RAVICTI- glycerol phenylbutyrate liquid
Ravicti by
Drug Labeling and Warnings
Ravicti by is a Prescription medication manufactured, distributed, or labeled by Horizon Therapeutics USA, Inc., HAS Healthcare Advanced Synthesis SA. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use RAVICTI safely and effectively. See full prescribing information for RAVICTI.
RAVICTI® (glycerol phenylbutyrate) oral liquid
Initial U.S. Approval: 1996RECENT MAJOR CHANGES
INDICATIONS AND USAGE
RAVICTI is a nitrogen-binding agent indicated for chronic management of patients with urea cycle disorders (UCDs) who cannot be managed by dietary protein restriction and/or amino acid supplementation alone. RAVICTI must be used with dietary protein restriction and, in some cases, dietary supplements. (1)
Limitations of Use:
DOSAGE AND ADMINISTRATION
- RAVICTI should be prescribed by a physician experienced in management of UCDs. For administration and preparation, see full prescribing information. (2.1, 2.6)
Switching From Sodium Phenylbutyrate Tablets or Powder to RAVICTI:
- Patients should receive the dosage of RAVICTI that contains the same amount of phenylbutyric acid, see full prescribing information for conversion. (2.2)
Initial Dosage in Phenylbutyrate-Naïve Patients (2.3):
- Recommended dosage range is 4.5 to 11.2 mL/m2/day (5 to 12.4 g/m2/day).
- For patients with some residual enzyme activity not adequately controlled with dietary restriction, the recommended starting dose is 4.5 mL/m2/day.
- Take into account patient's estimated urea synthetic capacity, dietary protein intake, and diet adherence.
Dosage Adjustment and Monitoring:
- Follow plasma ammonia levels to determine the need for dosage titration. (2.4)
Dosage Modifications in Patients with Hepatic Impairment:
DOSAGE FORMS AND STRENGTHS
Oral liquid: 1.1 g/mL. (3)
CONTRAINDICATIONS
Known hypersensitivity to phenylbutyrate. (4)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
Most common adverse reactions (≥10%) in adults are: diarrhea, flatulence, and headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Horizon at 1-866-479-6742 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Corticosteroids, valproic acid, or haloperidol: May increase plasma ammonia level; monitor ammonia levels closely. (7.1)
- Probenecid: May affect renal excretion of metabolites of RAVICTI, including phenylacetylglutamine (PAGN) and PAA. (7.2)
- CYP3A4 Substrates with narrow therapeutic index (e.g., alfentanil, quinidine, cyclosporine): RAVICTI may decrease exposure; monitor for decreased efficacy of the narrow therapeutic index drug. (7.3)
- Midazolam: Decreased exposure; monitor for suboptimal effect of midazolam. (7.3)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 11/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
2.2 Switching From Sodium Phenylbutyrate to RAVICTI
2.3 Initial Dosage in Phenylbutyrate-Naïve Patients
2.4 Dosage Adjustment and Monitoring
2.5 Dosage Modifications in Patients with Hepatic Impairment
2.6 Preparation for Nasogastric Tube or Gastrostomy Tube Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Neurotoxicity
5.2 Pancreatic Insufficiency or Intestinal Malabsorption
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Potential for Other Drugs to Affect Ammonia
7.2 Potential for Other Drugs to Affect RAVICTI
7.3 Potential for RAVICTI to Affect Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Clinical Studies in Adult Patients with UCDs
14.2 Clinical Studies in Pediatric Patients 2 Years to 17 Years of Age with UCDs
14.3 Clinical Studies in Pediatric Patients Less Than 2 Years of Age with UCDs
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
RAVICTI is indicated for use as a nitrogen-binding agent for chronic management of patients with urea cycle disorders (UCDs) who cannot be managed by dietary protein restriction and/or amino acid supplementation alone. RAVICTI must be used with dietary protein restriction and, in some cases, dietary supplements (e.g., essential amino acids, arginine, citrulline, protein-free calorie supplements).
Limitations of Use:
- RAVICTI is not indicated for the treatment of acute hyperammonemia in patients with UCDs because more rapidly acting interventions are essential to reduce plasma ammonia levels.
- The safety and efficacy of RAVICTI for the treatment of N-acetylglutamate synthase (NAGS) deficiency has not been established.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
RAVICTI should be prescribed by a physician experienced in the management of UCDs.
- Instruct patients to take RAVICTI with food or formula and to administer directly into the mouth via oral syringe or dosing cup.
- Instruct that RAVICTI should be administered just prior to breastfeeding in infants who are breastfeeding.
- For patients who cannot swallow, see the instructions on administration of RAVICTI by nasogastric tube or gastrostomy tube [see Dosage and Administration (2.6)].
- For patients who require a volume of less than 1 mL per dose via nasogastric or gastrostomy tube, the delivered dose may be less than anticipated. Closely monitor these patients using ammonia levels [see Dosage and Administration (2.6)].
- The recommended dosages for patients switching from sodium phenylbutyrate to RAVICTI and patients naïve to phenylbutyric acid are different [see Dosage and Administration (2.2, 2.3)]. For both subpopulations:
- Patients 2 years of age and older: Give RAVICTI in 3 equally divided dosages, each rounded up to the nearest 0.5 mL
- Patients less than 2 years: Give RAVICTI in 3 or more equally divided dosages, each rounded up to the nearest 0.1 mL.
- The maximum total daily dosage is 17.5 mL (19 g).
- RAVICTI must be used with dietary protein restriction and, in some cases, dietary supplements (e.g., essential amino acids, arginine, citrulline, protein-free calorie supplements).
2.2 Switching From Sodium Phenylbutyrate to RAVICTI
Patients switching from sodium phenylbutyrate to RAVICTI should receive the dosage of RAVICTI that contains the same amount of phenylbutyric acid. The conversion is as follows:
Total daily dosage of RAVICTI (mL) = total daily dosage of sodium phenylbutyrate tablets (g) × 0.86
Total daily dosage of RAVICTI (mL) = total daily dosage of sodium phenylbutyrate powder (g) × 0.81
2.3 Initial Dosage in Phenylbutyrate-Naïve Patients
The recommended dosage range, based upon body surface area, in patients naïve to phenylbutyrate (PBA) is 4.5 to 11.2 mL/m2/day (5 to 12.4 g/m2/day). For patients with some residual enzyme activity who are not adequately controlled with protein restriction, the recommended starting dosage is 4.5 mL/m2/day.
In determining the starting dosage of RAVICTI in treatment-naïve patients, consider the patient's residual urea synthetic capacity, dietary protein requirements, and diet adherence. Dietary protein is approximately 16% nitrogen by weight. Given that approximately 47% of dietary nitrogen is excreted as waste and approximately 70% of an administered PBA dose will be converted to urinary phenylacetylglutamine (U-PAGN), an initial estimated RAVICTI dose for a 24-hour period is 0.6 mL RAVICTI per gram of dietary protein ingested per 24-hour period. The total daily dosage should not exceed 17.5 mL.
2.4 Dosage Adjustment and Monitoring
During treatment with RAVICTI, patients should be followed clinically and with plasma ammonia levels to determine the need for dosage titration. Closely monitor plasma ammonia levels during treatment with RAVICTI and when changing the dosage of RAVICTI.
The methods used for measuring plasma ammonia levels vary among individual laboratories and values obtained using different assay methods may not be interchangeable. Normal ranges and therapeutic target levels for plasma ammonia depend upon the assay method used by the individual laboratory. During treatment with RAVICTI, refer to the assay-specific normal ranges and to the therapeutic target ranges for plasma ammonia.
Normal Plasma Ammonia
In patients treated with RAVICTI who experience neurologic symptoms (e.g. nausea, vomiting, headache, somnolence or confusion) in the absence of high plasma ammonia or other intercurrent illness to explain these symptoms, consider reducing the RAVICTI dosage and clinically monitor patients for potential neurotoxicity from high phenylacetate (PAA) concentrations. If available, obtain measurements of plasma PAA concentrations and plasma phenylacetylglutamine (PAGN) to calculate the ratio of plasma PAA to PAGN which may help to guide RAVICTI dosing. The PAA to PAGN ratio has generally been less than 1 in patients with UCDs who did not have significant plasma PAA accumulation. In general, a high PAA to PAGN ratio may indicate a slower or less efficient conjugation reaction to form PAGN, which may lead to increases in PAA without further conversion to PAGN [see Warnings and Precautions (5.1), Clinical Pharmacology (12.3)].
Elevated Plasma Ammonia
In patients 6 years and older, when plasma ammonia is elevated, increase the RAVICTI dosage to maintain fasting plasma ammonia to less than half the upper limit of normal (ULN). In infants and pediatric patients below 6 years of age, if obtaining fasting ammonia is problematic due to frequent feedings, adjust the RAVICTI dosage to keep the first ammonia of the morning below the ULN for age. If available, the ratio of PAA to PAGN in the same plasma sample may provide additional information to assist in dosage adjustment decisions [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
Dietary Protein Intake
If available, urinary phenylacetylglutamine (U-PAGN) measurements may be used to help guide RAVICTI dosage adjustment. Each gram of U-PAGN excreted over 24 hours covers waste nitrogen generated from 1.4 grams of dietary protein. If U-PAGN excretion is insufficient to cover daily dietary protein intake and the fasting ammonia is greater than half the ULN, the RAVICTI dosage should be increased. The amount of dosage adjustment should factor in the amount of dietary protein that has not been covered, as indicated by the 24-hour U-PAGN output, and the estimated RAVICTI dose needed per gram of dietary protein ingested and the maximum total daily dosage (i.e., 17.5 mL).
Consider a patient's use of concomitant medications, such as probenecid, when making dosage adjustment decisions based on U-PAGN. Probenecid may result in a decrease of the urinary excretion of PAGN [see Drug Interactions (7.2)].
2.5 Dosage Modifications in Patients with Hepatic Impairment
For patients with moderate to severe hepatic impairment, the recommended starting dosage is at the lower end of the recommended dosing range (4.5 mL/m2/day) and the dosage should be kept at the lowest necessary to control the patient's plasma ammonia [see Use in Specific Populations (8.7)].
2.6 Preparation for Nasogastric Tube or Gastrostomy Tube Administration
It is recommended that all patients who can swallow take RAVICTI orally, even those with nasogastric and/or gastrostomy tubes. However, for patients who cannot swallow, a nasogastric tube or gastrostomy tube may be used to administer RAVICTI as follows:
- Utilize an oral syringe to withdraw the prescribed dosage of RAVICTI from the bottle.
- Place the tip of the syringe into the nasogastric/gastrostomy tube.
- Utilizing the plunger of the syringe, administer RAVICTI into the tube.
- Flush once with 10 mL of water or formula and allow the flush to drain.
- If needed, flush a second time with an additional 10 mL of water or formula to clear the tube.
For patients who require a volume of less than 1 mL per dose via nasogastric or gastrostomy tube, the delivered dosage may be less than anticipated due to adherence of RAVICTI to the plastic tubing. Therefore, these patients should be closely monitored using ammonia levels following initiation of RAVICTI dosing or dosage adjustments.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Neurotoxicity
Increased exposure to PAA, the major metabolite of RAVICTI, may be associated with neurotoxicity in patients with UCDs. In a study of adult cancer patients, subjects received sodium phenylacetate administered as a 1-hour infusion twice daily at two dose levels of 125 and 150 mg/kg for a 2-week period. Of 18 subjects enrolled, 7 had a history of primary central nervous system tumor. Signs and symptoms of potential PAA neurotoxicity, which were reversible, were reported at plasma PAA concentrations above 500 micrograms/mL and included somnolence, fatigue, lightheadedness, headache, dysgeusia, hypoacusis, disorientation, impaired memory, and exacerbation of preexisting neuropathy. PAA concentrations were not measured when symptoms resolved.
In healthy subjects, after administration of 4 mL and 6 mL RAVICTI 3 times daily (13.2 g/day and 19.8 g/day, respectively) for 3 days, a dose-dependent increase in non-serious nervous system adverse reactions were observed. In subjects who had nervous system adverse reactions, plasma PAA concentrations, which were measured on Day 3 per protocol and not always at onset of symptoms, ranged from 8 to 56 micrograms/mL with 4 mL RAVICTI 3 times daily and from 31 to 242 micrograms/mL with 6 mL RAVICTI 3 times daily.
In clinical trials in patients with UCDs who had been on sodium phenylbutyrate prior to administration of RAVICTI, adverse reactions of headache, fatigue, symptoms of peripheral neuropathy, seizures, tremor and/or dizziness were reported. No correlation between plasma PAA concentration and neurologic symptoms was identified but plasma PAA concentrations were generally not consistently measured at the time of neurologic symptom occurrence [see Clinical Pharmacology (12.3)].
If symptoms of vomiting, nausea, headache, somnolence or confusion are present in the absence of high ammonia or other intercurrent illness which explains these symptoms, consider the potential for PAA neurotoxicity which may need reduction in the RAVICTI dosage [see Dosage and Administration (2.4)].
5.2 Pancreatic Insufficiency or Intestinal Malabsorption
Exocrine pancreatic enzymes hydrolyze RAVICTI in the small intestine, separating the active moiety, phenylbutyrate, from glycerol. This process allows phenylbutyrate to be absorbed into the circulation. Low or absent pancreatic enzymes or intestinal disease resulting in fat malabsorption may result in reduced or absent digestion of RAVICTI and/or absorption of phenylbutyrate and reduced control of plasma ammonia. Monitor ammonia levels closely in patients with pancreatic insufficiency or intestinal malabsorption.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Assessment of adverse reactions was based on exposure of 45 adult patients (31 female and 14 male) with UCD subtype deficiencies of ornithine transcarbamylase (OTC, n=40), carbamoyl phosphate synthetase (CPS, n=2), and argininosuccinate synthetase (ASS, n=1) in a randomized, double-blind, active-controlled (RAVICTI vs sodium phenylbutyrate), crossover, 4-week study (Study 1) that enrolled patients 18 years of age and older [see Clinical Studies (14.1)]. One of the 45 patients received only sodium phenylbutyrate prior to withdrawing on day 1 of the study due to an adverse reaction.
The most common adverse reactions (occurring in at least 10% of patients) reported during short-term treatment with RAVICTI were diarrhea, flatulence, and headache. Table 1 summarizes adverse reactions occurring in 2 or more patients treated with RAVICTI or sodium phenylbutyrate (incidence of at least 4% in either treatment arm).
Table 1: Adverse Reactions Reported in 2 or More Adult Patients with UCDs (at least 4% in Either Treatment Arm) in Study 1 Number (%) of Patients in Study 1 Sodium Phenylbutyrate
(N = 45)RAVICTI
(N = 44)Diarrhea 3 (7) 7 (16) Headache 4 (9) 6 (14) Flatulence 1 (2) 6 (14) Abdominal pain 2 (4) 3 (7) Vomiting 2 (4) 3 (7) Decreased appetite 2 (4) 3 (7) Fatigue 1 (2) 3 (7) Dyspepsia 3 (7) 2 (5) Nausea 3 (7) 1 (2) Dizziness 4 (9) 0 Abdominal discomfort 3 (7) 0 Other Adverse Reactions
RAVICTI has been evaluated in 77 patients with UCDs (51 adult and 26 pediatric patients ages 2 years to 17 years) in 2 open-label long-term studies, in which 69 patients completed 12 months of treatment with RAVICTI (median exposure = 51 weeks). During these studies there were no deaths.
Adverse reactions reported in at least 10% of adult patients were nausea, vomiting, diarrhea, decreased appetite, dizziness, headache, and fatigue.
Adverse reactions reported in at least 10% of pediatric patients ages 2 years to 17 years were upper abdominal pain, rash, nausea, vomiting, diarrhea, decreased appetite, and headache.
RAVICTI has been evaluated in 17 patients with UCDs ages 2 months to less than 2 years in 3 open-label studies. The median exposure was 6 months (range 0.2 to 20 months). Adverse reactions reported in at least 10% of pediatric patients aged 2 months to less than 2 years were neutropenia, vomiting, constipation, diarrhea, pyrexia, hypophagia, cough, nasal congestion, rhinorrhea, rash, and papule.
RAVICTI has been evaluated in 16 patients with UCDs less than 2 months of age (age range 0.1 to 2 months, median age 0.5 months) in a single, open-label study. The median exposure was 10 months (range 2 to 20 months). Adverse reactions reported in at least 10% of pediatric patients aged less than 2 months were vomiting, rash, gastroesophageal reflux, increased hepatic enzymes, feeding disorder (decreased appetite, hypophagia), anemia, cough, dehydration, metabolic acidosis, thrombocytosis, thrombocytopenia, neutropenia, lymphocytosis, diarrhea, flatulence, constipation, pyrexia, lethargy, and irritability/agitation.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of RAVICTI. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
- Abnormal body odor, including from skin, hair and urine
- Retching and gagging
- Dysgeusia or burning sensation in mouth
-
7 DRUG INTERACTIONS
7.1 Potential for Other Drugs to Affect Ammonia
7.3 Potential for RAVICTI to Affect Other Drugs
Drugs with narrow therapeutic index that are substrates of CYP3A4
RAVICTI is a weak inducer of CYP3A4 in humans. Concomitant use of RAVICTI may decrease the systemic exposure to drugs that are substrates of CYP3A4. Monitor for decreased efficacy of drugs with narrow therapeutic index (e.g., alfentanil, quinidine, cyclosporine) [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to RAVICTI during pregnancy. Healthcare providers are encouraged to report any prenatal exposure to RAVICTI by calling the Pregnancy Registry at 1-855-823-2595 or visiting www.ucdregistry.com.
Risk Summary
Limited available data with RAVICTI use in pregnant women are insufficient to inform a drug-associated risk of major birth defects and miscarriage. In an animal reproduction study, administration of oral glycerol phenylbutyrate to pregnant rabbits during organogenesis at doses up to 2.7–times the dose of 6.87 mL/m2/day in adult patients resulted in maternal toxicity, but had no effects on embryo-fetal development. In addition, there were no adverse developmental effects with administration of oral glycerol phenylbutyrate to pregnant rats during organogenesis at 1.9 times the dose of 6.87 mL/m2/day in adult patients; however, maternal toxicity, reduced fetal weights, and variations in skeletal development were observed in pregnant rats administered oral glycerol phenylbutyrate during organogenesis at doses greater than or equal to 5.7 times the dose of 6.87 mL/m2/day in adult patients [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
Oral administration of glycerol phenylbutyrate during the period of organogenesis up to 350 mg/kg/day in rabbits produced maternal toxicity, but no effects on embryo-fetal development. The dose of 350 mg/kg/day in rabbits is approximately 2.7 times the dose of 6.87 mL/m2/day in adult patients, based on combined area under the plasma concentration-time curve [AUCs] for PBA and PAA. In rats, at an oral dose of 300 mg/kg/day of glycerol phenylbutyrate (1.9 times the dose of 6.87 mL/m2/day in adult patients, based on combined AUCs for PBA and PAA) during the period of organogenesis, no effects on embryo-fetal development were observed. Doses of 650 mg/kg/day or greater produced maternal toxicity and adverse effects on embryo-fetal development including reduced fetal weights and cervical ribs at the 7th cervical vertebra. The dose of 650 mg/kg/day in rats is approximately 5.7 times the dose of 6.87 mL/m2/day in adult patients, based on combined AUCs for PBA and PAA. No developmental abnormalities, effects on growth, or effects on learning and memory were observed through maturation of offspring following oral administration in pregnant rats with up to 900 mg/kg/day of glycerol phenylbutyrate (8.5 times the dose of 6.87 mL/m2/day in adult patients, based on combined AUCs for PBA and PAA) during organogenesis and lactation.
8.2 Lactation
Risk Summary
There are no data on the presence of RAVICTI in human milk, the effects on the breastfed infant, or the effects on milk production. Because of the potential for serious adverse reactions, including neurotoxicity and tumorigenicity in a breastfed infant, advise patients that breastfeeding is not recommended during treatment with RAVICTI.
8.4 Pediatric Use
Patients 2 Years to 17 Years of Age
The safety and effectiveness of RAVICTI in patients 2 years to less than 18 years of age have been established in 3 clinical studies: 2 open-label, fixed-sequence, switchover clinical studies from sodium phenylbutyrate to RAVICTI, and 1 long-term, open label safety study [see Adverse Reactions (6.1), Clinical Studies (14.2)].
Patients Less Than 2 Years of Age
The safety and effectiveness of RAVICTI in patients with UCDs less than 2 years of age have been established in 3 open-label studies. Pharmacokinetics and pharmacodynamics (plasma ammonia), and safety were studied in 17 patients aged 2 months to less than 2 years of age and in 16 patients less than 2 months of age [see Adverse Reactions (6.1), Clinical Studies (14.3)].
Juvenile Animal Toxicity Data
In a juvenile rat study with daily oral dosing performed on postpartum day 2 through mating and pregnancy after maturation, terminal body weight was dose-dependently reduced by up to 16% in males and 12% in females at 900 mg/kg/day or higher (3 times the dose of 6.87 mL/m2/day in adult patients, based on combined AUCs for PBA and PAA). Learning, memory, and motor activity endpoints were not affected. However, fertility (number of pregnant rats) was decreased by up to 25% at 650 mg/kg/day or higher (2.6 times the dose of 6.87 mL/m2/day in adult patients, based on combined AUCs for PBA and PAA).
8.5 Geriatric Use
Clinical studies of RAVICTI did not include sufficient numbers of subjects 65 years of age and older to determine whether they respond differently than younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
The efficacy and safety of RAVICTI in patients with renal impairment are unknown. Monitor ammonia levels closely when starting patients with impaired renal function on RAVICTI.
8.7 Hepatic Impairment
No studies were conducted in patients with UCDs and hepatic impairment. Because conversion of PAA to PAGN occurs in the liver, patients with hepatic impairment may have reduced conversion capability and higher plasma PAA and PAA to PAGN ratio [see Clinical Pharmacology (12.3)]. Therefore, dosage for patients with moderate to severe hepatic impairment should be started at the lower end of the recommended dosing range and should be kept on the lowest dose necessary to control their ammonia levels [see Dosage and Administration (2.5)].
-
10 OVERDOSAGE
While there is no experience with overdosage in human clinical trials, PAA, a toxic metabolite of RAVICTI, can accumulate in patients who receive an overdose [see Warnings and Precautions (5.1)].
If over-exposure occurs, call your Poison Control Center at 1-800-222-1222 for current information on the management of poisoning or overdosage.
-
11 DESCRIPTION
RAVICTI (glycerol phenylbutyrate) is a clear, colorless to pale yellow oral liquid. It is insoluble in water and most organic solvents, and it is soluble in dimethylsulfoxide (DMSO) and greater than 65% acetonitrile.
Glycerol phenylbutyrate is a nitrogen-binding agent. It is a triglyceride containing 3 molecules of PBA linked to a glycerol backbone, the chemical name of which is benzenebutanoic acid, 1′, 1′ ′ –(1,2,3-propanetriyl) ester with a molecular weight of 530.67. It has a molecular formula of C33H38O6. The structural formula is:
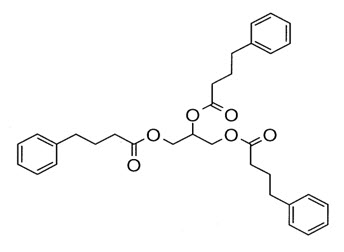
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
UCDs are inherited deficiencies of enzymes or transporters necessary for the synthesis of urea from ammonia (NH3, NH4+). Absence of these enzymes or transporters results in the accumulation of toxic levels of ammonia in the blood and brain of affected patients. RAVICTI is a triglyceride containing 3 molecules of PBA. PAA, the major metabolite of PBA, is the active moiety of RAVICTI. PAA conjugates with glutamine (which contains 2 molecules of nitrogen) via acetylation in the liver and kidneys to form PAGN, which is excreted by the kidneys (Figure 1). On a molar basis, PAGN, like urea, contains 2 moles of nitrogen and provides an alternate vehicle for waste nitrogen excretion.
Figure 1: RAVICTI Mechanism of Action
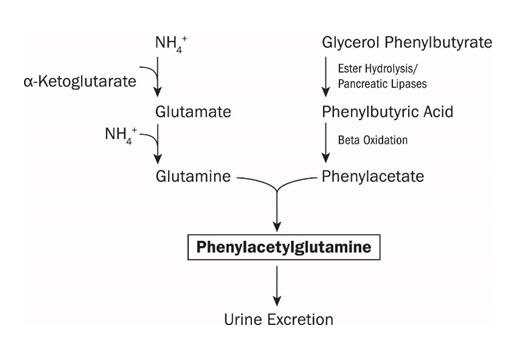
12.2 Pharmacodynamics
Pharmacological Effects
In clinical studies, total 24-hour area under the plasma concentration-time curve (AUC) of ammonia levels was comparable at steady state during the switchover period between RAVICTI and sodium phenylbutyrate [see Clinical Studies (14)].
Cardiac Electrophysiology
The effect of multiple doses of RAVICTI 13.2 g/day and 19.8 g/day (approximately 69% and 104% of the maximum recommended daily dosage) on QTc interval was evaluated in a randomized, placebo- and active-controlled (moxifloxacin 400 mg), four-treatment-arm, crossover study in 57 healthy subjects. The upper bound of the one-sided 95% CI for the largest placebo-adjusted, baseline-corrected QTc, based on individual correction method (QTcI) for RAVICTI, was below 10 ms.
12.3 Pharmacokinetics
Absorption
RAVICTI is a pro-drug of PBA. Upon oral ingestion, PBA is released from the glycerol backbone in the gastrointestinal tract by lipases. PBA derived from RAVICTI is further converted by β-oxidation to PAA.
In healthy, fasting adult subjects receiving a single oral dose of 2.9 mL/m2 of RAVICTI, peak plasma levels of PBA, PAA, and PAGN occurred at 2 hours, 4 hours, and 4 hours, respectively. Upon single-dose administration of RAVICTI, plasma concentrations of PBA were quantifiable in 15 of 22 participants at the first sample time postdose (0.25 hours). Mean maximum concentration (Cmax) for PBA, PAA, and PAGN was 37.0 micrograms/mL, 14.9 micrograms/mL, and 30.2 micrograms/mL, respectively. In healthy subjects, intact glycerol phenylbutyrate was detected in plasma. While the study was inconclusive, the incomplete hydrolysis of glycerol phenylbutyrate cannot be ruled out.
In healthy subjects, the systemic exposure to PAA, PBA, and PAGN increased in a dose-dependent manner. Following 4 mL of RAVICTI 3 times a day for 3 days, the mean Cmax and AUC were 66 micrograms/mL and 930 micrograms∙h/mL for PBA and 28 micrograms/mL and 942 micrograms∙h/mL for PAA, respectively. In the same study, following 6 mL of RAVICTI three times a day for 3 days, mean Cmax and AUC were 100 micrograms/mL and 1400 micrograms∙h/mL for PBA and 65 μg/mL and 2064 micrograms∙h/mL for PAA, respectively.
In adult patients with UCDs receiving multiple doses of RAVICTI, maximum plasma concentrations at steady state (Cmax,ss) of PBA, PAA, and PAGN occurred at 8 hours, 12 hours, and 10 hours, respectively, after the first dose in the day. Intact glycerol phenylbutyrate was not detectable in plasma in patients with UCDs.
In clinical studies of RAVICTI in patients with UCDs, the peak observed PAA concentrations by age group are shown in Table 2.
Table 2: Peak PAA Concentrations in Patients with UCDs Treated with RAVICTI in Clinical Trials Age Range RAVICTI Dose Mean Peak PAA Concentration*
(SD)Median Peak PAA Concentration *
(Range)- * micrograms/mL
Less than 2 months
(n=16)3.1 to 12.7 mL/m2/day
(3.4 to 14 g/m2/day)257 (162) 205 (96 to 707) 2 months to less than 2 years
(n=17)3.3 to 12.3 mL/m2/day
(3.7 to 13.5 g/m2/day)142 (299) 35 (1 to 1215) 2 years to 17 years
(n=53)1.4 to 13.7 mL/m2/day
(1.5 to 15.1 g/m2/day)70 (79) 50 (1 to 410) Adults
(n=43)0.6 to 14 mL/m2/day
(0.7 to 15.4 g/m2/day)39 (40) 25 (1.6 to 178) Distribution
In vitro, the extent of plasma protein binding for 14C-labeled metabolites was 81% to 98% for PBA (over 1 to 250 micrograms/mL), and 37% to 66% for PAA (over 5 to 500 micrograms/mL). The protein binding for PAGN was 7% to 12% and no concentration effects were noted.
Elimination
Metabolism
Upon oral administration, pancreatic lipases hydrolyze RAVICTI (i.e., glycerol phenylbutyrate), and release PBA. PBA undergoes β-oxidation to PAA, which is conjugated with glutamine in the liver and in the kidney through the enzyme phenylacetyl-CoA: L-glutamine-N-acetyltransferase to form PAGN. PAGN is subsequently eliminated in the urine.
Saturation of conjugation of PAA and glutamine to form PAGN was suggested by increases in the ratio of plasma PAA to PAGN with increasing dose and with increasing severity of hepatic impairment.
In healthy subjects, after administration of 4 mL, 6 mL, and 9 mL 3 times daily for 3 days, the ratio of mean AUC0-23h of PAA to PAGN was 1, 1.25, and 1.6, respectively. In a separate study, in patients with hepatic impairment (Child-Pugh B and C), the ratios of mean Cmax values for PAA to PAGN among all patients dosed with 6 mL and 9 mL twice daily were 3 and 3.7.
In in vitro studies, the specific activity of lipases for glycerol phenylbutyrate was in the following decreasing order: pancreatic triglyceride lipase, carboxyl ester lipase, and pancreatic lipase–related protein 2. Further, glycerol phenylbutyrate was hydrolyzed in vitro by esterases in human plasma. In these in vitro studies, a complete disappearance of glycerol phenylbutyrate did not produce molar equivalent PBA, suggesting the formation of mono- or bis-ester metabolites. However, the formation of mono- or bis-esters was not studied in humans.
Specific Populations
Age: Pediatric Population
Population pharmacokinetic modeling and dosing simulations suggest body surface area to be the most significant covariate explaining the variability of PAA clearance. PAA clearance was 10.9 L/h, 16.4 L/h, and 24.4 L/h, respectively, for patients ages 3 to 5, 6 to 11, and 12 to 17 years with UCDs.
In pediatric patients with UCDs (n = 14) ages 2 months to less than 2 years, PAA clearance was 6.8 L/h.
In pediatric patients with UCDs (n = 16) ages less than 2 months, PAA clearance was 3.8 L/h. The mean peak ratio of PAA to PAGN in UCD patients aged birth to less than 2 months was higher (mean: 1.6; range 0.1 to 7.1) than that of UCD patients aged 2 months to less than 2 years (mean 0.5; range 0.1 to 1.2).
Sex
In healthy adult subjects, a gender effect was found for all metabolites, with women generally having higher plasma concentrations of all metabolites than men at a given dose level. In healthy female subjects, mean Cmax for PAA was 51 and 120% higher than in male volunteers after administration of 4 mL and 6 mL 3 times daily for 3 days, respectively. The dose normalized mean AUC0-23h for PAA was 108% higher in females than in males.
Renal Impairment
The pharmacokinetics of RAVICTI in patients with impaired renal function, including those with end-stage renal disease (ESRD) or those on hemodialysis, have not been studied [see Use in Specific Populations (8.6)].
Hepatic Impairment
The effects of hepatic impairment on the pharmacokinetics of RAVICTI were studied in patients with mild, moderate and severe hepatic impairment of (Child-Pugh class A, B, and C, respectively) receiving 100 mg/kg of RAVICTI twice daily for 7 days.
Plasma glycerol phenylbutyrate was not measured in patients with hepatic impairment.
After multiple doses of RAVICTI in patients with hepatic impairment of Child-Pugh A, B, and C, geometric mean AUCt of PBA was 42%, 84%, and 50% higher, respectively, while geometric mean AUCt of PAA was 22%, 53%, and 94% higher, respectively, than in healthy subjects.
In patients with hepatic impairment of Child-Pugh A, B, and C, geometric mean AUCt of PAGN was 42%, 27%, and 22% lower, respectively, than that in healthy subjects.
The proportion of PBA excreted as PAGN in the urine in Child-Pugh A, B, and C was 80%, 58%, and 85%, respectively, and, in healthy volunteers, was 67%.
In another study in patients with moderate and severe hepatic impairment (Child-Pugh B and C), mean Cmax of PAA was 144 micrograms/mL (range: 14 to 358 micrograms/mL) after daily dosing of 6 mL of RAVICTI twice daily, while mean Cmax of PAA was 292 micrograms/mL (range: 57 to 655 micrograms/mL) after daily dosing of 9 mL of RAVICTI twice daily. The ratio of mean Cmax values for PAA to PAGN among all patients dosed with 6 mL and 9 mL twice daily were 3 and 3.7, respectively.
After multiple doses, a PAA concentration greater than 200 micrograms/mL was associated with a ratio of plasma PAA to PAGN concentrations higher than 2.5 [see Dosage and Administration (2.5)].
Drug Interaction Studies
In vitro PBA or PAA did not induce CYP1A2, suggesting that in vivo drug interactions via induction of CYP1A2 is unlikely.
In in vitro studies, PBA at a concentration of 800 micrograms/mL caused greater than 60% reversible inhibition of cytochrome P450 isoenzymes CYP2C9, CYP2D6, and CYP3A4/5 (testosterone 6β-hydroxylase activity). The in vitro study suggested that in vivo drug interactions with substrates of CYP2D6 cannot be ruled out. The inhibition of CYP isoenzymes 1A2, 2C8, 2C19, and 2D6 by PAA at the concentration of 2.8 mg/mL was observed in vitro. Clinical implication of these results is unknown.
Effects of RAVICTI on other drugs
Midazolam
In healthy subjects, when oral midazolam was administered after multiple doses of RAVICTI (4 mL three times a day for 3 days) under fed conditions, the mean Cmax and AUC for midazolam were 25% and 32% lower, respectively, compared to administration of midazolam alone. In addition, the mean Cmax and AUC for 1-hydroxy midazolam were 28% and 58% higher, respectively, compared to administration of midazolam alone [see Drug Interactions (7.3)].
Celecoxib
Concomitant administration of RAVICTI did not significantly affect the pharmacokinetics of celecoxib, a substrate of CYP2C9. When 200 mg of celecoxib was orally administered with RAVICTI after multiple doses of RAVICTI (4 mL three times a day for 6 days) under fed conditions (a standard breakfast was consumed 5 minutes after celecoxib administration), the mean Cmax and AUC for celecoxib were 13% and 8% lower than after administration of celecoxib alone.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 2-year study in Sprague-Dawley rats, glycerol phenylbutyrate caused a statistically significant increase in the incidence of pancreatic acinar cell adenoma, carcinoma, and combined adenoma or carcinoma at a dose of 650 mg/kg/day in males (4.7 times the dose of 6.9 mL/m2/day in adult patients, based on combined AUCs for PBA and PAA) and 900 mg/kg/day in females (8.4 times the dose of 6.9 mL/m2/day in adult patients, based on combined AUCs for PBA and PAA). The incidence of the following tumors was also increased in female rats at a dose of 900 mg/kg/day: thyroid follicular cell adenoma, carcinoma and combined adenoma or carcinoma, adrenal cortical combined adenoma or carcinoma, uterine endometrial stromal polyp, and combined polyp or sarcoma. The dose of 650 mg/kg/day in male rats is 3 times the dose of 7.5 mL/m2/day in pediatric patients, based on combined AUCs for PBA and PAA. The dose of 900 mg/kg/day in female rats is 5.5 times the dose of 7.5 mL/m2/day in pediatric patients, based on combined AUCs for PBA and PAA. In a 26-week study in transgenic (Tg.rasH2) mice, glycerol phenylbutyrate was not tumorigenic at doses up to 1000 mg/kg/day.
Mutagenesis
Glycerol phenylbutyrate was not genotoxic in the Ames test, the in vitro chromosomal aberration test in human peripheral blood lymphocytes, or the in vivo rat micronucleus test. The metabolites PBA, PAA, PAGN, and phenylacetylglycine were not genotoxic in the Ames test or in vitro chromosome aberration test in Chinese hamster ovary cells.
Impairment of Fertility
Glycerol phenylbutyrate had no effect on fertility or reproductive function in male and female rats at oral doses up to 900 mg/kg/day. At doses of 1200 mg/kg/day (approximately 7 times the dose of 6.9 mL/m2/day in adult patients, based on combined AUCs for PBA and PAA), maternal toxicity was observed and the number of nonviable embryos was increased.
-
14 CLINICAL STUDIES
14.1 Clinical Studies in Adult Patients with UCDs
Active-Controlled, 4-Week, Noninferiority Study (Study 1)
A randomized, double-blind, active-controlled, crossover, noninferiority study (Study 1) compared RAVICTI to sodium phenylbutyrate by evaluating ammonia levels in patients with UCDs who had been on sodium phenylbutyrate prior to enrollment for control of their UCD. Patients were required to have a confirmed diagnosis of UCD involving deficiencies of CPS, OTC, or ASS, confirmed via enzymatic, biochemical, or genetic testing. Patients had to have no clinical evidence of hyperammonemia at enrollment and were not allowed to receive drugs known to increase ammonia levels (e.g., valproate), increase protein catabolism (e.g., corticosteroids), or significantly affect renal clearance (e.g., probenecid).
The primary endpoint was the 24-hour AUC (a measure of exposure to ammonia over 24 hours) for venous ammonia on days 14 and 28 when the drugs were expected to be at steady state. Statistical noninferiority would be established if the upper limit of the 2-sided 95% CI for the ratio of the geometric means (RAVICTI/sodium phenylbutyrate) for the endpoint was 1.25 or less.
Forty-five patients were randomized 1:1 to 1 of 2 treatment arms to receive either
- – Sodium phenylbutyrate for 2 weeks → RAVICTI for 2 weeks; or
- – RAVICTI for 2 weeks → sodium phenylbutyrate for 2 weeks.
Sodium phenylbutyrate or RAVICTI were administered three times daily with meals. The dose of RAVICTI was calculated to deliver the same amount of PBA as the sodium phenylbutyrate dose the patients were taking when they entered the study. Forty-four patients received at least 1 dose of RAVICTI in the study.
Patients adhered to a low-protein diet and received amino acid supplements throughout the study. After 2 weeks of dosing, by which time patients had reached steady state on each treatment, all patients had 24 hours of ammonia measurements.
Demographic characteristics of the 45 patients enrolled in Study 1 were as follows: mean age at enrollment was 33 years (range: 18 to 75 years); 69% were female; 33% had adult-onset disease; 89% had OTC deficiency; 7% had ASS deficiency; 4% had CPS deficiency.
RAVICTI was non-inferior to sodium phenylbutyrate with respect to the 24-hour AUC for ammonia. Forty-four patients were evaluated in this analysis. Mean 24-hour AUCs for ammonia during steady-state dosing were 866 micromol∙h/L and 977 micromol∙h/L with RAVICTI and sodium phenylbutyrate, respectively. The ratio of geometric means was 0.91 [95% CI 0.8, 1.04].
The mean ammonia levels over 24-hours after 2 weeks of dosing (on day 14 and 28) in the double-blind short-term study (Study 1) are displayed in Figure 2 below. The mean and median maximum ammonia levels (Cmax) over 24 hours and 24-hour AUC for ammonia are summarized in Table 3. Ammonia values across different laboratories were normalized to a common normal range of 9 to 35 micromol/L using the following formula after standardization of the units to micromol/L:
Normalized ammonia (micromol/L) = ammonia readout in micromol/L × (35/ULN of a laboratory reference range specified for each assay)
Figure 2: Ammonia Levels in Adult Patients with UCDs in Short-Term Treatment Study 1
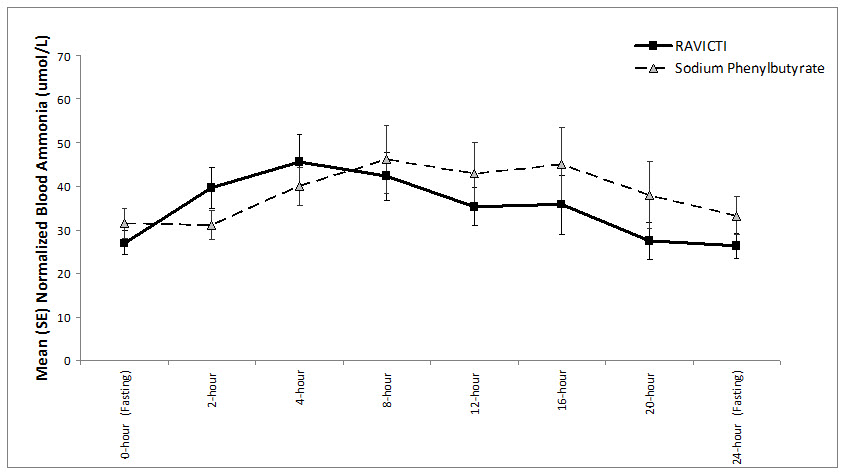
Table 3: Ammonia Levels in Adult Patients with UCDs in Short-Term Treatment Study 1 Timepoint Ammonia (n=44) Mean (SD) Median (min, max) Daily Cmax (micromol/L) RAVICTI 61 (46) 51 (12, 245) Sodium phenylbutyrate 71 (67) 46 (14, 303) 24-Hour AUC (micromol∙h/L) RAVICTI 866 (661) 673 (206, 3351) Sodium phenylbutyrate 977 (865) 653 (302, 4666) Open-Label, Uncontrolled, Extension Study in Adults
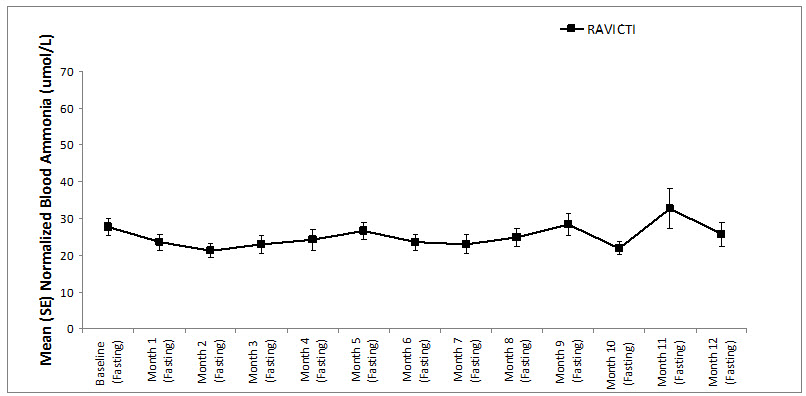
A long-term (12-month), uncontrolled, open-label study (Study 2) was conducted to assess monthly ammonia control and hyperammonemic crisis over a 12-month period. A total of 51 adults were in the study and all but 6 had been converted from sodium phenylbutyrate to RAVICTI. Venous ammonia levels were monitored monthly. Mean fasting ammonia values in adults in Study 2 were within normal limits during long-term treatment with RAVICTI (range: 6 to 30 micromol/L). Of 51 adult patients participating in the 12-month, open-label treatment with RAVICTI, 7 patients (14%) reported a total of 10 hyperammonemic crises. The fasting ammonia measured during Study 2 is displayed in Figure 3. Ammonia values across different laboratories were normalized to a common normal range of 9 to 35 micromol/L.
Figure 3: Ammonia Levels in Adult Patients with UCDs in Long-Term Treatment Study 2
Open-Label, Long-Term Study in Adults
An open-label long-term, study (Study 5) was conducted to assess ammonia control in adult patients with UCDs. The study enrolled patients with UCDs who had completed the safety extensions of Study 1, Study 3 or Study 4 (Study 2, 3E and 4E, respectively). A total of 43 adult patients between the ages of 19 and 61 years were in the study. The median length of study participation was 1.9 years (range 0 to 4.5 years). Venous ammonia levels were monitored at a minimum of every 6 months. Mean fasting ammonia values in adult patients in Study 5 were within normal limits during long-term (24 months) treatment with RAVICTI (range: 24.2 to 31.4 micromol/L). Of the 43 adult patients participating in the open-label treatment with RAVICTI, 9 patients (21%) reported a total of 21 hyperammonemic crises. Ammonia values across different laboratories were normalized to a common normal range of 10 to 35 micromol/L.
14.2 Clinical Studies in Pediatric Patients 2 Years to 17 Years of Age with UCDs
The efficacy of RAVICTI in pediatric patients 2 years to 17 years of age with UCDs was evaluated in 2 fixed-sequence, open-label, sodium phenylbutyrate to RAVICTI switchover studies (Studies 3 and 4). Study 3 was 7 days in duration and Study 4 was 10 days in duration.
These studies compared ammonia levels of patients on RAVICTI to ammonia levels of patients on sodium phenylbutyrate in 26 pediatric patients between 2 months and 17 years of age with UCDs. Four patients less than 2 years of age were excluded from this analysis due to insufficient data. The dose of RAVICTI was calculated to deliver the same amount of PBA as the dose of sodium phenylbutyrate that patients were taking when they entered the trial. Sodium phenylbutyrate or RAVICTI were administered in divided doses with meals. Patients adhered to a low-protein diet throughout the study. After a dosing period with each treatment, all patients underwent 24 hours of venous ammonia measurements, as well as blood and urine pharmacokinetic assessments.
UCD subtypes included OTC (n=12), ASL (n=8), and ASS deficiency (n=2), and patients received a mean RAVICTI dose of 8 mL/m2/day (8.8 g/m2/day), with doses ranging from 1.4 to 13.1 mL/m2/day (1.5 to 14.4 g/m2/day). Doses in these patients were based on previous dosing of sodium phenylbutyrate.
The 24-hour AUCs for ammonia (AUC0-24h) in 11 pediatric patients 6 years to 17 years of age with UCDs (Study 3) and 11 pediatric patients 2 years to 5 years of age with UCDs (Study 4) were similar between treatments. In pediatric patients 6 years to 17 years of age, the ammonia AUC0-24h was 604 micromol∙h/L vs 815 micromol∙h/L on RAVICTI vs sodium phenylbutyrate, respectively. In patients between 2 years and 5 years of age with UCDs, the ammonia AUC0-24h was 632 micromol∙h/L vs 720 micromol∙h/L on RAVICTI versus sodium phenylbutyrate, respectively.
The mean ammonia levels over 24 hours in open-label, short-term Studies 3 and 4 at common time points are displayed in Figure 4. Ammonia values across different laboratories were normalized to a common normal range of 9 to 35 micromol/L using the following formula after standardization of the units to micromol/L:
Normalized ammonia (micromol/L) = ammonia readout in micromol/L × (35/ULN of a laboratory reference range specified for each assay)
Figure 4: Ammonia Levels in Pediatric Patients 2 Years to 17 Years of Age with UCDs in Short-Term Treatment Studies 3 and 4
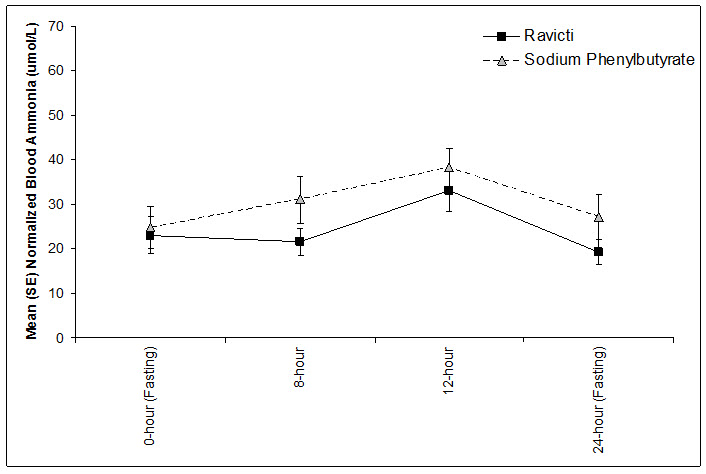
Open-Label, Uncontrolled, Extension Studies in Pediatric Patients 2 Years to 17 Years of Age
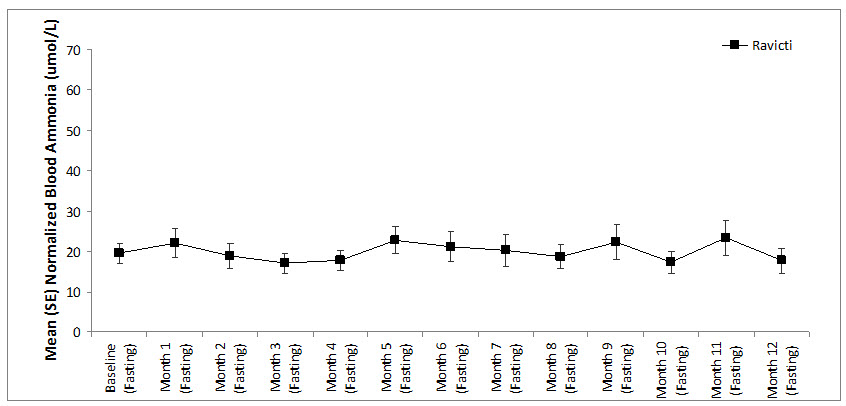
Long-term (12-month), uncontrolled, open-label studies were conducted to assess monthly ammonia control and hyperammonemic crises over a 12-month period. In two studies (Study 2, which also enrolled adults, and an extension of Study 3, referred to here as Study 3E), a total of 26 pediatric patients ages 6 years to 17 years were enrolled and all but 1 had been converted from sodium phenylbutyrate to RAVICTI. Mean fasting venous ammonia levels were within normal limits (range 17 to 23 micromol/L) during long-term treatment with RAVICTI. Of the 26 pediatric patients 6 years to 17 years of age participating in these two trials, 5 patients (19%) reported a total of 5 hyperammonemic crises. The fasting ammonia levels measured during these two extension studies in patients 6 years to 17 years are displayed in Figure 5. Ammonia values across different laboratories were normalized to a common normal range of 9 to 35 micromol/L.
Figure 5: Ammonia Levels in Pediatric Patients 2 Years to 17 Years of Age with UCDs in Long-Term Treatment Studies 2 and 3E
In an extension of Study 4 (referred to as Study 4E), after a median time on study of 4.5 months (range: 1 to 5.7 months), 2 of 16 pediatric patients ages 2 years to 5 years had experienced three hyperammonemic crises.
Open-Label, Long-Term Study in Pediatric Patients 1 Year to 17 Years of Age
An open-label, long-term study (Study 5) was conducted to assess ammonia levels in pediatric patients with UCD. The study enrolled patients with UCDs who had completed Studies 2, 3E and 4E. A total of 45 pediatric patients ages 1 year to 17 years were included in the study. The median length of treatment was 1.7 years (range 0.2 to 4.6 years). Venous ammonia levels were monitored at a minimum every 6 months. Mean ammonia values in pediatric patients in Study 5 were within normal limits during long-term (24 months) treatment with RAVICTI (range: 15.4 to 25.1 micromol/L). Of the 45 pediatric patients participating in the open-label treatment with RAVICTI, 11 patients (24%) reported a total of 22 hyperammonemic crises. Ammonia values across different laboratories were normalized to a common normal range of 10 to 35 micromol/L.
14.3 Clinical Studies in Pediatric Patients Less Than 2 Years of Age with UCDs
The efficacy of RAVICTI in pediatric patients less than 2 years of age with UCDs was evaluated in uncontrolled, open label studies (Studies 4/4E, 5 [see Clinical Studies (14.2)] and 6). A total of 17 pediatric patients with UCDs aged 2 months to less than 2 years participated in Studies 4/4E, 5 and 6. Study 6 enrolled 16 pediatric patients less than 2 months of age.
Uncontrolled, Open-Label Studies in Pediatric Patients Aged 2 Months to Less than 2 Years of Age (Studies 4/4E, 5)
A total of 7 patients with UCDs aged 2 months to less than 2 years participated in Studies 4/4E and 5. In these studies, there were 7, 6, 6, 6 and 3 pediatric patients who completed 1, 6, 9, 12 and 18 months, respectively (mean and median exposure of 15 and 17 months, respectively). Patients were converted from sodium phenylbutyrate to RAVICTI. The dosage of RAVICTI was calculated to deliver the same amount of PBA as the sodium phenylbutyrate dosage the patients were taking when they entered the study.
Patients received a mean RAVICTI dose of 7.5 mL/m2/day (8.2 g/m2/day), with doses ranging from 3.3 to 12.3 mL/m2/day (3.7 to 13.5 g/m2/day). Patients were dosed three times per day (n=3) or four times per day (n = 4).
Venous ammonia levels were monitored on days 1, 3, and 10 in Study 4 and at week 1 in Study 4E. Two patients had elevated ammonia values on day 1 of treatment (122 micromol/L and 111 micromol/L respectively) and neither had associated signs and symptoms of hyperammonemia. At day 10/week 1, six of the 7 patients had normal ammonia levels (less than 100 micromol/L) while the remaining patient had an elevated ammonia value on day 10 (168 micromol/L) and was asymptomatic.
During the extension period, venous ammonia levels were monitored monthly. Ammonia values across different laboratories were normalized (transformed) to a common normal pediatric range of 28 to 57 micromol/L for comparability. The mean ammonia levels in pediatric patients at month 1, 3, 6, 9 and 12 were 58, 49, 34, 65, and 31 micromol/L during treatment with RAVICTI, respectively.
Three patients reported a total of 3 hyperammonemic crises defined as having signs and symptoms consistent with hyperammonemia (such as frequent vomiting, nausea, headache, lethargy, irritability, combativeness, and/or somnolence) associated with high ammonia levels (greater than 100 micromol/L) and requiring medical intervention. Hyperammonemic crises were precipitated by gastroenteritis, vomiting, infection or no precipitating event (one patient). There were 4 patients who had one ammonia level that exceeded 100 micromol/L which was not associated with a hyperammonemic crisis.
Uncontrolled, Open-Label Study in Pediatric Patients Less Than 2 Years of Age (Study 6)
Study 6 was an uncontrolled, open label study in pediatric patients less than 2 years of age. The primary efficacy endpoint was successful transition to RAVICTI within a period of 4 days followed by 3 days of observation for a total of 7 days, where successful transition was defined as no signs and symptoms of hyperammonemia and a venous ammonia level less than 100 micromol/L. Ammonia levels were monitored for up to 4 days during transition and on day 7.
Pediatric Patients 2 Months to Less than 2 Years of Age
A total of 10 pediatric patients with UCDs aged 2 months to less than 2 years participated in Study 6, of which 6 patients converted from sodium phenylbutyrate to RAVICTI and 1 patient converted from sodium phenyl butyrate and sodium benzoate. The dosage of RAVICTI was calculated to deliver the same amount of PBA as the sodium phenylbutyrate dosage the patients were taking when they entered the trial. Two patients were treatment-naïve and received RAVICTI dosage of 7.5 mL/m2/day and 9.4 mL/m2/day, respectively. One additional patient was gradually discontinued from intravenous sodium benzoate and sodium phenylacetate while RAVICTI was initiated. The dosage of RAVICTI after transition was 8.5 mL/m2/day.
There were 9, 7, 7, 4, 1 and 4 pediatric patients who completed 1, 3, 6, 12, 18 and 24 months, respectively (mean and median exposure of 9 and 9 months, respectively).
Patients received a mean RAVICTI dose of 8 mL/m2/day (8.8 g/m2/day), with doses ranging from 4.8 to 11.5 mL/m2/day (5.3 to 12.6 g/m2/day). Patients were dosed three times a day (n=6), four times a day (n = 2), or five or more times a day (n=2).
Nine patients successfully transitioned as defined by the primary endpoint. One additional patient developed hyperammonemia on day 3 of dosing and experienced surgical complications (bowel perforation and peritonitis) following jejunal tube placement on day 4. This patient developed hyperammonemic crisis on day 6, and subsequently died of sepsis from peritonitis unrelated to drug. Although two patients had day 7 ammonia values of 150 micromol/L and 111 micromol/L respectively, neither had associated signs and symptoms of hyperammonemia.
During the extension phase, venous ammonia levels were monitored monthly. Ammonia values across different laboratories were normalized (transformed) to a common normal pediatric range of 28 to 57 micromol/L for comparability. The mean normalized ammonia levels in pediatric patients at months 1, 2, 3, 4, 5, 6, 9, 12, 15, 18 and 24 were 67, 53, 78, 93, 78, 67, 38, 38, 36, 48 and 53 micromol/L during treatment with RAVICTI, respectively. Three patients reported a total of 7 hyperammonemic crises as defined in Study 4/4E and 5. Hyperammonemic crises were precipitated by vomiting, upper respiratory tract infection, gastroenteritis, decreased caloric intake or had no identified precipitating event (3 events). There was one additional patient who had one ammonia level that exceeded 100 micromol/L which was not associated with a hyperammonemic crisis.
Pediatric Patients Less than 2 Months of Age
A total of 16 pediatric patients less than 2 months of age participated in Study 6. Median age at enrollment was 0.5 months (range: 0.1 to 2 months). Eight patients had OTC deficiency, 7 patients had ASS deficiency, and 1 patient had ASL deficiency. Ten of the 16 patients transitioned from sodium phenylbutyrate to RAVICTI within 3 days of treatment and their initial dosage of RAVICTI was calculated to deliver the same amount of phenylbutyrate as the sodium phenylbutyrate dosage administered prior to RAVICTI dosing. Three of the 16 patients were treatment-naïve and started RAVICTI at dosages of 9, 9.4, and 9.6 mL/m2/day. The remaining 3 of the 16 patients transitioned from intravenous sodium benzoate and sodium phenylacetate to RAVICTI within 3 days of treatment and their initial dosages of RAVICTI were 10.4, 10.9, and 10.9 mL/m2/day.
Of the 16 patients, 16, 14, 12, 6, and 3 patients were treated for 1, 3, 6, 12, and 18 months, respectively.
After the initial 7-day transition period, patients received a mean RAVICTI dosage of 8 mL/m2/day (8.8 g/m2/day), with doses ranging from 3.1 to 12.7 mL/m2/day (3.4 to 14 g/m2/day). The frequency of dosing varied throughout the study. The majority of patients were dosed three times per day with feeding. No patients discontinued during the 7-day transition phase. Ammonia values across different laboratories were normalized (transformed) to a common normal pediatric range of 28 to 57 micromol/L for comparability.
During the safety extension phase (months 1-24), venous ammonia levels were monitored monthly for the first 6 months of treatment and every 3 months thereafter until the patients terminated or completed the study. During the safety extension phase, 1 patient discontinued from the study due to an adverse event (increased hepatic enzymes), 2 patients were withdrawn from the study by their parent/guardian, and 4 patients discontinued from the study early to undergo a liver transplant (protocol-defined discontinuation criterion). The normalized ammonia levels in pediatric patients with available values (which varied by month of treatment) in Study 6 in patients less than 2 months of age are shown in Table 4.
Table 4: Ammonia* Levels in Pediatric Patients Less than 2 Months of Age with UCDs in Study 6 Month N (patients with available ammonia level) Normalized Ammonia (micromol/L)† Mean
(SD)Median
(Min, Max)- * normalized ammonia (micromol/L) = ammonia readout in micromol/L × (35/ULN of a laboratory reference range specified for each assay)
- † normal range: 28 to 57 micromol/L.
1 15 71 (52) 60 (18, 227) 2 11 58 (40) 50 (16, 168) 3 14 53 (34) 46 (11, 122) 4 11 94 (106) 64 (35, 407) 5 10 52 (18) 57 (27, 86) 6 9 49 (24) 42 (22, 91) 9 8 56 (34) 45 (22, 122) 12 6 35 (17) 36 (11, 60) 15 4 52 (12) 52 (39, 67) 18 3 64 (14) 63 (50, 78) 24 9 63 (29) 72 (23, 106) Five patients (all less than 1 month of age) experienced a total of 7 hyperammonemic crises defined as in Study 4/4E and 5. Hyperammonemic crises were precipitated by upper respiratory tract infection (2 events), change in diet (1 event), or had no identified precipitating event (4 events).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
RAVICTI® (glycerol phenylbutyrate) oral liquid 1.1 g/mL is supplied in multi-use, 25-mL glass bottles. The bottles are supplied in the following configurations:
- NDC: 75987-050-06: Single 25-mL bottle per carton
- NDC: 75987-050-07: Four 25-mL bottles per carton
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Neurotoxicity [see Warnings and Precautions (5.1)].
- Inform patients/caregivers that adverse reactions of RAVICTI are sometimes the same as symptoms of high blood ammonia. Neurological adverse reactions may also be associated with the major metabolite of RAVICTI, PAA, and may be reversible. Blood tests for PAA may be done to measure the amount of PAA in the blood. Instruct the patient/caregiver to contact the healthcare provider immediately if the patient experiences: nausea, vomiting, headache, fatigue, somnolence, lightheadedness, confusion, exacerbation of preexisting neuropathy, disorientation, impaired memory, dysgeusia, or hypoacusis.
Pregnancy Registry
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to RAVICTI during pregnancy [see Use in Specific Populations (8.1)].
Lactation
Advise patients that breastfeeding is not recommended during treatment with RAVICTI [see Use in Specific Populations (8.2)].
Administration
- Instruct patients to take RAVICTI with food or formula and to administer directly into the mouth via oral syringe or dosing cup.
- Instruct that RAVICTI should be administered just prior to breastfeeding in infants who are breastfeeding.
- Instruct patients to take RAVICTI orally, even if they have a nasogastric and/or gastrostomy tube. For patients who cannot swallow and who have a nasogastric tube or gastrostomy tube in place, instruct patients/caregivers to administer RAVICTI as follows:
- Utilize an oral syringe to withdraw the prescribed dosage of RAVICTI from the bottle.
- Place the tip of the syringe into the gastrostomy/nasogastric tube.
- Utilizing the plunger of the syringe, administer RAVICTI into the tube.
- Flush once with 10 mL of water or formula and allow the flush to drain.
- If needed, flush a second time with an additional 10 mL of water or formula to clear the tube.
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
-
PRINCIPAL DISPLAY PANEL - 25 mL Bottle Carton
NDC: 75987-050-06
RAVICTI®
(glycerol phenylbutyrate) Oral Liquid1.1 grams per mL
For Oral Use Only
25 mLPharmacist:
Dispense the enclosed
Medication Guide to
patient and/or caregiver.Rx Only
-
INGREDIENTS AND APPEARANCE
RAVICTI
glycerol phenylbutyrate liquidProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 75987-050 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength GLYCEROL PHENYLBUTYRATE (UNII: ZH6F1VCV7B) (GLYCEROL PHENYLBUTYRATE - UNII:ZH6F1VCV7B) GLYCEROL PHENYLBUTYRATE 1.1 g in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 75987-050-06 1 in 1 CARTON 02/28/2013 1 25 mL in 1 BOTTLE; Type 0: Not a Combination Product 2 NDC: 75987-050-07 4 in 1 CARTON 02/28/2013 2 25 mL in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203284 02/28/2013 Labeler - Horizon Therapeutics, Inc. (033470838)
Trademark Results [Ravicti]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 RAVICTI 85167410 4324829 Live/Registered |
HORIZON THERAPEUTICS, LLC 2010-11-02 |
 RAVICTI 85143421 not registered Dead/Abandoned |
Hyperion Therapeutics, Inc. 2010-10-01 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.