ZOKINVY- lonafarnib capsule
Lonafarnib by
Drug Labeling and Warnings
Lonafarnib by is a Prescription medication manufactured, distributed, or labeled by Eiger BioPharmaceuticals, Inc., Patheon, Inc., Bend Research, Inc., Corden Pharma Colorado, Inc., Eurofins Lancaster Laboratories, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ZOKINVY safely and effectively. See full prescribing information for ZOKINVY.
ZOKINVY™ (lonafarnib) capsules, for oral use
Initial U.S. Approval: 2020
INDICATIONS AND USAGE
ZOKINVY is a farnesyltransferase inhibitor indicated in patients 12 months of age and older with a body surface area of 0.39 m2 and above: (1)
- To reduce risk of mortality in Hutchinson-Gilford Progeria Syndrome.
- For treatment of processing-deficient Progeroid Laminopathies with either:
- Heterozygous LMNA mutation with progerin-like protein accumulation.
- Homozygous or compound heterozygous ZMPSTE24 mutations.
Limitations of Use
Not indicated for other Progeroid Syndromes or processing-proficient Progeroid Laminopathies. Based upon its mechanism of action, ZOKINVY would not be expected to be effective in these populations. (1)
DOSAGE AND ADMINISTRATION
- Start at 115 mg/m2 twice daily with morning and evening meals. (2.1)
- After 4 months, increase to 150 mg/m2 twice daily. (2.1)
- Round all total daily doses to nearest 25 mg increment. (2.1)
- See full prescribing information for dosage modifications due to adverse reactions. (2.2)
- See full prescribing information for preparation and administration instructions. (2.4)
DOSAGE FORMS AND STRENGTHS
Capsules: 50 mg and 75 mg. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- QTc Interval Prolongation: Increases the QTc interval. Avoid use in patients with symptomatic bradycardia, hypokalemia, or hypomagnesemia, and in combination with other drugs known to prolong the QTc interval. (5.1)
- Risk of Reduced Efficacy or Adverse Reactions Due to Drug Interactions: Prior to and during treatment, consider potential for drug interactions and review concomitant medications; monitor for adverse reactions. (5.2, 7)
- Laboratory Abnormalities: Monitor for changes in electrolytes, complete blood counts, and liver enzymes. (5.3)
- Nephrotoxicity: Caused nephrotoxicity in rats. Monitor renal function at regular intervals. (5.4, 13.2)
- Retinal Toxicity: Caused rod-dependent, low-light vision decline in monkeys. Perform ophthalmological evaluation at regular intervals and at the onset of any new visual changes. (5.5, 13.2)
- Impaired Fertility: Caused impaired fertility in female rats, impaired fertility and testicular toxicity in male rats, and toxicity in the male reproductive tract in monkeys. Advise females and males of reproductive potential of the animal fertility findings. (5.6, 13.1, 13.2)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the risk to a fetus and to use effective contraception. (5.7, 8.1, 8.3)
ADVERSE REACTIONS
The most common adverse reactions (incidence ≥25%) are vomiting, diarrhea, infection, nausea, decreased appetite, fatigue, upper respiratory tract infection, abdominal pain, musculoskeletal pain, electrolyte abnormalities, decreased weight, headache, myelosuppression, increased aspartate aminotransferase, decreased blood bicarbonate, cough, hypertension, and increased alanine aminotransferase. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Eiger BioPharmaceuticals, Inc. at 833-267-0545 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dosage Modifications Due to Adverse Reactions and Drug Interactions
2.3 Temporary Discontinuation for Midazolam Use
2.4 Preparation and Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 QTc Interval Prolongation
5.2 Risk of Reduced Efficacy or Adverse Reactions Due to Drug Interactions
5.3 Laboratory Abnormalities
5.4 Nephrotoxicity
5.5 Retinal Toxicity
5.6 Impaired Fertility
5.7 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on ZOKINVY
7.2 ZOKINVY’s Effect on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.6 Adult Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
ZOKINVY is indicated in patients 12 months of age and older with a body surface area (BSA) of 0.39 m2 and above:
- To reduce the risk of mortality in Hutchinson-Gilford Progeria Syndrome (HGPS)
- For the treatment of processing-deficient Progeroid Laminopathies with either:
- Heterozygous LMNA mutation with progerin-like protein accumulation
- Homozygous or compound heterozygous ZMPSTE24 mutations
Limitations of Use
ZOKINVY is not indicated for other Progeroid Syndromes or processing-proficient Progeroid Laminopathies. Based upon its mechanism of action, ZOKINVY would not be expected to be effective in these populations.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
- The starting dosage of ZOKINVY for patients with a BSA of 0.39 m2 and above is 115 mg/m2 twice daily with morning and evening meals (see Table 1) to reduce the risk of gastrointestinal adverse reactions [see Adverse Reactions (6.1)]. An appropriate dosage strength of ZOKINVY is not available for patients with a BSA of less than 0.39 m2 [see Indications and Usage (1)].
- After 4 months of treatment, increase the dosage to 150 mg/m2 twice daily with morning and evening meals (see Table 2).
- Round all total daily dosages to the nearest 25 mg increment (see Table 1 and Table 2).
- If a dose is missed, take the dose as soon as possible with food, up to 8 hours prior to the next scheduled dose. If less than 8 hours remain before the next scheduled dose, skip the missed dose, and resume taking ZOKINVY at the next scheduled dose.
Table 1 provides the BSA-based dosage recommendations for the starting dosage of 115 mg/m2 twice daily.
Table 1: Recommended Dosage and Administration for 115 mg/m2 Body Surface Area-Based Dosing BSA(m2) Total Daily
Dosage Rounded
to Nearest 25 mgMorning Dosing
Number of Capsule(s)Evening Dosing
Number of Capsule(s)ZOKINVY
50 mgZOKINVY
75 mgZOKINVY
50 mgZOKINVY
75 mg0.39 - 0.48 100 1 1 0.49 - 0.59 125 1 1 0.6 - 0.7 150 1 1 0.71 - 0.81 175 2 1 0.82 - 0.92 200 2 2 0.93 – 1 225 1 1 2 Table 2 provides the BSA-based dosage recommendations for the dosage of 150 mg/m2 twice daily.
Table 2: Recommended Dosage and Administration for 150 mg/m2 Body Surface Area-Based Dosing BSA(m2) Total Daily
Dosage Rounded
to Nearest 25 mgMorning Dosing
Number of Capsule(s)Evening Dosing
Number of Capsule(s)ZOKINVY
50 mgZOKINVY
75 mgZOKINVY
50 mgZOKINVY
75 mg0.39 - 0.45 125 1 1 0.46 - 0.54 150 1 1 0.55 - 0.62 175 2 1 0.63 - 0.7 200 2 2 0.71 - 0.79 225 1 1 2 0.8 - 0.87 250 1 1 1 1 0.88 - 0.95 275 2 1 1 0.96 – 1 300 2 2 2.2 Dosage Modifications Due to Adverse Reactions and Drug Interactions
Table 3: Recommended ZOKINVY Dosage Modifications
Adverse Reaction Severity Monitoring and Dose
Modifications for ZOKINVYQTc Interval Prolongation[see Warnings and Precautions (5.1), Drug Interactions (7.1)] If the QTc interval is greater than or equal to 500 msec Withhold ZOKINVY until QTc interval is less than 470 msec, then resume ZOKINVY at same dosage.
Monitor electrocardiograms (ECGs) prior to initiating ZOKINVY, during treatment, and as clinically indicated.Gastrointestinal Adverse Reactions [see Adverse Reactions (6.1)] For patients who have increased their dose of ZOKINVY to 150 mg/m2 twice daily and are experiencing repeated episodes of vomiting and/or diarrhea resulting in dehydration or weight loss The dose of ZOKINVY can be reduced to the starting dose of 115 mg/m2 twice daily (see Table 1).
Ensure ZOKINVY is taken with the morning and evening meals and with an adequate amount of water.CYP3A Drug Interactions [see Warnings and Precautions (5.2), Adverse Reactions (6.1), Drug Interactions (7.1)] When moderate CYP3A inhibitors are added for a patient already on steady state ZOKINVY No dosage adjustment for ZOKINVY is recommended. When initiating ZOKINVY in a patient who is concurrently on a moderate CYP3A inhibitor The patient may be at increased risk of adverse reactions.
Monitor the patient closely for adverse reactions for at least the first 7 days after initiating ZOKINVY.
If the patient experiences an adverse reaction during the first 7 days of the starting dose or thereafter, consider an alternative therapy that is not a moderate CYP3A inhibitor.2.3 Temporary Discontinuation for Midazolam Use
Temporarily discontinue ZOKINVY for 10 to 14 days before and 2 days after administration of midazolam [see Contraindications (4), Drug Interactions (7.2)].
2.4 Preparation and Administration Instructions
Administer ZOKINVY orally with the morning and evening meals.
Patients Able to Swallow Capsules
- Administer ZOKINVY capsules whole with a sufficient amount of water. Do not chew the capsules.
Patients Unable to Swallow Capsules
- The entire contents of ZOKINVY capsules can be mixed with Ora Blend SF® or Ora-Plus® or, for patients unable to access or tolerate Ora Blend SF or Ora-Plus, the contents of ZOKINVY capsules can be mixed with orange juice or applesauce (see preparation instructions below).
- Do not mix with juice containing grapefruit or Seville oranges [see Contraindications (4), Drug Interactions (7.1)].
- The mixture must be prepared fresh for each dose and be taken within approximately 10 minutes of mixing.
Preparation of Dose in Ora Blend SF, Ora-Plus, or Orange Juice
- For each capsule, empty contents of the capsule into a container containing 5 mL to 10 mL of the liquid.
- Mix thoroughly with a spoon.
- Consume entire serving.
Preparation of Dose in Applesauce
- For each capsule, empty contents of the capsule into a container containing 1 teaspoonful to 2 teaspoonfuls of applesauce.
- Mix thoroughly with a spoon.
- Consume entire serving.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 QTc Interval Prolongation
ZOKINVY prolongs the QTc interval. Prolongation of the QTc interval increases the risk of Torsade de pointes, other serious arrhythmias, and sudden death.
Avoid use of ZOKINVY in patients with a history of cardiac arrhythmias, as well as in other circumstances that may increase the risk of the occurrence of Torsade de pointes or sudden death, including symptomatic bradycardia, hypokalemia, or hypomagnesemia. Avoid use of ZOKINVY in combination with other drugs known or suspected to prolong the QTc interval [see Drug Interactions (7.1)].
Monitor ECGs prior to initiating ZOKINVY, during treatment, and as clinically indicated. If QTc interval is greater than 500 msec, withhold ZOKINVY until QTc interval is less than 470 msec, then resume ZOKINVY at same dosage.
Obtain serum electrolytes prior to initiating ZOKIVNY and during treatment as clinically indicated. Correct serum electrolyte abnormalities.
5.2 Risk of Reduced Efficacy or Adverse Reactions Due to Drug Interactions
Coadministration of ZOKINVY with other drugs may result in clinically significant drug interactions [see Dosage and Administration (2.2, 2.3), Contraindications (4), Drug Interactions (7.1, 7.2)]. These drug interactions can lead to:
- Reduced efficacy of ZOKINVY
- Increased risk of adverse reactions from ZOKINVY or co-administered drugs
See Table 5 and Table 6 for steps to prevent or manage these clinically significant drug interactions, including dosage recommendations [see Drug Interactions (7.1, 7.2)]. Consider the potential for drug interactions prior to and during ZOKINVY therapy; review concomitant medications during ZOKINVY therapy; and monitor for adverse reactions.
5.3 Laboratory Abnormalities
Some patients treated with ZOKINVY developed laboratory abnormalities [see Adverse Reactions (6.1)]. These included:
- Electrolyte abnormalities (43%), such as hyperkalemia, hypokalemia, hyponatremia, or hypercalcemia
- Myelosuppression (35%), such as reductions in absolute neutrophil count, white blood cell counts, lymphocytes, hemoglobin, or hematocrit
- Increased liver enzymes, such as aspartate aminotransferase (35%), or alanine aminotransferase (27%)
These laboratory abnormalities often improved while continuing ZOKINVY, but it is not possible to exclude ZOKINVY as a cause of the abnormalities. Periodically monitor electrolytes, complete blood counts, and liver enzymes, and manage abnormalities accordingly.
5.4 Nephrotoxicity
Lonafarnib caused nephrotoxicity in rats at plasma drug exposures approximately equal to that achieved with the human dose [see Nonclinical Toxicology (13.2)]. Monitor renal function at regular intervals during ZOKINVY therapy.
5.5 Retinal Toxicity
Lonafarnib caused rod-dependent, low-light vision decline in monkeys at plasma drug exposures similar to that achieved with the human dose [see Nonclinical Toxicology (13.2)]. Perform ophthalmological evaluation at regular intervals and at the onset of any new visual changes during ZOKINVY therapy.
5.6 Impaired Fertility
Lonafarnib caused impaired fertility in female rats at 1.2 times the human dose based on plasma drug exposure [see Nonclinical Toxicology (13.1)].
Lonafarnib caused impaired fertility and testicular toxicity in male rats at 1.5 times the human dose based on plasma drug exposure [see Nonclinical Toxicology (13.1)], and toxicity in the male reproductive tract in monkeys at doses lower than the human dose based on plasma drug exposure [see Nonclinical Toxicology (13.2)].
Advise females and males of reproductive potential of the animal fertility findings, and that the impact on pubertal development and the potential for impaired fertility with ZOKINVY therapy in humans have not been adequately evaluated [see Use in Specific Populations (8.3)].
5.7 Embryo-Fetal Toxicity
Based on findings from animal reproduction studies, ZOKINVY can cause embryo-fetal harm when administered to pregnant women. In animal reproduction studies, oral administration of lonafarnib in pregnant rats during organogenesis produced embryo-fetal toxicity at plasma drug exposures that were approximately equal to the recommended human dose. In pregnant rabbits, oral administration of lonafarnib during organogenesis produced skeletal malformations and variations at exposures lower than the human exposure. Advise pregnant women of the risk to a fetus. Advise females of reproductive potential to avoid becoming pregnant and to use appropriate effective contraception during treatment with ZOKINVY [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 84 subjects were treated with at least one dose of ZOKINVY with or without additional therapy, of which 8 were treated at a dosage of at least 115 mg/m2 twice daily for greater than or equal to 10 years.
The safety profile of ZOKINVY is based on 128 patient-years of treatment exposure (62 patients with HGPS and 1 patient with processing-deficient Progeroid Laminopathy with LMNA heterozygous mutation) and pooled results from two Phase 2 open-label, single-arm trials (n=63: 28 patients from Study 1 and 35 treatment naïve patients from Study 2). In Study 1, ZOKINVY treatment was initiated at 115 mg/m2 twice daily and increased to 150 mg/m2 twice daily after approximately 4 months for a total treatment duration of 24 to 30 months. Treatment naïve patients in Study 2 received ZOKINVY 150 mg/m2 twice daily for up to 36 months. In both studies, ZOKINVY was administered orally via capsules or the capsule contents were mixed with Ora Blend SF or Ora-Plus and administered orally as a suspension.
In these two studies, a total of 63 patients received ZOKINVY for a median duration of 2.2 years, with approximately 1.9 years at the recommended dose of 150 mg/m2 twice daily. The population was 2 to 17 years old, with a similar proportion of males (33 [52%] patients) and females (30 [48%] patients). Most patients had classic HGPS (60 [95%] patients) compared to non-classic HGPS (2 [3%] patients) and 1 (2%) patient had Progeroid Laminopathy with LMNA heterozygous mutation.
Table 4 summarizes adverse reactions reported in the clinical trials. The most common adverse reactions (≥25%) in the clinical trials were vomiting, diarrhea, infection, nausea, decreased appetite, fatigue, upper respiratory tract infection, abdominal pain, musculoskeletal pain, electrolyte abnormalities, decreased weight, headache, myelosuppression, increased aspartate aminotransferase, decreased blood bicarbonate, cough, hypertension, and increased alanine aminotransferase.
Table 4: Adverse Reactions in ≥5% of Patients in Study 1 and Treatment-Naïve Patients in Study 2 Receiving ZOKINVY 1Abdominal pain includes stomach pain and abdominal pain.
2Infection includes abdominal infection, candidiasis, chicken pox, Clostridium difficile colitis, colitis, croup, dengue fever, flu syndrome, flu-like symptoms, fungal infection, gastroenteritis, gastrointestinal infection, Helicobacter pylori infection, infection, infection viral, influenza, nail infection, otitis media, parotitis, perirectal abscess, pneumonia, small intestine infection, submandibular lymphadenitis, tonsillitis, viral infection.
3Upper respiratory infection includes bronchial infection, bronchitis, sinus infection, and upper respiratory infection.
4Electrolyte abnormalities include hypermagnesemia, hypokalemia, hyperkalemia, hyponatremia, hypercalcemia, hyperphosphatemia, hypocalcemia, and hypernatremia.
5Myelosuppression includes absolute neutrophil count decreased, low total white blood cells, lymphopenia, decreased hemoglobin, and hematocrit low.
6Musculoskeletal pain includes arthritis, back pain, bone pain, foot pain, intercostal pain, joint pain, knee pain, leg pain, musculoskeletal pain, pain in ankle/extremity/fingers/hip/leg/limb/lower limbs/left arm, shoulder pain, unilateral leg pain. Excludes musculoskeletal pain for abdomen.
7Cerebral ischemia includes cerebral ischemia, central nervous system hemorrhage, and ischemia cerebrovascular.
8Ocular changes include visual acuity change, corneal clouding, conjunctivitis, watering eyes, keratitis.Adverse Reactions ZOKINVY
n=63
n (%)Gastrointestinal disorders Vomiting 57 (90%) Diarrhea 51 (81%) Nausea 35 (56%) Abdominal Pain1 30 (48%) Constipation 14 (22%) Flatulence 4 (6%) General disorders and administration site conditions Fatigue 32 (51%) Pyrexia 9 (14%) Infections and Infestations Infection2 49 (78%) Upper Respiratory Tract Infection 32 (51%) Rhinitis 12 (19%) Investigations Decreased appetite (anorexia) 33 (53%) Electrolyte abnormalities4 27 (43%) Weight decreased 23 (37%) Myelosuppression5 22 (35%) Increased aspartate aminotransferase 22 (35%) Decreased blood bicarbonate 21 (33%) Hypertension 18 (29%) Increased alanine aminotransferase 17 (27%) Dehydration 3 (5%) Musculoskeletal and connective tissue disorders Musculoskeletal pain6 30 (48%) Nervous system disorders Headache 23 (37%) Cerebral ischemia7 7 (11%) Ophthalmic Ocular changes8 15 (24%) Psychiatric disorders Depressed mood 3 (5%) Respiratory, thoracic and mediastinal disorders Cough 21 (33%) Epistaxis 13 (21%) Skin and subcutaneous tissue disorders Rash 7 (11%) Pruritus 5 (8%) Mucositis 5 (8%) Gastrointestinal Adverse Reactions
As noted in Table 4, gastrointestinal adverse reactions were the most frequently reported adverse reactions. Of the 57 patients who experienced vomiting, 30 (53%) patients had mild vomiting (defined as no intervention required), 26 (46%) patients had moderate vomiting (defined as outpatient intravenous hydration; medical intervention required), and 1 (2%) patient had severe vomiting (defined as tube feeding, total parenteral nutrition, or hospitalization indicated). Of the 35 patients who experienced nausea, 34 (97%) patients had mild nausea (defined as loss of appetite without alteration in eating habits) and 1 (3%) patient had moderate nausea (defined as oral intake decreased without significant weight loss, dehydration, or malnutrition). During the first four months of treatment in Study 1, 19 (68%) patients had vomiting and 10 (36%) patients had nausea. By the end of therapy, 4 (14%) patients who were still on ZOKINVY required antiemetics or anti-nauseants. A total of 4 patients discontinued ZOKINVY, mostly due to nausea or vomiting.Of the 51 patients who experienced diarrhea, the majority of patients (approximately 92%) experienced mild or moderate diarrhea; 38 (75%) patients reported mild diarrhea (defined as an increase of less than 4 stools per day over baseline) and 9 (18%) patients reported moderate diarrhea (defined as increase of 4 to 6 stools per day over baseline; limiting instrumental activities of daily living). Four (8%) patients reported severe diarrhea (defined as increase of seven or more stools per day over baseline; hospitalization indicated; severe increase in ostomy output compared to baseline; limiting self-care activities of daily living). During the first four months of treatment in Study 1, 23 (82%) patients had diarrhea; by the end of therapy, 3 (11%) patients had diarrhea. Twelve (43%) patients were treated with loperamide.
Alanine Aminotransferase and Aspartate Aminotransferase Elevations
Increased alanine aminotransferase was commonly reported (17 [27%] patients). Of the 17 patients with increased alanine aminotransferase, 14 (82%) patients had mild increases (defined as greater than upper limit of normal (ULN) to 3.0 times ULN if baseline was normal; 1.5 to 3.0 times ULN if baseline was abnormal), 1 (6%) patient had moderate increases (defined as greater than 3.0 to 5.0 times ULN if baseline was normal or abnormal), and 2 (12%) patients had severe increases (defined as greater than 5.0 to 20.0 times ULN if baseline was normal or abnormal). Increased aspartate aminotransferase was also commonly reported (22 [35%] patients). Of the 22 patients with increased aspartate aminotransferase, 21 (95%) patients had mild increases (defined as greater than ULN to 3.0 times ULN if baseline was normal; 1.5 to 3.0 times ULN if baseline was abnormal) and 1 (5%) patient had a severe increase (defined as greater than 5.0 to 20.0 times ULN if baseline was normal or abnormal). One patient with alanine and aspartate aminotransferase elevations also experienced hypertriglyceridemia and hyperglycemia resulting in discontinuation of ZOKINVY.Hypertension
Increases in blood pressure have been documented in patients treated with ZOKINVY. At baseline, 22 (35%) patients had either a systolic blood pressure or a diastolic blood pressure or both above the 95th percentile. Over the course of the trials, 18 (29%) patients had hypertension based on systolic blood pressure or diastolic blood pressure measurements above the 95th percentile on 3 or more occasions. Five (8%) patients who were normotensive at baseline had either systolic blood pressure or diastolic blood pressure above the 95th percentile at the end of treatment. -
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on ZOKINVY
Table 5 presents clinically significant drug interactions involving drugs that affect ZOKINVY.
Table 5: Clinically Significant Drug Interactions (Drugs that Affect ZOKINVY) CYP3A Inhibitors Clinical Impact Lonafarnib is metabolized by CYP3A. Concomitant use of ZOKINVY with a strong CYP3A inhibitor may increase lonafarnib area under curve (AUC) and maximum concentration (Cmax) [see Clinical Pharmacology (12.3)], which may increase the incidence and severity of adverse reactions, including QTc interval prolongation. Prolongation of the QTc interval increases the risk of Torsade de pointes, other serious arrhythmias, and sudden death [see Warnings and Precautions (5.1)]. Prevention or Management Strong
CYP3A inhibitorsUse of ZOKINVY with strong CYP3A inhibitors is contraindicated [see Contraindications (4)]. Avoid consumption of grapefruit or Seville oranges. Moderate CYP3A
inhibitorsNo dosage adjustment for ZOKINVY is recommended when moderate CYP3A inhibitors are added to steady-state ZOKINVY [see Dosage and Administration (2.2)]. When initiating ZOKINVY in a patient who is currently on a moderate CYP3A inhibitor, the patient may be at increased risk of adverse reactions [see Dosage and Administration (2.2)]. CYP3A Inducers Clinical Impact Coadministration of ZOKINVY with a strong CYP3A inducer decreases lonafarnib Cmax and AUC [see Clinical Pharmacology (12.3)], which may reduce ZOKINVY efficacy. Prevention or Management Strong or moderate CYP3A inducers Use of ZOKINVY with strong or moderate CYP3A inducers is contraindicated [see Contraindications (4)]. Weak CYP3A inducers No ZOKINVY dosage adjustment is recommended. QTc Prolongation Drugs Clinical Implication ZOKINVY causes QTc interval prolongation [see Clinical Pharmacology (12.2)]. Concomitant use of ZOKINVY with other products that prolong the QTc interval may result in a greater increase of the QTc interval and adverse reactions associated with QTc interval prolongation, including Torsade de pointes, other serious arrythmias, and sudden death [see Warnings and Precautions (5.1)]. Prevention or Management Avoid concomitant use of ZOKINVY with other drugs with a known potential to prolong the QTc interval. If concomitant use cannot be avoided, obtain ECGs when initiating, during concomitant use, and as clinically indicated [see Warnings and Precautions (5.1)]. 7.2 ZOKINVY’s Effect on Other Drugs
Table 6 presents clinically significant drug interactions involving drugs affected by ZOKINVY.
Table 6: Clinically Significant Drug Interactions (Drugs Affected by ZOKINVY) CYP3A Substrates Clinical Impact Lonafarnib is a strong CYP3A mechanism-based inhibitor. Coadministration of ZOKINVY with a CYP3A substrate increases the AUC and Cmax of the CYP3A substrate [see Clinical Pharmacology (12.3)] which may increase the risk of the CYP3A substrate’s adverse reactions, including myopathy or rhabdomyolysis (with statins), or extreme sedation or respiratory depression (with midazolam). Prevention or Management HMG CoA reductase inhibitors (“Statins”) Coadministration of ZOKINVY with lovastatin, simvastatin, or atorvastatin is contraindicated [see Contraindications (4)]. Midazolam Coadministration of ZOKINVY with midazolam is contraindicated [see Contraindications (4)]. Temporarily discontinue ZOKINVY for 10-14 days before and 2 days after administration of midazolam [see Dosage and Administration (2.3)]. Other sensitive CYP3A substrates Avoid coadministration of ZOKINVY with sensitive CYP3A substrates. As noted above, use with lovastatin, simvastatin, or atorvastatin, and midazolam is contraindicated [see Contraindications (4)]). If coadministration of 11 other sensitive CYP3A substrates is unavoidable, monitor for adverse reactions and reduce the dosage of those sensitive CYP3A substrate(s) in accordance with their approved product labeling. Certain CYP3A substrates When ZOKINVY is coadministered with certain CYP3A substrates where minimal concentration changes may lead to serious or life-threatening toxicities, monitor for adverse reactions and reduce the dosage of the CYP3A substrate in accordance with its approved product labeling. Loperamide Clinical Impact Lonafarnib is a weak inhibitor of P-gp and strong inhibitor of CYP3A. Coadministration of ZOKINVY with loperamide increases the AUC and Cmax of loperamide [see Clinical Pharmacology (12.3)] which may increase the risk of loperamide’s adverse reactions. Prevention or Management Loperamide is contraindicated in patients less than 2 years of age. When ZOKINVY is coadministered with loperamide, do not exceed loperamide 1 mg once daily when first coadministered. Slowly increase loperamide dosage with caution in accordance with its approved product labeling. CYP2C19 Substrates Clinical Impact Lonafarnib is a moderate CYP2C19 inhibitor. Coadministration of ZOKINVY with a CYP2C19 substrate increases the AUC and Cmax of the CYP2C19 substrate [see Clinical Pharmacology (12.3)] which may increase the risk of the CYP2C19 substrate’s adverse reactions. Prevention or Management Avoid coadministration of ZOKINVY with CYP2C19 substrates. If coadministration is unavoidable, monitor for adverse reactions and reduce the dosage of the CYP2C19 substrate in accordance with its approved product labeling. P-gp Substrates Clinical Impact Lonafarnib is a weak P-gp inhibitor. Coadministration of ZOKINVY with a P-gp substrate increases the AUC and Cmax of the P-gp substrate [see Clinical Pharmacology (12.3)], which may increase the risk of the P-gp substrate’s adverse reactions. Prevention or Management When ZOKINVY is coadministered with P-gp substrates (e.g., digoxin, dabigatran) where minimal concentration changes may lead to serious or life-threatening toxicities, monitor for adverse reactions and reduce the dosage of the P-gp substrate in accordance with its approved product labeling. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies, ZOKINVY can cause embryofetal harm when administered to a pregnant woman. There are no human data on ZOKINVY use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. Advise pregnant women of the risk to a fetus.In animal reproduction studies, oral administration of lonafarnib to pregnant rats during organogenesis produced embryo-fetal toxicity at exposures that were 1.2-times the human exposure at the recommended dose of 150 mg/m2 twice daily. In pregnant rabbits, oral administration of lonafarnib during organogenesis produced skeletal malformations and variations at exposures lower than the human exposure at 150 mg/m2 twice daily, and maternal toxicity at 26 times the human exposure at 150 mg/m2 twice daily (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study in rats, oral administration of lonafarnib during organogenesis produced an increase in post-implantation loss (resorptions) and decreases in fetal body weight and number of live fetuses at 30 mg/kg/day (1.2 times the AUC [area under the plasma concentration-time curve] in humans at the recommended dose of 150 mg/m2 twice daily). No effects on embryo-fetal development in rats were observed at systemic exposures lower than the human AUC at 150 mg/m2 twice daily.In rabbits, oral administration of lonafarnib during organogenesis resulted in skeletal malformations and variations at systemic exposures lower than the human AUC at the recommended dose of 150 mg/m2 twice daily, and maternal toxicity (body weight loss and abortion) at 120 mg/kg/day (26 times the human AUC at 150 mg/m2 twice daily).
No effects in offspring were observed in a pre- and postnatal development study in rats with maternal administration of up to 20 mg/kg/day orally (AUC lower than the human AUC at 150 mg/m2 twice daily) during organogenesis through lactation.
8.2 Lactation
Risk Summary
There are no data on the presence of ZOKINVY in human milk, the effects on the breastfed infant, or the effects on milk production. Lonafarnib is excreted in rat milk (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk.The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ZOKINVY and any potential adverse effects of the breastfed infant from ZOKINVY or from the underlying maternal condition.
Data
Lonafarnib is excreted in milk following oral administration in lactating rats, with a mean milk to plasma concentration ratio of 1.5 at 12 hours.8.3 Females and Males of Reproductive Potential
Contraception
ZOKINVY can cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use appropriate effective contraception during treatment with ZOKINVY.Infertility
Based on findings in rats, ZOKINVY may reduce fertility in females and males of reproductive potential [see Warnings and Precautions (5.6), Nonclinical Toxicology (13.1)].8.4 Pediatric Use
The safety and effectiveness of ZOKINVY for the treatment of HGPS and processing-deficient Progeroid Laminopathies (with either heterozygous LMNA mutation with progerin-like protein accumulation or homozygous or compound heterozygous ZMPSTE24 mutations) have been established in pediatric patients 12 months of age and older. Use of ZOKINVY for these indications is supported by adequate and well-controlled studies in pediatric patients 2 years of age and older [see Clinical Studies (14)].
The safety and effectiveness of ZOKINVY in pediatric patients less than 12 months of age have not been established.
8.6 Adult Use
The safety and effectiveness of ZOKINVY for the treatment of HGPS and processing-deficient Progeroid Laminopathies (with either heterozygous LMNA mutation with progerin-like protein accumulation or homozygous or compound heterozygous ZMPSTE24 mutations) have been established in adults. Use of ZOKINVY in adults for these indications is based on adequate and well-controlled studies in pediatric patients 2 years of age and older [see Clinical Studies (14)].
-
11 DESCRIPTION
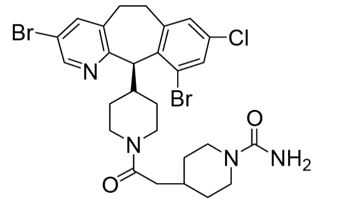
ZOKINVY (lonafarnib) is a farnesyltransferase inhibitor. The chemical name for lonafarnib is 4-[2-[4-[(11R)-3,10-dibromo-8-chloro-6,11-dihydro-5H- benzo[1,2] cyclohepta [2,4-b]pyridin-11-yl]piperidin-1-yl]-2- oxoethyl]piperidine-1-carboxamide. Its molecular formula is C27H31Br2ClN4O2, molecular mass is 638.8 g/mol, and its chemical structure is depicted below.
ZOKINVY (lonafarnib) capsules for oral administration contain 50 mg or 75 mg of lonafarnib as the active ingredient and the following inactive ingredients: croscarmellose sodium, magnesium stearate, poloxamer 188, povidone, and silicon dioxide. The capsule shells of both strengths contain gelatin, titanium dioxide, and yellow iron oxide; the 75 mg capsule also contains red iron oxide. The imprinting ink contains ammonia solution, black iron oxide, butyl alcohol, dehydrated alcohol, isopropyl alcohol, potassium hydroxide, propylene glycol, purified water, and shellac.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Lonafarnib inhibits farnesyltransferase to prevent farnesylation and subsequent accumulation of progerin and progerin-like proteins in the inner nuclear membrane.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of lonafarnib on the QTc interval was evaluated in a randomized, double-blind, placebo- and positive-controlled (moxifloxacin 400 mg single dose), multiple-dose, QTc study in 64 healthy subjects. Subjects received 200 mg lonafarnib twice daily for 9 consecutive days and a single 200 mg dose in the morning on Day 10, with food.The largest mean increase in QTc interval was 19 msec (upper bound of 90% confidence interval = 27 msec) on Day 10 at 48 hours after administration of the morning dose of lonafarnib 200 mg. The Cmax on Day 10 was 2233 ng/mL, which is similar to the mean Cmax of 2695 ng/mL observed in the HGPS patient population [see Clinical Pharmacology (12.3)].
12.3 Pharmacokinetics
The pharmacokinetics of lonafarnib at steady state in patients with HGPS following oral administration of lonafarnib twice daily with food are summarized in Table 7.
Table 7: Summary of Pharmacokinetic Parameters of Lonafarnib at Steady State after Oral Administration Twice Daily to Patients with HGPS Lonafarnib
DoseMedian (range)tmax
(hr)Mean (SD)
Cmax
(ng/mL)Mean (SD)
AUC0-8hr
(ng*hr/mL)Mean (SD)
AUCau
(ng*hr/mL)115 mg/m2 N 23 23 23 15 Results 2 (0, 6) 1777 (1083) 9869 (6327) 12365 (9135) 150 mg/m2 N 18 18 18 8 Results 4 (0, 12) 2695 (1090) 16020 (4978)) 19539 (6434) Absorption
The absolute bioavailability of lonafarnib following oral administration has not been determined. Following oral administration of lonafarnib 75 mg and 100 mg twice daily in healthy subjects under fasted conditions, the geometric mean (CV%) maximum peak plasma concentrations of lonafarnib were 834 (32%) ng/mL and 964 (32%) ng/mL, respectively.Effect of Food
Following a single oral dose of 75 mg lonafarnib in healthy subjects, the Cmax decreased 55% and AUC decreased 29% with a high-fat meal (approximately 43% fat of the total 952 calories) compared to fasted conditions. Cmax decreased 25% and AUC decreased 21% with a low-fat meal (approximately 12% fat of the total 421 calories) compared to fasted conditions.Distribution
In vitro plasma protein binding of lonafarnib was greater than or equal to 99% over the concentration range between 0.5 to 40.0 μg/mL. The apparent volumes of distribution were 87.8 L and 97.4 L, respectively, at steady state following oral administration of lonafarnib 100 mg and 75 mg twice daily in healthy subjects.Elimination
The mean half-life was approximately 4 to 6 hours following oral administration of lonafarnib 100 mg twice daily in healthy subjects.Metabolism
Lonafarnib is primarily metabolized by CYP3A and to a lesser extent by CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, and CYP2E1 in vitro.In a Phase 1 study in healthy subjects, autoinhibition activity of lonafarnib was demonstrated following repeated dosing of lonafarnib. Specifically, the clearance of lonafarnib following multiple-dose lonafarnib administration was reduced by 75% compared with that following single-dose lonafarnib administration.
Excretion
Following an oral administration of 104 mg [14C]-lonafarnib under fasted conditions in healthy subjects, approximately 62% of the total radiolabeled dose was recovered in feces and <1% of the total radiolabeled dose was recovered in urine up to 240 hours post-dose. The two most predominant metabolites were HM17 and HM21 (an active metabolite) accounting for 15% and 14% of plasma radioactivity, respectively.Specific Populations
Patients with Renal Impairment or Hepatic Impairment
ZOKINVY has not been studied in patients with renal impairment or in patients with hepatic impairment.Male and Female Patients
Following a single oral dose of 100 mg lonafarnib in healthy subjects, the plasma lonafarnib AUC and Cmax were 44% and 26% higher in female subjects, respectively, compared to male subjects. The observed exposure difference by sex in healthy subjects is not considered clinically meaningful.Geriatric Patients
Following a single oral dose of 100 mg lonafarnib in healthy subjects, the plasma lonafarnib exposure AUC and Cmax were 59% and 27% higher in subjects ≥65 years, respectively, compared to subjects 18 to 45 years of age. The observed higher exposure in geriatric subjects is not considered clinically relevant.Drug Interaction Studies
In Vitro Studies
Lonafarnib is a CYP3A substrate and a potent CYP3A time-dependent and mechanism-based inhibitor. Lonafarnib is an inhibitor of CYP2C8 and CYP2C19. Lonafarnib is not considered an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, or CYP2D6. Lonafarnib is unlikely to be an inducer of CYP1A2, CYP2B6, and CYP3A.Lonafarnib is not a substrate of transporters OATP1B1, OATP1B3, or BCRP, but is likely a marginal substrate of P-gp. Lonafarnib is an inhibitor of P-gp, OATP1B1, OATP1B3, and BCRP.
Clinical Studies: Effects of other Drugs on Lonafarnib
CYP3A inhibitors
Lonafarnib is a sensitive substrate for CYP3A. With coadministration of a single oral dose of 50 mg lonafarnib following 200 mg ketoconazole (a strong CYP3A inhibitor) once daily for 5 days, the Cmax and AUC of lonafarnib were increased by 270% and 425%, respectively, as compared to lonafarnib administered alone in healthy subjects [see Dosage and Administration (2.2), Contraindications (4), Drug Interactions (7.1)].Fluconazole
Coadministration of steady-state lonafarnib with multiple-dose fluconazole (a moderate inhibitor of CYP3A and CYP2C9) did not increase lonafarnib Cmax or AUC, as compared to lonafarnib administered alone in healthy subjects. [see Dosage and Administration (2.2), Drug Interactions (7.1)].CYP3A inducers
With coadministration of a single oral dose of 50 mg lonafarnib (combined with a single oral dose of 100 mg ritonavir) following 600 mg rifampin once daily for 8 days, the Cmax of lonafarnib was reduced by 92% and the AUC was reduced by 98%, as compared to without rifampin coadministration in healthy subjects [see Contraindications (4), Drug Interactions (7.1)].Clinical Studies: Effects of Lonafarnib on other Drugs
CYP3A Substrates
Lonafarnib is a strong inhibitor of CYP3A. With coadministration of a single oral dose of 3 mg midazolam with multiple oral doses of 100 mg lonafarnib twice daily for 5 days in healthy subjects, the Cmax and AUC of midazolam were increased by 180% and 639%, respectively [see Dosage and Administration (2.3), Contraindications (4), Drug Interactions (7.2)].Loperamide
With coadministration of a single oral 2 mg dose of loperamide (primarily metabolized by CYP2C8 and CYP3A and a substrate of P-gp) with multiple oral doses of lonafarnib 100 mg twice daily for 5 days in healthy subjects, the Cmax and AUC of loperamide were increased by 214% and 299%, respectively [see Drug Interactions (7.2)].CYP2C19 Substrates
Lonafarnib is a moderate CYP2C19 inhibitor. With coadministration of a single oral dose of 40 mg omeprazole with multiple oral doses of lonafarnib 75 mg twice daily for 5 days in healthy subjects, the Cmax and AUC of omeprazole were increased by 28% and 60%, respectively [see Drug Interactions (7.2)].P-gp and OATP1B Substrates
With coadministration of a single oral dose of 180 mg fexofenadine (a P-gp and OATP1B substrate) with multiple oral doses of 100 mg lonafarnib twice daily for 5 days in healthy subjects, the Cmax and AUC of fexofenadine were increased by 21% and 24%, respectively [see Drug Interactions (7.2)]. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies have not been conducted with lonafarnib.Mutagenesis
Lonafarnib was not genotoxic in the bacterial mutagenicity (Ames) assay, in vitro chromosomal aberration assay in mammalian cells, or in vivo micronucleus assay in mice.Impairment of Fertility
Lonafarnib produced impaired fertility in male rats at 90 mg/kg/day or higher (1.5 times the AUC in humans at the recommended dose of 150 mg/m2 twice daily), with a nearly complete loss of fertility at 180 mg/kg/day (3 times the AUC in humans). Male rats treated with 180 mg/kg/day exhibited small testes, flaccid testes, and discolored epididymis (84%, 56%, and 24% of males, respectively). No effects on fertility occurred in males at systemic exposures lower than the human AUC at 150 mg/m2 twice daily.Female rats treated with 30 mg/kg/day lonafarnib or higher (1.2 times the human AUC at the recommended human dose of 150 mg/m2 twice daily) showed a decrease in fertility, as indicated by reductions in the number of corpora lutea and implantation sites and increases in pre- and post-implantation loss. No effects on fertility occurred in females at systemic exposures lower than the human AUC at 150 mg/m2 twice daily [see Warnings and Precautions (5.6)].
13.2 Animal Toxicology and/or Pharmacology
Renal toxicity occurred in a 6-month oral toxicity study of lonafarnib in rats, with kidney lesions (interstitial necrosis and mineralization in the inner medulla) and correlating changes in clinical chemistry (e.g., hyperphosphatemia, hyperkalemia) and urinalysis parameters observed at systemic exposures approximately equal to the AUC in humans at the recommended dose of 150 mg/m2 twice daily. No evidence of renal toxicity was observed at systemic exposures lower than the human AUC at 150 mg/m2 twice daily [see Warnings and Precautions (5.4)].
Toxicity in the male reproductive tract occurred in a 1-year oral toxicity study in monkeys at 10 mg/kg/day or higher (AUC lower than the human AUC at the recommended dose of 150 mg/m2 twice daily). The lesions in the male reproductive tract included aspermia in the epididymis and atrophy of seminiferous tubules, seminal vesicle, and prostate gland. Testicular toxicity was also observed in rats, where severe impairment of male fertility occurred [see Warnings and Precautions (5.6), Nonclinical Toxicology (13.1)].
Ocular (retinal) toxicity occurred in a 1-year oral toxicity study in monkeys at 40 mg/kg/day (3.7 times the human AUC at the recommended dose of 150 mg/m2 twice daily). The retinal injury involved single cell necrosis of photoreceptor cells in the layer of rods and cones and the outer nuclear layer. No retinal toxicity was observed at 20 mg/kg/day (2.1 times the human AUC at 150 mg/m2 twice daily). However, in a follow-up study of lonafarnib effects on visual function in monkeys as evaluated by electroretinography, oral administration of 15 mg/kg/day for 13 weeks or 60 mg/kg/day for 6 weeks produced adverse effects on rod-dependent, low-light vision. The effects were observed at several time-points throughout the treatment period. No histological changes in the retina were observed at study termination [see Warnings and Precautions (5.5)].
-
14 CLINICAL STUDIES
The efficacy of ZOKINVY is based on results from the Observational Cohort Survival Study, which retrospectively compared survival data from two Phase 2 studies in patients with HGPS to those from a natural history cohort.
Study 1 (NCT00425607) was a Phase 2 open-label, single-arm trial that evaluated the efficacy of ZOKINVY in 28 patients (26 with classic HGPS, one with non-classic HGPS, and one with Progeroid Laminopathy with LMNA heterozygous mutation with progerin-like protein accumulation). Patients received ZOKINVY for 24 to 30 months. Patients initiated treatment with ZOKINVY 115 mg/m2 twice daily. After 4 months of treatment, patients who tolerated treatment had an increase in dose to 150 mg/m2 twice daily. Among the 28 patients treated, 27 patients with HGPS (16 females, 11 males) were included in the survival assessment. The median age at treatment initiation for the 27 patients was 7.5 years (range: 3 to 16 years). The body weight range was 6.6 to 17.6 kg and the BSA range was 0.38 to 0.75 m2 (ZOKINVY is not indicated in patients with a BSA less than 0.39 m2 because the appropriate dosage strength is not available for this population).
Following completion of Study 1, 26 patients enrolled in a second Phase 2 open label, single-arm trial (Study 2, NCT00916747) which consisted of two study phases. In the first phase of Study 2, patients received ZOKINVY with additional therapies for about 5 years. In the second phase of Study 2, patients received ZOKINVY 150 mg/m2 twice daily for a period of up to 3 years.
There were 35 treatment naïve patients with HGPS enrolled into the second phase of Study 2. Among the 35 treated patients (22 males, 13 females), 34 (97.1%) patients had classic HGPS and 1 (2.9%) patient had non-classic HGPS. The median age was 6 years (range: 2 to 17 years). The body weight range was 6.7 to 22 kg and the BSA range was 0.42 to 0.90 m2.
Throughout Study 1 and Study 2, ZOKINVY was administered orally via capsules or the capsule contents were mixed with Ora Blend SF or Ora-Plus and administered orally as a suspension.
The retrospective survival analysis was based on the mortality data from 62 treated patients (27 patients in Study 1 and 35 treatment-naïve patients in Study 2) and data from matched, untreated patients in a separate natural history cohort. The lifespan of HGPS patients treated with ZOKINVY increased by an average of 3 months through the first three years of follow-up and 2.5 years through the last follow-up time (11 years) compared to untreated patients. The survival analysis summary is provided in Table 8 and Figure 1.Table 8: Survival Analysis Summary for Patients with HGPS
1 Includes 27 patients in Study 1 and 35 treatment-naïve patients in Study 2. 2 Based on the area under the survival curves up to 11 years of follow-up. 3 Based on a Cox regression model (with treatment as the only covariate) stratified by continent of residency. Note: Treated patients were matched 1:1 to untreated patients (who were alive at the age when the treated patient began ZOKINVY) by mutation status (classic/unknown versus non-classic), sex and continent of residence using a fixed 50th percentile matching algorithm. In the fixed 50th percentile matching algorithm, candidate untreated patients were first sorted by decreasing last known age and the candidate in the 50th percentile was selected as the match. Follow-up time for a matched pair of treated and untreated patients began at the age of the treated patient at ZOKINVY initiation. Follow-up time censored
at 3-yearsLast follow-up time Summary Untreated
(n=62)ZOKINVY1
(n=62)Untreated
(n=62)ZOKINVY1
(n=62)Number of Deaths (%) 12 (19.4) 5 (8.1) 25 (40.3) 21 (33.9) Mean Survival Time (years) 2
(95% CI))2.6
(2.4, 2.8)2.8
(2.7, 3.0)5.5
(4.3, 6.8)8.0
(6.9, 9.1)Difference in Mean Survival Time
(years)
(95% CI)
--0.24
(-0.03, 0.50)
--2.5
(0.8, 4.1)Hazard Ratio for Risk of Death 3
(95% CI)
--0.30
(0.10, 0.89)
--0.40
(0.21, 0.77)Figure 1: Kaplan-Meier Survival Curves for Follow-up Time Censored at Last Follow-up for Patients with HGPS
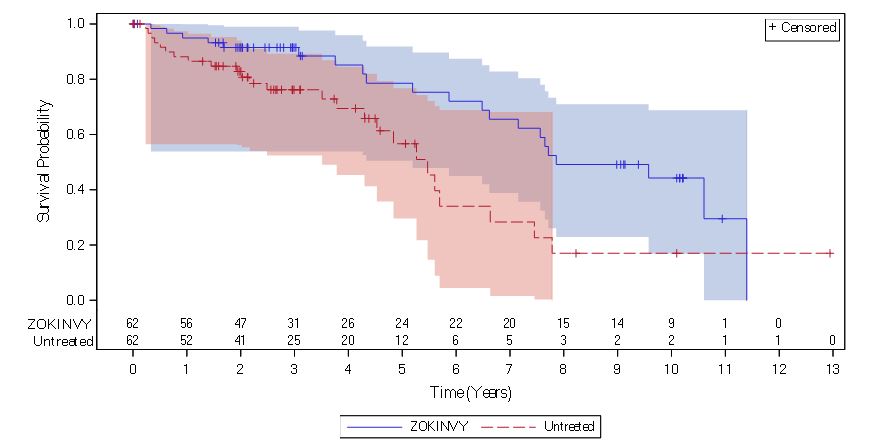
Note: The Kaplan-Meier (KM) survival curve for the ZOKINVY-treated patients is indicated with a solid line; the curve for the untreated patients is indicated with a dashed line. The shaded regions in blue and red represent the 95% confidence bands for the treated and untreated KM survival curves, respectively.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
ZOKINVY is supplied as:
- 50 mg capsules: Size 4 hard capsule, opaque yellow with “LNF” and “50” printed in black.
Bottles of 30 capsules each (NDC: 73079-050-30) - 75 mg capsules: Size 3 hard capsule, opaque light orange with “LNF and “75” printed in black.
Bottles of 30 capsules each (NDC: 73079-075-30)
Store at 20°C-25°C (68°F-77°F), excursions permitted to 15°C-30ºC (59°F-86°F) [see USP Controlled Room Temperature].
- 50 mg capsules: Size 4 hard capsule, opaque yellow with “LNF” and “50” printed in black.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Dosing [see Dosage and Administration (2.1)]
- Advise patients and caregivers that ZOKINVY should be taken twice daily with the morning and evening meals.
- Inform patients and caregivers that if a dose is missed, the next dose should be given as soon as possible up to 8 hours prior to the next scheduled dose. If less than 8 hours remain before the next scheduled dose, the patient should skip the missed dose and resume taking ZOKINVY at the next scheduled dose.
Preparation and Administration [see Dosage and Administration (2.4), Drug Interactions (7)]
- Advise patients to swallow the capsule whole with water. The capsules should not be chewed.
- For patients unable to swallow capsules, advise patients and caregivers that the contents of ZOKINVY can be mixed with Ora Blend SF or Ora-Plus. For patients unable to access or tolerate Ora Blend SF or Ora-Plus, the contents of ZOKINVY can be mixed with orange juice or applesauce. Advise patients not to mix the contents of ZOKINVY with juice containing grapefruit or Seville oranges. Advise patients and caregivers that the mixture must be prepared fresh for each dose and taken within approximately 10 minutes of mixing.
- Advise patients and caregivers to read and carefully follow the instructions for administering the capsule contents in Ora Blend SF, Ora-Plus, orange juice or applesauce [see Instructions for Use]. Advise patients and caregivers to call their healthcare provider or pharmacist if they have any questions.
QTc Interval Prolongation [see Warnings and Precautions (5.1), Drug Interactions (7.1), Clinical Pharmacology (12.2)]
- Inform patients and caregivers that ZOKINVY causes QTc interval prolongation and may increase the risk of Torsades de pointes, other ventricular arrhythmias, and sudden death.
- Instruct patients or caregivers to notify their healthcare provider if they experience symptoms such as dizziness, lightheadedness, heart palpitations, or loss of consciousness.
- Instruct patients to inform their healthcare provider if they are taking any other medications that may prolong the QTc interval.
Drug Interactions [see Dosage and Administration (2.2, 2.3), Contraindications (4), Warnings and Precautions (5.1, 5.2), Drug Interactions (7.1)]
Inform patients and caregivers that ZOKINVY may interact with several drugs. Advise patients and their caregivers to inform their healthcare provider before starting or discontinuing a prescription or non-prescription drug, supplement, or strong CYP3A inhibitor.Nephrotoxicity [see Warnings and Precautions (5.4), Nonclinical Toxicology (13.2)]
Inform the patient and caregiver of the risk of kidney damage.Retinal Toxicity [see Warnings and Precautions (5.5), Nonclinical Toxicology (13.2)]
Inform the patient and caregiver of the risk of developing difficulty with night vision. Advise patients and caregivers to contact their healthcare provider if they experience a change in vision.Gastrointestinal Adverse Reactions [see Dosage and Administration (2.2), Adverse Reactions (6.1)]
Inform patients and caregivers that gastrointestinal adverse reactions are common with ZOKINVY. These include, but are not limited to, vomiting, diarrhea, and nausea. Advise patients and caregivers to contact their healthcare provider if these adverse reactions persist.Hypertension [see Adverse Reactions (6.1)]
Inform patients and caregivers that blood pressure may increase while taking ZOKINVY. Symptoms of hypertension may include headaches, shortness of breath, nosebleeds, flushing, dizziness, or chest pain. Advise patients and caregivers to contact their healthcare provider if these adverse reactions occur.Impaired Fertility [see Warnings and Precautions (5.6), Nonclinical Toxicology (13.1)]
Inform females and males of reproductive potential that ZOKINVY may impact pubertal development and impair fertility.Embryo-Fetal Toxicity [see Warnings and Precautions (5.7), Use in Specific Populations (8.1, 8.3)]
Inform pregnant women and female patients of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ZOKINVY.
Manufactured for:
Eiger BioPharmaceuticals, Inc., 2155 Park Boulevard Palo Alto, CA 94306
ZOKINVY is a trademark of Eiger BioPharmaceuticals, Inc.
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration.
Revised: 03/2024
PATIENT INFORMATION
ZOKINVY™ (ZO-kinvy)
(lonafarnib)
capsules, for oral useWhat is ZOKINVY?
ZOKINVY is a prescription medicine used to:
- lower the risk of death in adults and children 12 months of age or older with Hutchinson-Gilford Progeria Syndrome (HGPS), who have a certain body surface area.
- treat adults and children 12 months of age or older with certain types of Progeroid Laminopathies, who have a certain body surface area.
ZOKINVY is not for use in people with non-HGPS Progeroid Syndromes or with Progeroid Laminopathies that are processing-proficient.
It is not known if ZOKINVY is safe and effective in children under 12 months of age.
Do not take ZOKINVY if you are taking:
- a strong CYP3A inhibitor
- a strong or moderate CYP3A inducer
- midazolam
- lovastatin, simvastatin, or atorvastatin
Ask your healthcare provider if you are not sure if your medicine may be one that is listed above.
Before taking ZOKINVY, tell your healthcare provider about all of your medical conditions, including if you:
- have a history of QTc interval prolongation or other abnormal heartbeat.
- have slow heartbeat.
- have low magnesium levels or low potassium levels.
- have kidney problems.
- have eye problems.
- are pregnant or plan to become pregnant. ZOKINVY can harm your unborn baby. Females who are able to become pregnant should use effective birth control (contraception) during treatment with ZOKINVY.
- are breastfeeding or plan to breastfeed. It is not known if ZOKINVY can pass into your breast milk. Talk to your healthcare provider about the best way to feed your baby during treatment with ZOKINVY.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Taking ZOKINVY with certain other medicines may affect how the other medicines work and other medicines may affect how ZOKINVY works, and may increase your risk of side effects, including a heart rhythm problem called QTc interval prolongation. Especially tell your healthcare provider if you take any other medicines that may cause QTc interval prolongation.
Know the medicines you take. Keep a list of them with you to show your healthcare provider and pharmacist when starting a new medicine.
How should I take ZOKINVY?
- Take ZOKINVY exactly as your healthcare provider tells you to.
- Do not stop taking ZOKINVY without talking to your healthcare provider. Your healthcare provider may change your dose of ZOKINVY if needed.
- Your healthcare provider will prescribe your dose of ZOKINVY based on your body size (height and weight).
- Take ZOKINVY 2 times a day with morning and evening meals.
- Swallow ZOKINVY capsules whole with an adequate amount of water. Do not chew ZOKINVY capsules.
- If you or your child cannot swallow the ZOKINVY capsule whole, see the detailed Instructions for Use that comes with ZOKINVY for information about how to prepare and give or take a dose of ZOKINVY by mixing the contents of a ZOKINVY capsule with either Ora Blend SF, Ora-Plus, orange juice, or applesauce.
- Each dose of ZOKINVY mixture must be prepared fresh and taken within about 10 minutes of mixing.
- Do not take ZOKINVY with any juice that contains grapefruit or Seville oranges. Seville oranges may also be called bitter or sour oranges.
- If you miss a dose of ZOKINVY, take it as soon as possible up to 8 hours before your next scheduled dose with food. If it is less than 8 hours before your next dose of ZOKINVY, skip the missed dose and take ZOKINVY at your next regularly scheduled dose.
What are the possible side effects of ZOKINVY?
ZOKINVY may cause serious side effects, including:
- A serious heart rhythm problem called QTc Interval Prolongation. This condition can cause an abnormal heartbeat in people who take ZOKINVY and may lead to death. Your healthcare provider should check your heart and do blood tests before and during treatment with ZOKINVY. Tell your healthcare provider right away if you feel dizzy, lightheaded, faint, or have a change in your heartbeat (a fast or irregular heartbeat).
- Severe kidney problems. ZOKINVY may cause severe kidney problems. Your healthcare provider will monitor your kidney function regularly during treatment with ZOKINVY.
- Eye problems. ZOVINKY may cause problems with night vision. Call your healthcare provider right away if you develop any new changes in your vision.
The most common side effects of ZOKINVY include:
- vomiting
- diarrhea
- infection
- nausea
- decreased appetite
- tiredness
- upper respiratory tract infection
- stomach-area (abdominal) pain
- muscle and joint (musculoskeletal) pain
- electrolyte abnormalities
- decreased weight
- headache
- myelosuppression
- increased liver enzyme blood test results
- decreased blood bicarbonate
- cough
- hypertension
Call your healthcare provider if you continue to have nausea, vomiting or diarrhea that leads to loss of appetite and weight loss during your treatment with ZOKINVY.
ZOKINVY may cause an increase in your blood pressure (hypertension). Your healthcare provider will check your blood pressure during treatment with ZOKINVY. Call your healthcare provider right away if you develop any symptoms of high blood pressure including headaches, shortness of breath, nosebleeds, flushing, tiredness, dizziness, vision problems, irregular heartbeat, or chest pain.
ZOKINVY may cause fertility problems in females and males, which may affect your ability to have children. Talk to your healthcare provider if you have concerns about fertility.
These are not all of the possible side effects of ZOKINVY.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to Eiger BioPharmaceuticals at 1-833-267-0545.
How should I store ZOKINVY?
- Store at room temperature between 68°F to 77°F (20°C to 25°C).
Keep ZOKINVY and all medicines out of reach of children.
General information about the safe and effective use of ZOKINVY
Medicines are sometimes prescribed for purposes other than those listed in the Patient Information. Do not use ZOKINVY for a condition for which it was not prescribed. Do not give ZOKINVY to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about ZOKINVY that is written for health professionals.
What are the ingredients in ZOKINVY?
Active ingredient: lonafarnib.
Inactive ingredients: croscarmellose sodium, magnesium stearate, poloxamer 188, povidone, and silicon dioxide.
The capsule shell (ZOKINVY 50 mg hard capsule) contains: gelatin, titanium dioxide and yellow iron oxide.
The capsule shell (ZOKINVY 75 mg hard capsule) contains: gelatin, titanium dioxide, yellow iron oxide and red iron oxide.
The printing ink contains: ammonia solution, black iron oxide, butyl alcohol, dehydrated alcohol, isopropyl alcohol, potassium hydroxide, propylene glycol, purified water, and shellac.
Manufactured for: Eiger BioPharmaceuticals, Inc., 2155 Park Boulevard Palo Alto, CA 94306
©2024 Eiger BioPharmaceuticals
For more information call Eiger BioPharmaceuticals at 650-282-6138 or visit www.zokinvy.com or www.eigerbio.com
-
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE
ZOKINVY™ (ZO-kinvy)
(lonafarnib)
capsules, for oral useThis Instructions for Use contains information on how to mix and give or take a dose of ZOKINVY if the capsules cannot be swallowed whole.
Read this Instructions for Use before you start giving or taking ZOKINVY and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about you or your child’s medical condition or treatment
Supplies needed to prepare and give or take a dose of ZOKINVYBefore mixing a dose of ZOKINVY, gather the following supplies:
- the prescribed number of ZOKINVY capsules for you or your child’s dose. Place the capsule or capsules on a clean flat surface.
- either Ora-Blend SF, Ora-Plus, orange juice or applesauce for mixing. Do not mix with juice that contains grapefruit or Seville oranges. Seville oranges may also be called bitter or sour oranges.
- if mixing with Ora-Blend SF, Ora-Plus or orange juice: a clean
medicine cup with 5 milliliter (mL) and 10 mL measurement levels,
OR if mixing with applesauce: a clean teaspoon.
- clean cup(s) for each ZOKINVY capsule to be mixed.
- a clean spoon for stirring the mixture.
How to open ZOKINVY capsules and mix the capsule contents
Step 1:
If mixing with Ora-Blend SF, Ora-Plus or orange
juice: Use a clean medicine cup to measure
either 5 mL or 10 mL of Ora-Blend SF, Ora-Plus
or orange juice.If mixing with applesauce: measure either 1 or 2
teaspoonfuls of applesauce.You can choose to use 5 mL or 10 mL of liquid or
1 or 2 teaspoonfuls of applesauce (See Figure A).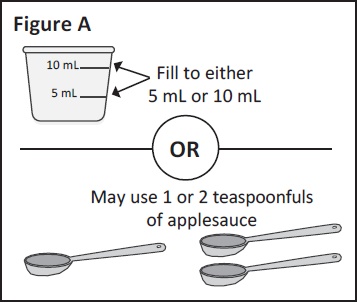
Step 2:
Place the Ora-Blend SF, Ora-Plus, orange juice or
applesauce measured in Step 1 into a clean cup
(See Figure B).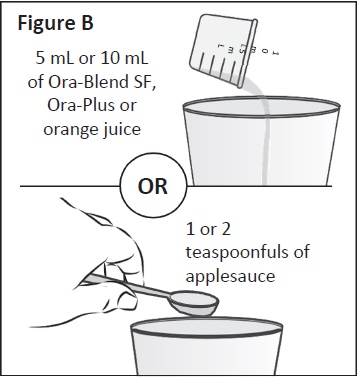
Step 3:
Hold a ZOKINVY capsule above the clean cup
containing the liquid or applesauce. Hold the
ZOKINVY capsule on both sides between your
thumb and forefinger. Gently twist and pull
apart the capsule (See Figure C). Empty the
contents of the capsule directly into the clean
cup (See Figure D).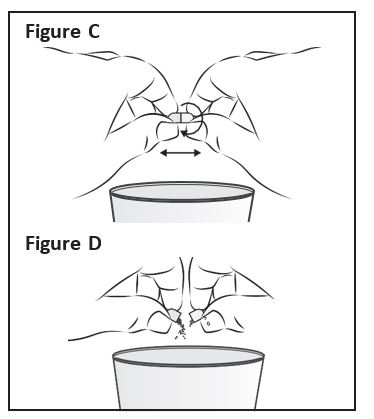
Step 4:
Using a clean spoon, mix the ZOKINVY
capsule contents well (See Figure E). If only 1
capsule is to be taken, skip to Step 6. If 2
capsules are to be taken, go to Step 5.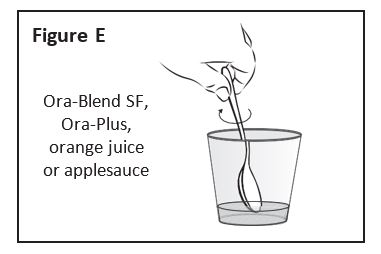
Step 5:
If 2 capsules will be taken, repeat Steps 1 through 4 for the second capsule. After completing the mixing process for the second capsule, the 2 servings can either be placed together in a single cup or remain in 2 serving cups for you or your child to take the full dose of ZOKINVY. After you finish, go to Step 6.
Step 6:
Give or take all of the ZOKINVY mixture with morning and evening meals within about 10 minutes of preparing.
How should I store ZOKINVY?
- Store at room temperature between 68°F to 77°F (20°C to 25°C)
Keep ZOKINVY and all medicines out of reach of children.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured for: Eiger BioPharmaceuticals, Inc., 2155 Park Boulevard Palo Alto, CA 94306
©2020 Eiger BioPharmaceuticals
Issued: 11/2020 - PRINCIPAL DISPLAY PANEL - NDC: 73079-050-30 - 50 mg 30-count Bottle Label
- PRINCIPAL DISPLAY PANEL - NDC: 73079-050-30 - 50 mg 30-count Bottle Carton Label
- PRINCIPAL DISPLAY PANEL - NDC: 73079-075-30 - 75 mg 30-count Bottle Label
- PRINCIPAL DISPLAY PANEL - NDC: 73079-075-30 - 75 mg 30-count Bottle Carton Label
-
INGREDIENTS AND APPEARANCE
ZOKINVY
lonafarnib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 73079-050 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LONAFARNIB (UNII: IOW153004F) (LONAFARNIB - UNII:IOW153004F) LONAFARNIB 50 mg Inactive Ingredients Ingredient Name Strength POVIDONE (UNII: FZ989GH94E) 50 mg POLOXAMER 188 (UNII: LQA7B6G8JG) 12.50 mg CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) 10.00 mg SILICON DIOXIDE (UNII: ETJ7Z6XBU4) 1.85 mg MAGNESIUM STEARATE (UNII: 70097M6I30) 0.616 mg Product Characteristics Color YELLOW (opaque yellow) Score no score Shape CAPSULE Size 12mm Flavor Imprint Code LNF;50 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 73079-050-30 1 in 1 CARTON 11/20/2020 1 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA213969 11/20/2020 ZOKINVY
lonafarnib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 73079-075 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LONAFARNIB (UNII: IOW153004F) (LONAFARNIB - UNII:IOW153004F) LONAFARNIB 75 mg Inactive Ingredients Ingredient Name Strength POVIDONE (UNII: FZ989GH94E) 75 mg POLOXAMER 188 (UNII: LQA7B6G8JG) 18.75 mg CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) 15.00 mg SILICON DIOXIDE (UNII: ETJ7Z6XBU4) 2.77 mg MAGNESIUM STEARATE (UNII: 70097M6I30) 0.924 mg Product Characteristics Color ORANGE (opaque light orange) Score no score Shape CAPSULE Size 14mm Flavor Imprint Code LNF;75 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 73079-075-30 1 in 1 CARTON 11/20/2020 1 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA213969 11/20/2020 Labeler - Eiger BioPharmaceuticals, Inc. (786243613) Establishment Name Address ID/FEI Business Operations Patheon, Inc. 240769596 ANALYSIS(73079-050, 73079-075) , MANUFACTURE(73079-050, 73079-075) , LABEL(73079-050, 73079-075) , PACK(73079-050, 73079-075) Establishment Name Address ID/FEI Business Operations Lonza Bend Inc. 966771631 MANUFACTURE(73079-050, 73079-075) Establishment Name Address ID/FEI Business Operations Lonza Bend Inc. 117222452 ANALYSIS(73079-050, 73079-075) Establishment Name Address ID/FEI Business Operations Corden Pharma Colorado, Inc. 076470525 ANALYSIS(73079-050, 73079-075) , API MANUFACTURE(73079-050, 73079-075) Establishment Name Address ID/FEI Business Operations Eurofins Lancaster Laboratories, Inc. 069777290 ANALYSIS(73079-050, 73079-075)




© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.