FACT SHEET FOR HEALTHCARE PROVIDERS EMERGENCY USE AUTHORIZATION (EUA) OF VEKLURY® (remdesivir) FOR THE TREATMENT OF CORONAVIRUS DISEASE 2019 (COVID-19) IN PEDIATRIC PATIENTS WEIGHING 3.5 KG TO LESS THAN 40 KG OR PEDIATRIC PATIENTS LESS THAN 12 YEARS OF AGE WEIGHING AT LEAST 3.5 KG, WITH POSITIVE RESULTS OF DIRECT SARS-CoV-2 VIRAL TESTING WHO ARE: HOSPITALIZED, OR NOT HOSPITALIZED AND HAVE MILD-TO-MODERATE COVID-19, AND ARE AT HIGH RISK FOR PROGRESSION TO SEVERE COVID-19, INCLUDING HOSPITALIZATION OR DEATH
remdesivir by
Drug Labeling and Warnings
remdesivir by is a Prescription medication manufactured, distributed, or labeled by Gilead Sciences, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
VEKLURY- remdesivir injection
VEKLURY- remdesivir injection, powder, lyophilized, for solution
Gilead Sciences, Inc.
Disclaimer: This drug has not been found by FDA to be safe and effective, and this labeling has not been approved by FDA. For further information about unapproved drugs, click here.
----------
FACT SHEET FOR HEALTHCARE PROVIDERS
EMERGENCY USE AUTHORIZATION (EUA) OF VEKLURY® (remdesivir) FOR THE TREATMENT OF CORONAVIRUS DISEASE 2019 (COVID-19) IN PEDIATRIC PATIENTS WEIGHING 3.5 KG TO LESS THAN 40 KG OR PEDIATRIC PATIENTS LESS THAN 12 YEARS OF AGE WEIGHING AT LEAST 3.5 KG, WITH POSITIVE RESULTS OF DIRECT SARS-CoV-2 VIRAL TESTING WHO ARE:
HOSPITALIZED, OR
NOT HOSPITALIZED AND HAVE MILD-TO-MODERATE COVID-19, AND ARE AT HIGH RISK FOR PROGRESSION TO SEVERE COVID-19, INCLUDING HOSPITALIZATION OR DEATH
| IMPORTANT PRESCRIBING INFORMATION | |
| Subject: | Updated Emergency Use Authorization (EUA) for treatment of coronavirus disease 2019 (COVID-19) in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg with positive results of direct severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral testing, who are: hospitalized, or not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death, and variations in carton and vial labeling of VEKLURY® (remdesivir) |
Dear Healthcare Provider:
Gilead Sciences, Inc., would like to clarify the appropriate use and the variable packaging and labeling of the antiviral VEKLURY® (remdesivir).
Gilead's remdesivir (brand name VEKLURY) was approved by the US Food and Drug Administration (FDA) on January 21, 2022 for adults and pediatric patients (12 years of age and older and weighing at least 40 kg) for the treatment of coronavirus disease 2019 (COVID-19) with positive results of direct severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral testing, who are:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
Healthcare providers should administer VEKLURY in these patients per the current US Prescribing Information available at www.gilead.com/science-and-medicine/medicines; please also see Important Safety Information at the end of this letter.
Emergency use of Gilead's remdesivir (brand name VEKLURY) for treatment of COVID-19 in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg with positive results of direct SARS-CoV-2 viral testing, who are: hospitalized or, not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
On January 21, 2022, FDA revised the Emergency Use Authorization (EUA) for VEKLURY, which now authorizes VEKLURY for use by healthcare providers to treat COVID-19 in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg with positive results of direct SARS-CoV-2 viral testing, who are: hospitalized, or not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death. Only Gilead's VEKLURY (remdesivir) for injection (supplied as 100 mg lyophilized powder in vial) is authorized for emergency use under the terms and conditions set forth in the Letter of Authorization for the EUA.
The safety and efficacy of VEKLURY to treat COVID-19 in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg with positive results of direct SARS-CoV-2 viral testing has not been established, and VEKLURY is not FDA approved for this use. For information about the authorized use of VEKLURY in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg, including dosing, administration, and preparation instructions, please review the EUA Fact Sheet for Healthcare Providers and FDA Letter of Authorization available at www.gilead.com/remdesivir.
Variations in packaging and labeling of Gilead's remdesivir (brand name VEKLURY)
Gilead's VEKLURY (remdesivir) has been manufactured for use under an EUA, and for commercial use. As such, VEKLURY has different packaging, labeling, and expiration dates depending on the date of manufacture. Packaging and labeling for Gilead's remdesivir EUA use may not necessarily include the brand name, VEKLURY.
To help avoid potential drug shortage, hospitals should continue to use all unexpired, unopened vials of Gilead's remdesivir – whether or not the vial includes the brand name VEKLURY or is labeled for use under EUA. Refer to the attached chart at the end of this letter that outlines what hospitals should do with unexpired, unopened vials of Gilead's remdesivir.
Authentic VEKLURY or remdesivir, manufactured by Gilead Sciences, Inc., will include the GILEAD name and logo on the carton and vial label. All packaging includes the drug name remdesivir. Current packaging variations for the two formulations are described below. Both formulations are for intravenous infusion only.
- VEKLURY® (remdesivir) for injection (supplied as 100 mg lyophilized powder in vial). The lyophilized powder formulation is always supplied with a red cap and the package and labeling may be marked "for use under Emergency Use Authorization (EUA)".
- VEKLURY® (remdesivir) injection (supplied as 100 mg/20 mL [5 mg/mL], solution in vial). The solution formulation is supplied with a blue cap and the package and labeling may be marked "for use under Emergency Use Authorization (EUA)". The solution should only be used in adults and pediatric patients 12 years of age and older and weighing at least 40 kg.
Veklury (remdesivir) injection (solution; left) and Veklury (remdesivir) for injection (lyophilized powder; right) now approved by FDA for use in accordance with the prescribing information (PI).
Veklury (remdesivir) injection (solution; left) and Veklury (remdesivir) for injection (lyophilized powder; right) previously authorized for emergency use.

Reporting Adverse Events and Medication Errors
Healthcare providers should direct questions on VEKLURY packaging or use to Gilead Sciences at 1-866-633-4474 or www.askgileadmedical.com.
Under the Emergency Use Authorization, the prescribing healthcare provider and/or the provider's designee are/is responsible for mandatory reporting of all serious adverse events and medication errors potentially related to VEKLURY treatment within 7 calendar days from the healthcare provider's awareness of the event using FDA Form 3500. Healthcare providers must report all serious adverse events and medication errors to FDA when utilizing VEKLURY under the EUA in accordance with the Healthcare Provider Fact Sheet.
For additional information about VEKLURY, including the full Prescribing Information, please visit www.vekluryhcp.com.
Information and reports of suspicious, counterfeit, or unregistered remdesivir can be submitted to Gilead anticounterfeiting@gilead.com and/or www.fraud.org/fakerx.
____________________
Signatory
Signatory Department
| Product | Remdesivir for injection, 100 mg, lyophilized powder | Remdesivir injection, 100 mg/20 mL (5 mg/mL), solution |
|---|---|---|
| Labeled "For use under Emergency Use Authorization (EUA)" (vials and cartons do not include the brand name, VEKLURY) | Continue use in:
| Continue use in:
|
| FDA-approved VEKLURY (carton and vials will include the brand name, VEKLURY) | Use in:
| Use in:
|
U.S. Indication and Important Safety Information for VEKLURY® (remdesivir)
Indication
VEKLURY is indicated for the treatment of COVID-19 in adults and pediatric patients ≥12 years old and weighing ≥40 kg with positive results of SARS-CoV-2 viral testing, who are:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19 and are at high risk for progression to severe COVID-19, including hospitalization or death.
- VEKLURY is contraindicated in patients with a history of clinically significant hypersensitivity reactions to VEKLURY or any of its components.
Warnings and precautions
- Hypersensitivity, including infusion-related and anaphylactic reactions: Hypersensitivity, including infusion-related and anaphylactic reactions, has been observed during and following administration of VEKLURY; most occurred within 1 hour. Monitor patients during infusion and observe for at least 1 hour after infusion is complete for signs and symptoms of hypersensitivity as clinically appropriate. Symptoms may include hypotension, hypertension, tachycardia, bradycardia, hypoxia, fever, dyspnea, wheezing, angioedema, rash, nausea, diaphoresis, and shivering. Slower infusion rates (maximum infusion time of up to120 minutes) can potentially prevent these reactions. If a severe infusion-related hypersensitivity reaction occurs, immediately discontinue VEKLURY and initiate appropriate treatment (see Contraindications).
- Increased risk of transaminase elevations: Transaminase elevations have been observed in healthy volunteers and in patients with COVID-19 who received VEKLURY; these elevations have also been reported as a clinical feature of COVID-19. Perform hepatic laboratory testing in all patients (see Dosage and administration). Consider discontinuing VEKLURY if ALT levels increase to >10× ULN. Discontinue VEKLURY if ALT elevation is accompanied by signs or symptoms of liver inflammation.
- Risk of reduced antiviral activity when coadministered with chloroquine or hydroxychloroquine: Coadministration of VEKLURY with chloroquine phosphate or hydroxychloroquine sulfate is not recommended based on data from cell culture experiments, demonstrating potential antagonism, which may lead to a decrease in the antiviral activity of VEKLURY.
Adverse reactions
- The most common adverse reaction (≥5% all grades) was nausea.
- The most common lab abnormalities (≥5% all grades) were increases in ALT and AST.
Drug interactions
- Drug interaction trials of VEKLURY and other concomitant medications have not been conducted in humans.
- Dosage: For adults and pediatric patients ≥12 years old and weighing ≥40 kg: 200 mg on Day 1, followed by once-daily maintenance doses of 100 mg from Day 2 administered only via intravenous infusion. VEKLURY should be initiated as soon as possible after diagnosis of symptomatic COVID-19.
-
Treatment duration:
- - For hospitalized patients requiring invasive mechanical ventilation and/or ECMO, the recommended total treatment duration is 10 days.
- - For hospitalized patients not requiring invasive mechanical ventilation and/or ECMO, the recommended treatment duration is 5 days. If a patient does not demonstrate clinical improvement, treatment may be extended for up to 5 additional days, for a total treatment duration of up to 10 days.
- - For non-hospitalized patients diagnosed with mild-to-moderate COVID-19 who are at high risk for progression to severe COVID-19, including hospitalization or death, the recommended total treatment duration is 3 days.
- Testing prior to and during treatment: Perform eGFR, hepatic laboratory, and prothrombin time testing prior to initiating VEKLURY and during use as clinically appropriate.
- Renal impairment: VEKLURY is not recommended in individuals with eGFR <30 mL/min.
- Dose preparation and administration: See full Prescribing Information.
Pregnancy and lactation
- Pregnancy: A pregnancy registry has been established. There are insufficient human data on the use of VEKLURY during pregnancy. COVID-19 is associated with adverse maternal and fetal outcomes, including preeclampsia, eclampsia, preterm birth, premature rupture of membranes, venous thromboembolic disease, and fetal death.
- Lactation: It is not known whether VEKLURY can pass into breast milk. Breastfeeding individuals with COVID-19 should follow practices according to clinical guidelines to avoid exposing the infant to COVID-19.
Please see full Prescribing Information for VEKLURY, available at www.gilead.com.
FACT SHEET FOR HEALTHCARE PROVIDERS
EMERGENCY USE AUTHORIZATION (EUA) OF VEKLURY® (remdesivir) FOR THE TREATMENT OF CORONAVIRUS DISEASE 2019 (COVID-19) IN PEDIATRIC PATIENTS WEIGHING 3.5 KG TO LESS THAN 40 KG OR PEDIATRIC PATIENTS LESS THAN 12 YEARS OF AGE WEIGHING AT LEAST 3.5 KG, WITH POSITIVE RESULTS OF DIRECT SARS-CoV-2 VIRAL TESTING WHO ARE:
HOSPITALIZED, OR
NOT HOSPITALIZED AND HAVE MILD-TO-MODERATE COVID-19, AND ARE AT HIGH RISK FOR PROGRESSION TO SEVERE COVID-19, INCLUDING HOSPITALIZATION OR DEATH
The U.S. Food and Drug Administration (FDA) has issued an Emergency Use Authorization (EUA) to permit the emergency use of VEKLURY for the treatment of coronavirus disease 2019 (COVID-19) in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg, with positive results of direct severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral testing, who are:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death. Refer to CDC website1 for additional details.
VEKLURY has been authorized by FDA for the emergency uses described above. VEKLURY is not FDA-approved for these uses.
VEKLURY is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of VEKLURY under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
This EUA is for the use of VEKLURY to treat COVID-19 in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg, with positive results of direct SARS-CoV-2 viral testing who are:
Hospitalized, or
Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
VEKLURY must be administered by intravenous (IV) infusion.
Healthcare providers must submit a report on all medication errors and ALL SERIOUS ADVERSE EVENTS related to VEKLURY. See Sections 8 and 9 of the Full EUA Prescribing Information for reporting requirements.
- See the Full EUA Prescribing Information for complete dosage, preparation, and administration instructions.
-
The only authorized dosage form of VEKLURY for pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg is VEKLURY for injection (supplied as 100 mg lyophilized powder in vial).
- The recommended dosage for pediatric patients weighing 3.5 kg to less than 40 kg is a single loading dose of VEKLURY 5 mg/kg on Day 1 followed by VEKLURY 2.5 mg/kg once daily from Day 2 [see Full EUA Prescribing Information, Recommended Dosage in Pediatric Patients (2.3)].
- The recommended dosage for pediatric patients less than 12 years of age and weighing 40 kg and higher is a single loading dose of 200 mg on Day 1 followed by once-daily maintenance doses of 100 mg from Day 2.
Hospitalized patients:
The treatment course of VEKLURY should be initiated as soon as possible after diagnosis of symptomatic COVID-19 has been made.
- The recommended total treatment duration for hospitalized patients requiring invasive mechanical ventilation and/or extracorporeal membrane oxygenation (ECMO) is 10 days.
- The recommended treatment duration for hospitalized patients not requiring invasive mechanical ventilation and/or ECMO is 5 days. If a patient does not demonstrate clinical improvement, treatment may be extended for up to 5 additional days for a total treatment duration of up to 10 days.
Non-hospitalized patients:
The treatment course of VEKLURY should be initiated as soon as possible after diagnosis of symptomatic COVID-19 has been made and within 7 days of symptom onset.
- The recommended total treatment duration for non-hospitalized patients diagnosed with mild-to-moderate COVID-19 who are at high risk for progression to severe COVID-19, including hospitalization or death, is 3 days.
Administer VEKLURY via intravenous infusion over 30 to 120 minutes.
For information on clinical trials that are testing the use of VEKLURY in COVID-19, please see www.clinicaltrials.gov.
AUTHORIZED USE
VEKLURY is a drug approved for the treatment of coronavirus disease 2019 (COVID-19) in adults and pediatric patients (12 years of age and older and weighing at least 40 kg) with positive results of direct severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral testing, who are:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
VEKLURY is not approved to treat pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg.
VEKLURY is authorized for use under an EUA for the treatment of COVID-19 in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg, with positive results of direct SARS-CoV-2 viral testing, who are:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death. Refer to CDC website2 for additional details.
For more information, see the long version of the "Fact Sheet for Healthcare Providers," available at https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization.
Contraindications
VEKLURY is contraindicated in patients with a history of clinically significant hypersensitivity reactions to VEKLURY or any components of the product.
Dosing
Patient Selection and Treatment Initiation
- Patients with positive results of direct SARS-CoV-2 viral testing.
- The treatment course of VEKLURY should be initiated as soon as possible after diagnosis of symptomatic COVID-19 has been made and for non-hospitalized patients, within 7 days of symptom onset.
- Pediatric patients (greater than 28 days old) must have an estimated glomerular filtration rate (eGFR) determined and full-term neonates (at least 7 days to less than or equal to 28 days old) must have serum creatinine determined before starting VEKLURY and be monitored during treatment as clinically appropriate.
- Perform hepatic laboratory testing in all patients before starting VEKLURY and during treatment as clinically appropriate.
- Determine prothrombin time in all patients before starting VEKLURY and monitor during treatment as clinically appropriate.
- The only authorized dosage form of VEKLURY for pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg is VEKLURY for injection (supplied as 100 mg lyophilized powder in vial).
- For pediatric patients weighing 3.5 kg to less than 40 kg, administer a body weight-based dosing regimen of VEKLURY.
- For pediatric patients less than 12 years of age and weighing 40 kg and higher, administer a single loading dose of VEKLURY 200 mg on Day 1 followed by once-daily maintenance doses of VEKLURY 100 mg from Day 2.
- Table 1 below provides the recommended dosage and dosage form in pediatric patients under this EUA [see Full EUA Prescribing Information, Recommended Dosage in Pediatric Patients (2.3)].
| Body weight | Recommended dosage form | Loading dose (on Day 1) | Maintenance dose (from Day 2) |
|---|---|---|---|
| 3.5 kg to less than 40 kg | VEKLURY for injection, lyophilized powder Only | 5 mg/kg | 2.5 mg/kg |
| 40 kg and higher | 200 mg | 100 mg |
Hospitalized patients:
The treatment course of VEKLURY should be initiated as soon as possible after diagnosis of symptomatic COVID-19 has been made.
- The recommended total treatment duration for hospitalized patients requiring invasive mechanical ventilation and/or extracorporeal membrane oxygenation (ECMO) is 10 days.
- The recommended treatment duration for hospitalized patients not requiring invasive mechanical ventilation and/or ECMO is 5 days. If a patient does not demonstrate clinical improvement, treatment may be extended for up to 5 additional days for a total treatment duration of up to 10 days.
Non-hospitalized patients:
The treatment course of VEKLURY should be initiated as soon as possible after diagnosis of symptomatic COVID-19 has been made and within 7 days of symptom onset.
- The recommended total treatment duration for non-hospitalized patients diagnosed with mild-to-moderate COVID-19 who are at high risk for progression to severe COVID-19, including hospitalization or death, is 3 days.
VEKLURY for injection must be reconstituted and further diluted prior to intravenous infusion.
Renal Impairment
VEKLURY is not recommended in pediatric patients (greater than 28 days old) with eGFR less than 30 mL/min or in full-term neonates (at least 7 days to less than or equal to 28 days old) with serum creatinine greater than or equal to 1 mg/dL.
Dose Preparation
See the Full EUA Prescribing Information for complete dosage, preparation, and administration instructions.
Care should be taken during admixture to prevent inadvertent microbial contamination. As there is no preservative or bacteriostatic agent present in this product, aseptic technique must be used in preparation of the final parenteral solution. It is always recommended to administer intravenous medication immediately after preparation when possible.
VEKLURY must be prepared and administered under the supervision of a healthcare provider. VEKLURY must be administered via intravenous infusion only. Do not administer by any other route.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. Prior to dilution in a 0.9% sodium chloride infusion bag, reconstituted VEKLURY for injection should be a clear, colorless to yellow solution, free of visible particles. Discard the vial if the lyophilized powder or reconstituted solution is discolored or contains particulate matter.
Important Preparation and Administration Instructions
- See the full EUA Prescribing Information for complete preparation and administration instructions.
- VEKLURY for Injection, 100 mg: Reconstitute VEKLURY for injection lyophilized powder with 19 mL of Sterile Water for Injection and further dilute in 0.9% sodium chloride prior to administration.
- Only use Sterile Water for Injection to reconstitute VEKLURY lyophilized powder.
- After reconstitution, use vials immediately to prepare diluted solution.
Administer diluted VEKLURY as an intravenous infusion over 30 to 120 minutes. - Discard any remaining reconstituted VEKLURY lyophilized powder and diluted solution.
Storage and Handling of Reconstituted Vial and Diluted Solution
After reconstitution, use VEKLURY for injection vial immediately to prepare diluted solution.
Store diluted VEKLURY solution for infusion for no more than 24 hours at room temperature (20°C to 25°C [68°F to 77°F]) or 48 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]) prior to administration.
Warnings
There are limited clinical data available for VEKLURY in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg. Serious and unexpected adverse events may occur that have not been previously reported with VEKLURY use.
Hypersensitivity Including Infusion-Related and Anaphylactic Reactions
Hypersensitivity reactions, including infusion-related and anaphylactic reactions, have been observed during and following administration of VEKLURY; most occurred within one hour. Signs and symptoms may include hypotension, hypertension, tachycardia, bradycardia, hypoxia, fever, dyspnea, wheezing, angioedema, rash, nausea, diaphoresis, and shivering. Slower infusion rates, with a maximum infusion time of up to 120 minutes, can be considered to potentially prevent these signs and symptoms. Monitor patients during infusion and observe patients for at least one hour after infusion is complete for signs and symptoms of hypersensitivity as clinically appropriate. If signs and symptoms of a clinically significant hypersensitivity reaction occur, immediately discontinue administration of VEKLURY and initiate appropriate treatment. The use of VEKLURY is contraindicated in patients with known hypersensitivity to VEKLURY or any components of the product [see Full EUA Prescribing Information, Contraindications (4), Warnings and Precautions (5.1)].
Increased Risk of Transaminase Elevations
Transaminase elevations have been observed in healthy volunteers who received 200 mg of VEKLURY followed by 100 mg doses up to 10 days; the transaminase elevations were mild (Grade 1) to moderate (Grade 2) in severity and resolved upon discontinuation of VEKLURY. Transaminase elevations have also been reported in patients with COVID-19 who received VEKLURY. Because transaminase elevations have been reported as a clinical feature of COVID-19, and the incidence was similar in patients receiving placebo versus VEKLURY in clinical trials of VEKLURY, discerning the contribution of VEKLURY to transaminase elevations in patients with COVID-19 can be challenging [see Full EUA Prescribing Information, Warnings and Precautions (5.2)].
Perform hepatic laboratory testing in all patients before starting VEKLURY and during treatment as clinically appropriate.
- Consider discontinuing VEKLURY if ALT levels increase to greater than 10 times the upper limit of normal.
- Discontinue VEKLURY if ALT elevation is accompanied by signs or symptoms of liver inflammation.
Risk of Reduced Antiviral Activity When Coadministered with Chloroquine Phosphate or Hydroxychloroquine Sulfate
Coadministration of VEKLURY and chloroquine phosphate or hydroxychloroquine sulfate is not recommended based on data from cell culture experiments demonstrating a potential antagonistic effect of chloroquine on the intracellular metabolic activation and antiviral activity of VEKLURY [see Full EUA Prescribing Information, Warnings and Precautions (5.3), Drug Interactions (10), Microbiology/Resistance Information (15)].
Serious Side Effects
Serious adverse reactions have been associated with VEKLURY [see Full EUA Prescribing Information, Overall Safety Summary (6.1)].
Additional serious adverse reactions associated with the drug may become apparent with more widespread use.
INSTRUCTIONS FOR HEALTHCARE PROVIDERS
As the healthcare provider, you must communicate to the parent/caregiver and to your patient, as age appropriate, information consistent with the "Fact Sheet for Parents and Caregivers" (and provide a copy of the Fact Sheet) prior to the pediatric patient receiving VEKLURY, including:
- That FDA has authorized the emergency use of VEKLURY for the treatment of coronavirus disease 2019 (COVID-19) in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg, with positive results of direct SARS-CoV-2 viral testing, who are:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
- The parent/caregiver has the option to accept or refuse VEKLURY.
- The significant known and potential risks and benefits of VEKLURY, and the extent to which such risks and benefits are unknown.
- Information on available alternative treatments and the risks and benefits of those alternatives.
If providing this information will delay the administration of VEKLURY to a degree that would endanger the lives of patients, the information must be provided to the parent/caregiver as soon as feasible after VEKLURY is administered.
For information on clinical trials that are testing the use of VEKLURY for COVID-19, please see www.clinicaltrials.gov.
MANDATORY REQUIREMENTS FOR VEKLURY ADMINISTRATION UNDER EMERGENCY USE AUTHORIZATION:
In order to mitigate the risks of using this product under EUA and to optimize the potential benefit of VEKLURY for this use, the following items are required. Use of VEKLURY under this EUA is limited to the following (all requirements must be met):
- VEKLURY is authorized for the treatment of COVID-19 in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg, with positive results of direct SARS-CoV-2 viral testing, who are:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death. Please refer to CDC website3 for additional details.
- As the healthcare provider, communicate to the parent/caregiver and your patient, as age appropriate, information consistent with the "Fact Sheet for Parents and Caregivers" prior to the patient receiving VEKLURY. Healthcare providers (to the extent practicable given the circumstances of the emergency) must document in the patient's medical record that the parent/caregiver has been:
- Given the "Fact Sheet for Parents and Caregivers,"
- Informed of alternatives to receiving VEKLURY, and
- Informed that VEKLURY is an approved drug that is authorized for this unapproved use under EUA.
- Pediatric patients (greater than 28 days old) must have an eGFR determined and full-term neonates (at least 7 days to less than or equal to 28 days old) must have serum creatinine determined before starting VEKLURY and monitored during treatment as clinically appropriate.
- Perform hepatic laboratory testing in all patients before starting VEKLURY and during treatment as clinically appropriate.
- Determine prothrombin time in all patients before starting VEKLURY and monitor during treatment as clinically appropriate.
- Patients with known hypersensitivity to any ingredient of VEKLURY must not receive VEKLURY.
- The prescribing healthcare provider and/or the provider's designee are/is responsible for mandatory reporting of all serious adverse events4 and medication errors potentially related to VEKLURY within 7 calendar days from the healthcare provider's awareness of the event, using FDA Form 3500 (for information on how to access this form, see below). The FDA requires that such reports, using FDA Form 3500, include the following:
- Patient demographics and baseline characteristics (e.g., patient identifier, age or date of birth, gender, weight, ethnicity, and race)
- A statement "Veklury (remdesivir) use for COVID-19 under Emergency Use Authorization (EUA)" under the "Describe Event, Problem, or Product Use/Medication Error" heading
- Information about the serious adverse event or medication error (e.g., signs and symptoms, test/laboratory data, complications, timing of drug initiation in relation to the occurrence of the event, duration of the event, treatments required to mitigate the event, evidence of event improvement/disappearance after stopping or reducing the dosage, evidence of event reappearance after reintroduction, clinical outcomes).
- Patient's preexisting medical conditions and use of concomitant products
- Information about the product (e.g., dosage, route of administration, NDC #).
- Complete and submit the report online: www.fda.gov/medwatch/report.htm
- Complete and submit a postage-paid FDA Form 3500 (https://www.fda.gov/media/76299/download) and return by:
- Mail to MedWatch, 5600 Fishers Lane, Rockville, MD 20852-9787, or
- Fax to 1-800-FDA-0178, or
- Call 1-800-FDA-1088 to request a reporting form
Gilead Global Patient Safety
Fax: 1-650-522-5477
E-mail: Safety_fc@gilead.com
Or call Gilead at 1-800-GILEAD-5 to report adverse events - The prescribing healthcare provider and/or the provider's designee is/are responsible for mandatory responses to requests from FDA for information about adverse events and medication errors following receipt of VEKLURY.
- death;
- a life-threatening adverse event;
- inpatient hospitalization or prolongation of existing hospitalization;
- a persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions;
- a congenital anomaly/birth defect;
- a medical or surgical intervention to prevent death, a life-threatening event, hospitalization, disability, or congenital anomaly.
APPROVED AVAILABLE ALTERNATIVES
There is no approved available alternative product for the treatment of COVID-19 in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg, with positive results of direct SARS-CoV-2 viral testing, and who are:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
There are EUAs for other COVID-19 treatments. Additional information on COVID-19 treatments can be found at https://www.covid19treatmentguidelines.nih.gov/. The healthcare provider should visit https://clinicaltrials.gov/ to determine whether the patient may be eligible for enrollment in a clinical trial.
AUTHORITY FOR ISSUANCE OF THE EUA
The Secretary of HHS has declared that circumstances exist that justify the emergency use of drugs and biological products during the COVID-19 pandemic. In response, the FDA has issued an EUA for the approved product, VEKLURY, for the unapproved use to treat COVID-19 in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg, with positive results of direct SARS-CoV-2 viral testing, and who are5:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death. Please refer to CDC website6 for additional details.
FDA has issued this EUA, requested by Gilead Sciences, Inc. and based on their submitted data. As a healthcare provider, you must comply with the mandatory requirements of the EUA (see above).
Although limited scientific information is available in the pediatric population, based on the totality of the scientific evidence available to date, it is reasonable to believe that VEKLURY may be effective for the treatment of COVID-19 in pediatric patients as specified in this Fact Sheet. You may be contacted and asked to provide information to help with the assessment of the use of the product during this emergency.
This EUA for VEKLURY will end when the Secretary determines that the circumstances justifying the EUA no longer exist or when there is a change in the approval status of the product such that an EUA is no longer needed.
FULL EUA PRESCRIBING INFORMATION
| FULL EUA PRESCRIBING INFORMATION: | 9 OTHER REPORTING REQUIREMENTS |
| CONTENTS* | 10 DRUG INTERACTIONS |
| 1 AUTHORIZED USE | 11 USE IN SPECIFIC POPULATIONS |
| 2 DOSAGE AND ADMINISTRATION | 11.1 Pregnancy |
| 2.1 Dosage and Administration Overview | 11.2 Lactation |
| 2.2 Important Testing Before and During | 11.3 Pediatric Use |
| Treatment | 11.4 Renal Impairment |
| 2.3 Recommended Dosage in Pediatric Patients | 11.5 Hepatic Impairment |
| 2.4 Renal Impairment | 12 OVERDOSAGE |
| 2.5 Dose Preparation and Administration, | 13 PRODUCT DESCRIPTION |
| VEKLURY for Injection | 13.1 Physical Appearance |
| 2.6 Storage of Prepared Dosages | 13.2 Inactive Ingredients |
| 3 DOSAGE FORMS AND STRENGTHS | 14 CLINICAL PHARMACOLOGY |
| 4 CONTRAINDICATIONS | 14.1 Mechanism of Action |
| 5 WARNINGS AND PRECAUTIONS | 14.2 Pharmacokinetics |
| 5.1 Hypersensitivity Including Infusion-Related and | 15 MICROBIOLOGY/RESISTANCE INFORMATION |
| Anaphylactic Reactions | 16 NONCLINICAL TOXICOLOGY |
| 5.2 Increased Risk of Transaminase Elevations | 17 ANIMAL PHARMACOLOGIC AND EFFICACY |
| 5.3 Risk of Reduced Antiviral Activity When | DATA |
| Coadministered with Chloroquine Phosphate or | 18 CLINICAL TRIAL RESULTS AND SUPPORTING |
| Hydroxychloroquine Sulfate | DATA FOR EUA |
| 6 OVERALL SAFETY SUMMARY | 19 HOW SUPPLIED/STORAGE AND HANDLING |
| 6.1 Clinical Trials Experience | 20 PATIENT COUNSELING INFORMATION |
| 7 PATIENT MONITORING RECOMMENDATIONS | 21 CONTACT INFORMATION |
| 8 ADVERSE REACTIONS AND MEDICATION | *Sections or subsections omitted from the full |
| ERRORS REPORTING REQUIREMENTS AND | prescribing information are not listed. |
| INSTRUCTIONS |
1. AUTHORIZED USE
VEKLURY is authorized for use under an EUA for the treatment of coronavirus disease 2019 (COVID-19) in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg, with positive results of direct severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral testing, who are:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death. Refer to CDC website7 for additional details.
2. DOSAGE AND ADMINISTRATION
2.1 Dosage and Administration Overview
- VEKLURY may only be administered in settings in which healthcare providers have immediate access to medications to treat a severe infusion or hypersensitivity reaction, such as anaphylaxis, and the ability to activate the emergency medical system (EMS), as necessary [see Warnings and Precautions (5.1)].
- Administer VEKLURY by intravenous infusion only. Do not administer by any other route.
2.2 Important Testing Before and During Treatment
- Pediatric patients (greater than 28 days old) must have an eGFR determined and full-term neonates (at least 7 days to less than or equal to 28 days old) must have serum creatinine determined before starting VEKLURY and during treatment as clinically appropriate [see Dosage and Administration (2.4), Use in Specific Populations (11.4)].
- Perform hepatic laboratory testing in all patients before starting VEKLURY and during treatment as clinically appropriate [see Warnings and Precautions (5.2), Use in Specific Populations (11.5)].
- Determine prothrombin time in all patients before starting VEKLURY and monitor during treatment as clinically appropriate [see Overall Safety Summary (6.1)].
2.3 Recommended Dosage in Pediatric Patients
The only authorized dosage form of VEKLURY for pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg is VEKLURY for injection (supplied as 100 mg lyophilized powder in vial).
For pediatric patients weighing 3.5 kg to less than 40 kg, administer a body weight-based dosing regimen of VEKLURY via intravenous (IV) infusion. The dosage should be calculated using the mg/kg dose according to the patient's weight.
For pediatric patients less than 12 years of age and weighing 40 kg and higher, administer a single loading dose of VEKLURY 200 mg on Day 1 followed by once-daily maintenance doses of VEKLURY 100 mg from Day 2.
Refer to Table 1 below for recommended dosage form and dosage in pediatric patients according to weight [see Dosage and Administration (2.5), Use in Specific Populations (11.3)].
| Body weight | Recommended dosage form | Loading dose (on Day 1) | Maintenance dose (from Day 2) |
|---|---|---|---|
| 3.5 kg to less than 40 kg | VEKLURY Lyophilized Powder for Injection Only | 5 mg/kg | 2.5 mg/kg |
| 40 kg and higher | 200 mg | 100 mg |
Hospitalized patients:
The treatment course of VEKLURY should be initiated as soon as possible after diagnosis of symptomatic COVID-19 has been made.
- The recommended total treatment duration for hospitalized patients requiring invasive mechanical ventilation and/or extracorporeal membrane oxygenation (ECMO) is 10 days.
- The recommended treatment duration for hospitalized patients not requiring invasive mechanical ventilation and/or ECMO is 5 days. If a patient does not demonstrate clinical improvement, treatment may be extended for up to 5 additional days for a total treatment duration of up to 10 days.
Non-hospitalized patients:
The treatment course of VEKLURY should be initiated as soon as possible after diagnosis of symptomatic COVID-19 has been made and within 7 days of symptom onset.
- The recommended total treatment duration for non-hospitalized patients diagnosed with mild-to-moderate COVID-19 who are at high risk for progression to severe COVID-19, including hospitalization or death, is 3 days.
VEKLURY for injection must be reconstituted and further diluted prior to administration via intravenous infusion.
2.4 Renal Impairment
VEKLURY is not recommended in pediatric patients (greater than 28 days old) with eGFR less than 30 mL/min or in full-term neonates (at least 7 days and less than or equal to 28 days old) with serum creatinine greater than or equal to 1 mg/dL.
2.5 Dose Preparation and Administration, VEKLURY for Injection
The authorized dosage form of VEKLURY for pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg is VEKLURY for injection (supplied as 100 mg lyophilized powder) only.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. Discard the vial if the lyophilized powder is discolored or contains particulate matter. Prior to dilution in 0.9% sodium chloride, reconstituted VEKLURY for injection should be a clear, colorless to yellow solution, free of visible particles.
- Care should be taken during admixture to prevent inadvertent microbial contamination. As there is no preservative or bacteriostatic agent present in this product, aseptic technique must be used in preparation of the final parenteral solution. It is always recommended to administer intravenous medication immediately after preparation when possible.
Reconstitution Instructions
Remove the required number of single-dose vial(s) from storage. For each vial:
- Aseptically reconstitute VEKLURY lyophilized powder by addition of 19 mL of Sterile Water for Injection using a suitably sized syringe and needle per vial.
- Only use Sterile Water for Injection to reconstitute VEKLURY lyophilized powder.
- Discard the vial if a vacuum does not pull the Sterile Water for Injection into the vial.
- Immediately shake the vial for 30 seconds.
- Allow the contents of the vial to settle for 2 to 3 minutes. A clear, colorless to yellow solution, free of visible particles, should result.
- If the contents of the vial are not completely dissolved, shake the vial again for 30 seconds and allow the contents to settle for 2 to 3 minutes. Repeat this procedure as necessary until the contents of the vial are completely dissolved. Discard the vial if the contents are not completely dissolved.
- Following reconstitution, each vial contains 100 mg/20 mL (5 mg/mL) of remdesivir solution.
- Use reconstituted VEKLURY for injection immediately to prepare the diluted solution.
Dilution and Administration Instructions, Pediatric Patients Weighing 3.5 kg to Less Than 40 kg
Dilution Instructions
- For pediatric patients weighing 3.5 kg to less than 40 kg, the 100 mg/20 mL (5 mg/mL) remdesivir reconstituted solution should be further diluted to a fixed concentration of 1.25 mg/mL using 0.9% sodium chloride.
- The final required infusion volume concentration of 1.25 mg/mL remdesivir diluted solution for infusion is based on the pediatric weight-based dosing regimens of 5 mg/kg for the Loading Dose and 2.5 mg/kg for each Maintenance Dose.
- Small 0.9% sodium chloride infusion bags (e.g., 25, 50, or 100 mL) or an appropriately sized syringe should be used for pediatric dosing. The recommended dose is administered via intravenous infusion in a total volume dependent on the dose to yield the target remdesivir concentration of 1.25 mg/mL.
- A syringe and syringe pump may be used for infusion volumes less than 50 mL.
- Refer to Table 2 for recommended rate of infusion.
Infusion with IV Bag
- Determine the total infusion volume needed to achieve a final infusion volume concentration of 1.25 mg/mL of remdesivir diluted solution based on the patient's calculated dose.
- Select an appropriately sized infusion bag (either prefilled with 0.9% sodium chloride or empty) to prepare VEKLURY diluted solution.
- If using a prefilled 0.9% sodium chloride infusion bag, withdraw and discard the amount of diluent equal to the volume of reconstituted VEKLURY solution needed per patient's dose plus a quantity sufficient to achieve a 1.25 mg/mL final volume concentration of remdesivir diluted solution.
- Withdraw the required volume of reconstituted VEKLURY solution into an appropriately sized syringe.
- Transfer the required volume of reconstituted VEKLURY solution to the 0.9% sodium chloride infusion bag.
- Gently invert the bag 20 times to mix the solution in the bag. Do not shake.
- If using an empty infusion bag, transfer the required volume of reconstituted VEKLURY solution to the bag, followed by a volume of 0.9% sodium chloride sufficient to achieve a 1.25 mg/mL final volume concentration of remdesivir diluted solution.
- The prepared infusion solution is stable for 24 hours at room temperature (20°C to 25°C [68°F to 77°F]) or 48 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]).
Infusion with Syringe
- Determine the total infusion volume needed to achieve a final infusion volume concentration of 1.25 mg/mL of remdesivir diluted solution based on patient's calculated dose.
- Select an appropriately sized syringe equal to or larger than the calculated total infusion volume of 1.25 mg/mL remdesivir solution needed.
- Withdraw the required volume of reconstituted VEKLURY solution from the vial into the syringe based on patient's calculated dose, followed by the required volume of 0.9% sodium chloride needed to achieve a 1.25 mg/mL final volume concentration of remdesivir diluted solution.
- Gently invert the syringe 20 times to mix the solution in the syringe. Do not shake.
The prepared diluted solution should be used immediately.
Administration Instructions
The prepared diluted solution should not be administered simultaneously with any other medication. The compatibility of VEKLURY with IV solutions and medications other than 0.9% sodium chloride injection, USP is not known.
Administer the diluted solution with the infusion rate described in Table 2.
| Infusion volume | Infusion time | Rate of infusion* |
|---|---|---|
|
|
||
| 100 mL | 30 min | 3.33 mL/min |
| 60 min | 1.67 mL/min | |
| 120 min | 0.83 mL/min | |
| 50 mL | 30 min | 1.67 mL/min |
| 60 min | 0.83 mL/min | |
| 120 min | 0.42 mL/min | |
| 25 mL | 30 min | 0.83 mL/min |
| 60 min | 0.42 mL/min | |
| 120 min | 0.21 mL/min | |
| 7 mL | 30 min | 0.23 mL/min |
| 60 min | 0.12 mL/min | |
| 120 min | 0.06 mL/min | |
Administration should be under conditions where management of severe hypersensitivity reactions, such as anaphylaxis, is possible. Monitor patients during infusion and observe patients for at least one hour after infusion is complete for signs and symptoms of hypersensitivity as clinically appropriate.
Dilution and Administration Instructions, Pediatric Patients Less Than 12 Years of Age and Weighing 40 kg and Higher
Dilution Instructions
For pediatric patients less than 12 years of age and weighing 40 kg and higher, refer to the dilution instructions in Table 3.
| VEKLURY dose | 0.9% sodium chloride infusion bag volume to be used | Volume to be withdrawn and discarded from 0.9% sodium chloride infusion bag | Required volume of reconstituted VEKLURY for injection |
|---|---|---|---|
| Loading dose 200 mg (2 vials) | 250 mL | 40 mL | 40 mL (2 × 20 mL) |
| 100 mL | 40 mL | 40 mL (2 × 20 mL) | |
| Maintenance dose 100 mg (1 vial) | 250 mL | 20 mL | 20 mL |
| 100 mL | 20 mL | 20 mL |
- Withdraw and discard the required volume of 0.9% sodium chloride from the infusion bag following instructions in Table 3, using an appropriately sized syringe and needle.
- Withdraw the required volume of reconstituted VEKLURY for injection from the VEKLURY vial following instructions in Table 3. Discard any unused portion remaining in the reconstituted vial.
- Transfer the required volume of reconstituted VEKLURY for injection to the selected infusion bag.
- Gently invert the bag 20 times to mix the solution in the bag. Do not shake.
- The prepared diluted solution is stable for 24 hours at room temperature (20°C to 25°C [68°F to 77°F]) or 48 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]).
Administration Instructions
The prepared diluted solution should not be administered simultaneously with any other medication. The compatibility of VEKLURY with IV solutions and medications other than 0.9% sodium chloride injection, USP is not known.
Administer the diluted solution with the infusion rate described in Table 4.
| Infusion volume | Infusion time | Rate of infusion |
|---|---|---|
| 250 mL | 30 min | 8.33 mL/min |
| 60 min | 4.17 mL/min | |
| 120 min | 2.08 mL/min | |
| 100 mL | 30 min | 3.33 mL/min |
| 60 min | 1.67 mL/min | |
| 120 min | 0.83 mL/min |
Administration should be under conditions where management of severe hypersensitivity reactions, such as anaphylaxis, is possible. Monitor patients during infusion and observe patients for at least one hour after infusion is complete for signs and symptoms of hypersensitivity as clinically appropriate.
2.6 Storage of Prepared Dosages
After reconstitution, use vials immediately to prepare diluted solution.
The diluted VEKLURY solution in syringe should be used immediately.
The diluted VEKLURY solution in the infusion bags can be stored up to 24 hours at room temperature (20°C to 25°C [68°F to 77°F]) or 48 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]) prior to administration.
IMPORTANT:
This product contains no preservative. Any unused portion of a single-dose VEKLURY vial should be discarded after a diluted solution is prepared. Maintain adequate records showing receipt, use, and disposition of VEKLURY. For unused intact vials, maintain adequate records showing disposition of VEKLURY; do not discard unused intact vials.
3. DOSAGE FORMS AND STRENGTHS
VEKLURY for injection,100 mg, available as a sterile, preservative-free white to off-white to yellow lyophilized powder in single-dose vial for reconstitution.
4. CONTRAINDICATIONS
VEKLURY is contraindicated in patients with a history of clinically significant hypersensitivity reactions to VEKLURY or any components of the product [see Warnings and Precautions (5.1)].
5. WARNINGS AND PRECAUTIONS
There are limited clinical data available for VEKLURY in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg. Serious and unexpected adverse events may occur that have not been previously reported with VEKLURY use.
5.1 Hypersensitivity Including Infusion-Related and Anaphylactic Reactions
Hypersensitivity reactions, including infusion-related and anaphylactic reactions, have been observed during and following administration of VEKLURY; most occurred within one hour. Signs and symptoms may include hypotension, hypertension, tachycardia, bradycardia, hypoxia, fever, dyspnea, wheezing, angioedema, rash, nausea, diaphoresis, and shivering. Slower infusion rates, with a maximum infusion time of up to 120 minutes, can be considered to potentially prevent these signs and symptoms. Monitor patients during infusion and observe patients for at least one hour after infusion is complete for signs and symptoms of hypersensitivity as clinically appropriate. If signs and symptoms of a clinically significant hypersensitivity reaction occur, immediately discontinue administration of VEKLURY and initiate appropriate treatment. The use of VEKLURY is contraindicated in patients with known hypersensitivity to VEKLURY or any components of the product [see Contraindications (4)].
5.2 Increased Risk of Transaminase Elevations
Transaminase elevations have been observed in healthy volunteers who received 200 mg of VEKLURY followed by 100 mg doses for up to 10 days; the transaminase elevations were mild (Grade 1) to moderate (Grade 2) in severity and resolved upon discontinuation of VEKLURY. Transaminase elevations have also been reported in patients with COVID-19 who received VEKLURY. Because transaminase elevations have been reported as a clinical feature of COVID-19, including in patients receiving placebo in clinical trials of VEKLURY, and the incidence was similar in patients receiving placebo versus VEKLURY in clinical trials of VEKLURY, discerning the contribution of VEKLURY to transaminase elevations in patients with COVID-19 can be challenging.
Perform hepatic laboratory testing in all patients before starting VEKLURY and while receiving VEKLURY as clinically appropriate.
- Consider discontinuing VEKLURY if ALT levels increase to greater than 10 times the upper limit of normal.
- Discontinue VEKLURY if ALT elevation is accompanied by signs or symptoms of liver inflammation.
5.3 Risk of Reduced Antiviral Activity When Coadministered with Chloroquine Phosphate or Hydroxychloroquine Sulfate
Coadministration of VEKLURY and chloroquine phosphate or hydroxychloroquine sulfate is not recommended based on data from cell culture experiments demonstrating a potential antagonistic effect of chloroquine on the intracellular metabolic activation and antiviral activity of VEKLURY [see Drug Interactions (10), Microbiology/Resistance Information (15)].
6. OVERALL SAFETY SUMMARY
Completion of FDA MedWatch Form to report all medication errors and adverse events occurring during VEKLURY treatment is mandatory. Please see the ADVERSE REACTIONS AND MEDICATION ERRORS REPORTING REQUIREMENTS AND INSTRUCTIONS section below for details on FDA MedWatch reporting.
6.1 Clinical Trials Experience
The safety of VEKLURY is based on data from three Phase 3 studies in 1,313 hospitalized adult subjects with COVID-19, one Phase 3 study in 279 non-hospitalized adult and pediatric subjects (12 years of age and older weighing at least 40 kg) with mild-to-moderate COVID-19, from four Phase 1 studies in 131 healthy adults, and from adult patients with COVID-19 who received VEKLURY under the Emergency Use Authorization or in a compassionate use program.
NIAID ACTT-1 was a randomized, double-blind, placebo-controlled clinical trial in hospitalized adult subjects with mild, moderate, and severe COVID-19 treated with VEKLURY (n=532) or placebo (n=516) for up to 10 days. Subjects treated with VEKLURY received 200 mg on Day 1 and 100 mg once daily on subsequent days. The collection of adverse event data in this trial was limited to severe (Grade 3) or potentially life-threatening (Grade 4) adverse events, serious adverse events, adverse events leading to study drug discontinuation, and moderate (Grade 2) severity or higher hypersensitivity reactions. Rates of adverse reactions (≥ Grade 3), serious adverse reactions, and adverse reactions leading to treatment discontinuation are presented in Table 5.
| Types of Adverse Reactions | VEKLURY N=532 n (%) | Placebo N=516 n (%) |
|---|---|---|
|
|
||
| Adverse reactions, Grades ≥3 | 41 (8%) | 46 (9%) |
| Serious adverse reactions | 2 (0.4%)* | 3 (0.6%) |
| Adverse reactions leading to treatment discontinuation | 11 (2%)† | 15 (3%) |
Study GS-US-540-5773 was a randomized, open-label clinical trial in hospitalized adult subjects with severe COVID-19 treated with VEKLURY 200 mg on Day 1 and 100 mg once daily for 5 (n=200) or 10 days (n=197). Adverse reactions were reported in 33 (17%) subjects in the 5-day group and 40 (20%) subjects in the 10-day group. The most common adverse reactions occurring in at least 5% of subjects in either the VEKLURY 5-day or 10-day group, respectively, were nausea (5% vs 3%), AST increased (3% vs 6%), and ALT increased (2% vs 7%). Rates of any adverse reaction, serious adverse reactions, and adverse reactions leading to treatment discontinuation are presented in Table 6.
| Types of Adverse Reactions | VEKLURY 5 Days N=200 n (%) | VEKLURY 10 Days N=197 n (%) |
|---|---|---|
|
|
||
| Any adverse reaction, all Grades | 33 (17%) | 40 (20%) |
| Serious adverse reactions | 3 (2%)* | 4 (2%)* |
| Adverse reactions leading to treatment discontinuation | 5 (3%)† | 9 (5%)† |
Study GS-US-540-5774 was a randomized, open-label clinical trial in hospitalized adult subjects with moderate COVID-19 treated with VEKLURY 200 mg on Day 1 and 100 mg daily for 5 (n=191) or 10 days (n=193), or standard of care (SOC) only (n=200). Adverse reactions were reported in 36 (19%) subjects in the 5-day group and 25 (13%) subjects in the 10-day group The most common adverse reaction occurring in at least 5% of subjects in the VEKLURY groups was nausea (7% in the 5-day group, 4% in the 10-day group). Rates of any adverse reaction, serious adverse reactions, and adverse reactions leading to treatment discontinuation are presented in Table 7.
| Types of Adverse Reactions | VEKLURY 5 Days N=191 n (%) | VEKLURY 10 Days N=193 n (%) |
|---|---|---|
|
|
||
| Any adverse reaction, all Grades | 36 (19%) | 25 (13%) |
| Serious adverse reactions | 1 (<1%)† | 0 |
| Adverse reactions leading to treatment discontinuation | 4 (2%)‡ | 4 (2%)‡ |
Study GS-US-540-9012 was a randomized, double-blind, placebo-controlled clinical trial in subjects who were non-hospitalized, were symptomatic for COVID-19 for ≤7 days, had confirmed SARS-CoV-2 infection, and had at least one risk factor for progression to hospitalization treated with VEKLURY (n=279; 276 adults and 3 pediatric subjects 12 years of age and older weighing at least 40 kg) or placebo (n=283; 278 adults and 5 pediatric subjects 12 years of age and older weighing at least 40 kg) for 3 days. Of the 279 subjects treated with VEKLURY, 227 subjects received at least one dose of VEKLURY at an outpatient facility, 44 subjects received at least one dose of VEKLURY in a home healthcare setting, and 8 subjects received at least one dose of VEKLURY at a skilled nursing facility. Subjects treated with VEKLURY received 200 mg on Day 1 and 100 mg once daily on subsequent days [see Clinical Trial Results and Supporting Data for EUA (18)]. Adverse reactions (all grades) were reported in 34 (12%) subjects in the VEKLURY group and 25 (9%) subjects in the placebo group. The most common adverse reaction occurring in at least 5% of subjects in the VEKLURY group was nausea (6%). There were no serious adverse reactions or adverse reactions leading to treatment discontinuation in either treatment group. Safety in subjects who received VEKLURY in a home healthcare setting was comparable to that observed in the overall GS-US-540-9012 study population, but these findings are based on limited data.
Less Common Adverse Reactions
Clinically significant adverse reactions that were reported in <2% of adult subjects exposed to VEKLURY in clinical trials are listed below:
- Hypersensitivity reactions [see Warnings and Precautions (5.1)].
- Generalized seizure
- Rash
Emergency Use Authorization Experience in Subjects with COVID-19
The following adverse reactions have been identified during use of VEKLURY primarily in adult subjects under Emergency Use Authorization:
- General disorders and administration site conditions: Administration site extravasation
- Skin and subcutaneous tissue disorders: Rash
- Immune system disorders: Anaphylaxis, angioedema, infusion-related reactions, hypersensitivity
- Investigations: Transaminase elevations
Laboratory Abnormalities
Study GS-US-399-5505 was a Phase 1, randomized, blinded, placebo-controlled clinical trial in healthy adult volunteers administered VEKLURY 200 mg on Day 1 and 100 mg for either 4 days or 9 days. Mild (Grade 1, n=8) to moderate (Grade 2, n=1) elevations in ALT were observed in 9 of 20 subjects receiving 10 days of VEKLURY; the elevations in ALT resolved upon discontinuation of VEKLURY. No subjects (0 of 9) who received 5 days of VEKLURY had graded increases in ALT.
The frequencies of laboratory abnormalities (Grades 3–4) occurring in at least 3% of adult subjects with COVID-19 receiving VEKLURY in Trials NIAID ACTT-1, 5773, and 5774 are presented in Table 8, Table 9, and Table 10, respectively.
| Laboratory Parameter Abnormality* | VEKLURY 10 Days N=532 | Placebo N=516 |
|---|---|---|
|
|
||
| ALT increased | 3% | 6% |
| AST increased | 6% | 8% |
| Bilirubin increased | 2% | 5% |
| Creatinine clearance decreased† | 18% | 20% |
| Creatinine increased | 15% | 16% |
| eGFR decreased | 18% | 24% |
| Glucose increased | 12% | 13% |
| Hemoglobin decreased | 15% | 22% |
| Lymphocytes decreased | 11% | 18% |
| Prothrombin time increased | 9% | 4% |
| Laboratory Parameter Abnormality* | VEKLURY 5 Days N=200 | VEKLURY 10 Days N=197 |
|---|---|---|
|
|
||
| ALT increased | 6% | 8% |
| AST increased | 7% | 6% |
| Creatinine clearance decreased† | 10% | 19% |
| Creatinine increased | 5% | 15% |
| Glucose increased | 11% | 8% |
| Hemoglobin decreased | 6% | 8% |
| Laboratory Parameter Abnormality* | VEKLURY 5 Days N=191 | VEKLURY 10 Days N=193 | SOC N=200 |
|---|---|---|---|
| SOC=Standard of care. | |||
|
|
|||
| ALT increased | 2% | 3% | 8% |
| Creatinine clearance decreased† | 2% | 5% | 8% |
| Glucose increased | 4% | 3% | 2% |
| Hemoglobin decreased | 3% | 1% | 6% |
The frequencies of laboratory abnormalities (Grades 3–4) occurring in at least 2% of subjects with COVID-19 receiving VEKLURY in Trial GS-US-540-9012 are presented in Table 11.
| Laboratory Parameter Abnormality* | VEKLURY 3 Days N=279 | Placebo N=283 |
|---|---|---|
|
|
||
| Creatinine clearance decreased† | 6% | 2% |
| Creatinine increased | 3% | 1% |
| Glucose increased | 6% | 6% |
| Lymphocytes decreased | 2% | 1% |
| Prothrombin time increased | 1% | 2% |
7. PATIENT MONITORING RECOMMENDATIONS
Patients should have appropriate clinical and laboratory monitoring to aid in early detection of any potential adverse events while receiving VEKLURY [see Dosage and Administration (2.2, 2.4)].
Administration should be under conditions where management of severe hypersensitivity reactions, such as anaphylaxis, is possible. Monitor patients during infusion and observe patients for at least one hour after infusion is complete for signs and symptoms of hypersensitivity as clinically appropriate.
Additionally, completion of FDA MedWatch Form to report all medication errors and serious adverse events is mandatory.
For mandatory reporting requirements, please see "MANDATORY REQUIREMENTS FOR VEKLURY ADMINISTRATION UNDER EMERGENCY USE AUTHORIZATION"above.
8. ADVERSE REACTIONS AND MEDICATION ERRORS REPORTING REQUIREMENTS AND INSTRUCTIONS
See Overall Safety Summary (Section 6) for additional information.
The prescribing healthcare provider and/or the provider's designee are/is responsible for the mandatory reporting of all serious adverse events* and medication errors potentially related to VEKLURY within 7 calendar days from the healthcare provider's awareness of the event, using FDA Form 3500 (for information on how to access this form, see below). The FDA requires that such reports, using FDA Form 3500, include the following:
- Patient demographics and baseline characteristics (e.g., patient identifier, age or date of birth, gender, weight, ethnicity, and race)
- A statement "Veklury (remdesivir) use for COVID-19 under Emergency Use Authorization (EUA)" under the "Describe Event, Problem, or Product Use/Medication Error" heading
- Information about the serious adverse event or medication error (e.g., signs and symptoms, test/laboratory data, complications, timing of drug initiation in relation to the occurrence of the event, duration of the event, treatments required to mitigate the event, evidence of event improvement/disappearance after stopping or reducing the dosage, evidence of event reappearance after reintroduction, clinical outcomes)
- Patient's preexisting medical conditions and use of concomitant products
- Information about the product (e.g., dosage, route of administration, NDC #).
Submit adverse event and medication error reports, using Form 3500, to FDA MedWatch using one of the following methods:
- Complete and submit the report online: www.fda.gov/medwatch/report.htm
- Complete and submit a postage-paid FDA Form 3500 (https://www.fda.gov/media/76299/download) and return by:
- Mail to MedWatch, 5600 Fishers Lane, Rockville, MD 20852-9787, or
- Fax to 1-800-FDA-0178, or
- Call 1-800-FDA-1088 to request a reporting form
The prescribing healthcare provider and/or the provider's designee is/are responsible for mandatory responses to requests from FDA for information about adverse events and medication errors following receipt of VEKLURY.
*Serious adverse events are defined as:
- Death or a life-threatening adverse event;
- A medical or surgical intervention to prevent death, a life-threatening event, hospitalization, disability, or congenital anomaly;
- Inpatient hospitalization or prolongation of existing hospitalization;
- A persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions; or
- A congenital anomaly/birth defect.
IMPORTANT: When reporting adverse events or medication errors to MedWatch, please complete the entire form with detailed information. It is important that the information reported to FDA be as detailed and complete as possible. Information to include:
- Patient demographics (e.g., patient initials, date of birth)
- Pertinent medical history
- Pertinent details regarding admission and course of illness
- Concomitant medications
- Timing of adverse event(s) in relationship to administration of VEKLURY
- Pertinent laboratory and virology information
- Outcome of the event and any additional follow-up information if it is available at the time of the MedWatch report. Subsequent reporting of follow-up information should be completed if additional details become available.
The following steps are highlighted to provide the necessary information for safety tracking:
- In section A, box 1, provide the patient's initials in the Patient Identifier
- In section A, box 2, provide the patient's date of birth
- In section B, box 5, description of the event:
- Write "Veklury (remdesivir) use for COVID-19 under Emergency Use Authorization (EUA)" as the first line
- Provide a detailed report of medication error and/or adverse event. It is important to provide detailed information regarding the patient and adverse event/medication error for ongoing safety evaluation of this unapproved drug. Please see information to include listed above.
- In section G, box 1, name and address:
- Provide the name and contact information of the prescribing healthcare provider or institutional designee who is responsible for the report.
- Provide the address of the treating institution (NOT the healthcare provider's office address).
9. OTHER REPORTING REQUIREMENTS
In addition, please provide a copy of all FDA MedWatch forms to:
Gilead Global Patient Safety
Fax: 1-650-522-5477
E-mail: Safety_fc@gilead.com
Or call Gilead at 1-800-GILEAD-5 to report adverse events
10. DRUG INTERACTIONS
Due to potential antagonism based on data from cell culture experiments, concomitant use of VEKLURY with chloroquine phosphate or hydroxychloroquine sulfate is not recommended [see Warnings and Precautions (5.3), Microbiology/Resistance Information (15)].
Clinical drug-drug interaction studies have not been performed with VEKLURY.
In vitro, remdesivir is a substrate for drug metabolizing enzyme CYP3A4, and is a substrate for Organic Anion Transporting Polypeptides 1B1 (OATP1B1) and P-glycoprotein (P-gp) transporters. In vitro, remdesivir is an inhibitor of CYP3A4, OATP1B1, OATP1B3, and MATE1. GS-704277 is a substrate for OATP1B1 and OATP1B3. The clinical relevance of these in vitro assessments has not been established.
Remdesivir is not a substrate for CYP1A1, 1A2, 2B6, 2C9, 2C19, or OATP1B3. GS-704277 and GS-441524 are not substrates for CYP1A1, 1A2, 2B6, 2C8, 2C9, 2D6, or 3A5. GS-441524 is also not a substrate for CYP2C19 or 3A4. GS-704277 and GS 441524 are not substrates for OAT1, OAT3, OCT1, OCT2, MATE1, or MATE2K. GS 441524 is also not a substrate for OATP1B1 or OATP1B3.
11. USE IN SPECIFIC POPULATIONS
11.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in individuals exposed to VEKLURY during pregnancy. Pregnant and recently pregnant individuals can go to https://covid-pr.pregistry.com to enroll or call 1-800-616-3791 to obtain information about the registry.
Risk Summary
Available data from published case reports and compassionate use of remdesivir in pregnant women are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In nonclinical reproductive toxicity studies, remdesivir demonstrated no adverse effect on embryo-fetal development when administered to pregnant animals at systemic exposures (AUC) of the predominant circulating metabolite of remdesivir (GS-441524) that were 4 times (rats and rabbits) the exposure in humans at the recommended human dose (RHD) (see Data). There are maternal and fetal risks associated with untreated COVID-19 in pregnancy (see Clinical Considerations).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Animal Data
Remdesivir was administered via intravenous injection to pregnant rats and rabbits (up to 20 mg/kg/day) on Gestation Days 6 through 17, and 7 through 20, respectively, and also to rats from Gestation Day 6 to Lactation/Post-partum Day 20. No adverse effects on embryo-fetal (rats and rabbits) or pre/postnatal (rats) development were observed in rats and rabbits at nontoxic doses in pregnant animals. During organogenesis, exposures to the predominant circulating metabolite (GS-441524) were 4 times higher (rats and rabbits) than the exposure in humans at the RHD. In a pre/postnatal development study, exposures to the predominant circulating metabolite of remdesivir (GS-441524) were similar to the human exposures at the RHD.
11.2 Lactation
Risk Summary
There are no available data on the presence of remdesivir in human milk, the effects on the breastfed infant, or the effects on milk production. In animal studies, remdesivir and metabolites have been detected in the nursing pups of mothers given remdesivir, likely due to the presence of remdesivir in milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for VEKLURY and any potential adverse effects on the breastfed child from VEKLURY or from the underlying maternal condition. Breastfeeding individuals with COVID-19 should follow practices according to clinical guidelines to avoid exposing the infant to COVID-19.
Animal Data
Remdesivir and its metabolites were detected in the plasma of nursing rat pups, likely due to the presence of remdesivir and/or its metabolites in milk, following daily intravenous administration of remdesivir to pregnant rats from Gestation Day 6 to Lactation Day 20. Exposures in nursing pups were approximately 1% that of maternal exposure on Lactation Day 10.
11.3 Pediatric Use
The safety and effectiveness of VEKLURY have not been established in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg, with positive results of direct SARS-CoV-2 viral testing, and who are:
- Hospitalized, or
- Not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
VEKLURY for injection (supplied as 100 mg lyophilized powder in vial) [see Dosage and Administration (2.2, 2.3, 2.4, 2.5)] is the only authorized dosage form of VEKLURY for pediatric patients in this age group.
Use in this age group is based on extrapolation of pediatric efficacy from adequate and well-controlled studies in adults [see Overall Safety Summary (6), Clinical Pharmacology (14), Clinical Trial Results and Supporting Data for EUA (18)].
Pediatric patients (older than 28 days) must have eGFR determined and full-term neonates (at least 7 days to less than or equal to 28 days) must have serum creatinine determined before dosing and daily while receiving VEKLURY. Pediatric patients should be monitored for renal function and consideration given for stopping therapy in the setting of substantial decline [see Dosage and Administration (2.2, 2.4)].
11.4 Renal Impairment
The pharmacokinetics of VEKLURY have not been evaluated in patients with renal impairment. Patients with eGFR greater than or equal to 30 mL/min have received VEKLURY for the treatment of COVID-19 with no dose adjustment of VEKLURY.
Pediatric patients (greater than 28 days old) must have eGFR determined and full-term neonates (at least 7 days to less than or equal to 28 days old) must have serum creatinine determined before dosing and while receiving VEKLURY. VEKLURY is not recommended in pediatric patients (at least 28 days old) with eGFR less than 30 mL/min or in full-term neonates (at least 7 days and less than or equal to 28 days old) with serum creatinine greater than or equal to 1 mg/dL [see Dosage and Administration (2.2, 2.4)].
11.5 Hepatic Impairment
The pharmacokinetics of VEKLURY have not been evaluated in patients with hepatic impairment [see Warnings and Precautions (5.2)].
Perform hepatic laboratory testing in all patients before starting VEKLURY and during treatment as clinically appropriate [see Dosage and Administration (2.2)].
12. OVERDOSAGE
There is no human experience of acute overdosage with VEKLURY. Treatment of overdose with VEKLURY should consist of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient. There is no specific antidote for overdose with VEKLURY.
13. PRODUCT DESCRIPTION
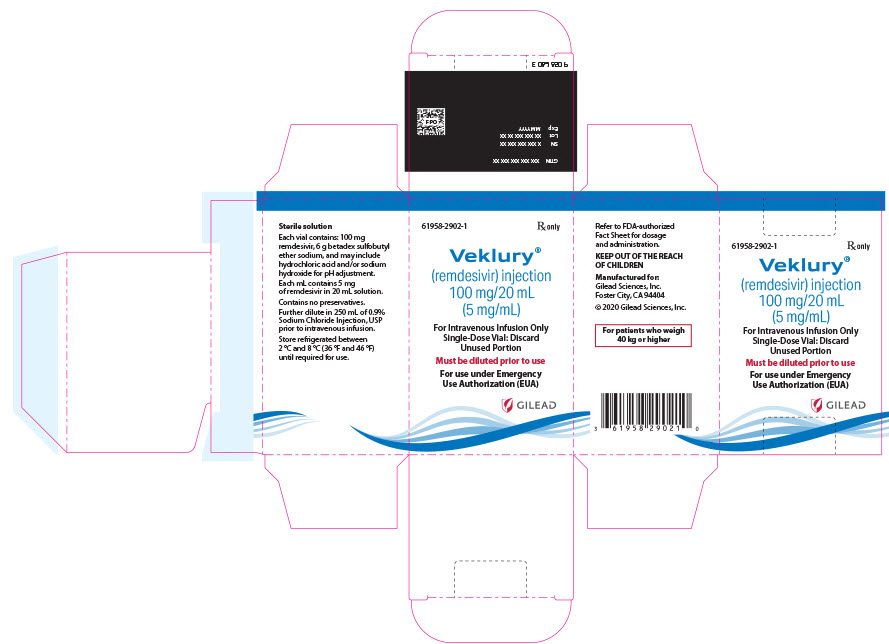
VEKLURY contains remdesivir, a SARS-CoV-2 nucleotide analog RNA polymerase inhibitor. The chemical name for remdesivir is 2-ethylbutyl N-{(S)-[2-C-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-2,5-anhydro-d-altrononitril-6-O-yl]phenoxyphosphoryl}-L-alaninate. It has a molecular formula of C27H35N6O8P and a molecular weight of 602.6 g/mol. Remdesivir has the following structural formula:
13.1 Physical Appearance
VEKLURY for injection contains 100 mg of remdesivir as a sterile, preservative-free lyophilized white to off-white to yellow powder in a single-dose clear glass vial. It requires reconstitution and then further dilution prior to administration by intravenous infusion [see Dosage and Administration (2.5, 2.6)].
14. CLINICAL PHARMACOLOGY
14.1 Mechanism of Action
Remdesivir is an inhibitor of the SARS-CoV-2 RNA-dependent RNA polymerase (RdRp), which is essential for viral replication. Remdesivir is an adenosine nucleotide prodrug that distributes into cells where it is metabolized to a nucleoside monophosphate intermediate by carboxyesterase 1 and/or cathepsin A, depending upon the cell type. The nucleoside monophosphate is subsequently phosphorylated by cellular kinases to form the pharmacologically active nucleoside triphosphate metabolite (GS-443902). Remdesivir triphosphate (RDV TP) acts as an analog of adenosine triphosphate (ATP) and competes with high selectivity (3.65-fold) over the natural ATP substrate for incorporation into nascent RNA chains by the SARS-CoV-2 RNA-dependent RNA polymerase, which results in delayed chain termination (position i+3) during replication of the viral RNA. In a biochemical assay assessing RDV-TP incorporation by the MERS-CoV RdRp complex, RDV-TP inhibited RNA synthesis with an IC50 value of 0.032 µM. RDV-TP can also inhibit viral RNA synthesis following its incorporation into the template viral RNA as a result of read-through by the viral polymerase that may occur at higher nucleotide concentrations. When remdesivir nucleotide is present in the viral RNA template, the efficiency of incorporation of the complementary natural nucleotide is compromised, thereby inhibiting viral RNA synthesis. Remdesivir triphosphate is a weak inhibitor of mammalian DNA and RNA polymerases, including human mitochondrial RNA polymerase.
14.2 Pharmacokinetics
The pharmacokinetic (PK) properties of remdesivir and metabolites have been evaluated in adults in several Phase 1 trials and are provided in Table 12. The multiple dose PK parameters of remdesivir and metabolites in healthy adults are provided in Table 13.
| Remdesivir | GS-441524 | GS-704277 | |
|---|---|---|---|
| ND=not detected | |||
|
|
|||
| Absorption | |||
| Tmax (h)* | 0.67–0.68 | 1.51–2.00 | 0.75–0.75 |
| Distribution | |||
| % bound to human plasma proteins | 88–93.6† | 2 | 1 |
| Blood-to-plasma ratio | 0.68–1.0 | 1.19 | 0.56 |
| Elimination | |||
| t1/2 (h)‡ | 1 | 27 | 1.3 |
| Metabolism | |||
| Metabolic pathway(s) | CES1 (80%) Cathepsin A (10%) CYP3A (10%) | Not significantly metabolized | HINT1 |
| Excretion | |||
| Major route of elimination | Metabolism | Glomerular filtration and active tubular secretion | Metabolism |
| % of dose excreted in urine§ | 10 | 49 | 2.9 |
| % of dose excreted in feces§ | ND | 0.5 | ND |
| Parameter Mean (CV%) | Remdesivir | GS-441524 | GS-704277 |
|---|---|---|---|
| CV=Coefficient of Variation; ND=Not detectable (at 24 hours post-dose) | |||
|
|
|||
| Cmax
(nanogram per mL) | 2229 (19.2) | 145 (19.3) | 246 (33.9) |
| AUCtau
(nanogram∙h per mL) | 1585 (16.6) | 2229 (18.4) | 462 (31.4) |
| Ctrough
(nanogram per mL) | ND | 69.2 (18.2) | ND |
Specific Populations
Pharmacokinetic differences based on sex, race, and age have not been evaluated.
The pharmacokinetics of VEKLURY in pediatric patients have not been evaluated.
Using modeling and simulation, the recommended dosing regimen is expected to result in comparable steady-state plasma exposures of remdesivir and metabolites in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg as observed in healthy adults.
15. MICROBIOLOGY/RESISTANCE INFORMATION
Antiviral Activity
Remdesivir exhibited cell culture antiviral activity against a clinical isolate of SARS-CoV-2 in primary human airway epithelial (HAE) cells with a 50% effective concentration (EC50) of 9.9 nM after 48 hours of treatment. Remdesivir inhibited the replication of SARS-CoV-2 in the continuous human lung epithelial cell lines Calu-3 and A549-hACE2 with EC50 values of 280 nM after 72 hours of treatment and 115 nM after 48 hours of treatment, respectively.
Remdesivir EC50 values for SARS-CoV-2 in A549-hACE2 cells were not different when combined with chloroquine phosphate or hydroxychloroquine sulfate at concentrations up to 2.5 μM. In a separate study, the antiviral activity of remdesivir was antagonized by chloroquine phosphate in a dose-dependent manner when the two drugs were co-incubated at clinically relevant concentrations in HEp-2 cells infected with respiratory syncytial virus (RSV). Higher remdesivir EC50 values were observed with increasing concentrations of chloroquine phosphate. Increasing concentrations of chloroquine phosphate or hydroxychloroquine sulfate reduced formation of remdesivir triphosphate in A549-hACE2, HEp-2, and normal human bronchial epithelial cells.
Based on cell culture susceptibility testing by plaque assay and/or N protein ELISA assay, remdesivir retained similar antiviral activity (≤1.5-fold change) against clinical isolates of SARS-CoV-2 variants containing the P323L substitution in the viral polymerase including the Alpha (B.1.1.7), Beta (B.1.351), Delta (B.1.617.2), Gamma (P.1), and Epsilon (B.1.429) variants compared to earlier lineage SARS-CoV-2 (lineage A) isolates.
SARS-CoV-2 RNA shedding results from GS-US-540-5776 (ACTT-1) indicate that remdesivir does not significantly reduce the amount of detectable SARS-CoV-2 RNA in oropharyngeal or nasopharyngeal swabs or plasma samples in hospitalized patients compared to placebo, and SARS-CoV-2 RNA shedding results from GS-US-540-9012 indicate that remdesivir does not significantly reduce the amount of detectable SARS-CoV-2 RNA in nasopharyngeal swabs in non-hospitalized patients compared to placebo.
Resistance
SARS-CoV-2 isolates with reduced susceptibility to remdesivir have been selected in cell culture. In a selection with GS-441524, the parent nucleoside of remdesivir, virus pools emerged expressing amino acid substitutions at V166A, N198S, S759A, V792I, C799F, and C799R in the viral RNA-dependent RNA polymerase (nsp12). When these substitutions were individually introduced into a wild-type recombinant virus by site-directed mutagenesis, 1.7- to 3.5-fold reductions in susceptibility to remdesivir were observed. In a cell culture resistance selection experiment with remdesivir, nsp12 amino acid substitution E802D emerged, resulting in a 2.5-fold reduction in susceptibility to remdesivir. In another selection with remdesivir using a SARS-CoV-2 isolate containing the P323L substitution in the viral polymerase, a single amino acid substitution at V166L emerged. Recombinant SARS-CoV-2 with substitutions at P323L alone or P323L+V166L in combination exhibited 1.3- and 1.5-fold reductions in remdesivir susceptibility, respectively.
Cell culture resistance profiling of remdesivir using the rodent CoV murine hepatitis virus identified two substitutions (F476L and V553L) in the viral RNA-dependent RNA polymerase at residues conserved across CoVs. Introduction of the corresponding substitutions (F480L and V557L) into SARS-CoV resulted in 6-fold reduction in susceptibility to remdesivir in cell culture and attenuated SARS-CoV pathogenesis in a mouse model. When individually introduced into a SARS-CoV-2 recombinant virus, the corresponding substitutions at F480L and V557L each conferred 2-fold reduced susceptibility to remdesivir. When individually introduced into a SARS-CoV-2 recombinant virus, the corresponding substitutions at F480L and V557L each conferred 2-fold reduced susceptibility to remdesivir.
SARS-CoV-2 nsp12 E802D substitution has emerged in one individual treated with remdesivir. The E802D substitution resulted in a 2.5-fold increase in the remdesivir EC50 value.
16. NONCLINICAL TOXICOLOGY
Carcinogenesis
Given the short-term administration of VEKLURY for the treatment of COVID-19, long-term animal studies to evaluate the carcinogenic potential of remdesivir were not conducted.
Mutagenesis
Remdesivir was not genotoxic in a battery of assays, including bacterial mutagenicity, chromosome aberration using human peripheral blood lymphocytes, and in vivo rat micronucleus assays.
Impairment of Fertility
Nonclinical toxicity studies in rats demonstrated no adverse effect on male fertility at exposures of the predominant circulating metabolite (GS-441524) approximately 2 times the exposure in humans at the RHD.
Reproductive toxicity, including decreases in corpora lutea, numbers of implantation sites, and viable embryos, was seen when remdesivir was administered intravenous daily at a systemically toxic dose (10 mg/kg) in female rats 14 days prior to mating and during conception; exposures of the predominant circulating metabolite (GS-441524) were 1.3 times the exposure in humans at the RHD.
Animal Toxicology and/or Pharmacology
Intravenous administration (slow bolus) of remdesivir to male rhesus monkeys at dosage levels of 5, 10, and 20 mg/kg/day for 7 days resulted, at all dose levels, in increased mean urea nitrogen and increased mean creatinine, renal tubular atrophy, and basophilia and casts.
Intravenous administration (slow bolus) of remdesivir to rats at dosage levels of ≥3 mg/kg/day for up to 4 weeks resulted in findings indicative of kidney injury and/or dysfunction.
Kidney-related effects in rats and monkeys were observed at exposures of the predominant circulating metabolite (GS-441524) that are lower than the exposure in humans at the RHD.
17. ANIMAL PHARMACOLOGIC AND EFFICACY DATA
- Remdesivir exhibited cell culture antiviral activity against a clinical isolate of SARS-CoV-2 in primary HAE cells (EC50 value= 9.9 nM) after 48 hours of treatment. Remdesivir inhibited the replication of SARS-CoV-2 in the continuous human lung epithelial cell line Calu-3 with an EC50 value of 280 nM after 72 hours of treatment.
- Remdesivir showed antiviral activity in SARS-CoV-2-infected rhesus monkeys. Administration of remdesivir at 10/5 mg/kg (10 mg/kg first dose, followed by 5 mg/kg once daily thereafter) using IV bolus injection initiated 12 hours post-inoculation with SARS-CoV-2 resulted in a reduction in clinical signs of respiratory disease, lung pathology and gross lung lesions, and lung viral RNA levels compared with vehicle-treated animals.
18. CLINICAL TRIAL RESULTS AND SUPPORTING DATA FOR EUA
VEKLURY is an antiviral drug with available data from four randomized clinical trials in adult patients with COVID-19. VEKLURY is approved for use to treat COVID-19 in adults and pediatric patients 12 years of age and older and weighing at least 40 kg. VEKLURY is not approved for use in pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg.
NIAID ACTT-1 Study in Hospitalized Subjects with Mild/Moderate and Severe COVID-19
A randomized, double-blind, placebo-controlled clinical trial (ACTT-1, NCT04280705) of hospitalized adult subjects with confirmed SARS-CoV-2 infection and mild, moderate, or severe COVID-19 compared treatment with VEKLURY for 10 days (n=541) with placebo (n=521). Mild/moderate disease was defined as SpO2 >94% and respiratory rate <24 breaths/minute without supplemental oxygen; severe disease was defined as an SpO2 ≤94% on room air, a respiratory rate ≥24 breaths/minute, an oxygen requirement, or a requirement for mechanical ventilation. Subjects had to have at least one of the following to be enrolled in the trial: radiographic infiltrates by imaging, SpO2 ≤94% on room air, a requirement for supplemental oxygen, or a requirement for mechanical ventilation. Subjects treated with VEKLURY received 200 mg on Day 1 and 100 mg once daily on subsequent days, for 10 days of treatment via intravenous infusion. Treatment with VEKLURY was stopped in subjects who were discharged from the hospital prior to the completion of 10 days of treatment.
At baseline, mean age was 59 years (with 36% of subjects aged 65 or older); 64% of subjects were male, 53% were White, 21% were Black, and 13% were Asian; 24% were Hispanic or Latino; 105 subjects had mild/moderate disease (10% in both treatment groups); 957 subjects had severe disease (90% in both treatment groups). Subjects in this trial were unvaccinated. A total of 285 subjects (27%) (n=131 received VEKLURY) were on invasive mechanical ventilation or ECMO. The most common comorbidities were hypertension (51%), obesity (45%), and type 2 diabetes mellitus (31%); the distribution of comorbidities was similar between the two treatment groups.
The primary clinical endpoint was time to recovery within 29 days after randomization. Recovery was defined as discharged from the hospital without limitations on activities, discharged from the hospital with limitations on activities and/or requiring home oxygen, or hospitalized but not requiring supplemental oxygen and no longer requiring ongoing medical care. The median time to recovery was 10 days in the VEKLURY group compared to 15 days in the placebo group (recovery rate ratio 1.29 [95% CI 1.12 to 1.49], p<0.001). Among subjects with mild/moderate disease at enrollment (n=105), the median time to recovery was 5 days in both the VEKLURY and placebo groups (recovery rate ratio 1.22 [95% CI 0.82 to 1.81]). Among subjects with severe disease at enrollment (n=957), the median time to recovery was 11 days in the VEKLURY group compared to 18 days in the placebo group (recovery rate ratio 1.31 [95% CI 1.12 to 1.52]).
A key secondary endpoint was clinical status on Day 15 assessed on an 8-point ordinal scale consisting of the following categories:
- not hospitalized, no limitations on activities;
- not hospitalized, limitation on activities and/or requiring home oxygen;
- hospitalized, not requiring supplemental oxygen - no longer requires ongoing medical care;
- hospitalized, not requiring supplemental oxygen - requiring ongoing medical care (COVID-19 related or otherwise);
- hospitalized, requiring supplemental oxygen;
- hospitalized, on noninvasive ventilation or high-flow oxygen devices;
- hospitalized, on invasive mechanical ventilation or ECMO; and
- death.
Overall, the odds of improvement in the ordinal scale were higher in the VEKLURY group at Day 15 when compared to the placebo group (odds ratio 1.54 [95% CI 1.25 to 1.91]).
Overall, 29-day mortality was 11% for the VEKLURY group vs 15% for the placebo group (hazard ratio 0.73 [95% CI 0.52 to 1.03]).
Study GS-US-540-5773 in Hospitalized Subjects with Severe COVID-19
A randomized, open-label multi-center clinical trial (Study 5773, NCT04292899) in adult subjects with confirmed SARS-CoV-2 infection, an SpO2 of ≤94% on room air, and radiological evidence of pneumonia compared 200 subjects who received VEKLURY for 5 days with 197 subjects who received VEKLURY for 10 days. Treatment with VEKLURY was stopped in subjects who were discharged from the hospital prior to completion of their protocol-defined duration of treatment. Subjects on mechanical ventilation at screening were excluded. All subjects received 200 mg of VEKLURY on Day 1 and 100 mg once daily on subsequent days via intravenous infusion, plus standard of care.
At baseline, the median age of subjects was 61 years (range, 20 to 98 years); 64% were male, 75% were White, 12% were Black, and 12% were Asian; 22% were Hispanic or Latino. More subjects in the 10-day group than the 5-day group required invasive mechanical ventilation or ECMO (5% vs 2%), or high-flow oxygen support (30% vs 25%) at baseline. Subjects in this trial were unvaccinated. Median duration of symptoms and hospitalization prior to first dose of VEKLURY were similar across treatment groups.
The primary endpoint was clinical status on Day 14 assessed on a 7-point ordinal scale consisting of the following categories:
- death;
- hospitalized, receiving invasive mechanical ventilation or ECMO;
- hospitalized, receiving noninvasive ventilation or high-flow oxygen devices;
- hospitalized, requiring low-flow supplemental oxygen;
- hospitalized, not requiring supplemental oxygen but receiving ongoing medical care (related or not related to COVID-19);
- hospitalized, requiring neither supplemental oxygen nor ongoing medical care (other than that specified in the protocol for remdesivir administration); and
- not hospitalized.
Overall, after adjusting for between-group differences at baseline, subjects receiving a 5-day course of VEKLURY had similar clinical status at Day 14 as those receiving a 10-day course (odds ratio for improvement 0.75 [95% CI 0.51 to 1.12]). There were no statistically significant differences in recovery rates or mortality rates in the 5-day and 10-day groups once adjusted for between-group differences at baseline. All-cause mortality at Day 28 was 12% vs 14% in the 5- and 10-day treatment groups, respectively.
Study GS-US-540-5774 in Hospitalized Subjects with Moderate COVID-19
A randomized, open-label multi-center clinical trial (Study 5774, NCT04292730) of hospitalized adult subjects with confirmed SARS-CoV-2 infection, SpO2 >94% and radiological evidence of pneumonia compared treatment with VEKLURY for 5 days (n=191) and treatment with VEKLURY for 10 days (n=193) with standard of care (n=200). Treatment with VEKLURY was stopped in subjects who were discharged from the hospital prior to completion of their protocol-defined duration of treatment. Subjects treated with VEKLURY received 200 mg on Day 1 and 100 mg once daily on subsequent days via intravenous infusion.
At baseline, the median age of subjects was 57 years (range, 12 to 95 years); 61% were male, 61% were White, 19% were Black, and 19% were Asian; 18% were Hispanic or Latino. Subjects in this trial were unvaccinated. Baseline clinical status, oxygen support status, and median duration of symptoms and hospitalization prior to first dose of VEKLURY were similar across treatment groups.
The primary endpoint was clinical status on Day 11 assessed on a 7-point ordinal scale consisting of the following categories:
- death;
- hospitalized, receiving invasive mechanical ventilation or ECMO;
- hospitalized, receiving noninvasive ventilation or high-flow oxygen devices;
- hospitalized, requiring low-flow supplemental oxygen;
- hospitalized, not requiring supplemental oxygen but receiving ongoing medical care (related or not related to COVID-19);
- hospitalized, requiring neither supplemental oxygen nor ongoing medical care (other than that specified in the protocol for remdesivir administration); and
- not hospitalized.
Overall, the odds of improvement in the ordinal scale were higher in the 5-day VEKLURY group at Day 11 when compared to those receiving only standard of care (odds ratio 1.65 [95% CI 1.09 to 2.48], p=0.017). The odds of improvement in clinical status with the 10-day treatment group when compared to those receiving only standard of care were not statistically significant (odds ratio 1.31 [95% CI 0.88 to 1.95]). All-cause mortality at Day 28 was ≤2% in all treatment groups.
Study GS-US-540-9012 in Non-Hospitalized Subjects with Mild-to-Moderate COVID-19 and at High Risk for Progression to Severe Disease
A randomized, double-blind, placebo-controlled, clinical trial (Study 9012, NCT04501952) evaluated VEKLURY 200 mg once daily for 1 day followed by VEKLURY 100 mg once daily for 2 days (for a total of 3 days of intravenously administered therapy) in 554 adult and 8 pediatric subjects (12 years of age and older and weighing at least 40 kg) who were non-hospitalized, had mild-to-moderate COVID-19, were symptomatic for COVID-19 for ≤7 days, had confirmed SARS-CoV-2 infection, and had at least one risk factor for progression to hospitalization. Risk factors for progression to hospitalization included age ≥60 years, obesity (BMI ≥30), chronic lung disease, hypertension, cardiovascular or cerebrovascular disease, diabetes mellitus, immunocompromised state, chronic mild or moderate kidney disease, chronic liver disease, current cancer, and sickle cell disease. Subjects who received, required, or were expected to require supplemental oxygen were excluded from the trial. Subjects were randomized in a 1:1 manner, stratified by residence in a skilled nursing facility (yes/no), age (<60 vs ≥60 years), and region (US vs ex-US) to receive VEKLURY (n=279) or placebo (n=283), plus standard of care.
At baseline, mean age was 50 years (with 30% of subjects aged 60 or older); 52% were male, 80% were White, 8% were Black, and 2% were Asian; 44% were Hispanic or Latino; median body mass index was 30.7 kg/m2. Subjects in this trial were unvaccinated. VEKLURY or placebo was first administered to subjects in outpatient facilities (84%), home healthcare settings (13%), or skilled nursing facilities (3%). The most common comorbidities were diabetes mellitus (62%), obesity (56%), and hypertension (48%). Median (Q1, Q3) duration of symptoms prior to treatment was 5 (3, 6) days; median viral load was 6.3 log10 copies/mL at baseline. The baseline demographics and disease characteristics were well balanced across the VEKLURY and placebo treatment groups.
The primary endpoint was the proportion of subjects with COVID-19 related hospitalization (defined as at least 24 hours of acute care) or all-cause mortality through Day 28. Events occurred in 2 (0.7%) subjects treated with VEKLURY compared to 15 (5.3%) subjects concurrently randomized to placebo (hazard ratio 0.134 [95% CI 0.031 to 0.586]; p=0.0076). No deaths were observed through Day 28.
19. HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
VEKLURY for injection, 100 mg, is supplied as a single-dose vial containing a sterile, preservative-free white to off-white to yellow lyophilized powder. It requires reconstitution and further dilution prior to administration by intravenous infusion [see Dosage and Administration (2.5)].
Discard unused portion.
The container closure is not made with natural rubber latex.
Storage and Handling
Do not reuse or save reconstituted or diluted VEKLURY for future use. This product contains no preservative; therefore, partially used vials should be discarded [see Dosage and Administration (2.6)].
Store VEKLURY for injection, 100 mg, vials below 30°C (below 86°F) until required for use.
After reconstitution, use vials immediately to prepare diluted solution. Dilute the reconstituted solution in 0.9% sodium chloride injection, USP within the same day as administration.
The diluted VEKLURY solution in syringe should be used immediately.
The diluted VEKLURY solution in the infusion bags can be stored up to 24 hours at room temperature (20°C to 25°C [68°F to 77°F]) or 48 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]) prior to administration.
20. PATIENT COUNSELING INFORMATION
SEE Fact Sheet for Parents and Caregivers
Hypersensitivity Reactions
Inform parents/caregivers that hypersensitivity reactions have been seen in patients receiving VEKLURY during and after infusion. Advise parents/caregivers to inform their healthcare provider if their child experiences any of the following: changes in heart rate; fever; shortness of breath, wheezing; swelling of the lips, face, or throat; rash; nausea; sweating; or shivering [see Warnings and Precautions (5.1)].
Increased Risk of Transaminase Elevations
Inform parents/caregivers that VEKLURY may increase the risk of hepatic laboratory abnormalities. Advise parents/caregivers to alert their healthcare provider immediately if their child experiences any symptoms of liver inflammation [see Warnings and Precaution (5.2)].
Drug Interactions
Inform parents/caregivers that VEKLURY may interact with other drugs. Advise patients to report to their healthcare provider the use of any other prescription or nonprescription medication or herbal products, including chloroquine phosphate or hydroxychloroquine sulfate [see Warnings and Precautions (5.3), Drug Interactions (10), Microbiology/Resistance Information (15)].
Pregnancy Registry
Advise patients that there is a pregnancy registry that monitors pregnancy outcomes in individuals exposed to VEKLURY during pregnancy [see Use in Specific Populations (11.1)].
Pregnancy
Inform patients to notify their healthcare provider immediately in the event of a pregnancy [see Use in Specific Populations (11.1)].
Lactation
Inform mothers that it is not known whether VEKLURY can pass into their breast milk [see Use in Specific Populations (11.2)].
21. CONTACT INFORMATION
If you have questions, please contact
www.askgileadmedical.com
1-866-633-4474
© 2022 Gilead Sciences, Inc. All rights reserved.
Revised: 01/2022
Fact Sheet for Parents and Caregivers
Emergency Use Authorization (EUA) of VEKLURY® (remdesivir) for Coronavirus Disease 2019 (COVID-19) for Children Weighing 8 pounds (3.5 kg) to Less Than 88 pounds (40 kg) or for Children Less Than 12 Years of Age Weighing at least 8 pounds (3.5 kg) who are: hospitalized, or not hospitalized and have mild-to-moderate COVID-19 and are at high risk for progression to severe COVID-19, including hospitalization or death
You are being given this Fact Sheet because your healthcare provider believes it is necessary to provide your child with VEKLURY for use for the treatment of coronavirus disease 2019 (COVID-19). The United States Food and Drug Administration (FDA) has issued an Emergency Use Authorization (EUA) for VEKLURY for use in children weighing 8 pounds (3.5 kg) to less than 88 pounds (40 kg) or children less than 12 years of age weighing at least 8 pounds (3.5 kg) with positive results of direct severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral testing, who are:
- hospitalized, or
- not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
This Fact Sheet contains information to help you understand the risks and benefits of your child receiving VEKLURY.
The FDA has issued an EUA to make VEKLURY available for this use during the COVID-19 pandemic (for more details about an EUA please see "What is an Emergency Use Authorization?" at the end of this document). VEKLURY is not approved for use as treatment for COVID-19 for the pediatric population covered under this EUA. Read this Fact Sheet for information about VEKLURY. Talk to your healthcare provider about your options or if you have any questions. It is your choice for your child to receive VEKLURY or stop it at any time.
What is COVID-19?
COVID-19 is caused by a virus called a coronavirus. You can get COVID-19 through close contact with another person who has the virus.
COVID-19 illnesses have ranged from very mild to severe, including illness with no reported symptoms and illness resulting in death. While information so far suggests that most COVID-19 illness is mild, serious illness can happen and may cause some of a child's other medical conditions to become worse. Older people and people of all ages with severe, long-lasting (chronic) medical conditions like heart disease, lung disease, and diabetes, for example, seem to be at higher risk of being hospitalized for COVID-19.
What is VEKLURY?
VEKLURY is a prescription medicine that is investigational for use for the treatment of COVID-19 in children weighing 8 pounds (3.5 kg) to less than 88 pounds (40 kg) or children less than 12 years of age weighing at least 8 pounds (3.5 kg) with positive results of direct SARS-CoV-2 viral testing, who are:
- hospitalized, or
- not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
VEKLURY is investigational for this use because it is still being studied and there is limited information about the safety and effectiveness of using VEKLURY for the treatment of COVID-19 in this population.
VEKLURY is an FDA-approved prescription medicine used to treat COVID-19 in adults and children (12 years of age and older and weighing at least 88 pounds (40 kg), with positive results of direct SARS-CoV-2 viral testing, who are:
- hospitalized, or
- not hospitalized and have mild-to-moderate COVID-19, and at high risk for progression to severe COVID-19, including hospitalization or death.
What should I tell my healthcare provider before my child receives VEKLURY?
Tell your healthcare provider about all of your child's medical conditions, including if your child:
- Has any allergies
- Has kidney or liver disease
- Has any serious illnesses
Tell your healthcare provider about all the medicines your child takes, including prescription and over-the-counter medicines, vitamins, and herbal supplements. VEKLURY may interact with other medicines and may cause serious side effects.
Especially tell your healthcare provider if your child is taking the medicines chloroquine phosphate or hydroxychloroquine sulfate.
How will my child receive VEKLURY?
- Hospitalized: VEKLURY is given to your child through a vein by intravenous (IV) infusion one time each day for up to 10 days. Your healthcare provider will decide how many doses your child needs.
- Not hospitalized: VEKLURY is given to your child through a vein by intravenous (IV) infusion one time each day for 3 days.
- Your healthcare provider will do certain blood tests before starting and during treatment with VEKLURY.
Who should generally not receive VEKLURY?
Your child should not receive VEKLURY if your child is allergic to remdesivir or any of the ingredients in VEKLURY.
What are the important possible side effects of VEKLURY?
Possible side effects of VEKLURY are:
-
Allergic reactions. Allergic reactions can happen during and after infusion with VEKLURY. Your healthcare provider will monitor your child for signs and symptoms of allergic reactions during their infusion and for at least 1 hour after their infusion. Tell your healthcare provider right away if your child gets any of the following signs and symptoms of allergic reactions:
- changes to heart rate
- fever
- shortness of breath or wheezing
- shivering
- swelling of the lips, face, or throat
- rash
- nausea
- sweating
- Increases in levels of liver enzymes. Increases in liver enzymes are common in people who have received VEKLURY and may be a sign of liver injury. Your healthcare provider will do blood tests to check your child's liver enzymes before receiving VEKLURY and as needed while receiving VEKLURY. Your healthcare provider may stop treatment with VEKLURY if your child develops liver problems.
The most common side effect of VEKLURY is nausea.
These are not all the possible side effects of VEKLURY. VEKLURY is still being studied so it is possible that all of the risks are not known at this time.
What other treatment choices are there?
Like VEKLURY, FDA may allow for the emergency use of other medicines to treat people with COVID-19. Go to https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization for information on the emergency use of other medicines that are not approved by the FDA to treat people with COVID-19. Your healthcare provider may talk with you about clinical trials your child may be eligible for.
It is your choice for your child to be treated or not to be treated with VEKLURY. Should you decide for your child not to receive it, it will not change your child's standard medical care.
How do I report side effects with VEKLURY?
Contact your healthcare provider if your child has any side effect that bothers them or does not go away.
Report side effects to FDA MedWatch at www.fda.gov/medwatch or call 1-800-FDA-1088 and to Gilead by calling 1-800-445-3235.
How can I learn more about COVID-19?
- Ask your healthcare provider.
- Visit https://www.cdc.gov/COVID19.
- Contact your local or state public health department.
What is an Emergency Use Authorization (EUA)?
The United States FDA has made VEKLURY available under an emergency access mechanism called an Emergency Use Authorization (EUA) for the treatment of coronavirus disease 2019 (COVID-19) in children weighing 8 pounds (3.5 kg) to less than 88 pounds (40 kg) or children less than 12 years of age weighing at least 8 pounds (3.5 kg), with positive results of direct severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral testing, who are:
- hospitalized, or
- not hospitalized and have mild-to-moderate COVID-19, and are at high risk for progression to severe COVID-19, including hospitalization or death.
The EUA is supported by a Secretary of Health and Human Service (HHS) declaration that circumstances exist to justify the emergency use of drugs and biological products during the COVID-19 pandemic.
VEKLURY for the authorized use has not undergone the same type of review as an FDA-approved product. In issuing an EUA under the COVID-19 public health emergency, the FDA has determined, among other things, that based on the total amount of scientific evidence available including data from adequate and well controlled clinical trials, it is reasonable to believe that the product may be effective for diagnosing, treating, or preventing COVID-19, or a serious or life threatening disease or condition caused by COVID-19; that the known and potential benefits of the product, when used to diagnose, treat, or prevent such disease or condition, outweigh the known and potential risks of such product; and that there are no adequate, approved, and available alternatives.
All of these criteria must be met to allow for the product to be used in the treatment of the authorized patient population during the COVID-19 pandemic. The EUA for VEKLURY is in effect for the duration of the COVID-19 declaration justifying emergency use of VEKLURY, unless terminated or revoked (after which VEKLURY may no longer be used under the EUA).
© 2022 Gilead Sciences, Inc. All rights reserved.
Revised: 01/2022
PRINCIPAL DISPLAY PANEL - 100 mg/20 mL Vial Label
remdesivir injection
100 mg/20 mL
(5 mg/mL)
For Intravenous Use Only
Single-Dose Vial: Discard
Unused Portion
For use under Emergency
Use Authorization (EUA)
90286902
Rx only

PRINCIPAL DISPLAY PANEL - 100 mg/20 mL Vial Carton
61958-2902-1
Rx only
remdesivir
injection
100 mg/20 mL
(5 mg/mL)
For Intravenous Use Only
Single-Dose Vial: Discard
Unused Portion
For use under Emergency
Use Authorization (EUA)
Each mL contains 5 mg
of remdesivir in 20 mL solution
GILEAD



| VEKLURY
remdesivir injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| VEKLURY
remdesivir injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Gilead Sciences, Inc. (185049848) |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.