NAFTIFINE HYDROCHLORIDE gel
Naftifine Hydrochloride by
Drug Labeling and Warnings
Naftifine Hydrochloride by is a Prescription medication manufactured, distributed, or labeled by Taro Pharmaceuticals U.S.A., Inc., Taro Pharmaceuticals Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NAFTIFINE HYDROCHLORIDE GEL safely and effectively. See full prescribing information for NAFTIFINE HYDROCHLORIDE GEL.
NAFTIFINE HYDROCHLORIDE gel, for topical use
Initial U.S. Approval: 1988INDICATIONS AND USAGE
Naftifine Hydrochloride Gel USP, 2% is an allylamine antifungal indicated for the treatment of interdigital tinea pedis caused by the organisms Trichophyton rubrum, Trichophyton mentagrophytes, and Epidermophyton floccosum. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Gel, 2%. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
If redness or irritation develops with the use of naftifine hydrochloride gel treatment should be discontinued. (5.1)
ADVERSE REACTIONS
The most common adverse reactions are application site reactions (2%). (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Taro Pharmaceuticals U.S.A., Inc., at 1-866-923-4914 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.See 17 for PATIENT COUNSELING INFORMATION.
Revised: 1/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Local Adverse Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
- 2 DOSAGE AND ADMINISTRATION
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
- 5 WARNINGS AND PRECAUTIONS
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
In two randomized, vehicle-controlled trials, 1143 subjects were treated with naftifine hydrochloride gel versus 571 subjects treated with the vehicle. The trial subjects were 12 to 92 years old, were primarily male (76%), and were 59% Caucasian, 38% Black or African American, and 23% Hispanic or Latino. Subjects received doses once daily, topically, for 2 weeks to cover the affected skin areas plus a ½-inch margin of surrounding healthy skin. The most common adverse reactions were application site reactions which occurred at the rate of 2% in naftifine hydrochloride gel arm versus 1% in vehicle arm. Most adverse reactions were mild in severity.
In an open-label pediatric pharmacokinetics and safety trial 22 pediatric subjects 12 to 17 years of age with interdigital tinea pedis received naftifine hydrochloride gel. The incidence of adverse reactions in the pediatric population was similar to that observed in adult population.
Cumulative irritancy testing revealed the potential for naftifine hydrochloride gel to cause irritation. There was no evidence that naftifine hydrochloride gel causes contact sensitization, phototoxicity, or photoallergenicity in healthy skin.
6.2 Postmarketing Experience
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. The following adverse reactions have been identified during post-approval use of naftifine hydrochloride: blisters, burning sensation, crusting, dryness, erythema/redness, inflammation, irritation, maceration, pain, pruritus [mild]/itching, rash and swelling.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
There are no adequate and well-controlled trials of naftifine hydrochloride gel in pregnant women. Because animal reproduction studies are not always predictive of human response, naftifine hydrochloride gel should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
The animal multiples of human exposure calculations were based on daily dose body surface area comparison (mg/m2) for the reproductive toxicology studies described in this section and in Section 13.1. The Maximum Recommended Human Dose (MRHD) was set at 4 g 2% gel per day (1.33 mg/kg/day for a 60 kg individual).
Systemic embryofetal development studies were conducted in rats and rabbits. Oral doses of 30 mg/kg/day, 100 mg/kg/day, and 300 mg/kg/day naftifine hydrochloride were administered during the period of organogenesis (gestational days 6 to 15) to pregnant female rats. No treatment-related effects on embryofetal toxicity or teratogenicity were noted at doses up to 300 mg/kg/day (36.5× MRHD). Subcutaneous doses of 10 mg/kg/day and 30 mg/kg/day naftifine hydrochloride were administered during the period of organogenesis (gestational days 6 to 15) to pregnant female rats. No treatment-related effects on embryofetal toxicity or teratogenicity were noted at 30 mg/kg/day (3.7× MRHD). Subcutaneous doses of 3 mg/kg/day, 10 mg/kg/day, and 30 mg/kg/day naftifine hydrochloride were administered during the period of organogenesis (gestational days 6 to 18) to pregnant female rabbits. No treatment-related effects on embryofetal toxicity or teratogenicity were noted at 30 mg/kg/day (7.3× MRHD).
A peri- and post-natal development study was conducted in rats. Oral doses of 30 mg/kg/day, 100 mg/kg/day, and 300 mg/kg/day naftifine hydrochloride were administered to female rats from gestational day 14 to lactation day 21. Reduced body weight gain of females during gestation and of the offspring during lactation was noted at 300 mg/kg/day (36.5× MRHD). No developmental toxicity was noted at 100 mg/kg/day (12.2× MRHD).
8.3 Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when naftifine hydrochloride gel is administered to a nursing woman.
8.4 Pediatric Use
The safety and effectiveness of naftifine hydrochloride gel have been established in the age group 12 to 18 with interdigital tinea pedis.
Use of naftifine hydrochloride gel in this age group is supported by evidence from adequate and well controlled studies in adults with additional safety and PK data from an open label trial, conducted in 22 adolescents ≥12 years of age who were exposed to naftifine hydrochloride gel at a dose of approximately 4 g/day [see Clinical Pharmacology (12.3)].
Safety and effectiveness in pediatric patients <12 years of age have not been established.
-
11 DESCRIPTION
Naftifine Hydrochloride Gel USP, 2% is a clear to yellow gel for topical use only. Each gram of naftifine hydrochloride gel contains 20 mg of naftifine hydrochloride, a synthetic allylamine antifungal compound.
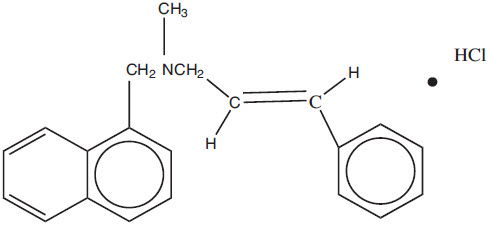
Chemically, naftifine HCl is (E)-N-Cinnamyl-N-methyl-1-napthalenemethylamine hydrochloride.
The molecular formula is C21H21N∙HCl with a molecular weight of 323.86.
The structural formula of naftifine hydrochloride is:
Naftifine Hydrochloride Gel USP, 2% contains the following inactive ingredients: alcohol (95% v/v), benzyl alcohol, edetate disodium, hydroxyethyl cellulose, polysorbate 20, propylene glycol, purified water and trolamine.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Naftifine hydrochloride gel is a topical antifungal drug [see Clinical Pharmacology (12.4)].
12.2 Pharmacodynamics
The pharmacodynamics of naftifine hydrochloride gel have not been established.
12.3 Pharmacokinetics
In vitro and in vivo bioavailability studies have demonstrated that naftifine penetrates the stratum corneum in sufficient concentration to inhibit the growth of dermatophytes.
Pharmacokinetic analysis of plasma samples from 32 subjects with tinea pedis treated with a mean dose of 3.9 grams naftifine hydrochloride gel applied once daily to both feet for 14 days showed increased exposure over the treatment period, with a geometric mean (CV%) AUC0-24 (area under plasma concentration-versus-time curve from time 0 to 24 hours) of 10.5 (118) ng∙hr/mL on Day 1 and an AUC0-24 of 70 (59) ng∙hr/mL on Day 14. The accumulation ratio based on AUC was approximately 6.
Maximum concentration (Cmax) also increased over the treatment period; geometric mean (CV%) Cmax after a single dose was 0.9 (92) ng/mL on Day 1; Cmax on Day 14 was 3.7 (64) ng/mL. Median Tmax was 20 hours (range: 8, 20 hours) after a single application on Day 1 and 8 hours (range: 0, 24 hours) on Day 14. Trough plasma concentrations increased during the trial period and reached steady state after 11 days. In the same pharmacokinetic trial, the fraction of dose excreted in urine during the treatment period was less than or equal to 0.01% of the applied dose.
In a second trial, the pharmacokinetics of naftifine hydrochloride gel was evaluated in 22 pediatric subjects 12 to 17 years of age with tinea pedis. Subjects were treated with a mean dose of 4.1 grams naftifine hydrochloride gel applied to the affected area once daily for 14 days. The results showed that the systemic exposure increased over the treatment period. Geometric mean (CV%) AUC0-24 was 15.9 (212) ng∙hr/mL on Day 1 and 60 (131) ng∙hr/mL on Day 14. Geometric mean (CV%) Cmax after a single dose was 1.40 (154) ng/mL on Day 1 and 3.81 (154) ng/mL on Day 14. The fraction of dose excreted in urine during the treatment period was less than or equal to 0.003% of the applied dose.
12.4 Microbiology
Mechanism of Action
Naftifine is an antifungal that belongs to the allylamine class. Although the exact mechanism of action against fungi is not known, naftifine hydrochloride appears to interfere with sterol biosynthesis by inhibiting the enzyme squalene 2, 3-epoxidase. The inhibition of enzyme activity by this allylamine results in decreased amounts of sterols, especially ergosterol, and a corresponding accumulation of squalene in the cells.
Mechanism of Resistance
To date, a mechanism of resistance to naftifine has not been identified.
Naftifine has been shown to be active against most isolates of the following fungi, both in vitro and in clinical infections, as described in the INDICATIONS AND USAGE section:
- Trichophyton rubrum
- Trichophyton mentagrophytes
- Epidermophyton floccosum
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 2-year dermal carcinogenicity study, naftifine hydrochloride cream was administered to Sprague- Dawley rats at topical doses of 1%, 2% and 3% (10 mg/kg/day, 20 mg/kg/day, and 30 mg/kg/day naftifine hydrochloride). No drug-related tumors were noted in this study up to the highest dose evaluated in this study of 30 mg/kg/day (36 times MRHD based on AUC comparison).
Naftifine hydrochloride revealed no evidence of mutagenic or clastogenic potential based on the results of two in vitro genotoxicity tests (Ames assay and Chinese hamster ovary cell chromosome aberration assay) and one in vivo genotoxicity test (mouse bone marrow micronucleus assay).
Oral administration of naftifine hydrochloride to rats, throughout mating, gestation, parturition, and lactation, demonstrated no effects on growth, fertility, or reproduction, at doses up to 100 mg/kg/day (12 times MRHD based on mg/m2 comparison).
-
14 CLINICAL STUDIES
Naftifine hydrochloride gel has been evaluated for efficacy in two randomized, double-blind, vehiclecontrolled, multicenter trials that included 1175 subjects with symptomatic and dermatophyte culturepositive interdigital tinea pedis. Subjects were randomized to receive naftifine hydrochloride gel or vehicle. Subjects applied naftifine hydrochloride gel 2% or vehicle to the affected area of the foot once daily for 2 weeks. Signs and symptoms of interdigital tinea pedis (presence or absence of erythema, pruritus, and scaling) were assessed and potassium hydroxide (KOH) examination and dermatophyte culture were performed 6 weeks after the first treatment.
The mean age of the study population was 45 years; 77% were male; and 60% were Caucasian, 35% were Black or African American, and 26% were Hispanic or Latino. At baseline, subjects were confirmed to have signs and symptoms of interdigital tinea pedis, positive KOH exam, and confirmed dermatophyte culture. The primary efficacy endpoint was the proportion of subjects with a complete cure at 6 weeks after the start of treatment (4 weeks after the last treatment). Complete cure was defined as both a clinical cure (absence of erythema, pruritus, and scaling) and mycological cure (negative KOH and dermatophyte culture).
The efficacy results at week 6, four weeks following the end of treatment, are presented in Table 1 below. Naftifine hydrochloride gel demonstrated complete cure in subjects with interdigital type tinea pedis.
Table 1 Interdigital Tinea Pedis: Number (%) of Subjects With Complete Cure, Effective Treatment, and Mycological Cure at Week 6 Following Treatment With Naftifine Hydrochloride Gel (Full Analysis Set, Missing Values Treated as Treatment Failure) Trial 1 Trial 2 Endpoint Naftifine Hydrochloride Gel, 2%
N=382
n (%)Vehicle
N=179
n (%)Naftifine Hydrochloride Gel, 2%
N=400
n (%)Vehicle
N=213
n (%)- * Complete cure is a composite endpoint of both mycological cure and clinical cure. Clinical cure is defined as the absence of erythema, pruritus, and scaling (grade of 0).
- † Effective treatment is a negative KOH preparation and negative dermatophyte culture, erythema, scaling, and pruritus grades of 0 or 1 (absent or nearly absent).
- ‡ Mycological cure is defined as negative KOH and dermatophyte culture.
Complete Cure* 64 (17%) 3 (2%) 104 (26%) 7 (3%) Treatment Effectiveness† 207 (54%) 11 (6%) 203 (51%) 15 (7%) Mycological Cure‡ 250 (65%) 25 (14%) 235 (59%) 22 (10%) -
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Naftifine Hydrochloride Gel USP, 2% is a colorless to yellow gel supplied in collapsible tubes in the following size:
45g – NDC: 51672-1376-6
60g - NDC: 51672-1376-3
- 17 PATIENT COUNSELING INFORMATION
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 60 g Tube Carton
NDC: 51672-1376-3
60 g
Naftifine Hydrochloride
Gel USP, 2%FOR TOPICAL USE ONLY. NOT FOR OPHTHALMIC USE, ORAL OR INTRAVAGINAL USE
Rx only
Keep this and all medications out of the reach of children.
TARO

-
INGREDIENTS AND APPEARANCE
NAFTIFINE HYDROCHLORIDE
naftifine hydrochloride gelProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 51672-1376 Route of Administration TOPICAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Naftifine Hydrochloride (UNII: 25UR9N9041) (Naftifine - UNII:4FB1TON47A) Naftifine Hydrochloride 20 mg in 1 g Inactive Ingredients Ingredient Name Strength alcohol (UNII: 3K9958V90M) benzyl alcohol (UNII: LKG8494WBH) edetate disodium (UNII: 7FLD91C86K) hydroxyethyl cellulose (2000 MPA.S at 1%) (UNII: S38J6RZN16) polysorbate 20 (UNII: 7T1F30V5YH) propylene glycol (UNII: 6DC9Q167V3) water (UNII: 059QF0KO0R) trolamine (UNII: 9O3K93S3TK) Product Characteristics Color YELLOW (colorless, to yellow) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 51672-1376-6 1 in 1 CARTON 04/10/2019 1 45 g in 1 TUBE; Type 0: Not a Combination Product 2 NDC: 51672-1376-3 1 in 1 CARTON 10/28/2019 2 60 g in 1 TUBE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA208201 04/10/2019 Labeler - Taro Pharmaceuticals U.S.A., Inc. (145186370) Establishment Name Address ID/FEI Business Operations Taro Pharmaceuticals Inc. 206263295 MANUFACTURE(51672-1376)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.