RIVAROXABAN tablet, coated
Rivaroxaban by
Drug Labeling and Warnings
Rivaroxaban by is a Prescription medication manufactured, distributed, or labeled by Sun Pharmaceutical Industries, Inc., Taro Pharmaceutical Industries, Ltd.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use RIVAROXABAN TABLETS safely and effectively. See full prescribing information for RIVAROXABAN TABLETS.
RIVAROXABAN tablets, for oral use
Initial U.S. Approval: 2011WARNING: (A) PREMATURE DISCONTINUATION OF RIVAROXABAN TABLETS INCREASES THE RISK OF THROMBOTIC EVENTS, (B) SPINAL/EPIDURAL HEMATOMA
See full prescribing information for complete boxed warning.
(A) Premature discontinuation of rivaroxaban increases the risk of thrombotic events
Premature discontinuation of any oral anticoagulant, including rivaroxaban, increases the risk of thrombotic events. To reduce this risk, consider coverage with another anticoagulant if rivaroxaban is discontinued for a reason other than pathological bleeding or completion of a course of therapy. ( 2.3, 5.1)
(B) Spinal/epidural hematoma
Epidural or spinal hematomas have occurred in patients treated with rivaroxaban who are receiving neuraxial anesthesia or undergoing spinal puncture. These hematomas may result in long-term or permanent paralysis. ( 5.2, 5.3, 6.2)
Monitor patients frequently for signs and symptoms of neurological impairment and if observed, treat urgently. Consider the benefits and risks before neuraxial intervention in patients who are or who need to be anticoagulated. ( 5.3)
RECENT MAJOR CHANGES
Warnings and Precautions (5.2) 06/2025
INDICATIONS AND USAGE
Rivaroxaban is a factor Xa inhibitor indicated:
- to reduce the risk of major cardiovascular events in patients with coronary artery disease (CAD) ( 1.7)
- to reduce the risk of major thrombotic vascular events in patients with peripheral artery disease (PAD), including patients after recent lower extremity revascularization due to symptomatic PAD ( 1.8)
DOSAGE AND ADMINISTRATION
- CAD or PAD: 2.5 mg orally twice daily with or without food, in combination with aspirin (75 to 100 mg) once daily ( 2.1)
DOSAGE FORMS AND STRENGTHS
- Tablets: 2.5 mg ( 3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Risk of bleeding: Rivaroxaban can cause serious and fatal bleeding. An agent to reverse the activity of rivaroxaban is available. ( 5.2)
- Pregnancy-related hemorrhage: Use rivaroxaban with caution in pregnant women due to the potential for obstetric hemorrhage and/or emergent delivery. ( 5.7, 8.1)
- Prosthetic heart valves: Rivaroxaban use not recommended. ( 5.8)
- Increased Risk of Thrombosis in Patients with Triple Positive Antiphospholipid Syndrome: Rivaroxaban use not recommended. ( 5.10)
ADVERSE REACTIONS
- The most common adverse reaction (>5%) in adult patients was bleeding. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Sun Pharmaceutical Industries, Inc. at 1-866-923-4914 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 11/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: (A) PREMATURE DISCONTINUATION OF RIVAROXABAN INCREASES THE RISK OF THROMBOTIC EVENTS, (B) SPINAL/EPIDURAL HEMATOMA
1 INDICATIONS AND USAGE
1.7 Reduction of Risk of Major Cardiovascular Events in Patients with Coronary Artery Disease (CAD)
1.8 Reduction of Risk of Major Thrombotic Vascular Events in Patients with Peripheral Artery Disease (PAD), Including Patients after Lower Extremity Revascularization due to Symptomatic PAD
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Adults
2.3 Switching to and from Rivaroxaban
2.4 Discontinuation for Surgery and other Interventions
2.5 Missed Dose
2.6 Administration Options
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Increased Risk of Thrombotic Events after Premature Discontinuation
5.2 Risk of Bleeding
5.3 Spinal/Epidural Anesthesia or Puncture
5.4 Use in Patients with Renal Impairment
5.5 Use in Patients with Hepatic Impairment
5.6 Use with P-gp and Strong CYP3A Inhibitors or Inducers
5.7 Risk of Pregnancy-Related Hemorrhage
5.8 Patients with Prosthetic Heart Valves
5.9 Acute PE in Hemodynamically Unstable Patients or Patients Who Require Thrombolysis or Pulmonary Embolectomy
5.10 Increased Risk of Thrombosis in Patients with Triple Positive Antiphospholipid Syndrome
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 General Inhibition and Induction Properties
7.2 Drugs that Inhibit Cytochrome P450 3A Enzymes and Drug Transport Systems
7.3 Drugs that Induce Cytochrome P450 3A Enzymes and Drug Transport Systems
7.4 Anticoagulants and NSAIDs/Aspirin
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 QT/QTc Prolongation
13 NON-CLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.6 Reduction of Risk of Major Cardiovascular Events in Patients with CAD
14.7 Reduction of Risk of Major Thrombotic Vascular Events in Patients with PAD, Including Patients after Lower Extremity Revascularization due to Symptomatic PAD
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: (A) PREMATURE DISCONTINUATION OF RIVAROXABAN INCREASES THE RISK OF THROMBOTIC EVENTS, (B) SPINAL/EPIDURAL HEMATOMA
A. Premature discontinuation of rivaroxaban increases the risk of thrombotic events
Premature discontinuation of any oral anticoagulant, including rivaroxaban, increases the risk of thrombotic events. If anticoagulation with rivaroxaban is discontinued for a reason other than pathological bleeding or completion of a course of therapy, consider coverage with another anticoagulant [see Dosage and Administration (2.3, 2.4), Warnings and Precautions (5.1)] .
B. Spinal/epidural hematoma
Epidural or spinal hematomas have occurred in patients treated with rivaroxaban who are receiving neuraxial anesthesia or undergoing spinal puncture. These hematomas may result in long-term or permanent paralysis. Consider these risks when scheduling patients for spinal procedures. Factors that can increase the risk of developing epidural or spinal hematomas in these patients include:
- use of indwelling epidural catheters
- concomitant use of other drugs that affect hemostasis, such as non-steroidal anti-inflammatory drugs (NSAIDs), platelet inhibitors, other anticoagulants
- a history of traumatic or repeated epidural or spinal punctures
- a history of spinal deformity or spinal surgery
- optimal timing between the administration of rivaroxaban and neuraxial procedures is not known [see Warnings and Precautions (5.2, 5.3)and Adverse Reactions (6.2)] .
Monitor patients frequently for signs and symptoms of neurological impairment. If neurological compromise is noted, urgent treatment is necessary [see Warnings and Precautions (5.3)] .
Consider the benefits and risks before neuraxial intervention in patients anticoagulated or to be anticoagulated for thromboprophylaxis [see Warnings and Precautions (5.3)] .
-
1 INDICATIONS AND USAGE
1.7 Reduction of Risk of Major Cardiovascular Events in Patients with Coronary Artery Disease (CAD)
Rivaroxaban tablets, in combination with aspirin, are indicated to reduce the risk of major cardiovascular events (cardiovascular death, myocardial infarction, and stroke) in adult patients with coronary artery disease.
1.8 Reduction of Risk of Major Thrombotic Vascular Events in Patients with Peripheral Artery Disease (PAD), Including Patients after Lower Extremity Revascularization due to Symptomatic PAD
Rivaroxaban tablets, in combination with aspirin, are indicated to reduce the risk of major thrombotic vascular events (myocardial infarction, ischemic stroke, acute limb ischemia, and major amputation of a vascular etiology) in adult patients with PAD, including patients who have recently undergone a lower extremity revascularization procedure due to symptomatic PAD.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Adults
Table 1: Recommended Dosage in Adults Indication Renal Considerations * Dosage Food/Timing - * Calculate CrCl based on actual weight. [See Warnings and Precautions (5.4) and Use in Specific Populations (8.6)]
Reduction of Risk of Major Cardiovascular Events (CV Death, MI, and Stroke) in CAD No dose adjustment needed based on CrCl 2.5 mg twice daily, plus aspirin (75 to 100 mg) once daily Take with or without food Reduction of Risk of Major Thrombotic Vascular Events in PAD, Including Patients after Lower Extremity Revascularization due to Symptomatic PAD No dose adjustment needed based on CrCl 2.5 mg twice daily, plus aspirin (75 to 100 mg) once daily
When starting therapy after a successful lower extremity revascularization procedure, initiate once hemostasis has been established.Take with or without food 2.3 Switching to and from Rivaroxaban
Switching from Warfarin to Rivaroxaban– When switching patients from warfarin to rivaroxaban, discontinue warfarin and start rivaroxaban as soon as the International Normalized Ratio (INR) is below 3.0 in adults and below 2.5 in pediatric patients to avoid periods of inadequate anticoagulation.
Switching from Rivaroxaban to Warfarin–
- Adults:
No clinical trial data are available to guide converting patients from rivaroxaban to warfarin. Rivaroxaban affects INR, so INR measurements made during coadministration with warfarin may not be useful for determining the appropriate dose of warfarin. One approach is to discontinue rivaroxaban and begin both a parenteral anticoagulant and warfarin at the time the next dose of rivaroxaban would have been taken.
Switching from Rivaroxaban to Anticoagulants other than Warfarin- For adult patients currently taking rivaroxaban and transitioning to an anticoagulant with rapid onset, discontinue rivaroxaban and give the first dose of the other anticoagulant (oral or parenteral) at the time that the next rivaroxaban dose would have been taken [see Drug Interactions (7.4)].
Switching from Anticoagulants other than Warfarin to Rivaroxaban- For adult patients currently receiving an anticoagulant other than warfarin, start rivaroxaban 0 to 2 hours prior to the next scheduled administration of the drug (e.g., low molecular weight heparin or non-warfarin oral anticoagulant) and omit administration of the other anticoagulant. For unfractionated heparin being administered by continuous infusion, stop the infusion and start rivaroxaban at the same time.
2.4 Discontinuation for Surgery and other Interventions
If anticoagulation must be discontinued to reduce the risk of bleeding with surgical or other procedures, rivaroxaban should be stopped at least 24 hours before the procedure to reduce the risk of bleeding [see Warnings and Precautions (5.2)] . In deciding whether a procedure should be delayed until 24 hours after the last dose of rivaroxaban, the increased risk of bleeding should be weighed against the urgency of intervention. Rivaroxaban should be restarted after the surgical or other procedures as soon as adequate hemostasis has been established, noting that the time to onset of therapeutic effect is short [see Warnings and Precautions (5.1)] . If oral medication cannot be taken during or after surgical intervention, consider administering a parenteral anticoagulant.
2.5 Missed Dose
2.6 Administration Options
For adult patients who are unable to swallow whole tablets, rivaroxaban tablets (2.5 mg) may be crushed and mixed with applesauce immediately prior to use and administered orally. Administration with food is not required for the 2.5 mg [see Clinical Pharmacology (12.3)] .
Administration of rivaroxaban tablets via nasogastric (NG) tube or gastric feeding tube:After confirming gastric placement of the tube, rivaroxaban tablets (2.5 mg) may be crushed and suspended in 50 mL of water and administered via an NG tube or gastric feeding tube. Since rivaroxaban absorption is dependent on the site of drug release, avoid administration of rivaroxaban distal to the stomach which can result in reduced absorption and thereby, reduced drug exposure. Enteral feeding is not required following administration of the 2.5 mg tablets [see Clinical Pharmacology (12.3)] .
Crushed rivaroxaban tablets (2.5 mg) are stable in water and in applesauce for up to 4 hours. An in vitrocompatibility study indicated that there is no adsorption of rivaroxaban from a water suspension of a crushed rivaroxaban tablet to PVC or silicone nasogastric (NG) tubing.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Rivaroxaban tablets are contraindicated in patients with:
- active pathological bleeding [see Warnings and Precautions (5.2)]
- severe hypersensitivity reaction to rivaroxaban (e.g., anaphylactic reactions) [see Adverse Reactions (6.2)]
-
5 WARNINGS AND PRECAUTIONS
5.1 Increased Risk of Thrombotic Events after Premature Discontinuation
Premature discontinuation of any oral anticoagulant, including rivaroxaban, in the absence of adequate alternative anticoagulation increases the risk of thrombotic events. An increased rate of stroke was observed during the transition from rivaroxaban to warfarin in clinical trials in another indication in patients. If rivaroxaban is discontinued for a reason other than pathological bleeding or completion of a course of therapy, consider coverage with another anticoagulant [see Dosage and Administration (2.3, 2.4)] .
5.2 Risk of Bleeding
Rivaroxaban increases the risk of bleeding and can cause serious or fatal bleeding. In deciding whether to prescribe rivaroxaban to patients at increased risk of bleeding, the risk of thrombotic events should be weighed against the risk of bleeding.
Promptly evaluate any signs or symptoms of blood loss and consider the need for blood replacement. Discontinue rivaroxaban in patients with active pathological hemorrhage. The terminal elimination half-life of rivaroxaban is 5 to 9 hours in healthy subjects aged 20 to 45 years.
Concomitant use of other drugs that impair hemostasis increases the risk of bleeding. These include aspirin, P2Y 12platelet inhibitors, dual antiplatelet therapy, other antithrombotic agents, fibrinolytic therapy, non-steroidal anti-inflammatory drugs (NSAIDs) [see Drug Interactions (7.4)] , selective serotonin reuptake inhibitors, and serotonin norepinephrine reuptake inhibitors.
Concomitant use of drugs that are known combined P-gp and strong CYP3A inhibitors increases rivaroxaban exposure and may increase bleeding risk [see Drug Interactions (7.2)] .
Risk of Hemorrhage in Acutely Ill Medical Patients at High Risk of Bleeding
Acutely ill medical patients with the following conditions are at increased risk of bleeding with the use of rivaroxaban for another indication: history of bronchiectasis, pulmonary cavitation, or pulmonary hemorrhage, active cancer (i.e., undergoing acute, in-hospital cancer treatment), active gastroduodenal ulcer in the three months prior to treatment, history of bleeding in the three months prior to treatment, or dual antiplatelet therapy. Rivaroxaban is not for use for another indication in these hospitalized, acutely ill medical patients at high risk of bleeding.
Reversal of Anticoagulant Effect
An agent to reverse the anti-factor Xa activity of rivaroxaban is available. Because of high plasma protein binding, rivaroxaban is not dialyzable [see Clinical Pharmacology (12.3)] . Protamine sulfate and vitamin K are not expected to affect the anticoagulant activity of rivaroxaban. Use of procoagulant reversal agents, such as prothrombin complex concentrate (PCC), activated prothrombin complex concentrate or recombinant factor VIIa, may be considered but has not been evaluated in clinical efficacy and safety studies. Monitoring for the anticoagulation effect of rivaroxaban using a clotting test (PT, INR or aPTT) or anti-factor Xa (FXa) activity is not recommended.
5.3 Spinal/Epidural Anesthesia or Puncture
When neuraxial anesthesia (spinal/epidural anesthesia) or spinal puncture is employed, patients treated with anticoagulant agents for prevention of thromboembolic complications are at risk of developing an epidural or spinal hematoma which can result in long-term or permanent paralysis [see Boxed Warning] .
To reduce the potential risk of bleeding associated with the concurrent use of rivaroxaban and epidural or spinal anesthesia/analgesia or spinal puncture, consider the pharmacokinetic profile of rivaroxaban [see Clinical Pharmacology (12.3)] . Placement or removal of an epidural catheter or lumbar puncture is best performed when the anticoagulant effect of rivaroxaban is low; however, the exact timing to reach a sufficiently low anticoagulant effect in each patient is not known.
An indwelling epidural or intrathecal catheter should not be removed before at least 2 half-lives have elapsed (i.e., 18 hours in young patients aged 20 to 45 years and 26 hours in elderly patients aged 60 to 76 years), after the last administration of rivaroxaban [see Clinical Pharmacology (12.3)] . The next rivaroxaban dose should not be administered earlier than 6 hours after the removal of the catheter. If traumatic puncture occurs, delay the administration of rivaroxaban for 24 hours.
Should the physician decide to administer anticoagulation in the context of epidural or spinal anesthesia/analgesia or lumbar puncture, monitor frequently to detect any signs or symptoms of neurological impairment, such as midline back pain, sensory and motor deficits (numbness, tingling, or weakness in lower limbs), bowel and/or bladder dysfunction. Instruct patients to immediately report if they experience any of the above signs or symptoms. If signs or symptoms of spinal hematoma are suspected, initiate urgent diagnosis and treatment including consideration for spinal cord decompression even though such treatment may not prevent or reverse neurological sequelae.
5.4 Use in Patients with Renal Impairment
Pediatric Patients
There are no clinical data in pediatric patients younger than 1 year with serum creatinine results above 97.5 thpercentile; therefore, avoid the use of rivaroxaban in these patients [see Use in Specific Populations (8.6)] .
5.5 Use in Patients with Hepatic Impairment
No clinical data are available for adult patients with severe hepatic impairment.
Avoid use of rivaroxaban in patients with moderate (Child-Pugh B) and severe (Child-Pugh C) hepatic impairment or with any hepatic disease associated with coagulopathy since drug exposure and bleeding risk may be increased [see Use in Specific Populations (8.7)].
No clinical data are available in pediatric patients with hepatic impairment.
5.6 Use with P-gp and Strong CYP3A Inhibitors or Inducers
Avoid concomitant use of rivaroxaban with known combined P-gp and strong CYP3A inhibitors [see Drug Interactions (7.2)].
Avoid concomitant use of rivaroxaban with drugs that are known combined P-gp and strong CYP3A inducers [see Drug Interactions (7.3)].
5.7 Risk of Pregnancy-Related Hemorrhage
In pregnant women, rivaroxaban should be used only if the potential benefit justifies the potential risk to the mother and fetus. Rivaroxaban dosing in pregnancy has not been studied. The anticoagulant effect of rivaroxaban cannot be monitored with standard laboratory testing. Promptly evaluate any signs or symptoms suggesting blood loss (e.g., a drop in hemoglobin and/or hematocrit, hypotension, or fetal distress) [see Warnings and Precautions (5.2)and Use in Specific Populations (8.1)] .
5.8 Patients with Prosthetic Heart Valves
On the basis of the GALILEO study, use of rivaroxaban is not recommended in patients who have had transcatheter aortic valve replacement (TAVR) because patients randomized to rivaroxaban experienced higher rates of death and bleeding compared to those randomized to an anti-platelet regimen. The safety and efficacy of rivaroxaban have not been studied in patients with other prosthetic heart valves or other valve procedures. Use of rivaroxaban is not recommended in patients with prosthetic heart valves.
5.9 Acute PE in Hemodynamically Unstable Patients or Patients Who Require Thrombolysis or Pulmonary Embolectomy
Initiation of rivaroxaban is not recommended acutely as an alternative to unfractionated heparin in patients with pulmonary embolism who present with hemodynamic instability or who may receive thrombolysis or pulmonary embolectomy.
5.10 Increased Risk of Thrombosis in Patients with Triple Positive Antiphospholipid Syndrome
Direct-acting oral anticoagulants (DOACs), including rivaroxaban, are not recommended for use in patients with triple-positive antiphospholipid syndrome (APS). For patients with APS (especially those who are triple positive [positive for lupus anticoagulant, anticardiolipin, and anti-beta 2-glycoprotein I antibodies]), treatment with DOACs has been associated with increased rates of recurrent thrombotic events compared with vitamin K antagonist therapy.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are also discussed in other sections of the labeling:
- Increased Risk of Stroke After Discontinuation in Another Indication [see Boxed Warningand Warnings and Precautions (5.1)]
- Bleeding Risk [see Warnings and Precautions (5.2, 5.4, 5.5, 5.6, 5.7)]
- Spinal/Epidural Hematoma [see Boxed Warningand Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Hemorrhage
The most common adverse reactions with rivaroxaban were bleeding complications [see Warnings and Precautions (5.2)] .
Reduction of Risk of Major Cardiovascular Events in Patients with CAD
In the COMPASS trial overall, the most frequent adverse reactions associated with permanent drug discontinuation
were bleeding events, with incidence rates of 2.7% for rivaroxaban 2.5 mg twice daily vs. 1.2% for placebo on
background therapy for all patients with aspirin 100 mg once daily. The incidences of important bleeding events in
the CAD and PAD populations in COMPASS were similar.
Table 10 shows the number of patients experiencing various types of major bleeding events in the COMPASS trial.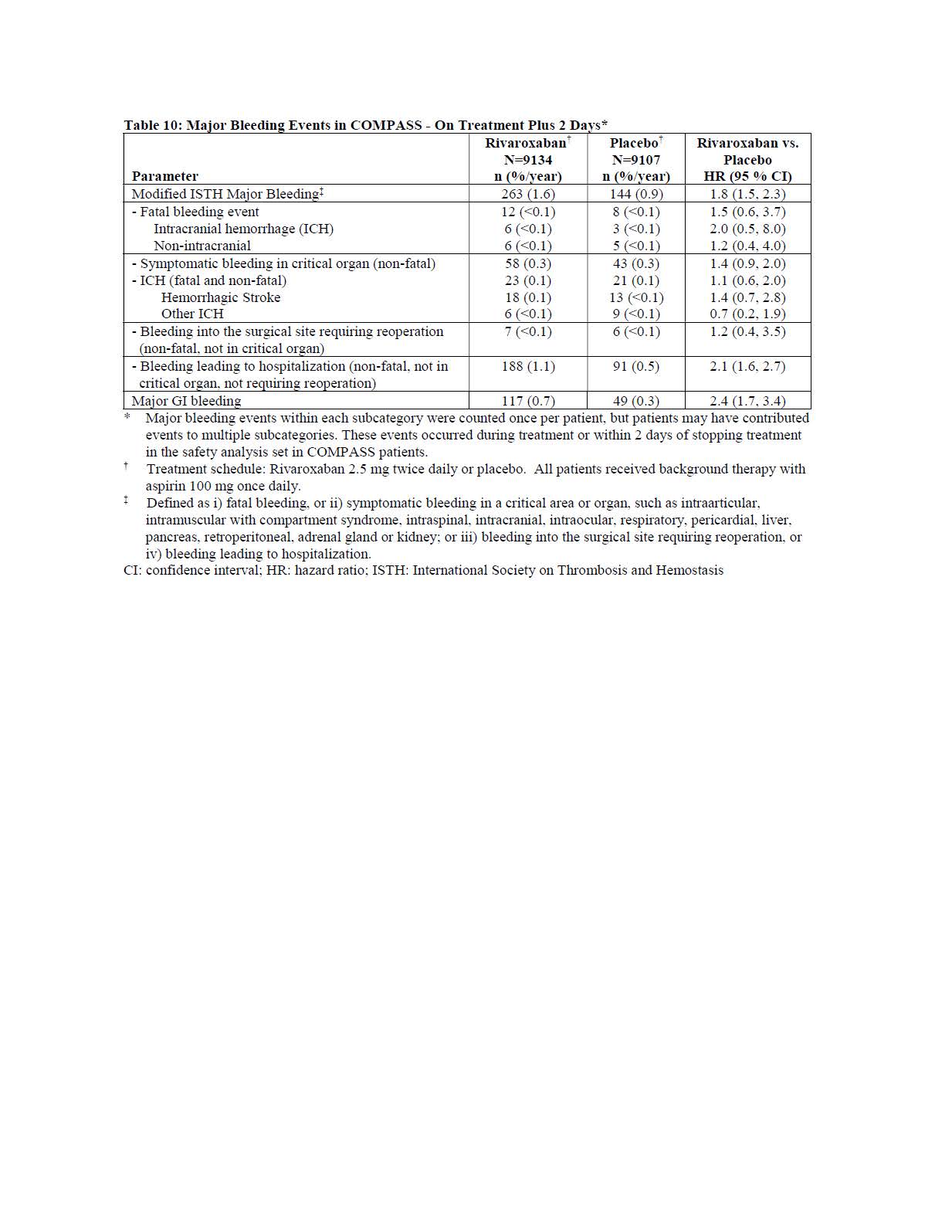
Reduction of Risk of Major Thrombotic Vascular Events in Patients with Peripheral Artery Disease (PAD), Including Patients after Lower Extremity Revascularization due to Symptomatic PAD
The incidence of premature permanent discontinuation due to bleeding events for rivaroxaban 2.5 mg twice daily vs. placebo on background therapy with aspirin 100 mg once daily in VOYAGER was 4.1% vs. 1.6% and in COMPASS PAD was 2.7% vs. 1.3%, respectively.
Table 11 shows the number of patients experiencing various types of TIMI (Thrombolysis in Myocardial Infarction) major bleeding events in the VOYAGER trial. The most common site of bleeding was gastrointestinal.
Table 11: Major Bleeding Events *in VOYAGER - On Treatment Plus 2 Days Rivaroxaban †
N=3256Placebo †
N=3248Rivaroxaban vs. Placebo
HR (95 % CI)Parameter n (%) Event rate
%/yearn (%) Event rate
%/yearCABG: Coronary artery bypass graft; CI: confidence interval; HR: hazard ratio; TIMI: Thrombolysis in Myocardial Infarction Bleeding Criteria - * Major bleeding events within each subcategory were counted once per patient, but patients may have contributed events to multiple subcategories.
- † Treatment schedule: Rivaroxaban 2.5 mg twice daily or placebo. All patients received background therapy with aspirin 100 mg once daily.
TIMI Major Bleeding
(CABG/non-CABG)62 (1.9) 0.96 44 (1.4) 0.67 1.4 (1.0, 2.1) Fatal bleeding 6 (0.2) 0.09 6 (0.2) 0.09 1.0 (0.3, 3.2) Intracranial bleeding 13 (0.4) 0.20 17 (0.5) 0.26 0.8 (0.4, 1.6) Clinically overt signs of hemorrhage associated with a drop in hemoglobin of ≥5 g/dL or drop in hematocrit of ≥15% 46 (1.4) 0.71 24 (0.7) 0.36 1.9 (1.2, 3.2) 6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of rivaroxaban. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic system disorders:agranulocytosis, thrombocytopenia
Hepatobiliary disorders:jaundice, cholestasis, hepatitis (including hepatocellular injury)
Immune system disorders:hypersensitivity, anaphylactic reaction, anaphylactic shock, angioedema
Nervous system disorders:hemiparesis
Renal disorders:Anticoagulant-related nephropathy
Respiratory, thoracic and mediastinal disorders:Eosinophilic pneumonia
Skin and subcutaneous tissue disorders:Stevens-Johnson syndrome, drug reaction with eosinophilia and systemic symptoms (DRESS)
Injury, poisoning and procedural complications:Atraumatic splenic rupture
-
7 DRUG INTERACTIONS
7.1 General Inhibition and Induction Properties
Rivaroxaban is a substrate of CYP3A4/5, CYP2J2, and the P-gp and ATP-binding cassette G2 (ABCG2) transporters. Combined P-gp and strong CYP3A inhibitors increase exposure to rivaroxaban and may increase the risk of bleeding. Combined P-gp and strong CYP3A inducers decrease exposure to rivaroxaban and may increase the risk of thromboembolic events.
7.2 Drugs that Inhibit Cytochrome P450 3A Enzymes and Drug Transport Systems
Interaction with Combined P-gp and Strong CYP3A Inhibitors
Avoid concomitant administration of rivaroxaban with known combined P-gp and strong CYP3A inhibitors (e.g., ketoconazole and ritonavir) [see Warnings and Precautions (5.6)and Clinical Pharmacology (12.3)] .
Although clarithromycin is a combined P-gp and strong CYP3A inhibitor, pharmacokinetic data suggests that no precautions are necessary with concomitant administration with rivaroxaban as the change in exposure is unlikely to affect the bleeding risk [see Clinical Pharmacology (12.3)] .
Interaction with Combined P-gp and Moderate CYP3A Inhibitors in Patients with Renal Impairment
Rivaroxaban should not be used in patients with CrCl 15 to <80 mL/min who are receiving concomitant combined P-gp and moderate CYP3A inhibitors (e.g., erythromycin) unless the potential benefit justifies the potential risk [see Warnings and Precautions (5.4)and Clinical Pharmacology (12.3)] .
7.3 Drugs that Induce Cytochrome P450 3A Enzymes and Drug Transport Systems
Avoid concomitant use of rivaroxaban with drugs that are combined P-gp and strong CYP3A inducers (e.g., carbamazepine, phenytoin, rifampin, St. John's wort) [see Warnings and Precautions (5.6)and Clinical Pharmacology (12.3)] .
7.4 Anticoagulants and NSAIDs/Aspirin
Coadministration of enoxaparin, warfarin, aspirin, clopidogrel and chronic NSAID use may increase the risk of bleeding [see Clinical Pharmacology (12.3)] .
Avoid concurrent use of rivaroxaban with other anticoagulants due to increased bleeding risk unless benefit outweighs risk. Promptly evaluate any signs or symptoms of blood loss if patients are treated concomitantly with aspirin, other platelet aggregation inhibitors, or NSAIDs [see Warnings and Precautions (5.2)] .
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The limited available data on rivaroxaban in pregnant women are insufficient to inform a drug-associated risk of adverse developmental outcomes. Use rivaroxaban with caution in pregnant patients because of the potential for pregnancy related hemorrhage and/or emergent delivery. The anticoagulant effect of rivaroxaban cannot be reliably monitored with standard laboratory testing. Consider the benefits and risks of rivaroxaban for the mother and possible risks to the fetus when prescribing rivaroxaban to a pregnant woman [see Warnings and Precautions (5.2, 5.7)] .
Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Pregnancy is a risk factor for venous thromboembolism and that risk is increased in women with inherited or acquired thrombophilias. Pregnant women with thromboembolic disease have an increased risk of maternal complications including pre-eclampsia. Maternal thromboembolic disease increases the risk for intrauterine growth restriction, placental abruption and early and late pregnancy loss.
Fetal/Neonatal Adverse Reactions
Based on the pharmacologic activity of Factor Xa inhibitors and the potential to cross the placenta, bleeding may occur at any site in the fetus and/or neonate.
Labor or Delivery
All patients receiving anticoagulants, including pregnant women, are at risk for bleeding and this risk may be increased during labor or delivery [see Warnings and Precautions (5.7)] . The risk of bleeding should be balanced with the risk of thrombotic events when considering the use of rivaroxaban in this setting.
Data
Human Data
There are no adequate or well-controlled studies of rivaroxaban in pregnant women, and dosing for pregnant women has not been established. Post-marketing experience is currently insufficient to determine a rivaroxaban-associated risk for major birth defects or miscarriage. In an in vitroplacenta perfusion model, unbound rivaroxaban was rapidly transferred across the human placenta.
Animal Data
Rivaroxaban crosses the placenta in animals. Rivaroxaban increased fetal toxicity (increased resorptions, decreased number of live fetuses, and decreased fetal body weight) when pregnant rabbits were given oral doses of ≥10 mg/kg rivaroxaban during the period of organogenesis. This dose corresponds to about 4 times the human exposure of unbound drug, based on AUC comparisons at the highest recommended human dose of 20 mg/day. Fetal body weights decreased when pregnant rats were given oral doses of 120 mg/kg during the period of organogenesis. This dose corresponds to about 14 times the human exposure of unbound drug. In rats, peripartal maternal bleeding and maternal and fetal death occurred at the rivaroxaban dose of 40 mg/kg (about 6 times maximum human exposure of the unbound drug at the human dose of 20 mg/day).
8.2 Lactation
Risk Summary
Rivaroxaban has been detected in human milk. There are insufficient data to determine the effects of rivaroxaban on the breastfed child or on milk production. Rivaroxaban and/or its metabolites were present in the milk of rats. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for rivaroxaban and any potential adverse effects on the breastfed infant from rivaroxaban or from the underlying maternal condition (see Data) .
Data
Animal Data
Following a single oral administration of 3 mg/kg of radioactive [ 14C]-rivaroxaban to lactating rats between Day 8 to 10 postpartum, the concentration of total radioactivity was determined in milk samples collected up to 32 hours post-dose. The estimated amount of radioactivity excreted with milk within 32 hours after administration was 2.1% of the maternal dose.
8.3 Females and Males of Reproductive Potential
Females of reproductive potential requiring anticoagulation should discuss pregnancy planning with their physician.
The risk of clinically significant uterine bleeding, potentially requiring gynecological surgical interventions, identified with oral anticoagulants including rivaroxaban should be assessed in females of reproductive potential and those with abnormal uterine bleeding.
8.4 Pediatric Use
For the rivaroxaban 2.5 mg tablets, there are no safety, efficacy, pharmacokinetic and pharmacodynamic data to support the use in pediatric patients. Therefore, rivaroxaban 2.5 mg tablets are not recommended for use in pediatric patients.
Although not all adverse reactions identified in the adult population have been observed in clinical trials of children and adolescent patients, the same warnings and precautions for adults should be considered for children and adolescents.
8.5 Geriatric Use
Of the total number of adult patients in clinical trials for the approved indications of rivaroxaban (N=64,943 patients), 64 percent were 65 years and over, with 27 percent 75 years and over. In clinical trials the efficacy of rivaroxaban in the elderly (65 years or older) was similar to that seen in patients younger than 65 years. Both thrombotic and bleeding event rates were higher in these older patients [see Clinical Pharmacology (12.3)and Clinical Studies (14)] .
8.6 Renal Impairment
In pharmacokinetic studies, compared to healthy adult subjects with normal creatinine clearance, rivaroxaban exposure increased by approximately 44 to 64% in adult subjects with renal impairment. Increases in pharmacodynamic effects were also observed [see Clinical Pharmacology (12.3)] .
Reduction of Risk of Major Cardiovascular Events in Patients with CAD and Reduction of Risk of Major Thrombotic Vascular Events in Patients with PAD, Including Patients After Recent Lower Extremity Revascularization due to Symptomatic PAD
Patients with Chronic Kidney Disease not on Dialysis
Patients with a CrCl <15 mL/min at screening were excluded from COMPASS and VOYAGER, and limited data are available for patients with a CrCl of 15 to 30 mL/min. In patients with CrCl <30 mL/min, a dose of 2.5 mg rivaroxaban twice daily is expected to give an exposure similar to that in patients with moderate renal impairment (CrCl 30 to <50 mL/min) [see Clinical Pharmacology (12.3)] , whose efficacy and safety outcomes were similar to those with preserved renal function.
Patients with End-Stage Renal Disease on Dialysis
No clinical outcome data is available for the use of rivaroxaban with aspirin in patients with ESRD on dialysis since these patients were not enrolled in COMPASS or VOYAGER. In patients with ESRD maintained on intermittent hemodialysis, administration of rivaroxaban 2.5 mg twice daily will result in concentrations of rivaroxaban and pharmacodynamic activity similar to those observed in moderate renal impaired patients in the COMPASS study [see Clinical Pharmacology (12.2, 12.3)] . It is not known whether these concentrations will lead to similar CV risk reduction and bleeding risk in patients with ESRD on dialysis as was seen in COMPASS.
Pediatric Use
There are no clinical data in pediatric patients younger than 1 year with serum creatinine results above 97.5 thpercentile; therefore, avoid the use of rivaroxaban in these patients [see Dosage and Administration (2.2)] .
8.7 Hepatic Impairment
In a pharmacokinetic study, compared to healthy adult subjects with normal liver function, AUC increases of 127% were observed in adult subjects with moderate hepatic impairment (Child-Pugh B).
The safety or PK of rivaroxaban in patients with severe hepatic impairment (Child-Pugh C) has not been evaluated [see Clinical Pharmacology (12.3)] .
Avoid the use of rivaroxaban in patients with moderate (Child-Pugh B) and severe (Child-Pugh C) hepatic impairment or with any hepatic disease associated with coagulopathy.
No clinical data are available in pediatric patients with hepatic impairment.
-
10 OVERDOSAGE
Overdose of rivaroxaban may lead to hemorrhage. Discontinue rivaroxaban and initiate appropriate therapy if bleeding complications associated with overdosage occur. Rivaroxaban systemic exposure is not further increased at single doses >50 mg due to limited absorption. The use of activated charcoal to reduce absorption in case of rivaroxaban overdose may be considered. Due to the high plasma protein binding, rivaroxaban is not dialyzable [see Warnings and Precautions (5.2)and Clinical Pharmacology (12.3)] . Partial reversal of laboratory anticoagulation parameters may be achieved with use of plasma products. An agent to reverse the anti-factor Xa activity of rivaroxaban is available.
-
11 DESCRIPTION
Rivaroxaban, USP, a factor Xa (FXa) inhibitor, is the active ingredient in rivaroxaban tablets, USP with the chemical name 5-Chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)phenyl]-1,3-oxazolidin-5-yl}methyl)-2-thiophenecarboxamide. The molecular formula of rivaroxaban, USP is C 19H 18ClN 3O 5S and the molecular weight is 435.89. The structural formula is:
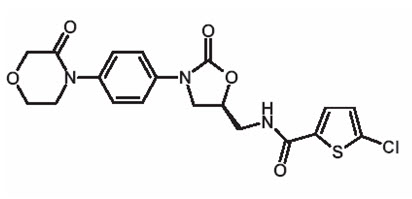
Rivaroxaban, USP is a pure ( S)-enantiomer. It is an odorless, non-hygroscopic, white to yellowish powder. Rivaroxaban, USP is only slightly soluble in organic solvents (e.g., acetone, polyethylene glycol 400) and is practically insoluble in water and aqueous media.
Each rivaroxaban tablet, USP contains 2.5 mg of rivaroxaban, USP. The inactive ingredients of rivaroxaban tablets, USP are: croscarmellose sodium, hydroxypropyl cellulose, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. Additionally, the film coating mixture for rivaroxaban 2.5 mg tablets, USP contains: ferric oxide yellow, ferrosoferric oxide, hypromellose, lactose monohydrate, polyethylene glycol 3350, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Rivaroxaban is a selective inhibitor of FXa. It does not require a cofactor (such as Anti-thrombin III) for activity. Rivaroxaban inhibits free FXa and prothrombinase activity. Rivaroxaban has no direct effect on platelet aggregation, but indirectly inhibits platelet aggregation induced by thrombin. By inhibiting FXa, rivaroxaban decreases thrombin generation.
12.2 Pharmacodynamics
Rivaroxaban produces dose-dependent inhibition of FXa activity. Clotting tests, such as prothrombin time (PT), activated partial thromboplastin time (aPTT) and HepTest ®, are also prolonged dose-dependently. In children treated with rivaroxaban, the correlation between anti-factor Xa to plasma concentrations is linear with a slope close to 1.
Monitoring for anticoagulation effect of rivaroxaban using anti-FXa activity or a clotting test is not recommended.
Specific Populations
Renal Impairment
The relationship between systemic exposure and pharmacodynamic activity of rivaroxaban was altered in adult subjects with renal impairment relative to healthy control subjects [see Use in Specific Populations (8.6)] .
Table 18: Percentage Increase in Rivaroxaban PK and PD Measures in Adult Subjects with Renal Impairment Relative to Healthy Subjects from Clinical Pharmacology Studies Measure Parameter Creatinine Clearance (mL/min) 50 to 79 30 to 49 15 to 29 ESRD
(on dialysis) *ESRD
(post-dialysis) *PT = Prothrombin time; FXa = Coagulation factor Xa; AUC = Area under the plasma concentration-time curve; AUEC = Area under the effect-time curve - * Separate stand-alone study.
Exposure AUC 44 52 64 47 56 FXa Inhibition AUEC 50 86 100 49 33 PT Prolongation AUEC 33 116 144 112 158 Hepatic Impairment
Anti-Factor Xa activity was similar in adult subjects with normal hepatic function and in mild hepatic impairment (Child-Pugh A class). There is no clear understanding of the impact of hepatic impairment beyond this degree on the coagulation cascade and its relationship to efficacy and safety.
12.3 Pharmacokinetics
Absorption
The absolute bioavailability of rivaroxaban is dose-dependent. For the 2.5 mg dose, it is estimated to be 80% to 100% and is not affected by food. Rivaroxaban 2.5 mg tablets can be taken with or without food.
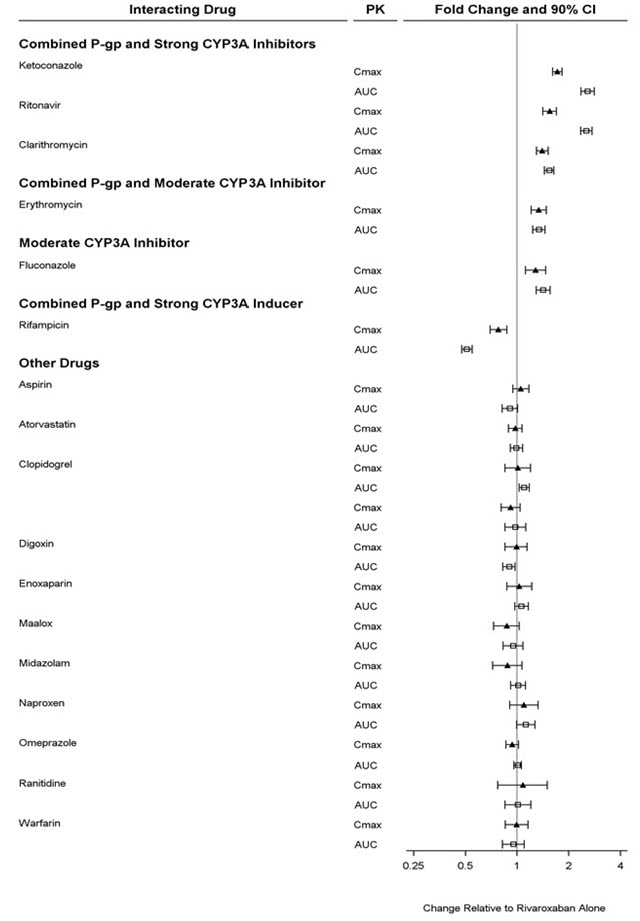
The maximum concentrations (C max) of rivaroxaban appear 2 to 4 hours after tablet intake. The pharmacokinetics of rivaroxaban were not affected by drugs altering gastric pH. Coadministration of rivaroxaban (30 mg single dose) with the H 2-receptor antagonist ranitidine (150 mg twice daily), the antacid aluminum hydroxide/magnesium hydroxide (10 mL) or rivaroxaban (20 mg single dose) with the PPI omeprazole (40 mg once daily) did not show an effect on the bioavailability and exposure of rivaroxaban (see Figure 3).
Absorption of rivaroxaban is dependent on the site of drug release in the GI tract. A 29% and 56% decrease in AUC and C maxcompared to tablet was reported when rivaroxaban granulate is released in the proximal small intestine. Exposure is further reduced when drug is released in the distal small intestine, or ascending colon. Avoid administration of rivaroxaban distal to the stomach which can result in reduced absorption and related drug exposure.
In a study with 44 healthy subjects, both mean AUC and C maxvalues for 20 mg rivaroxaban administered orally as a crushed tablet mixed in applesauce were comparable to that after the whole tablet. However, for the crushed tablet suspended in water and administered via an NG tube followed by a liquid meal, only mean AUC was comparable to that after the whole tablet, and C maxwas 18% lower.
Distribution
Protein binding of rivaroxaban in human plasma is approximately 92% to 95%, with albumin being the main binding component. The steady-state volume of distribution in healthy subjects is approximately 50 L.
Metabolism
Approximately 51% of an orally administered [ 14C]-rivaroxaban dose was recovered as inactive metabolites in urine (30%) and feces (21%). Oxidative degradation catalyzed by CYP3A4/5 and CYP2J2 and hydrolysis are the major sites of biotransformation. Unchanged rivaroxaban was the predominant moiety in plasma with no major or active circulating metabolites.
Excretion
In a Phase 1 study, following the administration of [ 14C]-rivaroxaban, approximately one-third (36%) was recovered as unchanged drug in the urine and 7% was recovered as unchanged drug in feces. Unchanged drug is excreted into urine, mainly via active tubular secretion and to a lesser extent via glomerular filtration (approximate 5:1 ratio). Rivaroxaban is a substrate of the efflux transporter proteins P-gp and ABCG2 (also abbreviated BCRP). Rivaroxaban's affinity for influx transporter proteins is unknown.
Rivaroxaban is a low-clearance drug, with a systemic clearance of approximately 10 L/hr in healthy volunteers following intravenous administration. The terminal elimination half-life of rivaroxaban is 5 to 9 hours in healthy subjects aged 20 to 45 years.
Specific Populations
The effects of level of renal impairment, age, body weight, and level of hepatic impairment on the pharmacokinetics of rivaroxaban are summarized in Figure 2.
Figure 2: Effect of Specific Adult Populations on the Pharmacokinetics of Rivaroxaban
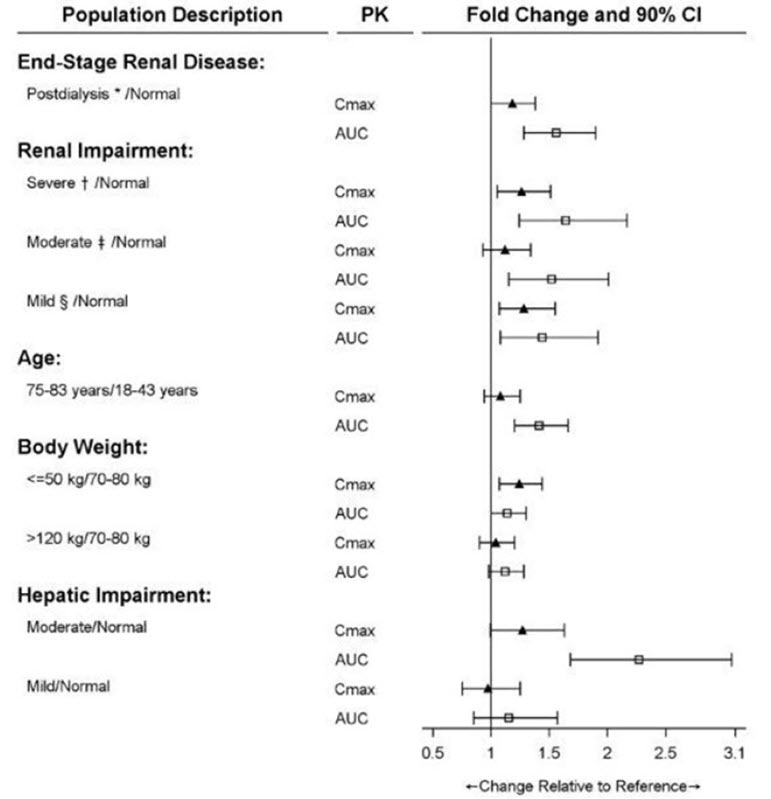
* ESRD subjects maintained with chronic and stable hemodialysis; reported PK findings are following single dose of rivaroxaban post hemodialysis.
†Creatinine clearance 15 to 29 mL/min.
‡Creatinine clearance 30 to 49 mL/min.
§Creatinine clearance 50 to 79 mL/min.
[See Dosage and Administration (2.1)]Gender
Gender did not influence the pharmacokinetics or pharmacodynamics of rivaroxaban.
Hemodialysis in ESRD subjects:Systemic exposure to rivaroxaban administered as a single 15 mg dose in ESRD subjects dosed 3 hours after the completion of a 4-hour hemodialysis session (post-dialysis) is 56% higher when compared to subjects with normal renal function (see Table 18). The systemic exposure to rivaroxaban administered 2 hours prior to a 4-hour hemodialysis session with a dialysate flow rate of 600 mL/min and a blood flow rate in the range of 320 mL/min to 400 mL/min is 47% higher compared to those with normal renal function. The extent of the increase is similar to the increase in patients with CrCl 15 mL/min to 50 mL/min taking rivaroxaban 15 mg. Hemodialysis had no significant impact on rivaroxaban exposure. Protein binding was similar (86% to 89%) in healthy controls and ESRD subjects in this study.
Pediatric Patients:Limited clinical data are available in children 1 year or older with moderate or severe renal impairment (eGFR <50 mL/min/1.73 m 2) or in children younger than 1 year with serum creatinine results above 97.5 thpercentile [see Use in Specific Populations (8.6)] .
Race
Healthy Japanese subjects were found to have 20 to 40% on average higher exposures compared to other ethnicities including Chinese. However, these differences in exposure are reduced when values are corrected for body weight.
Elderly
The terminal elimination half-life is 11 to 13 hours in the elderly subjects aged 60 to 76 years [see Use in Specific Populations (8.5)] .
Pediatric Patients
The rate and extent of absorption were similar between the tablet and suspension. After repeated administration of rivaroxaban for the treatment of another indication, the C maxof rivaroxaban in plasma was observed at median times of 1.5 to 2.2 hours in subjects who ranged from birth to less than 18 years of age.
In children who were 6 months to 9 years of age, in vitroplasma protein binding of rivaroxaban is approximately 90%.
The half-life of rivaroxaban in plasma of pediatric patients treated for another indication decreased with decreasing age. Mean half-life values were 4.2 hours in adolescents, 3 hours in children 2 to 12 years of age, 1.9 hours in children 0.5 to <2 years of age, and 1.6 hours in children <0.5 years of age.
An exploratory analysis in pediatric patients treated for another indication did not reveal relevant differences in rivaroxaban exposure based on gender or race.
Renal Impairment
The safety and pharmacokinetics of single-dose rivaroxaban (10 mg) were evaluated in a study in healthy subjects [CrCl ≥80 mL/min (n=8)] and in subjects with varying degrees of renal impairment (see Figure 2). Compared to healthy subjects with normal creatinine clearance, rivaroxaban exposure increased in subjects with renal impairment. Increases in pharmacodynamic effects were also observed [see Use in Specific Populations (8.6)] .
Hepatic Impairment
The safety and pharmacokinetics of single-dose rivaroxaban (10 mg) were evaluated in a study in healthy adult subjects (n=16) and adult subjects with varying degrees of hepatic impairment (see Figure 2). No patients with severe hepatic impairment (Child-Pugh C) were studied. Compared to healthy subjects with normal liver function, significant increases in rivaroxaban exposure were observed in subjects with moderate hepatic impairment (Child-Pugh B) (see Figure 2). Increases in pharmacodynamic effects were also observed [see Use in Specific Populations (8.7)] .
No clinical data are available in pediatric patients with hepatic impairment.
Drug Interactions
In vitrostudies indicate that rivaroxaban neither inhibits the major cytochrome P450 enzymes CYP1A2, 2C8, 2C9, 2C19, 2D6, 2J2, and 3A nor induces CYP1A2, 2B6, 2C19, or 3A. In vitrodata also indicates a low rivaroxaban inhibitory potential for P-gp and ABCG2 transporters.
The effects of coadministered drugs on the pharmacokinetics of rivaroxaban exposure are summarized in Figure 3 [see Drug Interactions (7)] .
Figure 3: Effect of Coadministered Drugs on the Pharmacokinetics of Rivaroxaban in Adults
Anticoagulants
In a drug interaction study, single doses of enoxaparin (40 mg subcutaneous) and rivaroxaban (10 mg) given concomitantly resulted in an additive effect on anti-factor Xa activity. In another study, single doses of warfarin (15 mg) and rivaroxaban (5 mg) resulted in an additive effect on factor Xa inhibition and PT. Neither enoxaparin nor warfarin affected the pharmacokinetics of rivaroxaban (see Figure 3).
NSAIDs/Aspirin
NSAIDs are known to increase bleeding, and bleeding risk may be increased when NSAIDs are used concomitantly with rivaroxaban. Neither naproxen nor aspirin affected the pharmacokinetics of rivaroxaban (see Figure 3).
Clopidogrel
In two drug interaction studies where clopidogrel (300 mg loading dose followed by 75 mg daily maintenance dose) and rivaroxaban (15 mg single dose) were coadministered in healthy subjects, an increase in bleeding time to 45 minutes was observed in approximately 45% and 30% of subjects in these studies, respectively. The change in bleeding time was approximately twice the maximum increase seen with either drug alone. There was no change in the pharmacokinetics of either drug.
Drug-Disease Interactions with Drugs that Inhibit Cytochrome P450 3A Enzymes and Drug Transport Systems
In a pharmacokinetic trial, rivaroxaban was administered as a single dose in subjects with mild (CrCl = 50 to 79 mL/min) or moderate renal impairment (CrCl = 30 to 49 mL/min) receiving multiple doses of erythromycin (a combined P-gp and moderate CYP3A inhibitor). Compared to rivaroxaban administered alone in subjects with normal renal function (CrCl >80 mL/min), subjects with mild and moderate renal impairment concomitantly receiving erythromycin reported a 76% and 99% increase in AUC infand a 56% and 64% increase in C max, respectively. Similar trends in pharmacodynamic effects were also observed.
-
13 NON-CLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Rivaroxaban was not carcinogenic when administered by oral gavage to mice or rats for up to 2 years. The systemic exposures (AUCs) of unbound rivaroxaban in male and female mice at the highest dose tested (60 mg/kg/day) were 1- and 2-times, respectively, the human exposure of unbound drug at the human dose of 20 mg/day. Systemic exposures of unbound drug in male and female rats at the highest dose tested (60 mg/kg/day) were 2- and 4-times, respectively, the human exposure.
Rivaroxaban was not mutagenic in bacteria (Ames-Test) or clastogenic in V79 Chinese hamster lung cells in vitroor in the mouse micronucleus test in vivo.
No impairment of fertility was observed in male or female rats when given up to 200 mg/kg/day of rivaroxaban orally. This dose resulted in exposure levels, based on the unbound AUC, at least 13 times the exposure in humans given 20 mg rivaroxaban daily.
-
14 CLINICAL STUDIES
14.6 Reduction of Risk of Major Cardiovascular Events in Patients with CAD
The evidence for the efficacy and safety of rivaroxaban for the reduction in the risk of stroke, myocardial infarction, or cardiovascular death in patients with coronary artery disease (CAD) or peripheral artery disease (PAD) was derived from the double-blind, placebo-controlled Cardiovascular Outco Mes for People using Anticoagulation Strategie Strial (COMPASS) [NCT10776424]. A total of 27,395 patients were evenly randomized to rivaroxaban 2.5 mg orally twice daily plus aspirin 100 mg once daily, rivaroxaban 5 mg orally twice daily alone, or aspirin 100 mg once daily alone. Because the 5 mg dose alone was not superior to aspirin alone, only the data concerning the 2.5 mg dose plus aspirin are discussed below.
Patients with established CAD or PAD were eligible. Patients with CAD who were younger than 65 years of age were also required to have documentation of atherosclerosis involving at least two vascular beds or to have at least two additional cardiovascular risk factors (current smoking, diabetes mellitus, an estimated glomerular filtration rate [eGFR] <60 mL per minute, heart failure, or non-lacunar ischemic stroke ≥1 month earlier). Patients with PAD were either symptomatic with ankle brachial index <0.90 or had asymptomatic carotid artery stenosis ≥50%, a previous carotid revascularization procedure, or established ischemic disease of one or both lower extremities. Patients were excluded for use of dual antiplatelet, other non-aspirin antiplatelet, or oral anticoagulant therapies, ischemic, non-lacunar stroke within 1 month, hemorrhagic or lacunar stroke at any time, or eGFR <15 mL/min.
The mean age was 68 years and 21% of the subject population were ≥75 years. Of the included patients, 91% had CAD (and will be referred to as the COMPASS CAD population), 27% had PAD (and will be referred to as the COMPASS PAD population), and 18% had both CAD and PAD. Of the patients with CAD, 69% had prior MI, 60% had prior percutaneous transluminal coronary angioplasty (PTCA)/atherectomy/ percutaneous coronary intervention (PCI), and 26% had history of coronary artery bypass grafting (CABG) prior to study. Of the patients with PAD, 49% had intermittent claudication, 27% had peripheral artery bypass surgery or peripheral percutaneous transluminal angioplasty, 26% had asymptomatic carotid artery stenosis > 50%, and 4% had limb or foot amputation for arterial vascular disease.
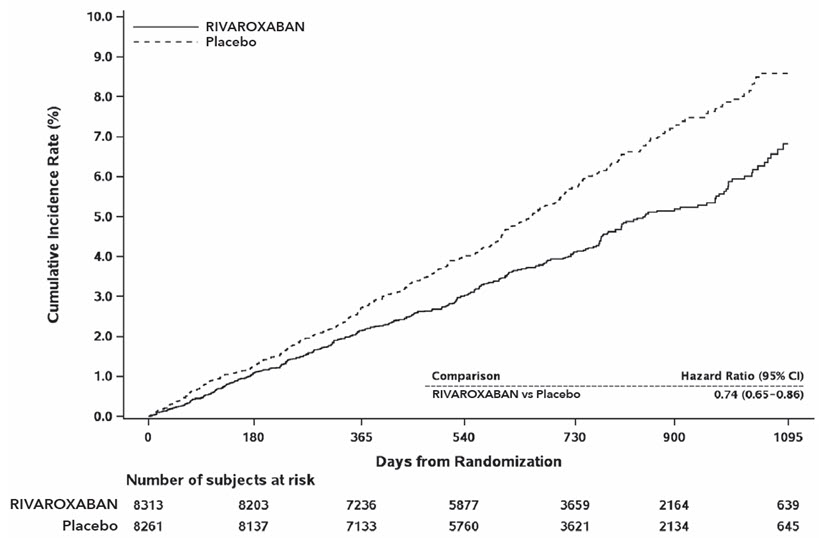
The mean duration of follow-up was 23 months. Relative to placebo, rivaroxaban reduced the rate of the primary composite outcome of stroke, myocardial infarction or cardiovascular death: HR 0.76 (95% CI: 0.66, 0.86; p=0.00004). In the COMPASS CAD population, the benefit was observed early with a constant treatment effect over the entire treatment period (see Table 26and Figure 10).
A benefit-risk analysis of the data from COMPASS was performed by comparing the number of CV events (CV deaths, myocardial infarctions and non-hemorrhagic strokes) prevented to the number of fatal or life-threatening bleeding events (fatal bleeds + symptomatic non-fatal bleeds into a critical organ) in the rivaroxaban group versus the placebo group. Compared to placebo, during 10,000 patient-years of treatment, rivaroxaban would be expected to result in 70 fewer CV events and 12 additional life-threatening bleeds, indicating a favorable balance of benefits and risks.
The results in the COMPASS CAD population were consistent across major subgroups (see Figure 9).
- * All patients received aspirin 100 mg once daily as background therapy.
Figure 9: Risk of Primary Efficacy Outcome by Baseline Characteristics in the COMPASS CAD Population (Intent-to-Treat Population) * 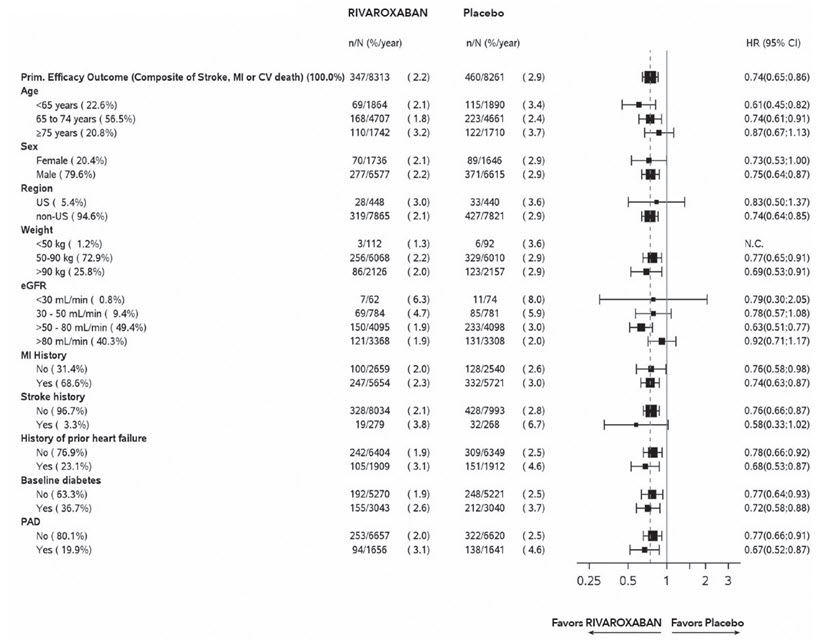
Table 26: Efficacy results from COMPASS CAD Population * Event Rivaroxaban †
N=8313Placebo †
N=8261Hazard Ratio (95% CI) ‡ n (%) Event Rate (%/year) n (%) Event Rate (%/year) CHD: coronary heart disease, CI: confidence interval; CV: cardiovascular; MI: myocardial infarction - * intention to treat analysis set, primary analyses.
- † Treatment schedule: Rivaroxaban 2.5 mg twice daily vs placebo. All patients received aspirin 100 mg once daily as background therapy.
- ‡ Rivaroxaban vs. placebo.
- § Coronary heart disease death: death due to acute MI, sudden cardiac death, or CV procedure.
- ¶ Acute limb ischemia is defined as limb-threatening ischemia leading to an acute vascular intervention (i.e., pharmacologic, peripheral arterial surgery/reconstruction, peripheral angioplasty/stent, or amputation).
- # CV death includes CHD death, or death due to other CV causes or unknown death.
Stroke, MI or CV death 347 (4.2) 2.2 460 (5.6) 2.9 0.74 (0.65, 0.86) - Stroke 74 (0.9) 0.5 130 (1.6) 0.8 0.56 (0.42, 0.75) - MI 169 (2.0) 1.1 195 (2.4) 1.2 0.86 (0.70, 1.05) - CV death 139 (1.7) 0.9 184 (2.2) 1.1 0.75 (0.60, 0.93) Coronary heart disease death, MI, ischemic stroke, acute limb ischemia 299 (3.6) 1.9 411 (5.0) 2.6 0.72 (0.62, 0.83) - Coronary heart disease death § 80 (1.0) 0.5 107 (1.3) 0.7 0.74 (0.55, 0.99) - Ischemic stroke 56 (0.7) 0.3 114 (1.4) 0.7 0.49 (0.35, 0.67) - Acute limb ischemia ¶ 13 (0.2) 0.1 27 (0.3) 0.2 0.48 (0.25, 0.93) CV death #, MI, ischemic stroke, acute limb ischemia 349 (4.2) 2.2 470 (5.7) 3.0 0.73 (0.64, 0.84) All-cause mortality 262 (3.2) 1.6 339 (4.1) 2.1 0.77 (0.65, 0.90) 14.7 Reduction of Risk of Major Thrombotic Vascular Events in Patients with PAD, Including Patients after Lower Extremity Revascularization due to Symptomatic PAD
The efficacy and safety of rivaroxaban 2.5 mg orally twice daily versus placebo on a background of aspirin 100 mg once daily in patients with PAD were evaluated in the COMPASS study (n=4996) and will be referred to as the COMPASS PAD population [see Clinical Studies (14.6)] .
The efficacy and safety of rivaroxaban were also evaluated for the reduction in the risk of the composite endpoint of myocardial infarction, ischemic stroke, cardiovascular death, acute limb ischemia (ALI), and major amputation of a vascular etiology in patients undergoing a lower extremity infrainguinal revascularization procedure due to symptomatic peripheral artery disease (PAD) in the double-blinded, placebo-controlled Vascular Outcomes stud Yof ASA alon Gwith rivaroxaban in Endovascular or surgical limb Revascularization for peripheral artery disease (PAD) trial (VOYAGER) [NCT02504216]. A total of 6,564 patients were equally randomized to rivaroxaban 2.5 mg orally twice daily vs placebo on a background therapy of aspirin 100 mg once daily.
Eligible patients included adults who were at least 50 years of age with documented moderate to severe symptomatic lower extremity atherosclerotic PAD who had a successful peripheral surgical procedure and/or endovascular procedure with or without clopidogrel (up to a maximum of 6 months was allowed; median duration of therapy was 31 days). Patients had either a prior history of limb revascularization with ankle brachial index ≤0.85 or no prior history of limb revascularization with ankle brachial index ≤0.80. Patients in need of dual antiplatelet for >6 months, or any additional antiplatelet other than aspirin and clopidogrel, or oral anticoagulant, as well as patients with a history of intracranial hemorrhage, stroke, or transient ischemic attack (TIA), or patients with eGFR <15 mL/min were excluded.
The mean age was 67 years and 20% of the subject population was ≥75 years. Of the included patients, 35% had surgical revascularization, 47% had endovascular revascularization with clopidogrel, and 18% endovascular revascularization without clopidogrel. The median duration of follow-up was 30.8 months.
Rivaroxaban 2.5 mg twice daily was superior to placebo in reducing the rate of the primary composite outcome of myocardial infarction, ischemic stroke, cardiovascular death, acute limb ischemia (ALI), and major amputation of a vascular etiology. The primary efficacy outcome and its components are provided in Table 27. The Kaplan-Meier plot for the primary efficacy outcome can be seen in Figure 11. The secondary efficacy outcomes were tested for superiority in a prespecified, hierarchical order and the first five of seven endpoints were significantly reduced in the rivaroxaban treatment arm (see Table 27). Compared to placebo during 10,000 patient-years of treatment, rivaroxaban would be expected to result in 181 fewer primary outcome events and 29 more TIMI major bleeding events, indicating a favorable balance of benefits and risks.
- * All patients received aspirin 100 mg once daily as background therapy.
Figure 11: Time to First Occurrence of Primary Efficacy Outcome (Myocardial Infarction, Ischemic Stroke, Cardiovascular Death, Acute Limb Ischemia, Major Amputation due to Vascular Origins) in VOYAGER * 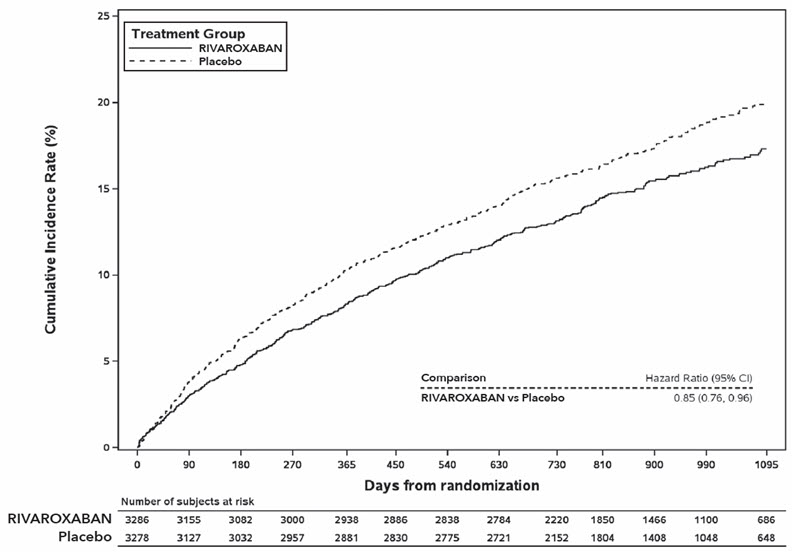
Figure 12 shows the risk of primary efficacy outcome across major subgroups. Subgroup analyses must be interpreted cautiously, as differences can reflect the play of chance among a large number of analyses. The primary efficacy endpoint generally shows homogeneous results across subgroups.
- * All patients received aspirin 100 mg once daily as background therapy.
Figure 12: Risk of Primary Efficacy Outcome by Baseline Characteristics in VOYAGER (Intent-to-Treat Population) * 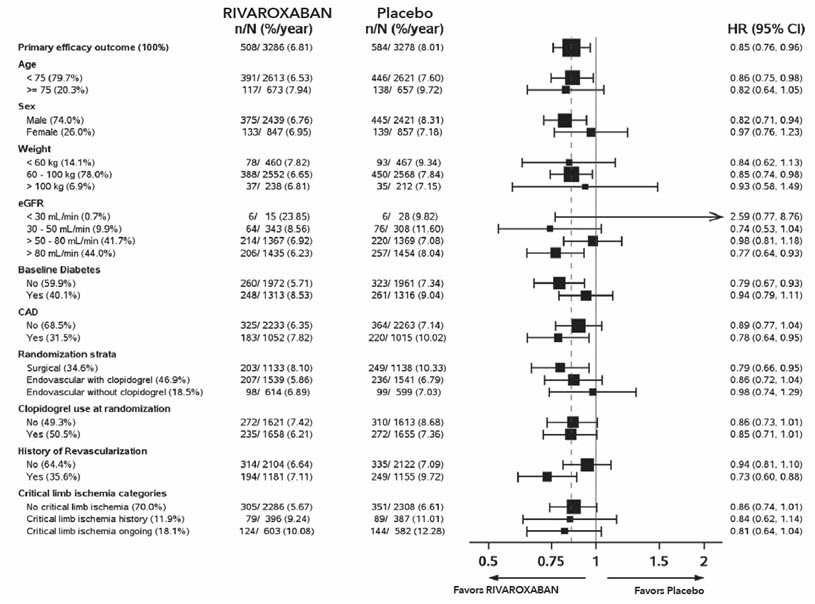
Table 27 provides the efficacy event rates for the prespecified endpoints in VOYAGER and similar endpoints in the COMPASS PAD population.
Table 27: Efficacy Results in VOYAGER (Intent-to-Treat Population) and COMPASS PAD VOYAGER COMPASS PAD Rivaroxaban
N=3286Placebo
N=3278Hazard Ratio
(95% CI) *
p-value †Rivaroxaban
N=2492Placebo
N=2504Hazard Ratio
(95% CI) *Outcome Components Event Rate
(%/year)Event Rate
(%/year)Efficacy endpoints in COMPASS PAD were analysed according to the pre-specified endpoints in VOYAGER when applicable.
ALI=acute limb ischemia, CHD=coronary heart disease; CI=confidence interval, CV=cardiovascular; MI=myocardial infarction, VTE=venous thromboembolism.- * Rivaroxaban vs. placebo.
- † Two-sided p-values
- ‡ Major thrombotic vascular event is the composite of MI, ischemic stroke, CV death, ALI, and major amputation of a vascular etiology.
- § Ischemic stroke for VOYAGER included stroke of uncertain/unknown etiology whereas COMPASS only included ischemic stroke.
- ¶ CV death includes Coronary Heart Disease death, or death due to other CV causes or sudden cardiac arrest and unknown death.
- # Adjudicated events in VOYAGER and investigator reported events in COMPASS
- Þ Secondary outcomes for VOYAGER were tested sequentially.
- ß CHD death includes death due to sudden cardiac death, MI, or coronary revascularization procedure
- à Unplanned index limb revascularization for recurrent limb ischemia was not captured in COMPASS study.
- è Investigator reported in VOYAGER and adjudicated events in COMPASS
5-Component Outcome (Major thrombotic vascular events) ‡ 6.8 8.0 0.85 (0.76, 0.96)
p=0.00853.4 4.8 0.71 (0.57, 0.87) MI 1.7 1.9 0.88 (0.70, 1.12) 1.1 1.5 0.76 (0.53, 1.09) Ischemic Stroke § 0.9 1.0 0.87 (0.63, 1.19) 0.5 0.9 0.55 (0.33, 0.93) CV death ¶ 2.5 2.2 1.14 (0.93, 1.40) 1.4 1.7 0.82 (0.59, 1.14) ALI 2.0 3.0 0.67 (0.55, 0.82) 0.4 0.8 0.56 (0.32, 0.99) Major amputation of a vascular etiology # 1.3 1.5 0.89 (0.68, 1.16) 0.2 0.6 0.40 (0.20, 0.79) VOYAGER Secondary Efficacy Outcomes Þ MI, ischemic stroke, CHD death, ßALI, and major amputation due to vascular etiology 5.8 7.3 0.80 (0.71, 0.91)
p=0.00082.8 4.2 0.66 (0.53, 0.83) Unplanned index limb revascularization for recurrent limb ischemia à 8.4 9.5 0.88 (0.79, 0.99)
p=0.028N/A N/A N/A Hospitalization for a coronary or peripheral cause of a thrombotic nature # 3.5 4.8 0.72 (0.62, 0.85)
p<0.00011.7 2.9 0.58 (0.44, 0.77) MI, ischemic stroke, all-cause mortality, ALI, and major amputation due to vascular etiology 8.2 9.3 0.89 (0.79, 0.99)
p=0.0294.8 6.0 0.80 (0.67, 0.96) MI, all-cause stroke, CV death, ALI, and major amputation due to vascular etiology 6.9 8.1 0.86 (0.76, 0.96)
p=0.0103.4 4.9 0.70 (0.57, 0.86) All-cause mortality 4.0 3.7 1.08 (0.92, 1.27) 2.8 3.1 0.91 (0.72, 1.16) VTE events è 0.3 0.5 0.61 (0.37, 1.00) 0.2 0.3 0.67 (0.30, 1.49) -
16 HOW SUPPLIED/STORAGE AND HANDLING
Rivaroxaban Tablets, USP are available in the strengths and packages listed below:
2.5 mg tablets are round, light yellow, film-coated and debossed on one side with "◻" and "2.5" on the other side. The tablets are supplied in the packages listed:
NDC: 51672-4228-4 Bottle containing 60 tablets
NDC: 51672-4228-9 Bottle containing 180 tablets
NDC: 51672-4228-1 Blister package containing 100 tablets (10 blister cards containing 10 tablets each) -
17 PATIENT COUNSELING INFORMATION
For the tablets, advise the patient and/or caregiver to read the FDA-approved patient labeling (Medication Guide).
Instructions for Patient Use
- Advise patients to take rivaroxaban tablets only as directed.
- Remind patients to not discontinue rivaroxaban tablets without first talking to their healthcare professional.
Adults
- Advise patients who cannot swallow the tablet whole to crush rivaroxaban tablets and combine with a small amount of applesauce followed by food [see Dosage and Administration (2.6)] .
- For patients requiring an NG tube or gastric feeding tube, instruct the patient or caregiver to crush the rivaroxaban tablet and mix it with a small amount of water before administering via the tube [see Dosage and Administration (2.6)] .
- If a dose is missed, advise the patient according to the instructions in the Full Prescribing Information based on their dosing schedule [see Dosage and Administration (2.5)] .
Bleeding Risks
- Advise patients to report any unusual bleeding or bruising to their physician. Inform patients that it might take them longer than usual to stop bleeding, and that they may bruise and/or bleed more easily when they are treated with rivaroxaban tablets [see Warnings and Precautions (5.2)] .
- If patients have had neuraxial anesthesia or spinal puncture, and particularly, if they are taking concomitant NSAIDs or platelet inhibitors, advise patients to watch for signs and symptoms of spinal or epidural hematoma, such as back pain, tingling, numbness (especially in the lower limbs), muscle weakness, and stool or urine incontinence. If any of these symptoms occur, advise the patient to contact his or her physician immediately [see Boxed Warning] .
Invasive or Surgical Procedures
Instruct patients to inform their healthcare professional that they are taking rivaroxaban before any invasive procedure (including dental procedures) is scheduled.
Concomitant Medication and Herbals
Advise patients to inform their physicians and dentists if they are taking, or plan to take, any prescription or over-the-counter drugs or herbals, so their healthcare professionals can evaluate potential interactions [see Drug Interactions (7)] .
Pregnancy and Pregnancy-Related Hemorrhage
- Advise patients to inform their physician immediately if they become pregnant or intend to become pregnant during treatment with rivaroxaban tablets [see Use in Specific Populations (8.1)] .
- Advise pregnant women receiving rivaroxaban tablets to immediately report to their physician any bleeding or symptoms of blood loss [see Warnings and Precautions (5.7)] .
Lactation
Advise patients to discuss with their physician the benefits and risks of rivaroxaban tablets for the mother and for the child if they are nursing or intend to nurse during anticoagulant treatment [see Use in Specific Populations (8.2)] .
Females and Males of Reproductive Potential
Advise patients who can become pregnant to discuss pregnancy planning with their physician [see Use in Specific Populations (8.3)] .
- SPL UNCLASSIFIED SECTION
- MEDICATION GUIDE
-
PRINCIPAL DISPLAY PANEL - 2.5 mg Tablet Bottle Label
NDC: 51672-4228-4
60 TabletsRivaroxaban Tablets, USP
2.5 mgDispense with the accompanying Medication Guide
Rx only

-
INGREDIENTS AND APPEARANCE
RIVAROXABAN
rivaroxaban tablet, coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 51672-4228 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RIVAROXABAN (UNII: 9NDF7JZ4M3) (RIVAROXABAN - UNII:9NDF7JZ4M3) RIVAROXABAN 2.5 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM LAURYL SULFATE (UNII: 368GB5141J) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERROSOFERRIC OXIDE (UNII: XM0M87F357) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) Product Characteristics Color yellow (Light yellow) Score no score Shape ROUND Size 6mm Flavor Imprint Code o;2 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 51672-4228-4 60 in 1 BOTTLE; Type 0: Not a Combination Product 10/15/2025 2 NDC: 51672-4228-9 180 in 1 BOTTLE; Type 0: Not a Combination Product 10/15/2025 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA208557 10/15/2025 Labeler - Sun Pharmaceutical Industries, Inc. (146974886) Establishment Name Address ID/FEI Business Operations Taro Pharmaceutical Industries, Ltd. 600072078 manufacture(51672-4228)



© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.