Calcium Acetate by Chartwell RX LLC / Chartwell Pharmaceuticals Congers, LLC. CALCIUM ACETATE tablet
Calcium Acetate by
Drug Labeling and Warnings
Calcium Acetate by is a Prescription medication manufactured, distributed, or labeled by Chartwell RX LLC, Chartwell Pharmaceuticals Congers, LLC.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use CALCIUM ACETATE TABLETS safely and effectively. See full prescribing information for CALCIUM ACETATE TABLETS.
CALCIUM ACETATE tablets, for oral use
Initial U.S. Approval: 1990INDICATIONS AND USAGE
- Calcium acetate is a phosphate binder indicated for the reduction of serum phosphorus in patients with end stage renal disease. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
- Tablets: 667 mg calcium acetate per tablet. (3)
CONTRAINDICATIONS
- Hypercalcemia. (4)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
- The most common (> 10%) adverse reactions are hypercalcemia, nausea and vomiting. (6.1)
- In clinical studies, patients have occasionally experienced nausea during calcium acetate therapy. (6)
To report SUSPECTED ADVERSE REACTIONS, contactChartwell RX, LLC., at 845-232-1683 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 5/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypercalcemia
5.2 Concomitant Use with Medications
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Ciprofloxacin
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Labor and Delivery
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment and Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
- 2 DOSAGE AND ADMINISTRATION
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypercalcemia
Patients with end stage renal disease may develop hypercalcemia when treated with calcium, including calcium acetate. Avoid the use of calcium supplements, including calcium based nonprescription antacids, concurrently with calcium acetate.
An overdose of calcium acetate may lead to progressive hypercalcemia, which may require emergency measures. Therefore, early in the treatment phase during the dosage adjustment period, monitor serum calcium levels twice weekly. Should hypercalcemia develop, reduce the calcium acetate dosage, or discontinue the treatment, depending on the severity of hypercalcemia.
More severe hypercalcemia (Ca >12 mg/dL) is associated with confusion, delirium, stupor and coma. Severe hypercalcemia can be treated by acute hemodialysis and discontinuing calcium acetate therapy.
Mild hypercalcemia (10.5 to 11.9 mg/dL) may be asymptomatic or manifest as constipation, anorexia, nausea, and vomiting. Mild hypercalcemia is usually controlled by reducing the calcium acetate dose or temporarily discontinuing therapy. Decreasing or discontinuing Vitamin D therapy is recommended as well.
Chronic hypercalcemia may lead to vascular calcification and other soft-tissue calcification. Radiographic evaluation of suspected anatomical regions may be helpful in early detection of soft tissue calcification. The long-term effect of calcium acetate on the progression of vascular or soft tissue calcification has not been determined.
Hypercalcemia (>11 mg/dL) was reported in 16% of patients in a 3-month study of solid dose formulation of calcium acetate; all cases resolved upon lowering the dose or discontinuing treatment.
Maintain the serum calcium-phosphorus (Ca × P) product below 55 mg2/dL2.
-
6 ADVERSE REACTIONS
Hypercalcemia is discussed elsewhere [see Warnings and Precautions (5.1)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In clinical studies, calcium acetate has been generally well tolerated.
Calcium acetate was studied in a 3-month, open-label, non-randomized study of 98 enrolled ESRD hemodialysis patients and an alternate liquid formulation of calcium acetate was studied in a two week double-blind, placebo-controlled, cross-over study with 69 enrolled ESRD hemodialysis patients. Adverse reactions (>2% on treatment) from these trials are presented in Table 1.
Table 1: Adverse Reactions in Patients with End-Stage renal Disease undergoing Hemodialysis Preferred Term Total adverse reactions reported for calcium acetate
n=167
n (%)3-mo, open-label study of calcium acetate
n=98
n (%)Double-blind, placebo-controlled, cross-over study of liquid calcium acetate
n=69Calcium acetate Placebo n (%) n (%) Nausea 6 (3.6) 6 (6.1) 0 (0.0) 0 (0.0) Vomiting 4 (2.4) 4 (4.1) 0 (0.0) 0 (0.0) Hypercalcemia 21 (12.6) 16 (16.3) 5 (7.2) 0 (0.0) Mild hypercalcemia may be asymptomatic or manifest itself as constipation, anorexia, nausea, and vomiting. More severe hypercalcemia is associated with confusion, delirium, stupor, and coma. Decreasing dialysate calcium concentration could reduce the incidence and severity of calcium acetate-induced hypercalcemia. Isolated cases of pruritus have been reported, which may represent allergic reactions.
6.2 Postmarketing Experience
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate their frequency or to establish a causal relationship to drug exposure.
The following additional adverse reactions have been identified during post-approval of calcium acetate: dizziness, edema, and weakness.
-
7 DRUG INTERACTIONS
The drug interaction of calcium acetate is characterized by the potential of calcium to bind to drugs with anionic functions (e.g., carboxyl, and hydroxyl groups). Calcium acetate may decrease the bioavailability of tetracyclines or fluoroquinolones via this mechanism.
There are no empirical data on avoiding drug interactions between calcium acetate and most concomitant drugs. When administering an oral medication with calcium acetate where a reduction in the bioavailability of that medication would have a clinically significant effect on its safety or efficacy, administer the drug one hour before or three hours after calcium acetate. Monitor blood levels of the concomitant drugs that have a narrow therapeutic range.
Patients taking anti-arrhythmic medications for the control of arrhythmias and anti-seizure medications for the control of seizure disorders were excluded from the clinical trials with all forms of calcium acetate.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C
Calcium acetate tablets contain calcium acetate. Animal reproduction studies have not been conducted with calcium acetate, and there are no adequate and well controlled studies of calcium acetate use in pregnant women. Patients with end stage renal disease may develop hypercalcemia with calcium acetate treatment [see Warnings and Precautions (5.1)]. Maintenance of normal serum calcium levels is important for maternal and fetal well being. Hypercalcemia during pregnancy may increase the risk for maternal and neonatal complications such as stillbirth, preterm delivery, and neonatal hypocalcemia and hypoparathyroidism. Calcium acetate treatment, as recommended, is not expected to harm a fetus if maternal calcium levels are properly monitored during and following treatment.
8.3 Nursing Mothers
Calcium acetate tablets contain calcium acetate and is excreted in human milk. Human milk feeding by a mother receiving calcium acetate is not expected to harm an infant, provided maternal serum calcium levels are appropriately monitored.
8.5 Geriatric Use
Clinical studies of calcium acetate did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other clinical experience has not identified differences in responses between elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
-
10 OVERDOSAGE
Administration of calcium acetate in excess of the appropriate daily dosage may result in hypercalcemia [see Warnings and Precautions (5.1)].
-
11 DESCRIPTION
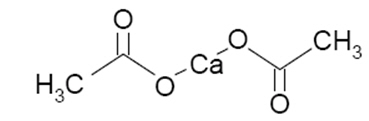
Calcium acetate tablets act as a phosphate binder. Its chemical name is calcium acetate. Its molecular formula is C4H6CaO4, and its molecular weight is 158.17. Its structural formula is:
Each tablet contains 667 mg calcium acetate, USP (anhydrous; Ca(CH3COO)2; MW=158.17 grams) equal to 169 mg (8.45 mEq) calcium. Inactive Ingredients: polyethylene glycol 8000, sodium lauryl sulfate, crospovidone and sodium stearyl fumarate.
Calcium acetate tablets are administered orally for the control of hyperphosphatemia in end stage renal failure.
-
12 CLINICAL PHARMACOLOGY
Patients with ESRD retain phosphorus and can develop hyperphosphatemia. High serum phosphorus can precipitate serum calcium resulting in ectopic calcification. Hyperphosphatemia also plays a role in the development of secondary hyperparathyroidism in patients with ESRD.
12.1 Mechanism of Action
Calcium acetate, when taken with meals, combines with dietary phosphate to form an insoluble calcium phosphate complex, which is excreted in the feces, resulting in decreased serum phosphorus concentration.
12.2 Pharmacodynamics
Orally administered calcium acetate from pharmaceutical dosage forms is systemically absorbed up to approximately 40% under fasting conditions and up to approximately 30% under nonfasting conditions. This range represents data from both healthy subjects and renal dialysis patients under various conditions.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
Effectiveness of calcium acetate in decreasing serum phosphorus has been demonstrated in two studies of the calcium acetate solid oral dosage form.
Ninety-one patients with end-stage renal disease who were undergoing hemodialysis and were hyperphosphatemic (serum phosphorus >5.5 mg/dL) following a 1-week phosphate binder washout period contributed efficacy data to an open-label, non-randomized study.
The patients received calcium acetate 667 mg tablets at each meal for a period of 12 weeks. The initial starting dose was 2 tablets per meal for 3 meals a day, and the dose was adjusted as necessary to control serum phosphorus levels. The average final dose after 12 weeks of treatment was 3.4 tablets per meal. Although there was a decrease in serum phosphorus, in the absence of a control group the true magnitude of effect is uncertain.
The data presented in Table 2 demonstrate the efficacy of calcium acetate in the treatment of hyperphosphatemia in end-stage renal disease patients. The effects on serum calcium levels are also presented.
Table 2: Average Serum Phosphorous and Calcium Levels at Pre-Study, Interim, and Study Completion Time points Parameter Pre-Study Week 4 * Week 8 Week 12 p-value† - * Ninety-one patients completed at least 6 weeks of the study.
- † ANOVA of difference in values at pre-study and study completion.
- ‡ Values expressed as mean ± SE.
Phosphorus (mg/dL)‡ 7.4 ± 0.17 5.9 ± 0.16 5.6 ± 0.17 5.2 ± 0.17 ≤0.01 Calcium (mg/dL)‡ 8.9 ± 0.09 9.5 ± 0.10 9.7 ± 0.10 9.7 ± 0.10 ≤0.01 There was a 30% decrease in serum phosphorus levels during the 12 week study period (p<0.01).
Two-thirds of the decline occurred in the first month of the study. Serum calcium increased 9% during the study mostly in the first month of the study.
Treatment with the phosphate binder was discontinued for patients from the open-label study, and those patients whose serum phosphorus exceeded 5.5 mg/dL were eligible for entry into a double-blind, placebo-controlled, cross-over study. Patients were randomized to receive calcium acetate or placebo, and each continued to receive the same number of tablets as had been individually established during the previous study. Following 2 weeks of treatment, patients switched to the alternative therapy for an additional 2 weeks.
The phosphate binding effect of calcium acetate is shown in the Table 3.
Table 3: Serum Phosphorous and Calcium Levels at Study Initiation and After Completion of Each Treatment Arm Parameter Pre-Study Post-Treatment p-value* Calcium Acetate Placebo - * ANOVA of calcium acetate vs. placebo after 2 weeks of treatment.
- † Values expressed as mean ± SEM
Phosphorus (mg/dL)† 7.3 ± 0.18 5.9 ± 0.24 7.8 ± 0.22 <0.01 Calcium (mg/dL)† 8.9 ± 0.11 9.5 ± 0.13 8.8 ± 0.12 <0.01 Overall, 2 weeks of treatment with calcium acetate statistically significantly (p<0.01) decreased serum phosphorus by a mean of 19% and increased serum calcium by a statistically significant (p<0.01) but clinically unimportant mean of 7%.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Each tablet for oral administration is debossed "CE" over "2" on one side and plain on the other side. Each white round tablet contains 667 mg calcium acetate (anhydrous; Ca(CH3 COO)2; MW=158.17 grams) equal to 169 mg (8.45 mEq) calcium.
NDC: 62135-192-22 Bottles of 200 -
17 PATIENT COUNSELING INFORMATION
Inform patients to take calcium acetate with meals, adhere to their prescribed diets, and avoid the use of calcium supplements including nonprescription antacids. Inform the patients about the symptoms of hypercalcemia [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
Advise patients who are taking an oral medication where reduction in the bioavailability of that medication would have clinically significant effect on its safety or efficacy to take the drug one hour before or three hours after calcium acetate.
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 667 mg Tablet Bottle Label
-
INGREDIENTS AND APPEARANCE
CALCIUM ACETATE
calcium acetate tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 62135-192 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CALCIUM ACETATE (UNII: Y882YXF34X) (CALCIUM CATION - UNII:2M83C4R6ZB) CALCIUM ACETATE 667 mg Product Characteristics Color WHITE Score no score Shape ROUND Size 12mm Flavor Imprint Code CE2 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 62135-192-22 200 in 1 BOTTLE; Type 0: Not a Combination Product 07/01/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA202420 07/01/2019 Labeler - Chartwell RX LLC (079394054)

© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.