DAKLINZA- daclatasvir tablet
DAKLINZA by
Drug Labeling and Warnings
DAKLINZA by is a Prescription medication manufactured, distributed, or labeled by E.R. Squibb & Sons, L.L.C.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DAKLINZA safely and effectively. See full prescribing information for DAKLINZA.
DAKLINZA® (daclatasvir) tablets, for oral use
Initial U.S. Approval: 2015WARNING: RISK OF HEPATITIS B VIRUS REACTIVATION IN PATIENTS COINFECTED WITH HCV AND HBV
See full prescribing information for complete boxed warning.
Hepatitis B virus (HBV) reactivation has been reported, in some cases resulting in fulminant hepatitis, hepatic failure, and death. (5.1)
INDICATIONS AND USAGE
DAKLINZA is a hepatitis C virus (HCV) NS5A inhibitor indicated for use with sofosbuvir, with or without ribavirin, for the treatment of chronic HCV genotype 1 or 3 infection. (1)
Limitations of Use:
- Sustained virologic response (SVR12) rates are reduced in genotype 3 patients with cirrhosis receiving DAKLINZA in combination with sofosbuvir for 12 weeks. (14)
DOSAGE AND ADMINISTRATION
- Testing prior to the initiation of therapy:
- 60 mg taken orally once daily with or without food in combination with sofosbuvir with or without ribavirin. (2.2)
- Recommended treatment duration: 12 weeks. (2.2)
- Dose modification: Reduce dosage to 30 mg once daily with strong CYP3A inhibitors and increase dosage to 90 mg once daily with moderate CYP3A inducers. (2.3)
DOSAGE FORMS AND STRENGTHS
- Tablets: 60 mg, 30 mg, and 90 mg (3)
CONTRAINDICATIONS
- Strong inducers of CYP3A, including phenytoin, carbamazepine, rifampin, and St. John’s wort. (4)
WARNINGS AND PRECAUTIONS
- Risk of Hepatitis B Virus Reactivation: Test all patients for evidence of current or prior HBV infection before initiation of HCV treatment. Monitor HCV/HBV coinfected patients for HBV reactivation and hepatitis flare during HCV treatment and post-treatment follow-up. Initiate appropriate patient management for HBV infection as clinically indicated. (5.1)
- Bradycardia When Coadministered with Sofosbuvir and Amiodarone: Serious symptomatic bradycardia may occur in patients taking amiodarone with a sofosbuvir-containing regimen, particularly in patients also receiving beta blockers or those with underlying cardiac comorbidities and/or advanced liver disease. Coadministration of amiodarone with DAKLINZA in combination with sofosbuvir is not recommended. In patients with no alternative treatment options, cardiac monitoring is recommended. (5.3, 6.2, 7.3)
ADVERSE REACTIONS
Most common adverse reactions (≥10%) observed with DAKLINZA in combination with sofosbuvir were headache and fatigue. (6.1)
Most common adverse reactions (≥10%) observed with DAKLINZA in combination with sofosbuvir and ribavirin were headache, anemia, fatigue, and nausea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Bristol-Myers Squibb at 1-800-721-5072 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Drug Interactions: Coadministration of DAKLINZA can alter the concentration of other drugs and other drugs may alter the concentration of daclatasvir. Consult the full prescribing information before use for contraindicated drugs and other potential drug-drug interactions. (2.3, 4, 5.2, 7, 12.3)
- Clearance of HCV infection with direct acting antivirals may lead to changes in hepatic function, which may impact safe and effective use of concomitant medications. Frequent monitoring of relevant laboratory parameters (INR or blood glucose) and dose adjustments of certain concomitant medications may be necessary (7.3)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: RISK OF HEPATITIS B VIRUS REACTIVATION IN PATIENTS COINFECTED WITH HCV AND HBV
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Testing Prior to the Initiation of Therapy
2.2 Recommended Dosage
2.3 Dosage Modification Due to Drug Interactions
2.4 Discontinuation of Therapy
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Hepatitis B Virus Reactivation in Patients Coinfected with HCV and HBV
5.2 Risk of Adverse Reactions or Loss of Virologic Response Due to Drug Interactions
5.3 Serious Symptomatic Bradycardia When Coadministered with Sofosbuvir and Amiodarone
5.4 Risks Associated with Ribavirin Combination Treatment
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Potential for Other Drugs to Affect DAKLINZA
7.2 Potential for DAKLINZA to Affect Other Drugs
7.3 Established and Other Potentially Significant Drug Interactions
7.4 Drugs without Clinically Significant Interactions with DAKLINZA
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Description of Clinical Trials
14.2 Clinical Trials in HCV Genotype 3 (ALLY-3)
14.3 Clinical Trials in HCV/HIV Coinfected Subjects (ALLY-2)
14.4 Clinical Trials in Subjects with Child-Pugh A, B, or C Cirrhosis or with HCV Recurrence after Liver Transplantation (ALLY-1)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: RISK OF HEPATITIS B VIRUS REACTIVATION IN PATIENTS COINFECTED WITH HCV AND HBV
Test all patients for evidence of current or prior hepatitis B virus (HBV) infection before initiating treatment with DAKLINZA. HBV reactivation has been reported in HCV/HBV coinfected patients who were undergoing or had completed treatment with HCV direct-acting antivirals and were not receiving HBV antiviral therapy. Some cases have resulted in fulminant hepatitis, hepatic failure, and death. Monitor HCV/HBV coinfected patients for hepatitis flare or HBV reactivation during HCV treatment and post-treatment follow-up. Initiate appropriate management for HBV infection as clinically indicated [see Warnings and Precautions (5.1)].
-
1 INDICATIONS AND USAGE
DAKLINZA is indicated for use with sofosbuvir, with or without ribavirin, for the treatment of patients with chronic hepatitis C virus (HCV) genotype 1 or genotype 3 infection [see Dosage and Administration (2) and Clinical Studies (14)].
Limitations of Use:
- Sustained virologic response (SVR12) rates are reduced in HCV genotype 3-infected patients with cirrhosis receiving DAKLINZA in combination with sofosbuvir for 12 weeks [see Clinical Studies (14.2)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Testing Prior to the Initiation of Therapy
Testing for HBV infection: Test all patients for evidence of current or prior HBV infection by measuring hepatitis B surface antigen (HBsAg) and hepatitis B core antibody (anti-HBc) before initiating HCV treatment with DAKLINZA [see Warnings and Precautions (5.1)].
NS5A Resistance Testing in HCV Genotype 1a-Infected Patients with Cirrhosis: Consider screening for the presence of NS5A polymorphisms at amino acid positions M28, Q30, L31, and Y93 in patients with cirrhosis who are infected with HCV genotype 1a prior to the initiation of treatment with DAKLINZA and sofosbuvir with or without ribavirin [see Microbiology (12.4), Table 11].
2.2 Recommended Dosage
The recommended dosage of DAKLINZA is 60 mg, taken orally, once daily, with or without food [see Clinical Pharmacology (12.3)].
Table 1 provides the recommended DAKLINZA-containing treatment regimens and duration based on HCV genotype and patient population. The optimal duration of DAKLINZA and sofosbuvir with or without ribavirin has not been established for HCV genotype 3 patients with cirrhosis or for HCV genotype 1 patients with Child-Pugh C cirrhosis [see Clinical Studies (14.2, 14.4)].
For patients with HCV/HIV-1 coinfection, follow the dosage recommendations in Table 1 [see Clinical Studies (14)]. Refer to Drug Interactions (7) for dosage recommendations for concomitant HIV-1 antiviral drugs.
For specific dosage recommendations for sofosbuvir, refer to the prescribing information.
For HCV genotype 1 or 3 patients with Child-Pugh B or C cirrhosis or post-transplantation patients, the starting dose of ribavirin is 600 mg once daily, increasing up to 1000 mg daily as tolerated. The starting dose and on-treatment dose of ribavirin can be decreased based on hemoglobin and creatinine clearance.
For HCV genotype 3 patients with compensated cirrhosis (Child-Pugh A), the recommended dosing of ribavirin is based on weight (1000 mg for patients weighing less than 75 kg and 1200 mg for those weighing at least 75 kg administered orally in two divided doses with food).
Table 1: Recommended Treatment Regimen and Duration for DAKLINZA in Patients with Genotype 1 or 3 HCV Patient Population Treatment and Duration Genotype 1
Without cirrhosis
DAKLINZA + sofosbuvir for 12 weeks
Compensated (Child-Pugh A) cirrhosis
Decompensated (Child-Pugh B or C) cirrhosis
DAKLINZA + sofosbuvir + ribavirin
for 12 weeksPost-transplant
Genotype 3
Without cirrhosis
DAKLINZA + sofosbuvir for 12 weeks
Compensated (Child-Pugh A) or
decompensated (Child-Pugh B or C) cirrhosisDAKLINZA + sofosbuvir + ribavirin
for 12 weeksPost-transplant
2.3 Dosage Modification Due to Drug Interactions
Refer to the drug interactions and contraindications sections for other drugs before coadministration with DAKLINZA.
Table 2: Recommended DAKLINZA Dosage Modification with CYP3A Inhibitors and Inducers Concomitant Drugs DAKLINZA Dosage Strong CYP3A inhibitors and certain HIV antiviral agents
[see Drug Interactions (7.3)]30 mg once daily
Moderate CYP3A inducers and nevirapine
[see Drug Interactions (7.3)]90 mg once daily
Strong CYP3A inducers
[see Contraindications (4)]Contraindicated
Dosage reduction of DAKLINZA for adverse reactions is not recommended.
-
3 DOSAGE FORMS AND STRENGTHS
Tablets:
- 60 mg: 60 mg of daclatasvir (equivalent to 66 mg daclatasvir dihydrochloride), light green, biconvex, pentagonal, and debossed with “BMS” on one side and “215” on the other side.
- 30 mg: 30 mg of daclatasvir (equivalent to 33 mg daclatasvir dihydrochloride), green, biconvex, pentagonal, and debossed with “BMS” on one side and “213” on the other side.
- 90 mg: 90 mg of daclatasvir (equivalent to 99 mg daclatasvir dihydrochloride), light green, biconvex, round, and embossed with “BMS” on one side and “011” on the other side.
-
4 CONTRAINDICATIONS
- When DAKLINZA is used in combination with other agents, the contraindications applicable to those agents are applicable to the combination regimen. Refer to the respective prescribing information for a list of contraindications.
- DAKLINZA is contraindicated in combination with drugs that strongly induce CYP3A and, thus, may lead to lower exposure and loss of efficacy of DAKLINZA. Contraindicated drugs include, but are not limited to those listed in Table 3 [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
Table 3: Drugs that are Contraindicated with DAKLINZA Drug Class Drugs Within Class that are Contraindicated with DAKLINZAa Clinical Comments a This table is not a comprehensive list of all drugs that strongly induce CYP3A. Anticonvulsants
phenytoin, carbamazepine
May lead to loss of virologic response to DAKLINZA
Antimycobacterial agents
rifampin
Herbal products
St. John’s wort (Hypericum perforatum)
-
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Hepatitis B Virus Reactivation in Patients Coinfected with HCV and HBV
Hepatitis B virus (HBV) reactivation has been reported in HCV/HBV coinfected patients who were undergoing or had completed treatment with HCV direct-acting antivirals, and who were not receiving HBV antiviral therapy. Some cases have resulted in fulminant hepatitis, hepatic failure, and death. Cases have been reported in patients who are HBsAg positive and also in patients with serologic evidence of resolved HBV infection (i.e., HBsAg negative and anti-HBc positive). HBV reactivation has also been reported in patients receiving certain immunosuppressant or chemotherapeutic agents; the risk of HBV reactivation associated with treatment with HCV direct-acting antivirals may be increased in these patients.
HBV reactivation is characterized as an abrupt increase in HBV replication manifesting as a rapid increase in serum HBV DNA level. In patients with resolved HBV infection, reappearance of HBsAg can occur. Reactivation of HBV replication may be accompanied by hepatitis, i.e., increases in aminotransferase levels and, in severe cases, increases in bilirubin levels, liver failure, and death can occur.
Test all patients for evidence of current or prior HBV infection by measuring HBsAg and anti-HBc before initiating HCV treatment with DAKLINZA. In patients with serologic evidence of HBV infection, monitor for clinical and laboratory signs of hepatitis flare or HBV reactivation during HCV treatment with DAKLINZA and during post-treatment follow-up. Initiate appropriate patient management for HBV infection as clinically indicated.
5.2 Risk of Adverse Reactions or Loss of Virologic Response Due to Drug Interactions
The concomitant use of DAKLINZA and other drugs may result in known or potentially significant drug interactions, some of which may lead to [see Contraindications (4) and Drug Interactions (7)]:
- loss of therapeutic effect of DAKLINZA and possible development of resistance,
- dosage adjustments of concomitant medications or DAKLINZA,
- possible clinically significant adverse reactions from greater exposures of concomitant drugs or DAKLINZA.
See Table 3 for drugs contraindicated with DAKLINZA due to loss of efficacy and possible development of resistance [see Contraindications (4)]. See Table 7 for steps to prevent or manage other possible and known significant drug interactions [see Drug Interactions (7)]. Consider the potential for drug interactions before and during DAKLINZA therapy, review concomitant medications during DAKLINZA therapy, and monitor for the adverse reactions associated with the concomitant drugs.
5.3 Serious Symptomatic Bradycardia When Coadministered with Sofosbuvir and Amiodarone
Postmarketing cases of symptomatic bradycardia and cases requiring pacemaker intervention have been reported when amiodarone was coadministered with a sofosbuvir-containing regimen. A fatal cardiac arrest was reported in a patient receiving a sofosbuvir-containing regimen (ledipasvir/sofosbuvir). Bradycardia has generally occurred within hours to days, but cases have been observed up to 2 weeks after initiating HCV treatment. Patients also taking beta blockers or those with underlying cardiac comorbidities and/or advanced liver disease may be at increased risk for symptomatic bradycardia with coadministration of amiodarone. Bradycardia generally resolved after discontinuation of HCV treatment. The mechanism for this bradycardia effect is unknown.
Coadministration of amiodarone with DAKLINZA in combination with sofosbuvir is not recommended. For patients taking amiodarone who have no alternative treatment options and who will be coadministered DAKLINZA and sofosbuvir:
- Counsel patients about the risk of serious symptomatic bradycardia.
- Cardiac monitoring in an inpatient setting for the first 48 hours of coadministration is recommended, after which outpatient or self-monitoring of the heart rate should occur on a daily basis through at least the first 2 weeks of treatment.
Patients who are taking sofosbuvir in combination with DAKLINZA who need to start amiodarone therapy due to no other alternative treatment options should undergo similar cardiac monitoring as outlined above.
Due to amiodarone’s long elimination half-life, patients discontinuing amiodarone just prior to starting sofosbuvir in combination with DAKLINZA should also undergo similar cardiac monitoring as outlined above.
Patients who develop signs or symptoms of bradycardia should seek medical evaluation immediately. Symptoms may include near-fainting or fainting, dizziness or lightheadedness, malaise, weakness, excessive tiredness, shortness of breath, chest pain, confusion, or memory problems [see Adverse Reactions (6.2) and Drug Interactions (7.3), Table 7].
5.4 Risks Associated with Ribavirin Combination Treatment
If DAKLINZA and sofosbuvir are administered with ribavirin, the warnings and precautions for ribavirin, in particular the pregnancy avoidance warning, apply to this combination regimen. Refer to the ribavirin prescribing information for a full list of the warnings and precautions for ribavirin.
-
6 ADVERSE REACTIONS
If DAKLINZA and sofosbuvir are administered with ribavirin, refer to the prescribing information for ribavirin regarding ribavirin-associated adverse reactions.
The following serious adverse reaction is described below and elsewhere in the labeling:
- Serious Symptomatic Bradycardia When Coadministered with Sofosbuvir and Amiodarone [see Warnings and Precautions (5.3)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Approximately 2400 subjects with chronic HCV infection have been treated with the recommended dose of DAKLINZA in combination with other anti-HCV drugs in clinical trials. Six hundred seventy-nine subjects have received a DAKLINZA and sofosbuvir-based regimen. Safety experience from three clinical trials of DAKLINZA and sofosbuvir with or without ribavirin is presented.
DAKLINZA and Sofosbuvir
In the ALLY-3 trial, 152 treatment-naive and treatment-experienced subjects with HCV genotype 3 infection were treated with DAKLINZA 60 mg once daily in combination with sofosbuvir for 12 weeks. The most common adverse reactions (frequency of 10% or greater) were headache and fatigue. All adverse reactions were mild to moderate in severity. No subjects discontinued therapy for adverse events.
In the ALLY-2 trial, 153 treatment-naive and treatment-experienced subjects with HCV/HIV-1 coinfection were treated with DAKLINZA 60 mg once daily (dose-adjusted for concomitant antiretroviral use) in combination with sofosbuvir for 12 weeks. The most common adverse reaction (frequency of 10% or greater) was fatigue. The majority of adverse reactions were mild to moderate in severity. No subjects discontinued therapy for adverse events. Adverse reactions considered at least possibly related to treatment and occurring at a frequency of 5% or greater in ALLY-3 or ALLY-2 are presented in Table 4.
Table 4: Adverse Reactions (All Severity) Reported at ≥5% Frequency, DAKLINZA + Sofosbuvir, Studies ALLY-3 and ALLY-2 Adverse Reaction ALLY-3: HCV Genotype 3
n=152ALLY-2: HCV/HIV-1 Coinfection
n=153Headache
14%
8%
Fatigue
14%
15%
Nausea
8%
9%
Diarrhea
5%
7%
DAKLINZA, Sofosbuvir, and Ribavirin
In the ALLY-1 trial, 113 subjects with chronic HCV infection, including 60 subjects with Child-Pugh A, B, or C cirrhosis and 53 subjects with recurrence of HCV after liver transplantation, were treated with DAKLINZA 60 mg once daily in combination with sofosbuvir and ribavirin for 12 weeks. The most common adverse reactions (frequency of 10% or greater) among the 113 subjects were headache, anemia, fatigue, and nausea. The majority of adverse reactions were mild to moderate in severity. Of the 15 (13%) subjects who discontinued study drug for adverse events, 13 (12%) subjects discontinued ribavirin only and 2 (2%) subjects discontinued all study drugs. During treatment, 4 subjects in the cirrhotic cohort underwent liver transplantation. Adverse reactions considered at least possibly related to treatment and occurring at a frequency of 5% or greater in either treatment cohort in ALLY-1 are presented in Table 5.
Table 5: Adverse Reactions (All Severity) Reported at ≥5% Frequency in Either Treatment Cohort, DAKLINZA + Sofosbuvir + Ribavirin, Study ALLY-1 Adverse Reaction Child-Pugh A, B, or C Cirrhosis
n=60Recurrence after Liver Transplantation
n=53Headache
12%
30%
Anemia
20%
19%
Fatigue
15%
17%
Nausea
15%
6%
Rash
8%
2%
Diarrhea
3%
6%
Insomnia
3%
6%
Dizziness
0
6%
Somnolence
5%
0
Laboratory Abnormalities
Selected Grade 3 and 4 treatment-emergent laboratory abnormalities observed in clinical trials of DAKLINZA in combination with sofosbuvir with or without ribavirin are presented in Table 6.
Table 6: Selected Grade 3 and 4 Laboratory Abnormalities in Clinical Trials of DAKLINZA + Sofosbuvir ± Ribavirin, Studies ALLY-3, ALLY-2, and ALLY-1 Parameter Percent with Abnormality ALLY-3: HCV
Genotype 3
DAKLINZA +
Sofosbuvir
n=152ALLY-2: HCV/HIV-1
Coinfection
DAKLINZA +
Sofosbuvir
n=153ALLY-1: Child-Pugh A,
B, or C with Cirrhosis
and Post-transplant
DAKLINZA +
Sofosbuvir + Ribavirin
n=113a In the ALLY-2 trial, Grade 3 and 4 increases in total bilirubin were observed only in subjects receiving concomitant atazanavir. Hemoglobin (≤8.9 g/dL)
0
0
6%
Alanine aminotransferase (ALT) increased (≥5.1 × ULN)
0
0
2%
Aspartate aminotransferase (AST) increased (≥5.1 × ULN)
0
0
3%
Total bilirubin increased (≥2.6 × ULN)
0
5%a
8%
Lipase increased (≥3.1 × ULN)
2%
4%
4%
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of DAKLINZA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiac Disorders: Serious symptomatic bradycardia has been reported in patients taking amiodarone who initiate treatment with a sofosbuvir-containing regimen [see Warnings and Precautions (5.3) and Drug Interactions (7.3)].
-
7 DRUG INTERACTIONS
7.1 Potential for Other Drugs to Affect DAKLINZA
Daclatasvir is a substrate of CYP3A. Therefore, moderate or strong inducers of CYP3A may decrease the plasma levels and therapeutic effect of daclatasvir [see Dosage and Administration (2.3), Contraindications (4), and Table 7]. Strong inhibitors of CYP3A (eg, clarithromycin, itraconazole, ketoconazole, ritonavir) may increase the plasma levels of daclatasvir [see Dosage and Administration (2.3) and Table 7].
7.2 Potential for DAKLINZA to Affect Other Drugs
Daclatasvir is an inhibitor of P-glycoprotein transporter (P-gp), organic anion transporting polypeptide (OATP) 1B1 and 1B3, and breast cancer resistance protein (BCRP). Administration of DAKLINZA may increase systemic exposure to medicinal products that are substrates of P-gp, OATP 1B1 or 1B3, or BCRP, which could increase or prolong their therapeutic effect or adverse reactions (see Table 7).
7.3 Established and Other Potentially Significant Drug Interactions
Clearance of HCV infection with direct acting antivirals may lead to changes in hepatic function, which may impact the safe and effective use of concomitant medications. For example, altered blood glucose control resulting in serious symptomatic hypoglycemia has been reported in diabetic patients in postmarketing case reports and published epidemiological studies. Management of hypoglycemia in these cases required either discontinuation or dose modification of concomitant medications used for diabetes treatment.
Frequent monitoring of relevant laboratory parameters (e.g. International Normalized Ratio [INR] in patients taking warfarin, blood glucose levels in diabetic patients) or drug concentrations of concomitant medications such as cytochrome P450 substrates with a narrow therapeutic index (e.g. certain immunosuppressants) is recommended to ensure safe and effective use. Dose adjustments of concomitant medications may be necessary.
Refer to the prescribing information for other agents in the regimen for drug interaction information. The most conservative recommendation should be followed.
Please also refer to Section 4 (Contraindications) and Section 12.3 (Pharmacokinetics) for complete information on all drug interactions.
Table 7 provides clinical recommendations for established or potentially significant drug interactions between DAKLINZA and other drugs [see Contraindications (4)]. Clinically relevant increase in concentration is indicated as “↑” and clinically relevant decrease as “↓” for drug interaction data [see Clinical Pharmacology (12.3)].
Table 7: Established and Other Potentially Significant Drug Interactions Concomitant Drug Class:
Drug NameEffect on Concentrationa Clinical Comment a The direction of the arrow (↑ = increase, ↓ = decrease) indicates the direction of the change in pharmacokinetic parameters.
b These interactions have been studied [see Clinical Pharmacology (12.3), Tables 9 and 10].HIV antiviral agents
Protease inhibitors:
Atazanavir with ritonavirb
Indinavir
Nelfinavir
Saquinavir
↑ Daclatasvir
Decrease DAKLINZA dose to 30 mg once daily.Other antiretrovirals:
Cobicistat-containing antiretroviral regimens
Examples: atazanavir/cobicistat, elvitegravir/cobicistat/emtricitabine/
tenofovir disoproxil fumarate
↑ Daclatasvir
Decrease DAKLINZA dose to 30 mg once daily except with darunavir combined with cobicistat.Non-nucleoside reverse transcriptase inhibitors (NNRTI):
EfavirenzbEtravirine
Nevirapine
↓ Daclatasvir
Increase DAKLINZA dose to 90 mg once daily.Strong CYP3A inhibitors (see also HIV antiviral agents)
Examples: clarithromycin, itraconazole, ketoconazole,b nefazodone, posaconazole, telithromycin, voriconazole
↑ Daclatasvir
Decrease DAKLINZA dose to 30 mg once daily when coadministered with strong inhibitors of CYP3A.
Moderate CYP3A inducers (see also HIV antiviral agents)
Examples: bosentan, dexamethasone, modafinil, nafcillin, rifapentine
↓ Daclatasvir
Increase DAKLINZA dose to 90 mg once daily when coadministered with moderate inducers of CYP3A.
Anticoagulants
Dabigatran etexilate mesylate
↑ Dabigatran
Use of DAKLINZA with dabigatran etexilate is not recommended in specific renal impairment groups, depending on the indication. Please see the dabigatran prescribing information for specific recommendations.
Cardiovascular agents
Antiarrhythmic:
Amiodarone
Amiodarone: effects unknown
Coadministration of amiodarone with DAKLINZA in combination with sofosbuvir is not recommended because it may result in serious symptomatic bradycardia. The mechanism of this effect is unknown. If coadministration is required, cardiac monitoring is recommended. [See Warnings and Precautions (5.3) and Adverse Reactions (6.2).]Antiarrhythmic:
Digoxinb
↑ Digoxin
Patients already receiving daclatasvir initiating digoxin: Initiate treatment using the lowest appropriate digoxin dosage. Monitor digoxin concentrations; adjust digoxin doses if necessary and continue monitoring.Patients already receiving digoxin prior to initiating daclatasvir: Measure serum digoxin concentrations before initiating daclatasvir. Reduce digoxin concentrations by decreasing digoxin dosage by approximately 15% to 30% or by modifying the dosing frequency and continue monitoring.
Lipid-lowering agents
HMG-CoA reductase inhibitors:
Atorvastatin
Fluvastatin
Pitavastatin
PravastatinRosuvastatinb
Simvastatin
↑ Atorvastatin
↑ Fluvastatin
↑ Pitavastatin
↑ Pravastatin
↑ Rosuvastatin
↑ Simvastatin
Monitor for HMG-CoA reductase inhibitor-associated adverse events such as myopathy.Narcotic Analgesic/Treatment of Opioid Dependence
buprenorphine
buprenorphine/naloxone↑ buprenorphine
↑ norbuprenorphineFor buprenorphine or buprenorphine/naloxone, no adjustment is needed, but clinical monitoring for buprenorphine-associated adverse events is recommended.
7.4 Drugs without Clinically Significant Interactions with DAKLINZA
Please see Section 12.3 (Pharmacokinetics) for information regarding anticipated interactions that are not clinically relevant.
Based on the results of drug interaction trials [see Clinical Pharmacology (12.3)], no clinically relevant changes in exposure were observed for cyclosporine, darunavir (with ritonavir), dolutegravir, escitalopram, ethinyl estradiol/norgestimate, lopinavir (with ritonavir), methadone, midazolam, tacrolimus, or tenofovir with concomitant use of daclatasvir. No clinically relevant changes in daclatasvir exposure were observed with cyclosporine, darunavir (with ritonavir), dolutegravir, escitalopram, famotidine, lopinavir (with ritonavir), omeprazole, sofosbuvir, tacrolimus, or tenofovir. No dosage adjustment for daclatasvir is necessary with darunavir/cobicistat or moderate CYP3A inhibitors, including atazanavir (unboosted), fosamprenavir, ciprofloxacin, diltiazem, erythromycin, fluconazole, or verapamil.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
No adequate human data are available to determine whether or not DAKLINZA poses a risk to pregnancy outcomes. In animal reproduction studies in rats and rabbits, no evidence of fetal harm was observed with oral administration of daclatasvir during organogenesis at doses that produced exposures up to 6 and 22 times, respectively, the recommended human dose (RHD) of 60 mg of DAKLINZA. However, embryofetal toxicity was observed in rats and rabbits at maternally toxic doses that produced exposures of 33 and 98 times the human exposure, respectively, at the RHD of 60 mg of DAKLINZA [see Data]. In rat pre- and postnatal developmental studies, no developmental toxicity was observed at maternal systemic exposure (AUC) to daclatasvir approximately 3.6 times higher than the RHD of DAKLINZA.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
If DAKLINZA and sofosbuvir are administered with ribavirin, the combination regimen is contraindicated in pregnant women and in men whose female partners are pregnant. Refer to the ribavirin prescribing information for more information on use in pregnancy.
Data
Animal Data
Daclatasvir was administered orally to pregnant rats at doses of 0, 50, 200, or 1000 mg/kg/day on gestation days 6 to 15. Maternal toxicity (mortality, adverse clinical signs, body-weight losses, and reduced food consumption) was noted at doses of 200 and 1000 mg/kg/day. In the offspring, malformations of the fetal brain, skull, eyes, ears, nose, lip, palate, or limbs were observed at doses of 200 and 1000 mg/kg. The dose of 1000 mg/kg was associated with profound embryolethality and lower fetal body weight. No malformations were noted at 50 mg/kg/day. Systemic exposure (AUC) at 50 mg/kg/day in pregnant females was 6 times higher than exposures at the RHD.
In rabbits, daclatasvir was initially administered at doses of 0, 40, 200, or 750 mg/kg/day during the gestation days 7 to 19. Daclatasvir dosing was modified due to vehicle toxicity during the study to doses of 20, 99, and 370 mg/kg/day, respectively. Maternal toxicity was noted at doses of 200/99 and 750/370 mg/kg/day with adverse clinical signs and severe reductions in body weight and food consumption. Mortality and euthanasia occurred in multiple dams at 750/370 mg/kg/day. At 200/99 mg/kg/day, fetal effects included increased embryofetal lethality, reduced fetal body weights, and increased incidences of fetal malformations of the ribs as well as head and skull. No malformations were noted in rabbits at 40/20 mg/kg/day. Systemic exposures (AUC) at 40/20 mg/kg/day were 22 times higher than exposures at the RHD.
In a pre- and postnatal developmental study, daclatasvir was administered orally at 0, 25, 50, or 100 mg/kg/day from gestation day 6 to lactation day 20. At 100 mg/kg/day, maternal toxicity included mortality and dystocia; developmental toxicity included slight reductions in offspring viability in the perinatal and neonatal periods and reductions in birth weight that persisted into adulthood. There was neither maternal nor developmental toxicity at doses up to 50 mg/kg/day. Systemic exposures (AUC) at this dose were 3.6 times higher than the RHD.
8.2 Lactation
Risk Summary
It is not known whether DAKLINZA is present in human milk, affects human milk production, or has effects on the breastfed infant. Daclatasvir was present in the milk of lactating rats (see Data).
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for DAKLINZA and any potential adverse effects on the breastfed child from DAKLINZA or from the underlying maternal condition.
If DAKLINZA is administered with ribavirin, the nursing mothers information for ribavirin also applies to this combination regimen. Refer to ribavirin prescribing information for additional information.
Data
Milk concentrations of daclatasvir were evaluated on lactation day 10 as part of the rat pre- and postnatal development study (see Data in 8.1). Daclatasvir was present in rat milk with concentrations 1.7 to 2 times maternal plasma levels.
8.3 Females and Males of Reproductive Potential
If DAKLINZA and sofosbuvir are administered with ribavirin, the information for ribavirin with regard to pregnancy testing, contraception, and infertility also applies to this combination regimen. Refer to ribavirin prescribing information for additional information.
8.4 Pediatric Use
Safety and effectiveness of DAKLINZA in pediatric patients younger than 18 years of age have not been established.
8.5 Geriatric Use
Of 1184 subjects treated with the recommended dose of DAKLINZA in ten clinical trials, 7% of subjects were 65 years of age or older. Safety was similar across older and younger subjects and there were no safety findings unique to subjects 65 years and older. SVR12 rates were comparable among older and younger subjects. No dosage adjustment of DAKLINZA is required for elderly patients [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
No dosage adjustment of DAKLINZA is required for patients with any degree of renal impairment [see Clinical Pharmacology (12.3)]. Refer also to the sofosbuvir and ribavirin prescribing information for information regarding use in patients with renal impairment.
8.7 Hepatic Impairment
Based on a hepatic impairment study in non–HCV-infected subjects, no dosage adjustment of DAKLINZA is required for patients with mild (Child-Pugh A), moderate (Child-Pugh B), or severe (Child-Pugh C) hepatic impairment [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There is no known antidote for overdose of DAKLINZA. Treatment of overdose with DAKLINZA should consist of general supportive measures, including monitoring of vital signs and observation of the patient’s clinical status. Because daclatasvir is highly protein bound (>99%), dialysis is unlikely to significantly reduce plasma concentrations of the drug.
-
11 DESCRIPTION
DAKLINZA (daclatasvir) is an inhibitor of HCV nonstructural protein 5A (NS5A). The chemical name for drug substance daclatasvir dihydrochloride is carbamic acid, N,N′-[[1,1′-biphenyl]-4,4′-diylbis[1H-imidazole-5,2-diyl-(2S)-2,1-pyrrolidinediyl[(1S)-1-(1-methylethyl)-2-oxo-2,1-ethanediyl]]]bis-, C,C′-dimethyl ester, hydrochloride (1:2). Its molecular formula is C40H50N8O62HCl, and its molecular weight is 738.88 (free base). Daclatasvir dihydrochloride has the following structural formula:
Daclatasvir dihydrochloride drug substance is white to yellow. Daclatasvir is freely soluble in water (>700 mg/mL).
DAKLINZA 60 mg tablets contain 60 mg daclatasvir (equivalent to 66 mg daclatasvir dihydrochloride) and the inactive ingredients anhydrous lactose (116 mg), microcrystalline cellulose, croscarmellose sodium, silicon dioxide, magnesium stearate, and Opadry green.
DAKLINZA 30 mg tablets contain 30 mg daclatasvir (equivalent to 33 mg daclatasvir dihydrochloride) and the inactive ingredients anhydrous lactose (58 mg), microcrystalline cellulose, croscarmellose sodium, silicon dioxide, magnesium stearate, and Opadry green.
DAKLINZA 90 mg tablets contain 90 mg daclatasvir (equivalent to 99 mg daclatasvir dihydrochloride) and the inactive ingredients anhydrous lactose (173 mg), microcrystalline cellulose, croscarmellose sodium, silicon dioxide, magnesium stearate, and Opadry green.
Opadry green contains hypromellose, titanium dioxide, polyethylene glycol 400, FD&C blue #2/indigo carmine aluminum lake, and yellow iron oxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Daclatasvir is a direct-acting antiviral agent (DAA) against the hepatitis C virus [see Microbiology (12.4)].
12.3 Pharmacokinetics
The pharmacokinetic properties of daclatasvir were evaluated in healthy adult subjects and in subjects with chronic HCV. Administration of daclatasvir tablets in HCV-infected subjects resulted in approximately dose-proportional increases in Cmax, AUC, and Cmin up to 60 mg once daily. Steady state is anticipated after approximately 4 days of once-daily daclatasvir administration. Exposure of daclatasvir was similar between healthy and HCV-infected subjects. Population pharmacokinetic estimates for daclatasvir 60 mg once daily in chronic HCV-infected subjects are shown in Table 8.
Table 8: Population Pharmacokinetic Estimates for Daclatasvir in Chronic HCV-Infected Subjects Receiving Daclatasvir 60 mg Once Daily and Sofosbuvir 400 mg Once Daily Parameters Daclatasvir 60 mg once daily
(n=152)AUC0-24h (ngh/mL)
Mean ± standard deviation
10973 ± 5288
Median (range)
9680 (3807-41243)
C24h (ng/mL)
Mean ± standard deviation
182 ± 137
Median (range)
148 (21-1050)
Absorption and Bioavailability
In HCV-infected subjects following multiple oral doses of daclatasvir tablet ranging from 1 mg to 100 mg once daily, peak plasma concentrations occurred within 2 hours post dose.
In vitro studies with human Caco-2 cells indicated that daclatasvir is a substrate of P-gp. The absolute bioavailability of the tablet formulation is 67%.
Effect of Food on Oral Absorption
In healthy subjects, administration of a daclatasvir 60 mg tablet after a high-fat, high-caloric meal (approximately 951 total kcal, 492 kcal from fat, 312 kcal from carbohydrates, 144 kcal from protein) decreased daclatasvir Cmax and AUC(0-inf) by 28% and 23%, respectively, compared with fasted conditions. A food effect was not observed with administration of a daclatasvir 60 mg tablet after a low-fat, low-caloric meal (approximately 277 total kcal, 41 kcal from fat, 190 kcal from carbohydrates, 44 kcal from protein) compared with fasted conditions [see Dosage and Administration (2)].
Distribution
With multiple dosing, protein binding of daclatasvir in HCV-infected subjects was approximately 99% and independent of dose at the dose range studied (1-100 mg). In subjects who received daclatasvir 60 mg tablet orally followed by 100 μg [13C,15N]-daclatasvir intravenous dose, estimated volume of distribution at steady state was 47 L.
Metabolism
Daclatasvir is a substrate of CYP3A, with CYP3A4 being the primary CYP isoform responsible for metabolism. Following single-dose oral administration of 25 mg 14C-daclatasvir in healthy subjects, the majority of radioactivity in plasma was predominately attributed to parent drug (97% or greater).
Elimination
Following single-dose oral administration of 25 mg 14C-daclatasvir in healthy subjects, 88% of total radioactivity was recovered in feces (53% of the dose as unchanged daclatasvir) and 6.6% of the dose was excreted in the urine (primarily as unchanged daclatasvir). Following multiple-dose administration of daclatasvir in HCV-infected subjects, with doses ranging from 1 mg to 100 mg once daily, the terminal elimination half-life of daclatasvir ranged from approximately 12 to 15 hours. In subjects who received daclatasvir 60 mg tablet orally followed by 100 μg [13C,15N]-daclatasvir intravenous dose, the total clearance was 4.2 L/h.
Specific Populations
Renal Impairment
The pharmacokinetics of daclatasvir following a single 60 mg oral dose was studied in non–HCV-infected subjects with renal impairment. Using a regression analysis, the predicted AUC(0-inf) of daclatasvir was estimated to be 26%, 60%, and 80% higher in subjects with creatinine clearance (CLcr) values of 60, 30, and 15 mL/min, respectively, relative to subjects with normal renal function (CLcr of 90 mL/min, defined using the Cockcroft-Gault CLcr formula), and daclatasvir unbound AUC(0-inf) was predicted to be 18%, 39%, and 51% higher for subjects with CLcr values of 60, 30, and 15 mL/min, respectively, relative to subjects with normal renal function. Using observed data, subjects with end-stage renal disease requiring hemodialysis had a 27% increase in daclatasvir AUC(0-inf) and a 20% increase in unbound AUC(0-inf) compared to subjects with normal renal function as defined using the Cockcroft-Gault CLcr formula [see Use in Specific Populations (8.6)].
Daclatasvir is highly protein bound to plasma proteins and is unlikely to be removed by dialysis.
Hepatic Impairment
The pharmacokinetics of daclatasvir following a single 30 mg oral dose was studied in non–HCV-infected subjects with mild (Child-Pugh A), moderate (Child-Pugh B), and severe (Child-Pugh C) hepatic impairment compared to a corresponding matched control group. The Cmax and AUC(0-inf) of total daclatasvir (free and protein-bound drug) were lower by 46% and 43%, respectively, in Child-Pugh A subjects; by 45% and 38%, respectively, in Child-Pugh B subjects; and by 55% and 36%, respectively, in Child-Pugh C subjects. The Cmax and AUC(0‑inf) of unbound daclatasvir were lower by 43% and 40%, respectively, in Child-Pugh A subjects; by 14% and 2%, respectively, in Child-Pugh B subjects; and by 33% and 5%, respectively, in Child-Pugh C subjects [see Use in Specific Populations (8.7)].
Pediatric Patients
The pharmacokinetics of daclatasvir in pediatric patients has not been evaluated.
Geriatric Patients
Population pharmacokinetic analysis in HCV-infected subjects showed that within the age range (18-79 years) analyzed, age did not have a clinically relevant effect on the pharmacokinetics of daclatasvir [see Use in Specific Populations (8.5)].
Drug Interactions
Cytochrome P450 (CYP) Enzymes
Daclatasvir is a substrate of CYP3A. In vitro, daclatasvir did not inhibit (IC50 greater than 40 microM) CYP enzymes 1A2, 2B6, 2C8, 2C9, 2C19, or 2D6. Daclatasvir did not have a clinically relevant effect on the exposure of midazolam, a sensitive CYP3A substrate.
Transporters
Daclatasvir is a substrate of P-gp. However, cyclosporine, which inhibits multiple transporters including P-gp, did not have a clinically relevant effect on the pharmacokinetics of daclatasvir. Daclatasvir, in vitro, did not inhibit OCT2 and did not have a clinically relevant effect on the pharmacokinetics of tenofovir, an OAT substrate. Daclatasvir demonstrated inhibitory effects on digoxin (a P-gp substrate) and rosuvastatin (an OATP 1B1, OATP 1B3, and BCRP substrate) in drug-drug interaction trials.
Drug interaction studies were conducted with daclatasvir and other drugs likely to be coadministered or drugs used as probes to evaluate potential drug-drug interactions. The effects of daclatasvir on the Cmax, AUC, and Cmin of the coadministered drug are summarized in Table 9, and the effects of the coadministered drug on the Cmax, AUC, and Cmin of daclatasvir are summarized in Table 10. For information regarding clinical recommendations, see Contraindications (4) and Drug Interactions (7.3). Drug interaction studies were conducted in healthy adults unless otherwise noted.
Table 9: Effect of DAKLINZA on the Pharmacokinetics of Concomitant Drugs Concomitant Drug Coadministered Drug Dose DAKLINZA Dose Ratio of Pharmacokinetic Parameters of Coadministered Drug Combination/No Combination (90% CI) Cmax AUC Cmina Note: In Table 9, for the concomitant medication, drug-drug interaction data were not included if 90% CIs for Cmax, AUC, and Cmin (if applicable for Cmin) were within 80% to 125%. These concomitant medications include cyclosporine, escitalopram, ethinyl estradiol/norgestimate, midazolam, tacrolimus, and tenofovir disoproxil fumarate.
a Cmin was defined as either the Ctau or the Ctrough concentration value.
b The buprenorphine and norbuprenorphine pharmacokinetic parameters were dose normalized to 8 mg.
c Samples up to 6 hours collected; C0h substituted for C12h concentration value.
d The methadone pharmacokinetic parameters were dose normalized to 40 mg.
NA = Not available.Buprenorphine
/NaloxoneStable maintenance 8/2 mg to 24/6 mg QD
60 mg QD
Buprenorphineb
1.30
(1.03, 1.64)
Norbuprenorphineb
1.65
(1.38, 1.99)Buprenorphineb
1.37
(1.24, 1.52)
Norbuprenorphineb
1.62
(1.30, 2.02)Buprenorphineb
1.17
(1.03, 1.32)
Norbuprenorphineb
1.46
(1.12, 1.89)Darunavirc
600 mg BID with ritonavir 100 mg BID
30 mg QD
0.97
(0.80, 1.17)0.90
(0.73, 1.11)0.98
(0.67, 1.44)Digoxin
0.125 mg QD
60 mg QD
1.65
(1.52, 1.80)1.27
(1.20, 1.34)1.18
(1.09, 1.28)Dolutegravir
50 mg QD
60 mg QD
1.29
(1.07, 1.57)1.33
(1.11, 1.59)1.45
(1.25, 1.68)Lopinavirc
400 mg BID with ritonavir 100 mg BID
30 mg QD
1.22
(1.06, 1.41)1.15
(0.77, 1.72)1.54
(0.46, 5.07)Methadone
Stable maintenance
40-120 mg QD60 mg QD
Total
methadoned:
1.09 (0.99, 1.21)
R-methadoned:
1.07 (0.97, 1.18)Total
methadoned:
1.11 (0.97, 1.26)
R-methadoned:
1.08 (0.94, 1.24)Total
methadoned:
1.12 (0.96, 1.29)
R-methadoned:
1.08 (0.93, 1.26)Rosuvastatin
10 mg single dose
60 mg QD
2.04
(1.83, 2.26)1.58
(1.44, 1.74)NA
Simeprevir
150 mg QD
60 mg QD
1.39
(1.27, 1.52)1.44
(1.32, 1.56)1.49
(1.33, 1.67)Table 10: Effect of Coadministered Drugs on DAKLINZA Pharmacokinetics Concomitant Drug Coadministered Drug Dose DAKLINZA Dose Ratio of Pharmacokinetic Parameters of Daclatasvir
Combination/No Combination (90% CI)Cmax AUC Cmina Note: In Table 10, drug-drug interaction data for daclatasvir were not included for a study with tacrolimus because the 90% CIs for Cmax, AUC, and Cmin were within 80% to 125%.
a Cmin was defined as either the Ctau or the Ctrough daclatasvir concentration value.
b Observed, non-dose normalized data. For the reference arm, a 60 mg QD dose of daclatasvir was administered without the HIV comedications (boosted protease inhibitors, efavirenz) in order to compare the effect on daclatasvir exposures.
NA = Not available.Atazanavir/
ritonavir300 mg/100 mg QD
20 mg QD
(test arm)0.45
(0.41, 0.49)b0.70
(0.65, 0.75)b1.22
(1.08, 1.37)bCyclosporine
400 mg single
dose60 mg QD
1.04
(0.94, 1.15)1.40
(1.29, 1.53)1.56
(1.41, 1.71)Darunavir/
ritonavir800 mg/100 mg QD
30 mg QD
(test arm)0.38
(0.35, 0.42)b0.70
(0.66, 0.75)bNA
Dolutegravir
50 mg QD
60 mg QD
1.03
(0.84, 1.25)0.98
(0.83, 1.15)1.06
(0.88, 1.29)Efavirenz
600 mg QD
120 mg QD
(test arm)1.67
(1.51, 1.84)b1.37
(1.21, 1.55)b0.83
(0.69, 1.00)bEscitalopram
10 mg QD
60 mg QD
1.14
(0.98, 1.32)1.12
(1.01, 1.26)1.23
(1.09, 1.38)Famotidine
40 mg single
dose60 mg single dose
(2 hours after famotidine administration)0.56
(0.46, 0.67)0.82
(0.70, 0.96)0.89
(0.75, 1.06)Ketoconazole
400 mg QD
10 mg single dose
1.57
(1.31, 1.88)3.00
(2.62, 3.44)NA
Lopinavir/
ritonavir400 mg/100 mg BID
30 mg QD
(test arm)0.34
(0.31, 0.37)b0.58
(0.54, 0.62)bNA
Omeprazole
40 mg single dose
60 mg single dose
0.64
(0.54, 0.77)0.84
(0.73, 0.96)0.92
(0.80, 1.05)Rifampin
600 mg QD
60 mg single dose
0.44
(0.40, 0.48)0.21
(0.19, 0.23)NA
Simeprevir
150 mg QD
60 mg QD
1.50
(1.39, 1.62)1.96
(1.84, 2.10)2.68
(2.42, 2.98)Tenofovir disoproxil fumarate
300 mg QD
60 mg QD
1.06
(0.98, 1.15)1.10
(1.01, 1.21)1.15
(1.02, 1.30)No clinically relevant interaction is anticipated for daclatasvir or the following concomitant medications: peginterferon alfa, ribavirin, or antacids. No clinically relevant interaction is anticipated for daclatasvir with concomitant use of rilpivirine.
12.4 Microbiology
Mechanism of Action
Daclatasvir is an inhibitor of NS5A, a nonstructural protein encoded by HCV. Daclatasvir binds to the N-terminus of NS5A and inhibits both viral RNA replication and virion assembly. Characterization of daclatasvir-resistant viruses, biochemical studies, and computer modeling data indicate that daclatasvir interacts with the N-terminus within Domain 1 of the protein, which may cause structural distortions that interfere with NS5A functions.
Antiviral Activity
Daclatasvir had median EC50 values of 0.008 nM (range, 0.002-0.03 nM; n=35), 0.002 nM (range, 0.0007-0.006 nM; n=30), and 0.2 nM (range, 0.006-3.2 nM; n=17) against hybrid replicons containing genotypes 1a, 1b, and 3a subject-derived NS5A sequences, respectively, without detectable daclatasvir resistance-associated polymorphisms at NS5A amino acid positions 28, 30, 31, or 93. Daclatasvir activity was reduced against genotypes 1a, 1b, and 3a subject-derived replicons with resistance-associated polymorphisms at positions 28, 30, 31, or 93, with median EC50 values of 76 nM (range, 4.6-2409 nM; n=5), 0.05 nM (range, 0.002-10 nM; n=12), and 13.5 nM (range, 1.3-50 nM; n=4), respectively. Similarly, the EC50 values of daclatasvir against 3 genotype 3b and 1 genotype 3i subject-derived NS5A sequences with polymorphisms (relative to a genotype 3a reference) at positions 30+31 (genotype 3b) or 30+62 (genotype 3i) were ≥3620 nM.
Daclatasvir was not antagonistic with interferon alfa, HCV NS3/4A protease inhibitors, HCV NS5B nucleoside analog inhibitors, and HCV NS5B non-nucleoside inhibitors in cell culture combination antiviral activity studies using the cell-based HCV replicon system.
Resistance
In Cell Culture
HCV genotype 1a, 1b, and 3a replicon variants with reduced susceptibility to daclatasvir were selected in cell culture, and the genotype and phenotype of daclatasvir-resistant NS5A amino acid variants were characterized. Phenotypic analysis of genotype 1a replicons expressing single NS5A M28T, Q30E, Q30H, Q30R, L31V, Y93C, Y93H, and Y93N substitutions exhibited 500-, 18500-, 1083-, 900-, 2500-, 1367-, 8500-, and 34833-fold reduced susceptibility to daclatasvir, respectively. For genotype 1b, L31V and Y93H single substitutions and L31M/Y93H and L31V/Y93H combinations exhibited 33-, 30-, 16000-, and 33667-fold reduced susceptibility to daclatasvir, respectively. A P32-deletion (P32X) in genotype 1b reduced daclatasvir susceptibility by >1,000,000-fold. For genotype 3a, single A30K, L31F, L31I, and Y93H substitutions exhibited 117-, 320-, 240-, and 3733-fold reduced susceptibility to daclatasvir, respectively.
In Clinical Studies
Among subjects with HCV genotype 1 or genotype 3 infection and treated in the ALLY-1, -2, and -3 trials with DAKLINZA and sofosbuvir with or without ribavirin for 12 weeks, 31 subjects (11 with genotype 1a, 1 with genotype 1b, and 19 with genotype 3) qualified for resistance analysis due to virologic failure. Post-baseline NS5A and NS5B population-based nucleotide sequence analysis results were available for 31 and 28 subjects, respectively.
Virus from all 31 subjects at the time of virologic failure harbored one or more of the following NS5A resistance-associated substitutions (including pre-existing amino acid polymorphisms or treatment-emergent substitutions): M28T, Q30H/K/R, L31M/V, H54R, H58D/P, or Y93C/N for genotype 1a subjects, P32-deletion (P32X) for the genotype 1b subject, and A30K/S, L31I, S62A/L/P/R/T, or Y93H for genotype 3 subjects. Among HCV genotype 1a virologic failure subjects, the most common NS5A amino acid substitutions occurred at position Q30 (Q30H/K/R; 73% [8/11], all treatment-emergent). Among HCV genotype 3 virologic failure subjects, the most common NS5A amino acid polymorphism or treatment-emergent substitution was Y93H (89% [17/19], treatment-emergent in 11 of 17 subjects).
For NS5B, 6 of 28 subjects at the time of virologic failure had virus with NS5B substitutions possibly associated with sofosbuvir resistance or exposure: A112T, L159F, E237G, or Q355H (genotype 1a subjects), or S282T+Q355H (genotype 3 subject).
Persistence of Resistance-Associated Substitutions
In a long-term follow-up study that included HCV genotype 1- and genotype 3-infected subjects treated with daclatasvir-containing regimens in phase 2/3 clinical trials, viral populations with treatment-emergent NS5A resistance-associated substitutions persisted at detectable levels for more than 1 year in most subjects.
Effect of Baseline HCV Amino Acid Polymorphisms on Treatment Response
Genotype 1a NS5A polymorphisms: In HCV genotype 1a-infected subjects with cirrhosis, the presence of an NS5A amino acid polymorphism at position M28, Q30, L31, or Y93 (defined as any change from reference identified by population-based nucleotide sequencing) was associated with reduced efficacy of DAKLINZA and sofosbuvir with or without ribavirin for 12 weeks in the ALLY-1 and ALLY-2 trials (see Table 11). Due to the limited sample size, insufficient data are available to determine the impact of specific NS5A polymorphisms at these positions on SVR12 rates in subjects with cirrhosis. Six of 54 subjects (11%) with cirrhosis had one of the following specific NS5A polymorphisms at baseline: M28V/T (n=2), Q30R (n=1), L31M (n=2), or Y93N (n=1); 2 subjects with M28V or Q30R achieved SVR12 while 4 subjects with M28T, L31M, or Y93N did not achieve SVR. Eleven of 112 subjects (10%) without cirrhosis had one or more of the following specific NS5A polymorphisms at baseline: M28T/V (n=3), Q30H/L/R (n=5), L31M (n=1), and Y93C/H/S (n=4); all noncirrhotic subjects with these baseline NS5A polymorphisms achieved SVR12. Based on an analysis of 1026 HCV genotype 1a NS5A amino acid sequences from pooled clinical trials, the prevalence of polymorphisms at these positions was 11% overall, and 11% in the U.S.
Genotype 1b NS5A polymorphisms: In a pooled analysis of 43 subjects infected with HCV genotype 1b with available baseline nucleotide sequence data in ALLY-1 and -2, virus from 21% (n=9) of subjects receiving DAKLINZA and sofosbuvir with or without ribavirin had one of the following baseline NS5A amino acid polymorphisms: R30K/M/Q (n=4), L31M (n=2), or Y93H (n=3). All 9 subjects with NS5A polymorphisms achieved SVR12, including 5 who were noncirrhotic and 4 who were in the post-transplant period.
Genotype 3 NS5A polymorphisms: In the ALLY-3 trial in which HCV genotype 3-infected subjects received DAKLINZA and sofosbuvir for 12 weeks, the presence of an NS5A Y93H polymorphism was associated with a reduced SVR12 rate (see Table 11). In a pooled analysis of 175 subjects infected with HCV genotype 3 with available baseline nucleotide sequence data in the ALLY-1, -2, and -3 trials, virus from 7% (13/175) of subjects had the NS5A Y93H polymorphism, and all 13 of these subjects were in the ALLY-3 trial. Phylogenetic analysis of NS5A sequences indicated that all genotype 3 subjects with available data in the ALLY-1, -2, and -3 trials (n=175) were infected with HCV subtype 3a.
Table 11: Impact of NS5A Amino Acid Polymorphisms on SVR12 Rates in Subjects with HCV Genotype 1a or Genotype 3 Infection in Phase 3 Trials of DAKLINZA + Sofosbuvir ± Ribavirin NS5A Polymorphisms SVR12 Rates after 12 Weeks of Treatment with
DAKLINZA + Sofosbuvir ± RibavirinaWith NS5A
Polymorphism(s)
% (n/N)Without NS5A Polymorphism(s)
% (n/N)ba HCV genotype 1a-infected subjects received DAKLINZA + sofosbuvir ± ribavirin for 12 weeks in the ALLY-1 and ALLY-2 trials. HCV genotype 3-infected subjects received DAKLINZA + sofosbuvir for 12 weeks in the ALLY-3 trial; no data on the impact of Y93H are available for HCV genotype 3-infected subjects treated with DAKLINZA + sofosbuvir ± ribavirin in ALLY-1 and ALLY-2 trials.
b None of the 11 subjects with Child-Pugh C cirrhosis had an indicated NS5A polymorphism; 5 achieved SVR (genotype 1a: 4/9; genotype 3a: 1/2).
c Any change from genotype 1a reference.
d Includes subjects who were post-transplant with undefined cirrhosis status.HCV genotype 1a-infected subjects: M28,c Q30,c L31,c or Y93c
76% (13/17)
95% (142/149)
Without cirrhosisd
100% (11/11)
99% (100/101)
With cirrhosis (Child-Pugh A, B, or C)
33% (2/6)
88% (42/48)
HCV genotype 3-infected subjects: Y93H
54% (7/13)
92% (149/162)
Without cirrhosisd
67% (6/9)
98% (125/128)
With cirrhosis (Child-Pugh A, B, or C)
25% (1/4)
71% (24/34)
Cross-Resistance
Based on resistance patterns observed in cell culture replicon studies and HCV-infected subjects, cross-resistance between daclatasvir and other NS5A inhibitors is expected. Cross-resistance between daclatasvir and other classes of direct-acting antivirals is not expected. The impact of prior daclatasvir treatment experience on the efficacy of other NS5A inhibitors has not been studied. Conversely, the efficacy of DAKLINZA in combination with sofosbuvir has not been studied in subjects who have previously failed treatment with regimens that include an NS5A inhibitor.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis and Mutagenesis
A 2-year carcinogenicity study in Sprague Dawley rats and a 6-month study in transgenic (Tg rasH2) mice were conducted with daclatasvir. In the 2-year study in rats, no drug-related increase in tumor incidence was observed at doses up to 50 mg/kg/day (both sexes). Daclatasvir exposures at these doses were approximately 6-fold (males and females) the human systemic exposure at the therapeutic daily dose of DAKLINZA. In transgenic mice no drug-related increase in tumor incidence was observed at doses of 300 mg/kg/day (both sexes).
Daclatasvir was not genotoxic in a battery of in vitro or in vivo assays, including bacterial mutagenicity (Ames) assays, mammalian mutation assays in Chinese hamster ovary cells, or in an in vivo oral micronucleus study in rats.
If DAKLINZA and sofosbuvir are administered in a regimen containing ribavirin, the information for ribavirin on carcinogenesis and mutagenesis also applies to this combination regimen (see prescribing information for ribavirin).
Impairment of Fertility
Daclatasvir had no effects on fertility in female rats at any dose tested. Daclatasvir exposures at these doses in females were approximately 24-fold the human systemic exposure at the therapeutic daily dose of DAKLINZA. In male rats, effects on reproductive endpoints at 200 mg/kg/day included reduced prostate/seminal vesicle weights, minimally increased dysmorphic sperm, as well as increased mean pre-implantation loss in litters sired by treated males. Daclatasvir exposures at the 200 mg/kg/day dose in males were approximately 26-fold the human systemic exposure at the therapeutic daily dose of DAKLINZA. Exposures at 50 mg/kg/day in males produced no notable effects and was 4.7-fold the exposure in humans at the recommended daily dose of DAKLINZA.
If DAKLINZA and sofosbuvir are administered with ribavirin, the information for ribavirin on impairment of fertility also applies to this combination regimen (see prescribing information for ribavirin).
-
14 CLINICAL STUDIES
14.1 Description of Clinical Trials
The efficacy of DAKLINZA in combination with sofosbuvir and with or without ribavirin was evaluated in three phase 3 clinical trials, as summarized in Table 12 [see Clinical Studies (14.2, 14.3, 14.4)]. HCV RNA levels were measured during these clinical trials using the COBAS® TaqMan® HCV test (version 2.0), for use with the High Pure System. The assay had a lower limit of quantification (LLOQ) of 25 IU per mL. Sustained virologic response was the primary endpoint and was defined as HCV RNA below the LLOQ at post-treatment week 12 (SVR12).
Table 12: Genotype 1 and 3 Patient Populations from DAKLINZA Trials Trial Population Study Arms and Duration
(Number of Subjects Treated)ALLY-3
(AI444218)Genotype 3, treatment-naive and treatment-experienced, with or without cirrhosis
DAKLINZA and sofosbuvir for 12 weeks
(N=152)ALLY-2
(AI444216)Genotype 1 and 3, treatment-naive and treatment-experienced, with or without cirrhosis, HCV/HIV-1 coinfection
DAKLINZA and sofosbuvir for 12 weeks
(N=137)ALLY-1
(AI444215)Genotype 1 and 3, treatment-naive or treatment-experienced, with or without cirrhosis, including decompensated cirrhosis and post-transplant
DAKLINZA and sofosbuvir and ribavirin for 12 weeks
(N=103)14.2 Clinical Trials in HCV Genotype 3 (ALLY-3)
ALLY-3 was an open-label trial that included 152 subjects with chronic HCV genotype 3 infection and compensated liver disease who were treatment naive (n=101) or treatment experienced (n=51). Most treatment-experienced subjects had failed prior treatment with peginterferon/ribavirin, but 7 subjects had been treated previously with a sofosbuvir regimen and 2 subjects with a regimen containing an investigational agent. Previous exposure to NS5A inhibitors was prohibited. Subjects received DAKLINZA 60 mg plus sofosbuvir 400 mg once daily for 12 weeks and were monitored for 24 weeks post treatment.
The 152 treated subjects in ALLY-3 had a median age of 55 years (range, 24-73); 59% of the subjects were male; 90% were white, 5% were Asian, and 4% were black. Most subjects (76%) had baseline HCV RNA levels greater than or equal to 800,000 IU per mL; 21% of the subjects had compensated cirrhosis, and 40% had the IL28B rs12979860 CC genotype.
SVR12 and outcomes in subjects without SVR12 in ALLY-3 are shown by patient population in Table 13. SVR12 rates were comparable regardless of HCV treatment history, age, gender, IL28B allele status, or baseline HCV RNA level. For SVR outcomes related to baseline NS5A amino acid polymorphisms, see Microbiology (12.4).
Table 13: ALLY-3: SVR12 in Treatment-Naive and Treatment-Experienced Subjects with or without Cirrhosis with Genotype 3 HCV Treated with DAKLINZA in Combination with Sofosbuvir for 12 Weeks Treatment Outcomes Total
n=152a Includes 11 subjects with missing or inconclusive cirrhosis status.
b One subject had quantifiable HCV RNA at end of treatment.
c Relapse rates are calculated with a denominator of subjects with HCV RNA not detected at the end of treatment.SVR12
All
89% (135/152)No cirrhosisa
96% (115/120)
With cirrhosis
63% (20/32)
Outcomes for subjects without SVR12
On-treatment virologic failureb
0.7% (1/152)
Relapsec
11% (16/151)
14.3 Clinical Trials in HCV/HIV Coinfected Subjects (ALLY-2)
ALLY-2 was an open-label trial that included 153 subjects with chronic hepatitis C and HIV coinfection who received DAKLINZA and sofosbuvir for 12 weeks. Subjects with HCV genotype 1, 2, 3, 4, 5, or 6 infection were eligible to enroll. Subjects were HCV treatment-naive (n=101) or HCV treatment-experienced (n=52). Prior exposure to NS5A inhibitors was prohibited. The dose of DAKLINZA was 60 mg once daily (dose-adjusted for concomitant antiretroviral use) and the dose of sofosbuvir was 400 mg once daily [see Drug Interactions (7.3)].
The 153 treated subjects had a median age of 53 years (range, 24-71); 88% of subjects were male; 63% were white, 33% were black, and 1% were Asian. Sixty-eight percent of subjects had HCV genotype 1a, 15% had HCV genotype 1b, 8% had genotype 2, 7% had genotype 3, and 2% had genotype 4. Most subjects (80%) had baseline HCV RNA levels greater than or equal to 800,000 IU per mL; 16% of the subjects had compensated cirrhosis, and 73% had IL28B rs12979860 non-CC genotype. Concomitant HIV therapy included PI-based regimens (darunavir + ritonavir, atazanavir + ritonavir, or lopinavir/ritonavir) for 46% of subjects, NNRTI-based regimens (efavirenz, nevirapine, or rilpivirine) for 26%, integrase-based regimens (raltegravir or dolutegravir) for 26%, and nucleoside-only regimens (abacavir + emtricitabine + zidovudine) for 1%. Two patients were not receiving treatment for HIV.
SVR and outcomes in subjects with HCV genotype 1 without SVR12 in ALLY-2 are shown by patient population in Table 14. Available data on subjects with HCV genotype 2, 4, 5, or 6 infection are insufficient to provide recommendations for these genotypes; therefore, these results are not presented in Table 14. SVR12 rates were comparable regardless of antiretroviral therapy, HCV treatment history, age, race, gender, IL28B allele status, HCV genotype 1 subtype, or baseline HCV RNA level. For SVR outcomes related to baseline NS5A amino acid polymorphisms, see Microbiology (12.4).
No subjects switched their antiretroviral therapy regimen due to loss of plasma HIV-1 RNA suppression. There was no change in absolute CD4+ T-cell counts at the end of 12 weeks of treatment.
Table 14: ALLY-2: SVR12 in Subjects with Genotype 1 and 3 HCV/HIV Coinfection Treated with DAKLINZA in Combination with Sofosbuvir for 12 Weeks Treatment Outcomes Total
n=137a Includes 5 subjects with inconclusive cirrhosis status.
b One subject with cirrhosis.
c One subject had detectable HCV RNA at end of treatment.
d Relapse rates are calculated with a denominator of subjects with HCV RNA not detected at the end of treatment.SVR12
Genotype 1
97% (123/127)No cirrhosisa
98% (103/105)
With cirrhosis
91% (20/22)
Genotype 3b
100% (10/10)
Outcomes for genotype 1 subjects without SVR12
On-treatment virologic failurec
0.8% (1/127)
Relapsed
1.6% (2/126)
Missing post-treatment data
0.8% (1/126)
14.4 Clinical Trials in Subjects with Child-Pugh A, B, or C Cirrhosis or with HCV Recurrence after Liver Transplantation (ALLY-1)
ALLY-1 was an open-label trial of DAKLINZA, sofosbuvir, and ribavirin that included 113 subjects with chronic HCV infection and Child-Pugh A, B, or C cirrhosis (n=60) or HCV recurrence after liver transplantation (n=53). Subjects with HCV genotype 1, 2, 3, 4, 5, or 6 infection were eligible to enroll. Subjects could be HCV treatment-naive or treatment-experienced, although prior exposure to NS5A inhibitors was prohibited. Subjects received DAKLINZA 60 mg once daily, sofosbuvir 400 mg once daily, and ribavirin for 12 weeks and were monitored for 24 weeks post treatment. Subjects received an initial ribavirin dose of 600 mg or less daily with food; the initial and on-treatment dosing of ribavirin was modified based on hemoglobin and creatinine clearance measurements. If tolerated, the ribavirin dose was titrated up to 1000 mg per day. A high proportion of reductions in ribavirin dosing occurred in the trial. By week 6, approximately half of the subjects received 400 mg per day or less of ribavirin. In total, 16 subjects (15%) completed less than 12 weeks and 11 subjects (10%) completed less than 6 weeks of ribavirin therapy, respectively. For the cohort of patients with cirrhosis (Child-Pugh A, B, or C), the median time to discontinuation of ribavirin was 43 days (range, 8-82, n=9). For the post-transplant cohort, the median time to discontinuation of ribavirin was 20 days (range, 3-57, n=7).
The 113 treated subjects in ALLY-1 had a median age of 59 years (range, 19-82); 67% of the subjects were male; 96% were white, 4% were black, and 1% Asian. Most subjects (59%) were treatment-experienced, and most (71%) had baseline HCV RNA levels greater than or equal to 800,000 IU per mL. Fifty-eight percent of subjects had HCV genotype 1a, 19% had HCV genotype 1b, 4% had genotype 2, 15% had genotype 3, 4% had genotype 4, and 1% had genotype 6, 77% had IL28B rs12979860 non-CC genotype. Among the 60 subjects in the cirrhosis cohort, 20% were Child-Pugh A, 53% were Child-Pugh B, and 27% were Child-Pugh C, and 35% had a Baseline Model for End-Stage Liver Disease (MELD) score of 15 or greater. Most (55%) of the 53 subjects in the post-transplant cohort had F3 or F4 fibrosis (based on FibroSURE® results).
SVR12 and outcomes in subjects without SVR12 in ALLY-1 are shown for subjects with HCV genotype 1 by patient population in Table 15. Available data on subjects with HCV genotype 2, 4, 5, or 6 infection are insufficient to provide recommendations; therefore, these results are not presented in Table 15.
SVR12 rates were comparable regardless of age, gender, IL28B allele status, or baseline HCV RNA level. For SVR12 outcomes related to baseline NS5A amino acid polymorphisms, see Microbiology (12.4). No HCV genotype 1 or genotype 3 subjects with Child-Pugh C cirrhosis had baseline resistance-associated NS5A amino acid polymorphisms. SVR12 rates were comparable between genotype 3 (5/6 with Child-Pugh B or C cirrhosis and 10/11 post-liver transplant) and genotype 1 subjects with or without decompensated cirrhosis.
Table 15: ALLY-1: SVR12 in Genotype 1 Subjects with Child-Pugh A, B, or C Cirrhosis or with HCV Genotype 1 Recurrence after Liver Transplantation Treated with DAKLINZA in Combination with Sofosbuvir and Ribavirin for 12 Weeks Treatment Outcomes Child-Pugh A, B, or C Cirrhosis
n=45Post-Liver Transplant
n=41a One subject had detectable HCV RNA at end of treatment.
b Relapse rates are calculated with a denominator of subjects with HCV RNA not detected at end of treatment.SVR12
Genotype 1
82% (37/45)
95% (39/41)
Child-Pugh A
91% (10/11)
-
Child-Pugh B
92% (22/24)
-
Child-Pugh C
50% (5/10)
-
Genotype 1a
76% (26/34)
97% (30/31)
Genotype 1b
100% (11/11)
90% (9/10)
Outcomes for subjects without SVR12
On-treatment virologic failure
2% (1/45)a
0
Relapseb
16% (7/44)
5% (2/41)
-
16 HOW SUPPLIED/STORAGE AND HANDLING
DAKLINZA is packaged in bottles as described in the table.
Tablet Strength
Tablet Color/Shape
Tablet Markings
Package Size
NDC Code
60 mg
Light green, biconvex, pentagonal
Debossed with “BMS” on one side and “215” on the other side
Bottles of 28
0003-0215-01
30 mg
Green, biconvex, pentagonal
Debossed with “BMS” on one side and “213” on the other side
Bottles of 28
0003-0213-01
90 mg
Light green, biconvex, round
Embossed with “BMS” on one side and “011” on the other side
Bottles of 28
0003-0011-01
Store DAKLINZA tablets at 25°C (77°F), with excursions permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Risk of Hepatitis B Virus Reactivation in Patients Coinfected with HCV and HBV
Inform patients that HBV reactivation can occur in patients coinfected with HBV during or after treatment of HCV infection. Advise patients to tell their healthcare provider if they have a history of HBV infection [see Warnings and Precautions (5.1)].
Drug Interactions
Inform patients of the potential for drug interactions with DAKLINZA, and that some drugs should not be taken with DAKLINZA [see Contraindications (4), Drug Interactions (7), and Clinical Pharmacology (12.3)].
Symptomatic Bradycardia When Used in Combination with Sofosbuvir and Amiodarone
Advise patients to seek medical evaluation immediately for symptoms of bradycardia, such as near-fainting or fainting, dizziness or lightheadedness, malaise, weakness, excessive tiredness, shortness of breath, chest pain, confusion or memory problems [see Warnings and Precautions (5.3), Adverse Reactions (6.2), and Drug Interactions (7.3)].
DAKLINZA Combination Therapy with Sofosbuvir
Inform patients that DAKLINZA should not be used alone. DAKLINZA should be used in combination with sofosbuvir with or without ribavirin for the treatment of HCV genotype 1 or HCV genotype 3 infection [see Indications and Usage (1)].
Missed Doses
Advise patients to take DAKLINZA every day at the regularly scheduled time with or without food. Inform patients that it is important not to miss or skip doses and to take DAKLINZA for the duration that is recommended by the physician. For instructions for missed doses of other agents in the regimen, refer to the respective prescribing information.
Pregnancy
Advise patients to avoid pregnancy during combination treatment with DAKLINZA and sofosbuvir with ribavirin for 6 months after completion of treatment. Inform patients to notify their healthcare provider immediately in the event of a pregnancy [see Use in Specific Populations (8.1)].
- SPL UNCLASSIFIED SECTION
-
Patient Information
DAKLINZA® (dak lin za)
(daclatasvir)
tabletsImportant: DAKLINZA is used in combination with other antiviral medicines. When taking DAKLINZA in combination with sofosbuvir, you should read the Patient Information leaflet for sofosbuvir. When taking DAKLINZA in combination with sofosbuvir and ribavirin, you should also read the Medication Guide for ribavirin.
What is DAKLINZA?
- DAKLINZA is a prescription medicine used to treat chronic (lasting a long time) hepatitis C genotype 1 or genotype 3 infection in adults.
- Take DAKLINZA with sofosbuvir or with sofosbuvir and ribavirin. You should not take DAKLINZA by itself.
It is not known if DAKLINZA is safe and effective in children under 18 years of age.
What is the most important information I should know about DAKLINZA?
DAKLINZA can cause serious side effects, including:
- Hepatitis B virus reactivation: Before starting treatment with DAKLINZA, your healthcare provider will do blood tests to check for hepatitis B virus infection. If you have ever had hepatitis B virus infection, the hepatitis B virus could become active again during or after treatment of hepatitis C virus with DAKLINZA. Hepatitis B virus becoming active again (called reactivation) may cause serious liver problems, including liver failure and death. Your healthcare provider will monitor you if you are at risk for hepatitis B virus reactivation during treatment and after you stop taking DAKLINZA.
For more information about side effects, see the section “What are the possible side effects of DAKLINZA?”
Before taking DAKLINZA, tell your healthcare provider about all of your medical conditions, including if you:
- have ever had hepatitis B virus infection
- have diabetes
- have liver problems other than hepatitis C infection
- have had a liver transplant
- have heart problems
-
are pregnant or plan to become pregnant. It is not known if DAKLINZA will harm your unborn baby.
- ∘ When taking DAKLINZA in combination with sofosbuvir and ribavirin, tell your healthcare provider right away if you or your female sexual partner becomes pregnant.
- ∘ Males and females who take DAKLINZA with sofosbuvir and ribavirin should also read the ribavirin Medication Guide for important pregnancy, contraception, and infertility information.
-
are breastfeeding or plan to breastfeed. It is not known if DAKLINZA passes into your breast milk.
- ∘ Talk to your healthcare provider about the best way to feed your baby during treatment with DAKLINZA.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
DAKLINZA and other medicines may affect each other. This can cause you to have too much or not enough DAKLINZA or other medicines in your body. This may affect the way DAKLINZA or your other medicines work or may cause side effects. Keep a list of your medicines to show your healthcare provider and pharmacist.
- You can ask your healthcare provider or pharmacist for a list of medicines that interact with DAKLINZA.
- Do not start taking a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to take DAKLINZA with other medicines.
How should I take DAKLINZA?
- Take DAKLINZA exactly how you are told to by your healthcare provider.
- Do not change your dose unless you are told to by your healthcare provider.
- Do not stop taking DAKLINZA without first talking with your healthcare provider.
- Take DAKLINZA one time each day with or without food.
- If you miss a dose, call your healthcare provider or pharmacist. It is important that you do not miss or skip doses of DAKLINZA during treatment.
- If you take too much DAKLINZA, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of DAKLINZA?
DAKLINZA can cause serious side effects, including:
- Hepatitis B virus reactivation. See “What is the most important information I should know about DAKLINZA?”
DAKLINZA in combination with sofosbuvir and amiodarone may cause serious side effects, including:
- Slow heart rate (bradycardia). DAKLINZA combination treatment with sofosbuvir may result in slowing of the heart rate (pulse) along with other symptoms when taken with amiodarone, a medicine used to treat certain heart problems. Get medical help right away if you take amiodarone with sofosbuvir and DAKLINZA and get any of the following symptoms:
- o fainting or near-fainting
- o dizziness or lightheadedness
- o not feeling well
- o weakness
- o tiredness
- o shortness of breath
- o chest pain
- o confusion
- o memory problems
The most common side effects of DAKLINZA when used in combination with sofosbuvir include:
- headache
- tiredness
The most common side effects of DAKLINZA when used in combination with sofosbuvir and ribavirin include:
- headache
- low red blood cell count (anemia)
- tiredness
- nausea
These are not all the possible side effects of DAKLINZA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store DAKLINZA?
- Store DAKLINZA at room temperature between 68°F and 77°F (20°C and 25°C).
Keep DAKLINZA and all medicines out of the reach of children.
General information about the safe and effective use of DAKLINZA
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use DAKLINZA for a condition for which it was not prescribed. Do not give DAKLINZA to other people, even if they have the same symptoms that you have. It may harm them.
You can ask your pharmacist or healthcare provider for information about DAKLINZA that is written for health professionals.
What are the ingredients in DAKLINZA?
Active ingredient: daclatasvir
Inactive ingredients: anhydrous lactose, microcrystalline cellulose, croscarmellose sodium, silicon dioxide, magnesium stearate, and Opadry green. Opadry green contains hypromellose, titanium dioxide, polyethylene glycol 400, FD&C blue #2/indigo carmine aluminum lake, and yellow iron oxide.
Manufactured for: Bristol-Myers Squibb Company, Princeton, NJ 08543, USA
DAKLINZA is a trademark of Bristol-Myers Squibb Company. For more information, go to www.patientsupportconnect.com or call 1-844-442-6663.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Revised October 2019 -
DAKLINZA 90 mg Tablets Representative Packaging
See How Supplied section for a complete list of available packages of DAKLINZA.
28 Tablets
NDC: 0003-0011-01
Daklinza®
(daclatasvir) tablets
90 mg
Note to pharmacist: Do not cover ALERT box with pharmacy label.
ALERT: Find out about medicines that should NOT be taken with Daklinza®
Rx only
Bristol-Myers Squibb -
DAKLINZA 60 mg Tablets Representative Packaging
28 Tablets
NDC: 0003-0215-01
Daklinza®
(daclatasvir) tablets
60 mg
Note to pharmacist: Do not cover ALERT box with pharmacy label.
ALERT: Find out about medicines that should NOT be taken with Daklinza®
Rx only
Bristol-Myers Squibb -
DAKLINZA 30 mg Tablets Representative Packaging
28 Tablets
NDC: 0003-0213-01
Daklinza®
(daclatasvir) tablets
30 mg
Note to pharmacist: Do not cover ALERT box with pharmacy label.
ALERT: Find out about medicines that should NOT be taken with Daklinza®
Rx only
Bristol-Myers Squibb -
INGREDIENTS AND APPEARANCE
DAKLINZA
daclatasvir tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0003-0011 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DACLATASVIR DIHYDROCHLORIDE (UNII: 50ZO25C11D) (DACLATASVIR - UNII:LI2427F9CI) DACLATASVIR 90 mg Inactive Ingredients Ingredient Name Strength ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) INDIGOTINDISULFONATE SODIUM (UNII: D3741U8K7L) ALUMINUM OXIDE (UNII: LMI26O6933) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color GREEN (Light Green) Score no score Shape ROUND Size 10mm Flavor Imprint Code BMS;011 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0003-0011-01 28 in 1 BOTTLE; Type 0: Not a Combination Product 05/18/2016 04/30/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA206843 05/18/2016 04/30/2019 DAKLINZA
daclatasvir tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0003-0215 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DACLATASVIR DIHYDROCHLORIDE (UNII: 50ZO25C11D) (DACLATASVIR - UNII:LI2427F9CI) DACLATASVIR 60 mg Inactive Ingredients Ingredient Name Strength ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) INDIGOTINDISULFONATE SODIUM (UNII: D3741U8K7L) ALUMINUM OXIDE (UNII: LMI26O6933) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color GREEN (Light Green) Score no score Shape PENTAGON (5 sided) Size 9mm Flavor Imprint Code BMS;215 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0003-0215-01 28 in 1 BOTTLE; Type 0: Not a Combination Product 07/27/2015 12/31/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA206843 07/27/2015 12/31/2019 DAKLINZA
daclatasvir tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0003-0213 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DACLATASVIR DIHYDROCHLORIDE (UNII: 50ZO25C11D) (DACLATASVIR - UNII:LI2427F9CI) DACLATASVIR 30 mg Inactive Ingredients Ingredient Name Strength ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) INDIGOTINDISULFONATE SODIUM (UNII: D3741U8K7L) ALUMINUM OXIDE (UNII: LMI26O6933) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color GREEN Score no score Shape PENTAGON (5 sided) Size 7mm Flavor Imprint Code BMS;213 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0003-0213-01 28 in 1 BOTTLE; Type 0: Not a Combination Product 07/27/2015 06/30/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA206843 07/27/2015 06/30/2020 Labeler - E.R. Squibb & Sons, L.L.C. (011550092)
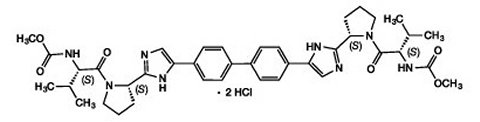



Trademark Results [DAKLINZA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 DAKLINZA 85871477 5171750 Live/Registered |
Bristol-Myers Squibb Company 2013-03-08 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.