopurine with doxorubicin was reported to be hepatotoxic in 19 of 20 patients undergoing remission-induction therapy for leukemia resistant to previous therapy.
Immunosuppression
Mercaptopurine recipients may manifest decreased cellular hypersensitivities and decreased allograft rejection. Induction of immunity to infectious agents or vaccines will be subnormal in these patients; the degree of immunosuppression will depend on antigen dose and temporal relationship to drug. This immunosuppressive effect should be carefully considered with regard to intercurrent infections and risk of subsequent neoplasia.
Pregnancy
Pregnancy Category D
Mercaptopurine can cause fetal harm when administered to a pregnant woman. Women receiving mercaptopurine in the first trimester of pregnancy have an increased incidence of abortion; the risk of malformation in offspring surviving first trimester exposure is not accurately known. In a series of 28 women receiving mercaptopurine after the first trimester of pregnancy, 3 mothers died undelivered, 1 delivered a stillborn child, and 1 aborted; there were no cases of macroscopically abnormal fetuses. Since such experience cannot exclude the possibility of fetal damage, mercaptopurine should be used during pregnancy only if the benefit clearly justifies the possible risk to the fetus, and particular caution should be given to the use of mercaptopurine in the first trimester of pregnancy.
There are no adequate and well-controlled studies in pregnant women. If this drug is used during pregnancy or if the patient becomes pregnant while taking the drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant.
-
PRECAUTIONS
General
The safe and effective use of mercaptopurine demands close monitoring of the CBC and patient clinical status. After selection of an initial dosage schedule, therapy will frequently need to be modified depending upon the patient’s response and manifestations of toxicity. It is probably advisable to start with lower dosages in patients with impaired renal function, due to slower elimination of the drug and metabolites and a greater cumulative effect.
Information for Patients
Patients should be informed that the major toxicities of mercaptopurine are related to myelosuppression, hepatotoxicity, and gastrointestinal toxicity. Patients should never be allowed to take the drug without medical supervision and should be advised to consult their physician if they experience fever, sore throat, jaundice, nausea, vomiting, signs of local infection, bleeding from any site, or symptoms suggestive of anemia. Women of childbearing potential should be advised to avoid becoming pregnant.
Laboratory Tests
(Also see WARNINGS: Bone Marrow Toxicity.) It is recommended that evaluation of the hemoglobin or hematocrit, total white blood cell count and differential count, and quantitative platelet count be obtained weekly while the patient is on therapy with mercaptopurine. Bone marrow examination may also be useful for the evaluation of marrow status. The decision to increase, decrease, continue, or discontinue a given dosage of mercaptopurine must be based upon the degree of severity and rapidity with which changes are occurring. In many instances, particularly during the induction phase of acute leukemia, complete blood counts will need to be done more frequently than once weekly in order to evaluate the effect of the therapy. Consider testing for TPMT and NUDT15 deficiency in patients who experience severe bone marrow toxicities or repeated episodes of myelosuppression.
Drug Interactions
When allopurinol and mercaptopurine are administered concomitantly, the dose of mercaptopurine must be reduced to one third to one quarter of the usual dose to avoid severe toxicity.
There is usually complete cross-resistance between mercaptopurine and thioguanine.
The dosage of mercaptopurine may need to be reduced when this agent is combined with other drugs whose primary or secondary toxicity is myelosuppression. Enhanced marrow suppression has been noted in some patients also receiving trimethoprim-sulfamethoxazole.
Inhibition of the anticoagulant effect of warfarin, when given with mercaptopurine, has been reported.
As there is in vitro evidence that aminosalicylate derivatives (e.g., olsalazine, mesalazine, or sulphasalazine) inhibit the TPMT enzyme, they should be administered with caution to patients receiving concurrent mercaptopurine therapy (see WARNINGS).
Carcinogenesis, Mutagenesis, Impairment of Fertility
Mercaptopurine causes chromosomal aberrations in animals and humans and induces dominant-lethal mutations in male mice. In mice, surviving female offspring of mothers who received chronic low doses of mercaptopurine during pregnancy were found sterile, or if they became pregnant, had smaller litters and more dead fetuses as compared to control animals. Carcinogenic potential exists in humans, but the extent of the risk is unknown.
The effect of mercaptopurine on human fertility is unknown for either males or females.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, and because of the potential for serious adverse reactions in nursing infants from mercaptopurine, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Geriatric Use
Clinical studies of mercaptopurine did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
-
ADVERSE REACTIONS
The principal and potentially serious toxic effects of mercaptopurine are bone marrow toxicity and hepatotoxicity (see WARNINGS and PRECAUTIONS).
Hematologic
The most frequent adverse reaction to mercaptopurine is myelosuppression. The induction of complete remission of acute lymphatic leukemia frequently is associated with marrow hypoplasia. Patients without TPMT enzyme activity (homozygous-deficient) are particularly susceptible to hematologic toxicity, and some patients with low or intermediate TPMT enzyme activity are more susceptible to hematologic toxicity than patients with normal TPMT activity (see WARNINGS: Bone Marrow Toxicity), although the latter can also experience severe toxicity. Maintenance of remission generally involves multiple-drug regimens whose component agents cause myelosuppression. Anemia, leukopenia, and thrombocytopenia are frequently observed. Dosages and also schedules are adjusted to prevent life-threatening cytopenias.
Renal
Hyperuricemia and/or hyperuricosuria may occur in patients receiving mercaptopurine as a consequence of rapid cell lysis accompanying the antineoplastic effect. Renal adverse effects can be minimized by increased hydration, urine alkalinization, and the prophylactic administration of a xanthine oxidase inhibitor such as allopurinol. The dosage of mercaptopurine should be reduced to one third to one quarter of the usual dose if allopurinol is given concurrently.
Gastrointestinal
Intestinal ulceration has been reported. Nausea, vomiting, and anorexia are uncommon during initial administration, but may increase with continued administration. Mild diarrhea and sprue-like symptoms have been noted occasionally, but it is difficult at present to attribute these to the medication. Oral lesions are rarely seen, and when they occur they resemble thrush rather than antifolic ulcerations.
Miscellaneous
The administration of mercaptopurine has been associated with skin rashes and hyperpigmentation. Alopecia has been reported.
Drug fever has been very rarely reported with mercaptopurine. Before attributing fever to mercaptopurine, every attempt should be made to exclude more common causes of pyrexia, such as sepsis, in patients with acute leukemia.
Oligospermia has been reported.
-
OVERDOSAGE
Signs and symptoms of overdosage may be immediate (anorexia, nausea, vomiting, and diarrhea); or delayed (myelosuppression, liver dysfunction, and gastroenteritis). Dialysis cannot be expected to clear mercaptopurine. Hemodialysis is thought to be of marginal use due to the rapid intracellular incorporation of mercaptopurine into active metabolites with long persistence. The oral LD 50 of mercaptopurine was determined to be 480 mg/kg in the mouse and 425 mg/kg in the rat.
There is no known pharmacologic antagonist of mercaptopurine. The drug should be discontinued immediately if unintended toxicity occurs during treatment. If a patient is seen immediately following an accidental overdosage of the drug, it may be useful to induce emesis.
-
DOSAGE AND ADMINISTRATION
Maintenance Therapy
Once a complete hematologic remission is obtained, maintenance therapy is considered essential. Maintenance doses will vary from patient to patient. The usual daily maintenance dose of mercaptopurine is 1.5 to 2.5 mg/kg/day as a single dose. It is to be emphasized that in pediatric patients with acute lymphatic leukemia in remission, superior results have been obtained when mercaptopurine has been combined with other agents (most frequently with methotrexate) for remission maintenance. Mercaptopurine should rarely be relied upon as a single agent for the maintenance of remissions induced in acute leukemia.
Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published. 1-8 There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.
Dosage with Concomitant Allopurinol
When allopurinol and mercaptopurine are administered concomitantly, the dose of mercaptopurine must be reduced to one-third to one-quarter of the usual dose to avoid severe toxicity.
Dosage in patients with TPMT and/or NUDT15 Deficiency
Homozygous deficiency in either TPMT or NUDT15
Patients with homozygous deficiency of either enzyme typically require 10% or less of the standard PURIXAN dosage. Reduce initial dosage in patients who are known to have homozygous TPMT or NUDT15 deficiency.
Heterozygous deficiency in TPMT and/or NUDT15Reduce the PURIXAN dosage based on tolerability. Most patients with heterozygous TPMT or NUDT15 deficiency tolerate recommended mercaptopurine doses, but some require dose reduction based on toxicities. Patients who are heterozygous for both TPMT and NUDT15 may require more substantial dosage reductions.
Dosage in Renal and Hepatic Impairment
It is probably advisable to start with lower dosages in patients with impaired renal function, due to slower elimination of the drug and metabolites and a greater cumulative effect. Consideration should be given to reducing the dosage in patients with impaired hepatic function.
-
HOW SUPPLIED
Pale yellow to buff, scored tablets containing 50 mg mercaptopurine, imprinted with “9|3”; bottles of 25 (NDC: 69076-913-02) and bottles of 250 (NDC: 69076-913-25).
Store at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature]. Store in a dry place. Dispense in tight container as defined in the USP.
-
REFERENCES
1. ONS Clinical Practice Committee. Cancer Chemotherapy Guidelines and Recommendations for Practice. Pittsburgh, PA: Oncology Nursing Society;1999:32-41.
2. Recommendations for the safe handling of parenteral antineoplastic drugs. Washington, DC: Division of Safety; Clinical Center Pharmacy Department and Cancer Nursing Services, National Institutes of Health; 1992. US Dept of Health and Human Services. Public Health Service publication NIH 92-2621.
3. AMA Council on Scientific Affairs. Guidelines for handling parenteral antineoplastics. JAMA. 1985;253:1590-1591.
4. National Study Commission on Cytotoxic Exposure. Recommendations for handling cytotoxic agents. 1987. Available from Louis P. Jeffrey, Chairman, National Study Commission on Cytotoxic Exposure. Massachusetts College of Pharmacy and Allied Health Sciences, 179 Longwood Avenue, Boston, MA 02115.
5. Clinical Oncological Society of Australia. Guidelines and recommendations for safe handling of antineoplastic agents. Med J Australia. 1983;1:426-428.
6. Jones RB, Frank R, Mass T. Safe handling of chemotherapeutic agents: a report from the Mount Sinai Medical Center. CA-A Cancer J for Clinicians. 1983;33:258-263.
7. American Society of Hospital Pharmacists. ASHP technical assistance bulletin on handling cytotoxic and hazardous drugs. Am J Hosp Pharm. 1990;47:1033-1049.
8. Controlling Occupational Exposure to Hazardous Drugs. (OSHA Work-Practice Guidelines.) Am J Health-Syst Pharm. 1996;53:1669-1685.
Manufactured for:
Quinn Pharmaceuticals, LLC
Coral Springs, FL
www.quinnrx.com
PH: 844-477-8466Rev. 05/2018
Made in U.S.A.
-
PRINCIPAL DISPLAY PANEL
NDC: 69076-913-02
Mecaptopurine
Tablets, USP
50 mg
Each tablet contains 50 mg
mercaptopurine.
Cytotoxic Agent
Carefully read warning and
accompanying package
insert for dosage
information.Quinn Pharmaceuticals Rx only
25 Tablets

NDC: 69076-913-25
250 Tablets

-
INGREDIENTS AND APPEARANCE
MERCAPTOPURINE
mercaptopurine tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 69076-913 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MERCAPTOPURINE (UNII: E7WED276I5) (MERCAPTOPURINE ANHYDROUS - UNII:PKK6MUZ20G) MERCAPTOPURINE 50 mg Inactive Ingredients Ingredient Name Strength STARCH, CORN (UNII: O8232NY3SJ) STARCH, POTATO (UNII: 8I089SAH3T) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) STEARIC ACID (UNII: 4ELV7Z65AP) Product Characteristics Color yellow (Pale yellow to buff) Score 2 pieces Shape ROUND Size 9mm Flavor Imprint Code 9;3 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 69076-913-25 250 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 10/05/2016 2 NDC: 69076-913-02 25 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 10/05/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA authorized generic NDA009053 10/05/2016 Labeler - Quinn Pharmaceuticals, LLC (079421782) Registrant - Stason Pharmaceuticals, Inc. (807437553) Establishment Name Address ID/FEI Business Operations Stason Pharmaceuticals, Inc. 807437553 manufacture(69076-913)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.