LEUKERAN- chlorambucil tablet, film coated
LEUKERAN by
Drug Labeling and Warnings
LEUKERAN by is a Prescription medication manufactured, distributed, or labeled by Aspen Global Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
- BOXED WARNING (What is this?)
-
DESCRIPTION
LEUKERAN (chlorambucil) was first synthesized by Everett et al. It is a bifunctional alkylating agent of the nitrogen mustard type that has been found active against selected human neoplastic diseases. Chlorambucil is known chemically as 4-[bis(2-chlorethyl)amino]benzenebutanoic acid and has the following structural formula:
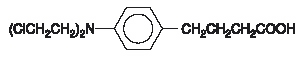
Chlorambucil hydrolyzes in water and has a pKa of 5.8.
LEUKERAN (chlorambucil) is available in tablet form for oral administration. Each film-coated tablet contains 2 mg chlorambucil and the inactive ingredients colloidal silicon dioxide, hypromellose, lactose (anhydrous), macrogol/PEG 400, microcrystalline cellulose, red iron oxide, stearic acid, titanium dioxide, and yellow iron oxide. -
CLINICAL PHARMACOLOGY
Mechanism of Action
Chlorambucil, an aromatic nitrogen mustard derivative, is an alkylating agent. Chlorambucil interferes with DNA replication and induces cellular apoptosis via the accumulation of cytosolic p53 and subsequent activation of Bax, an apoptosis promoter.
Pharmacokinetics
In a study of 12 patients given single oral doses of 0.2 mg/kg of LEUKERAN, the mean dose-adjusted (±SD) plasma chlorambucil Cmax was 492 ± 160 ng/mL, the AUC was 883 ± 329 ng.h/mL, the mean elimination half-life (t½) was 1.3 ± 0.5 hours, and the Tmax was 0.83 ± 0.53 hours. For the major metabolite, phenylacetic acid mustard (PAAM), the mean dose-adjusted (± SD) plasma Cmax was 306 ± 73 ng/mL, the AUC was 1204 ± 285 ng.h/mL, mean t½ was 1.8 ± 0.4 hours, and the Tmax was 1.9 ± 0.7 hours.
After single oral doses of 0.6 to 1.2 mg/kg, peak plasma chlorambucil levels (Cmax) are reached within 1 hour and the terminal elimination half-life (t½) of the parent drug is estimated at 1.5 hours.
Absorption: Chlorambucil is rapidly and completely (>70%) absorbed from the gastrointestinal tract. Consistent with the rapid, predictable absorption of chlorambucil, the inter-individual variability in the plasma pharmacokinetics of chlorambucil has been shown to be relatively small following oral dosages of between 15 and 70 mg (2-fold intra-patient variability, and a 2 to 4 fold interpatient variability in AUC). The absorption of chlorambucil is reduced when taken after food. In a study of ten patients, food intake increased the median Tmax by 2-fold and reduced the dose-adjusted Cmax and AUC values by 55% and 20%, respectively.
Distribution: The apparent volume of distribution averaged 0.31 L/kg following a single 0.2 mg/kg oral dose of chlorambucil in 11 cancer patients with chronic lymphocytic leukemia.
Chlorambucil and its metabolites are extensively bound to plasma and tissue proteins. In vitro, chlorambucil is 99% bound to plasma proteins, specifically albumin. Cerebrospinal fluid levels of chlorambucil have not been determined.
Metabolism: Chlorambucil is extensively metabolized in the liver primarily to phenylacetic acid mustard, which has antineoplastic activity. Chlorambucil and its major metabolite undergo oxidative degradation to monohydroxy and dihydroxy derivatives.
Excretion: After a single dose of radiolabeled chlorambucil (14C), approximately 20% to 60% of the radioactivity appears in the urine after 24 hours. Again, less than 1% of the urinary radioactivity is in the form of chlorambucil or phenylacetic acid mustard. - INDICATIONS AND USAGE
- CONTRAINDICATIONS
-
WARNINGS
Because of its carcinogenic properties, chlorambucil should not be given to patients with conditions other than chronic lymphatic leukemia or malignant lymphomas. Convulsions, infertility, leukemia, and secondary malignancies have been observed when chlorambucil was employed in the therapy of malignant and non-malignant diseases.
There are many reports of acute leukemia arising in patients with both malignant and non-malignant diseases following chlorambucil treatment. In many instances, these patients also received other chemotherapeutic agents or some form of radiation therapy. The quantitation of the risk of chlorambucil-induction of leukemia or carcinoma in humans is not possible. Evaluation of published reports of leukemia developing in patients who have received chlorambucil (and other alkylating agents) suggests that the risk of leukemogenesis increases with both chronicity of treatment and large cumulative doses. However, it has proved impossible to define a cumulative dose below which there is no risk of the induction of secondary malignancy. The potential benefits from chlorambucil therapy must be weighed on an individual basis against the possible risk of the induction of a secondary malignancy.
Chlorambucil has been shown to cause chromatid or chromosome damage in humans. Both reversible and permanent sterility have been observed in both sexes receiving chlorambucil.
A high incidence of sterility has been documented when chlorambucil is administered to prepubertal and pubertal males. Prolonged or permanent azoospermia has also been observed in adult males. While most reports of gonadal dysfunction secondary to chlorambucil have related to males, the induction of amenorrhea in females with alkylating agents is well documented and chlorambucil is capable of producing amenorrhea. Autopsy studies of the ovaries from women with malignant lymphoma treated with combination chemotherapy including chlorambucil have shown varying degrees of fibrosis, vasculitis, and depletion of primordial follicles.
Rare instances of skin rash progressing to erythema multiforme, toxic epidermal necrolysis, or Stevens-Johnson syndrome have been reported. Chlorambucil should be discontinued promptly in patients who develop skin reactions.Pregnancy
Pregnancy Category D. Chlorambucil can cause fetal harm when administered to a pregnant woman. Unilateral renal agenesis has been observed in 2 offspring whose mothers received chlorambucil during the first trimester. Urogenital malformations, including absence of a kidney, were found in fetuses of rats given chlorambucil. There are no adequate and well-controlled studies in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant.
-
PRECAUTIONS
General
Many patients develop a slowly progressive lymphopenia during treatment. The lymphocyte count usually rapidly returns to normal levels upon completion of drug therapy. Most patients have some neutropenia after the third week of treatment and this may continue for up to 10 days after the last dose. Subsequently, the neutrophil count usually rapidly returns to normal. Severe neutropenia appears to be related to dosage and usually occurs only in patients who have received a total dosage of 6.5 mg/kg or more in one course of therapy with continuous dosing. About one quarter of all patients receiving the continuous-dose schedule, and one third of those receiving this dosage in 8 weeks or less may be expected to develop severe neutropenia.
While it is not necessary to discontinue chlorambucil at the first evidence of a fall in neutrophil count, it must be remembered that the fall may continue for 10 days after the last dose, and that as the total dose approaches 6.5 mg/kg, there is a risk of causing irreversible bone marrow damage. The dose of chlorambucil should be decreased if leukocyte or platelet counts fall below normal values and should be discontinued for more severe depression.
Chlorambucil should not be given at full dosages before 4 weeks after a full course of radiation therapy or chemotherapy because of the vulnerability of the bone marrow to damage under these conditions. If the pretherapy leukocyte or platelet counts are depressed from bone marrow disease process prior to institution of therapy, the treatment should be instituted at a reduced dosage.
Persistently low neutrophil and platelet counts or peripheral lymphocytosis suggest bone marrow infiltration. If confirmed by bone marrow examination, the daily dosage of chlorambucil should not exceed 0.1 mg/kg. Chlorambucil appears to be relatively free from gastrointestinal side effects or other evidence of toxicity apart from the bone marrow depressant action. In humans, single oral doses of 20 mg or more may produce nausea and vomiting.
Children with nephrotic syndrome and patients receiving high pulse doses of chlorambucil may have an increased risk of seizures. As with any potentially epileptogenic drug, caution should be exercised when administering chlorambucil to patients with a history of seizure disorder or head trauma, or who are receiving other potentially epileptogenic drugs.
Administration of live vaccines to immunocompromised patients should be avoided.Information for Patients
Patients should be informed that the major toxicities of chlorambucil are related to hypersensitivity, drug fever, myelosuppression, hepatotoxicity, infertility, seizures, gastrointestinal toxicity, and secondary malignancies. Patients should never be allowed to take the drug without medical supervision and should consult their physician if they experience skin rash, bleeding, fever, jaundice, persistent cough, seizures, nausea, vomiting, amenorrhea, or unusual lumps/masses. Women of childbearing potential should be advised to avoid becoming pregnant.
Laboratory Tests
Patients must be followed carefully to avoid life-endangering damage to the bone marrow during treatment. Weekly examination of the blood should be made to determine hemoglobin levels, total and differential leukocyte counts, and quantitative platelet counts. Also, during the first 3 to 6 weeks of therapy, it is recommended that white blood cell counts be made 3 or 4 days after each of the weekly complete blood counts. Galton et al have suggested that in following patients it is helpful to plot the blood counts on a chart at the same time that body weight, temperature, spleen size, etc., are recorded. It is considered dangerous to allow a patient to go more than 2 weeks without hematological and clinical examination during treatment.
Carcinogenesis, Mutagenesis, Impairment of Fertility
See WARNINGS section for information on carcinogenesis, mutagenesis, and impairment of fertility.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from chlorambucil, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Geriatric Use
Clinical studies of chlorambucil did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Use in Patients with Renal Impairment: The impact of renal impairment on chlorambucil elimination has not been formally studied. The renal elimination of unchanged chlorambucil and its major active metabolites, phenylacetic acid mustard, represents less than 1% of the administered dose. In addition, no dose adjustment was required in 2 dialysis patients on chlorambucil. Therefore, renal impairment is not expected to significantly impact the elimination of chlorambucil.
Use in Patients with Hepatic Impairment: No formal studies have been conducted in patients with hepatic impairment. As chlorambucil is primarily metabolized in the liver, patients with hepatic impairment should be closely monitored for toxicity and dose reduction may be considered in patients with hepatic impairment when treated with LEUKERAN (see DOSAGE AND ADMINISTRATION). -
ADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact Aspen Global Inc. Toll-Free at 1-855-800-8165 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Hematologic
The most common side effect is bone marrow suppression, anemia, leukopenia, neutropenia, thrombocytopenia, or pancytopenia. Although bone marrow suppression frequently occurs, it is usually reversible if the chlorambucil is withdrawn early enough. However, irreversible bone marrow failure has been reported.
Gastrointestinal
Gastrointestinal disturbances such as nausea and vomiting, diarrhea, and oral ulceration occur infrequently.
CNS
Tremors, muscular twitching, myoclonia, confusion, agitation, ataxia, flaccid paresis, and hallucinations have been reported as rare adverse experiences to chlorambucil which resolve upon discontinuation of drug. Rare, focal and/or generalized seizures have been reported to occur in both children and adults at both therapeutic daily doses and pulse-dosing regimens, and in acute overdose (see PRECAUTIONS: General).
Dermatologic
Allergic reactions such as urticaria and angioneurotic edema have been reported following initial or subsequent dosing. Skin hypersensitivity (including rare reports of skin rash progressing to erythema multiforme, toxic epidermal necrolysis, and Stevens-Johnson syndrome) has been reported (see WARNINGS).
-
OVERDOSAGE
Reversible pancytopenia was the main finding of inadvertent overdoses of chlorambucil. Neurological toxicity ranging from agitated behavior and ataxia to multiple grand mal seizures has also occurred. As there is no known antidote, the blood picture should be closely monitored and general supportive measures should be instituted, together with appropriate blood transfusions, if necessary. Chlorambucil is not dialyzable.
Oral LD50 single doses in mice are 123 mg/kg. In rats, a single intraperitoneal dose of 12.5 mg/kg of chlorambucil produces typical nitrogen-mustard effects; these include atrophy of the intestinal mucous membrane and lymphoid tissues, severe lymphopenia becoming maximal in 4 days, anemia, and thrombocytopenia. After this dose, the animals begin to recover within 3 days and appear normal in about a week, although the bone marrow may not become completely normal for about 3 weeks. An intraperitoneal dose of 18.5 mg/kg kills about 50% of the rats with development of convulsions. As much as 50 mg/kg has been given orally to rats as a single dose, with recovery. Such a dose causes bradycardia, excessive salivation, hematuria, convulsions, and respiratory dysfunction. -
DOSAGE AND ADMINISTRATION
The usual oral dosage is 0.1 to 0.2 mg/kg body weight daily for 3 to 6 weeks as required. This usually amounts to 4 to 10 mg per day for the average patient. The entire daily dose may be given at one time. These dosages are for initiation of therapy or for short courses of treatment. The dosage must be carefully adjusted according to the response of the patient and must be reduced as soon as there is an abrupt fall in the white blood cell count. Patients with Hodgkin’s disease usually require 0.2 mg/kg daily, whereas patients with other lymphomas or chronic lymphocytic leukemia usually require only 0.1 mg/kg daily. When lymphocytic infiltration of the bone marrow is present, or when the bone marrow is hypoplastic, the daily dose should not exceed 0.1 mg/kg (about 6 mg for the average patient).
Alternate schedules for the treatment of chronic lymphocytic leukemia employing intermittent, biweekly, or once-monthly pulse doses of chlorambucil have been reported. Intermittent schedules of chlorambucil begin with an initial single dose of 0.4 mg/kg. Doses are generally increased by 0.1 mg/kg until control of lymphocytosis or toxicity is observed. Subsequent doses are modified to produce mild hematologic toxicity. It is felt that the response rate of chronic lymphocytic leukemia to the biweekly or once-monthly schedule of chlorambucil administration is similar or better to that previously reported with daily administration and that hematologic toxicity was less than or equal to that encountered in studies using daily chlorambucil.
Radiation and cytotoxic drugs render the bone marrow more vulnerable to damage, and chlorambucil should be used with particular caution within 4 weeks of a full course of radiation therapy or chemotherapy. However, small doses of palliative radiation over isolated foci remote from the bone marrow will not usually depress the neutrophil and platelet count. In these cases chlorambucil may be given in the customary dosage.
It is presently felt that short courses of treatment are safer than continuous maintenance therapy, although both methods have been effective. It must be recognized that continuous therapy may give the appearance of “maintenance” in patients who are actually in remission and have no immediate need for further drug. If maintenance dosage is used, it should not exceed 0.1 mg/kg daily and may well be as low as 0.03 mg/kg daily. A typical maintenance dose is 2 mg to 4 mg daily, or less, depending on the status of the blood counts. It may, therefore, be desirable to withdraw the drug after maximal control has been achieved, since intermittent therapy reinstituted at time of relapse may be as effective as continuous treatment.
Procedures for proper handling and disposal of anticancer drugs should be used. Several guidelines on this subject have been published.1-4 There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.Special Populations
Hepatic Impairment: Patients with hepatic impairment should be closely monitored for toxicity. As chlorambucil is primarily metabolized in the liver, dose reduction may be considered in patients with hepatic impairment when treated with LEUKERAN. However, there are insufficient data in patients with hepatic impairment to provide a specific dosing recommendation.
-
HOW SUPPLIED
LEUKERAN is supplied as brown, film-coated, round, biconvex tablets containing 2 mg chlorambucil in amber glass bottles with child-resistant closures. One side is engraved with “GX EG3” and the other side is engraved with an “L.”
Bottle of 25 (NDC: 76388-635-25)Store in a refrigerator, 2° to 8°C (36° to 46°F).
-
REFERENCES
1. NIOSH Alert: Preventing occupational exposures to antineoplastic and other hazardous drugs in healthcare settings. 2004. U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, DHHS (NIOSH) Publication No. 2004-165.
2. OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling Occupational Exposure to Hazardous Drugs. OSHA, 1999. http://www.osha.gov/dts/osta/otm/otm_vi/otm_vi_2.html
3. American Society of Health-System Pharmacists. ASHP guidelines on handling hazardous drugs. Am J Health-Syst Pharm. (2006) 63:1172-1193.
4. Polovich, M., White, J. M., & Kelleher, L.O. (eds.) 2005. Chemotherapy and biotherapy guidelines and recommendations for practice (2nd. ed.) Pittsburgh, PA: Oncology Nursing Society.
- SPL UNCLASSIFIED SECTION
-
PACKAGE LABEL PRINCIPAL DISPLAY PANEL
Principal Display Panel
NDC: 76388-635-25
LEUKERAN®
(chlorambucil) Tablets
2 mg
25 Tablets
Each tablet contains 2 mg chlorambucil.
Rx Only.

Leukeran 2mg 25 ct. Carton

Leukeran 2mg 25 ct. Label
-
INGREDIENTS AND APPEARANCE
LEUKERAN
chlorambucil tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 76388-635 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CHLORAMBUCIL (UNII: 18D0SL7309) (CHLORAMBUCIL - UNII:18D0SL7309) CHLORAMBUCIL 2 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) HYPROMELLOSES (UNII: 3NXW29V3WO) ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) FERRIC OXIDE RED (UNII: 1K09F3G675) STEARIC ACID (UNII: 4ELV7Z65AP) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color BROWN Score no score Shape ROUND Size 7mm Flavor Imprint Code GX;EG3;L Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 76388-635-25 25 in 1 BOTTLE; Type 0: Not a Combination Product 02/13/1985 2 NDC: 76388-635-50 50 in 1 BOTTLE; Type 0: Not a Combination Product 02/13/1985 10/31/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA010669 02/13/1985 Labeler - Aspen Global Inc. (850491625)

Trademark Results [LEUKERAN]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 LEUKERAN 72008531 0639954 Live/Registered |
BURROUGHS WELLCOME & CO. (U. S. A.) INC. 1956-05-18 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.