VANRAFIA- atrasentan tablet, film coated
VANRAFIA by
Drug Labeling and Warnings
VANRAFIA by is a Prescription medication manufactured, distributed, or labeled by Novartis Pharmaceuticals Corporation. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VANRAFIA safely and effectively. See full prescribing information for VANRAFIA.
VANRAFIA™ (atrasentan) tablets, for oral use
Initial U.S. Approval: 2025
WARNING: EMBRYO-FETAL TOXICITY
See full prescribing information for complete boxed warning.
- VANRAFIA may cause major birth defects if used during pregnancy (4.1, 5.1, 8.1)
- Exclude pregnancy before start of treatment. (2.1, 4.1, 5.1, 8.3)
- Use effective contraception before start of treatment, during treatment and two weeks after treatment. (4.1, 5.1, 8.3)
- Discontinue VANRAFIA if pregnancy occurs. (4.1, 5.1)
INDICATIONS AND USAGE
VANRAFIA is an endothelin receptor antagonist indicated to reduce proteinuria in adults with primary immunoglobulin A nephropathy (IgAN) at risk of rapid disease progression, generally a urine protein-to-creatinine ratio (UPCR) ≥ 1.5 g/g. (1)
This indication is approved under accelerated approval based on a reduction of proteinuria. It has not been established whether VANRAFIA slows kidney function decline in patients with IgAN. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory clinical trial. (1)
DOSAGE AND ADMINISTRATION
- 0.75 mg orally once daily with or without food (2.2)
DOSAGE FORMS AND STRENGTHS
- Tablets: 0.75 mg (3)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥ 5%) were peripheral edema and anemia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
- Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 4/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: EMBRYO-FETAL TOXICITY
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Pregnancy Testing
2.2 Recommended Dosage
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Pregnancy
4.2 Hypersensitivity
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
5.2 Hepatotoxicity
5.3 Fluid Retention
5.4 Decreased Sperm Counts
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on VANRAFIA
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 IgA Nephropathy
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: EMBRYO-FETAL TOXICITY
VANRAFIA is contraindicated for use in pregnant patients; it may cause major birth defects based on animal data [see Contraindications (4.1), Warnings and Precautions (5.1), Use in Specific Populations (8.1)]. Exclude pregnancy prior to initiation of treatment with VANRAFIA. Advise use of effective contraception before the initiation of treatment, during treatment, and for two weeks after discontinuation of treatment with VANRAFIA. Stop VANRAFIA as soon as possible if the patient becomes pregnant [see Dosage and Administration (2.1), Contraindications (4.1), Warnings and Precautions (5.1), Use in Specific Populations (8.1, 8.3)].
-
1 INDICATIONS AND USAGE
VANRAFIA is indicated to reduce proteinuria in adults with primary immunoglobulin A nephropathy (IgAN) at risk of rapid disease progression, generally a urine protein-to-creatinine ratio (UPCR) ≥ 1.5 g/g.
This indication is approved under accelerated approval based on a reduction of proteinuria [see Clinical Studies (14.1)]. It has not been established whether VANRAFIA slows kidney function decline in patients with IgAN. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory clinical trial.
-
2 DOSAGE AND ADMINISTRATION
2.1 Pregnancy Testing
Exclude pregnancy before initiating VANRAFIA [see Warnings and Precautions (5.1), Use in Specific Populations (8.1, 8.3)].
2.2 Recommended Dosage
The recommended dose of VANRAFIA is 0.75 mg administered orally once daily with or without food [see Clinical Pharmacology (12.3)].
Swallow tablets whole. Do not cut, crush, or chew.
If a dose or doses are missed, take the prescribed dose at the next scheduled time. Do not double the dose to make up for a missed dose.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
Based on data from animal reproduction studies, VANRAFIA may cause fetal harm when administered to a pregnant patient and is contraindicated during pregnancy. The available human data for endothelin receptor antagonists do not establish the presence or absence of major birth defects related to the use of VANRAFIA. Counsel patients who can become pregnant of the potential risk to a fetus. Exclude pregnancy prior to initiation of treatment with VANRAFIA. Advise patients to use effective contraception prior to initiation of treatment, during treatment, and for two weeks after discontinuation of treatment with VANRAFIA [see Dosage and Administration (2.1) and Use in Specific Populations (8.1, 8.3)]. When pregnancy is detected, discontinue VANRAFIA as soon as possible [see Dosage and Administration (2.1), Contraindications (4.1), Use in Specific Populations (8.1, 8.3)].
5.2 Hepatotoxicity
Some endothelin receptor antagonists (ERAs) have caused elevations of aminotransferases, hepatotoxicity, and liver failure. Asymptomatic and transient transaminase elevations have been observed with VANRAFIA [see Adverse Reactions (6.1)]. Obtain liver enzyme testing before initiating VANRAFIA and repeat during treatment as clinically indicated. In patients with elevated aminotransferases at baseline (>3 × upper limit of normal [ULN]), consider periodic liver test monitoring. Do not initiate VANRAFIA in patients with severe hepatic impairment.
Advise patients to report symptoms suggesting hepatic injury (nausea, vomiting, right upper quadrant pain, fatigue, anorexia, jaundice, dark urine, fever, or itching). If clinically relevant aminotransferase elevations occur, or if elevations are accompanied by an increase in bilirubin >2 x ULN, or by clinical symptoms of hepatotoxicity, discontinue VANRAFIA. Consider re-initiation of VANRAFIA when hepatic enzyme levels normalize in patients who have not experienced clinical symptoms of hepatotoxicity or jaundice.
5.3 Fluid Retention
Fluid retention may occur with ERAs and has been observed in clinical studies with VANRAFIA [see Adverse Reactions (6.1)]. VANRAFIA has not been evaluated in IgAN patients with heart failure. If clinically significant fluid retention develops, consider initiating or increasing diuretic treatment and interrupting VANRAFIA treatment.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Embryo-fetal Toxicity [see Warnings and Precautions (5.1)]
- Hepatotoxicity [see Warnings and Precautions (5.2)]
- Fluid Retention [see Warnings and Precautions (5.3)]
- Decreased Sperm Counts [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of VANRAFIA was evaluated in ALIGN (NCT04573478), a randomized, double-blind, placebo controlled clinical study in 403 adults with IgAN [see Clinical Studies (14.1)]. The median duration of treatment was 47 weeks (range: 0 to 128 weeks). The most common adverse reactions (≥ 5%) with VANRAFIA were peripheral edema and anemia. Table 1 describes the adverse reactions that occurred in ≥ 2% of patients treated with VANRAFIA and higher than placebo in the ALIGN study.
Table 1: Adverse Reactions Reported in ≥ 2% of Adult Patients with IgAN Treated with VANRAFIA and Higher than Placebo in ALIGN *Includes related terms
**Elevations in ALT or AST > 3-fold upper limit of normal (ULN)Adverse Reaction VANRAFIA
(N = 201)
n (%)Placebo
(N = 202)
n (%)Peripheral edema* 21 (10%) 14 (7%) Anemia* 12 (6%) 2 (1%) Liver transaminase elevation** 4 (2%) 2 (1%) Laboratory Tests and Vital Signs
Hemoglobin Decrease
At Week 36, the mean change in hemoglobin from baseline for those patients receiving VANRAFIA in the ALIGN study was -0.7 g/dL compared to -0.2 g/dL for those receiving placebo. The incidence of a hemoglobin decrease > 2 g/dL compared to baseline and below the lower limit of normal was greater for the VANRAFIA arm (12%) compared to the placebo arm (4%). These decreases are thought to be in part due to hemodilution. There were no treatment discontinuations due to anemia or hemoglobin decrease in the ALIGN study.
Blood Pressure Decrease
At Week 36, the mean change from baseline in systolic and diastolic blood pressure (BP) for patients receiving VANRAFIA in the ALIGN study was -4 mmHg and -4 mmHg, respectively, compared to +3 mmHg and +2 mmHg, respectively, in patients receiving placebo. Hypotension observed in VANRAFIA treated patients was mild or moderate in severity, rarely symptomatic, and did not necessitate treatment discontinuation.
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on VANRAFIA
Strong or Moderate CYP3A Inducers
Avoid concomitant use with a strong or moderate CYP3A inducer.
Atrasentan is a CYP3A substrate [see Clinical Pharmacology (12.3)]. Concomitant use with a strong and moderate CYP3A inducer is expected to decrease atrasentan exposure [see Clinical Pharmacology (12.3)], which may reduce VANRAFIA efficacy.
OATP1B1/1B3 Inhibitors
Avoid concomitant use with organic anion transporting polypeptides 1B1/1B3 (OATP1B1/1B3) inhibitors.
Atrasentan is a OATP1B1/1B3 substrate [see Clinical Pharmacology (12.3)]. Concomitant use with a OATP1B1/1B3 inhibitor increases atrasentan exposure [see Clinical Pharmacology (12.3)], which may increase the risk of VANRAFIA adverse reactions.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on data from animal reproductive toxicity studies, VANRAFIA may cause fetal harm, including birth defects and fetal death, when administered to a pregnant patient and is contraindicated during pregnancy [see Contraindications (4.1)]. There are no available data on VANRAFIA use in pregnancy to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Available data from published literature and post-marketing surveillance over decades of use with products in the same pharmacologic class (ERA) have not identified an increased risk of major birth defects. However, these data are limited and do not establish the presence or absence of a drug-associated risk of major birth defects. Methodological limitations of these post marketing reports and published literature include lack of a control group; limited information regarding dose, duration, and timing of exposure; and missing data. These limitations preclude establishing a reliable estimate of the risk of adverse fetal and neonatal outcomes with maternal endothelin receptor antagonist use.
In animal reproduction studies, oral administration of atrasentan to pregnant rats and rabbits throughout organogenesis at doses that were below the maximum recommended human dose (MRHD) based on area under the curve (AUC) caused teratogenic effects in rats and rabbits (see Data). Advise pregnant patients of the potential risk to the fetus [see Contraindications (4.1)].
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defects, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In embryo-fetal development studies in pregnant rats and rabbits, teratogenicity and/or embryo-fetal toxicity were observed.
In pregnant rats, oral administration of atrasentan throughout organogenesis at doses of 0.1, 0.3, 1.0, and 3.0 mg/kg/day resulted in developmental abnormalities primarily including the ear, lower jaw, or skull in all treated groups with detectable plasma exposures to atrasentan. The no adverse effect level of atrasentan plasma exposure was not determined. In pregnant rabbits, oral administration of atrasentan throughout organogenesis at doses of 0.1, 0.3, 1.0 and 3.0 mg/kg/day resulted in visceral malformations including deformities in the cardiovascular system in all atrasentan-treated groups. The lowest detectable plasma exposures to atrasentan were approximately 0.2 times the AUC at the MRHD.
In the pre- and postnatal development study in rats, atrasentan was orally administered to pregnant rats at doses of 1, 10, or 100 mg/kg/day during the period from gestation Day 15 through lactation Day 20. No adverse effects on pre- and postnatal development were observed at doses up to 10 mg/kg/day which resulted in maternal exposure approximately 55 times the AUC at the MRHD. Higher exposure to atrasentan (dose of 100 mg/kg/day) increased pup mortality during the pre-weaning period, and increased heart weight which correlated histologically with myocardial hypertrophy.
8.2 Lactation
Risk Summary
There are no data on the presence of atrasentan in human milk, the effects on the breastfed infant, or the effect on milk production. Because of the potential for adverse reactions, such as fluid retention in breastfed infants, advise patients not to breastfeed during treatment with VANRAFIA.
8.3 Females and Males of Reproductive Potential
Based on data from animal reproductive toxicity studies, VANRAFIA may cause fetal harm, including birth defects and fetal death, when administered to a pregnant patient and is contraindicated during pregnancy [see Contraindications (4.1), Use in Specific Populations (8.1)].
Pregnancy Testing
Exclude pregnancy before initiating VANRAFIA in females of reproductive potential. The patient should contact their physician immediately for pregnancy testing if onset of menses is delayed or pregnancy is suspected. If pregnancy is confirmed, the physician should discuss with the patient the risks to the pregnancy and the fetus.
Contraception
Patients who can become pregnant while using VANRAFIA should use effective contraception prior to initiation of treatment, during treatment, and for two weeks after discontinuation of treatment with VANRAFIA to prevent pregnancy [see Warnings and Precautions (5.1)].
Infertility
Decreased sperm counts have been observed in some patients with diabetic kidney disease (DKD) receiving VANRAFIA 0.75 mg once daily with return to normal levels within approximately 3 months after drug discontinuation. This effect has not been studied in patients with IgAN [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and efficacy of VANRAFIA in pediatric patients have not been established.
8.5 Geriatric Use
There were 29 (7%) patients 65 years of age and older in the ALIGN study of VANRAFIA. Of the total number of VANRAFIA-treated patients, 15 (7%) were 65 years to 75 years of age, and 3 (2%) were 75 years of age and older. No overall differences in safety and effectiveness were observed between these patients and younger patients.
-
10 OVERDOSAGE
There is no experience with overdose of VANRAFIA. Atrasentan has been given in a single dose up to 139.5 mg and multiple doses up to 40 mg/day in healthy volunteers. Overdose of VANRAFIA may result in headache or vasodilation. In the event of an overdose, standard supportive measures should be taken, as required. Dialysis is unlikely to be effective because atrasentan is highly protein-bound.
-
11 DESCRIPTION
VANRAFIA contains atrasentan, an endothelin type A (ETA) receptor antagonist. The chemical name of atrasentan hydrochloride is (2R, 3R, 4S)-4-(1,3-benzodioxol-5-yl)-1-[2-(dibutylamino)-2-oxoethyl]-2-(4-methoxyphenyl)-3-pyrrolidinecarboxylic acid hydrochloride. Atrasentan hydrochloride has a molecular weight of 547.09 g/mol, a molecular formula of C29H38N2O6HCl and the following structural formula.
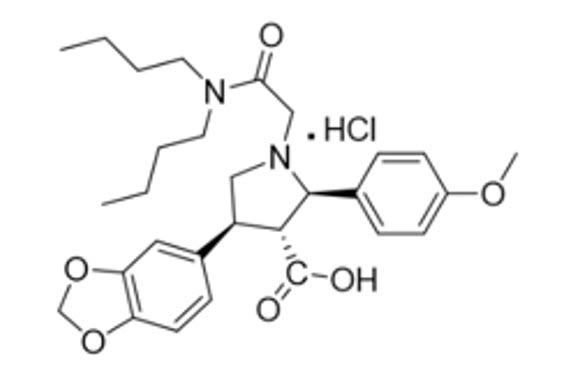
Atrasentan is a slightly hygroscopic white to off-white powder that is slightly soluble in water.
VANRAFIA is available as a film-coated tablet for oral administration. Each VANRAFIA tablet contains 0.75 mg atrasentan (equivalent to 0.803 mg of atrasentan hydrochloride) and contains the following excipients: crospovidone, glyceryl dibehenate, hypromellose, lactose monohydrate, L-cysteine hydrochloride monohydrate, polyethylene glycol, and silicon dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Atrasentan is an ETA receptor antagonist (Ki = 0.034 nM) with >1800-fold selectivity for the ETA receptor compared to the endothelin type B receptor (Ki = 63.3 nM). Endothelin (ET)-1 is thought to contribute to the pathogenesis of IgAN via the ETAR.
12.2 Pharmacodynamics
Dose-response information is not available. At the recommended dose regimen, no statistically significant exposure-response (E-R) relationship was identified between atrasentan exposure and the percentage reduction from baseline in UPCR at Week 36 over the observed atrasentan exposure range. Increased atrasentan exposure was associated with an increased incidence of anemia, but no association was observed between atrasentan exposure and hypotension or peripheral edema.
Cardiac Electrophysiology
At exposures 40% higher than the clinical exposure at the maximum recommended dose, clinically significant QTc interval prolongation was not observed.
12.3 Pharmacokinetics
Atrasentan area under the time concentration curve (AUC) is dose proportional across the 0.35 mg to 30 mg (0.47 to 40 times the approved recommended dosage) dose range. Atrasentan steady state plasma concentrations are reached within 7 days with 2- to 3-fold accumulation.
Absorption
Atrasentan time to peak plasma concentration (Tmax) is approximately 0.5 hour.
Effect of Food
No clinically significant differences in atrasentan pharmacokinetics were observed following administration with a high-fat meal (800 to 1000 Kcal, > 50% fat) in healthy subjects.
Distribution
Atrasentan steady-state apparent (oral) volume of distribution (Vd/F) is 1180 L. Atrasentan is > 99% bound to human plasma proteins, in vitro.
Elimination
Atrasentan effective half-life is approximately 24 to 41 hours with an apparent (oral) clearance (CL/F) of 19 L/h.
Metabolism
Atrasentan is extensively metabolized by CYP3A and multiple uridine 5'-diphospho-glucuronosyltransferases (UGTs) with approximately half via CYP3A and the remaining half via glucuronidation by multiple UGTs.
Excretion
After a single dose of radiolabeled atrasentan 10 mg to healthy subjects, approximately 86% of the dose was recovered in feces (5.5% as parent atrasentan). Renal excretion was minimal, with < 4% recovered in urine (negligible amounts of parent atrasentan).
Specific Populations
No clinically significant differences in the pharmacokinetics of atrasentan were observed based on age (19 to 77 years), sex, race, mild to severe renal impairment (eGFR 15 to 90 mL/min/1.73 m2, estimated by CKD-EPI), or mild to moderate hepatic impairment (Child-Pugh class A or B). The effect of severe hepatic impairment (Child-Pugh class C) or end-stage renal disease (eGFR < 15 mL/min/1.73 m2) on atrasentan pharmacokinetics is unknown.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Strong and moderate CYP3A inducers: Atrasentan Ctrough decreased by 90% following coadministration of a single dose of 10 mg VANRAFIA with rifampin (strong CYP3A inducer).
OATP1B1/1B3 inhibitors: Atrasentan Cmax was 4.3 times as high and AUC was 3.8 times as high following coadministration of a single dose of 0.75 mg VANRAFIA with cyclosporine (OATP1B1/1B3 inhibitor).
Strong CYP3A inhibitors: Atrasentan AUC increased by 90% following coadministration of a single dose of 10 mg VANRAFIA with ketoconazole (strong CYP3A inhibitor).
Other Drugs: No clinically significant differences in the pharmacokinetics of midazolam (CYP3A4 substrate), losartan (CYP2C9 and CYP3A4 substrate) or fexofenadine (P-gp substrate) were observed or expected when used concomitantly with VANRAFIA.
In Vitro Studies
CYP450 Enzymes: Atrasentan is a CYP3A substrate. Atrasentan inhibits in vitro CYP3A, CYP2B6, CYP2C8 and CYP2C9 and induces CYP3A and CYP2B6, but is not expected to cause clinically significant interactions with these CYP450 enzymes in the liver. Atrasentan does not inhibit CYP1A2, CYP2C19, or CYP2D6 and is not an inducer of CYP1A2.
Transporter Systems: Atrasentan is a substrate of P-gp and OATP1B1/1B3 but not a substrate of BCRP, MRP2/4, NTCP, OCT1, or OATP2B1. Atrasentan inhibits P-gp, OATP1B1, and OATP1B3, but not expected to cause clinically significant interactions. Atrasentan does not inhibit MRP, NTCP, OCT, OAT1, MATE1, or MATE2K.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In female rats orally administered atrasentan for 2 years, no atrasentan-related tumor findings were observed at exposures approximately 12 times the AUC at the MRHD. At a higher exposure (approximately 24 times the AUC at the MRHD, or 2 mg/kg/day), an increased incidence of benign uterine stromal polyps considered of low human relevance was observed. In male rats, no carcinogenic effects were observed at exposures approximately 100 times the AUC at the MRHD.
There were no atrasentan-related tumor findings observed in male and female transgenic mice administered atrasentan for 6 months at doses up to 60 mg/kg/day.
Mutagenesis
There was no evidence of mutagenicity or clastogenicity for atrasentan in in vitro bacteria reverse mutation and chromosomal aberration assays, or in an in vivo mouse micronucleus study.
Impairment of Fertility
In a fertility study, male rats were treated with atrasentan at doses of 5, 20, and 60 mg/kg/day and mated with untreated female rats. No adverse effects on male fertility were observed at 5 mg/kg/day, approximately 53 times the AUC at the MRHD. At higher doses, decreased numbers of implantation sites, reduced viable fetuses, and increased pre-implantation loss were observed in the untreated female rats mated with the treated male rats. These effects were reversible at 20 mg/kg/day, approximately 422 times the AUC at MRHD. In rats and dogs, testicular degeneration, dilatation of seminiferous tubules, interstitial edema of the testes, and prostate inflammation were observed at all doses tested, and a no adverse effect dose level could not be determined.
In a female rat fertility study, no reproductive toxicity was observed following oral administration during premating, mating, and early gestation at doses up to 100 mg/kg/day.
In female rats, cystic endometrial hyperplasia was observed at the lowest dose tested of 0.8 mg/kg/day.
-
14 CLINICAL STUDIES
14.1 IgA Nephropathy
The effect of VANRAFIA on proteinuria was assessed in a randomized, double-blind, placebo-controlled, multicenter, global study (ALIGN, NCT04573478) in adults with biopsy-proven primary IgAN, an eGFR ≥ 30 mL/min/1.73 m2, and urine protein ≥ 1 g/day on a stable dose of maximally tolerated renin angiotensin system inhibitor. The study included two cohorts: a main cohort of 340 patients and an exploratory cohort of 64 patients who were also on a stable dose of sodium glucose co-transporter 2 inhibitor (SGLT2i) at baseline. Patients with chronic kidney disease due to another condition in addition to IgAN or those who had been recently treated with systemic immunosuppressants were excluded. Patients were randomized (1:1) to receive either VANRAFIA 0.75 mg or placebo once daily. RAS inhibitor therapy was continued throughout the study. Rescue immunosuppressive treatment could be initiated per investigator discretion during the trial.
The efficacy analysis included the first 270 patients in the main cohort who reached the Week 36 visit. At baseline, the mean age was 45 years (range: 19 to 77 years); 59% were male, 36% White, 57% Asian, 2% Black or African American and 5% other or not specified. At baseline, 60% had a history of hypertension, 1.5% had a history of type 2 diabetes, and 45% had hematuria based on urine dipstick. The mean baseline eGFR was 59 mL/min/1.73 m2. The geometric mean baseline UPCR was 1.5 g/g sampled from a 24-hour urine and 15% of patients had proteinuria > 3.5 g/day.
The primary endpoint was the percent reduction in UPCR at Week 36 relative to baseline (see Table 2).
Table 2: Percent Reduction in UPCR at Week 36 Relative to Baseline in ALIGN aSupportive care: primarily a stable dose of maximally-tolerated RAS inhibitor therapy.
bLeast squares geometric mean ratio in UPCR (sampled from a 24-hr urine collection) to baseline was reported as a percent reduction along with the respective 95% confidence interval.
cThe estimate of the ratio of least squares geometric mean ratio in UPCR (sampled from a 24-hr urine collection) to baseline comparing VANRAFIA with placebo was reported as a relative percent reduction along with the respective 95% confidence interval and 2-sided p-value.
dMixed model repeated measures analysis included all observed UPCR data except for subjects with intercurrent events (i.e., restricted medication use, chronic dialysis, kidney transplant). These subjects had UPCR data excluded beginning at the start date of the earliest event. The only intercurrent events observed were restricted medication use, which occurred in 3.0% and 5.2% of VANRAFIA and placebo treated subjects, respectively.
eTwo-sided p-value statistically significant at the 0.01 level.
Abbreviations: CI, confidence interval; N, number of randomized subjects in each group in the main cohort; UPCR, urine protein-to-creatinine ratio; RAS, renin-angiotensin system.VANRAFIA
on top of supportive carea
(N = 135)Placebo
on top of supportive carea
(N = 135)% Reduction in UPCR (95% CI) at Week 36 relative to baselineb,d 38% (32%, 44%) 3% (-7%, 12%) VANRAFIA versus placebo: % reduction in UPCR (95% CI) at Week 36 relative to baseline compared on a relative scalec,d 36% (26%, 45%) p-valuee < 0.0001 The adjusted geometric mean percent change from baseline in UPCR over time is displayed in Figure 1.
Figure 1: Geometric Mean Percent Change from Baseline in UPCR by Visit in ALIGN
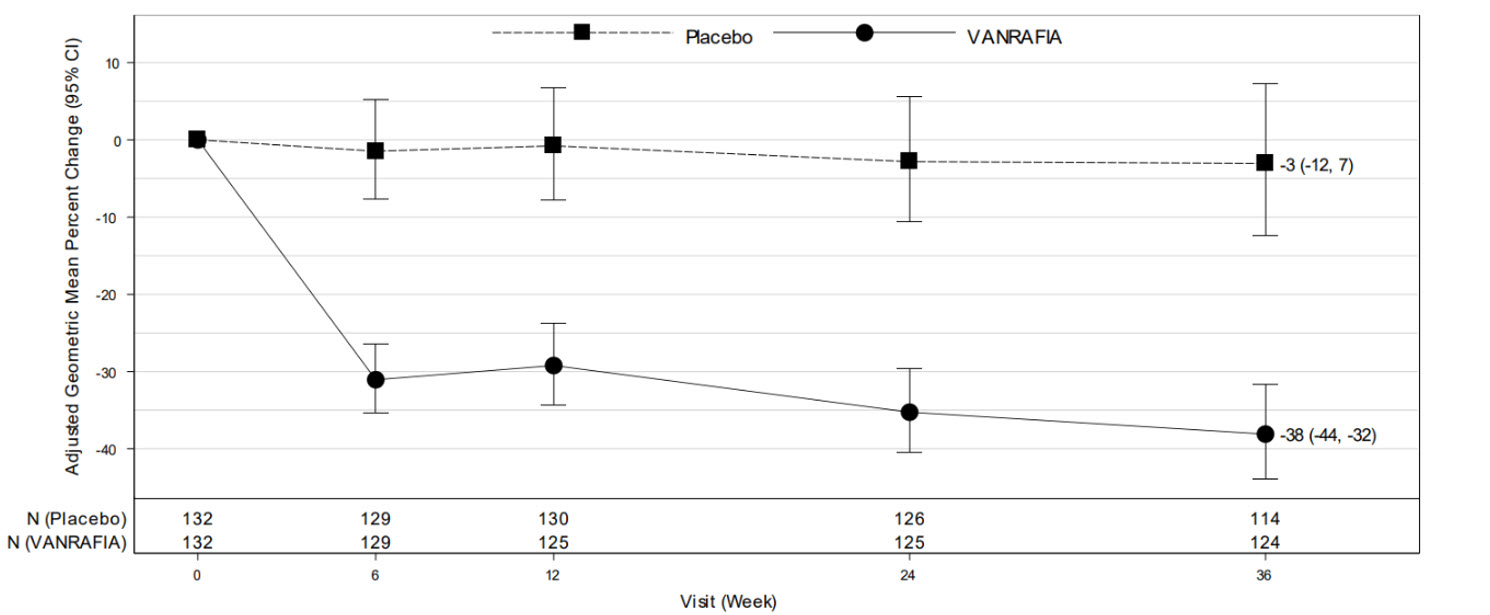
Adjusted % change relative to baseline in UPCR (sampled from a 24-hr urine collection) was estimated based on the MMRM analysis in Table 2.
N represents the number of evaluable subjects included in the analysis (i.e., with non-missing UPCR values and baseline covariates, and did not initiate restricted medication use, chronic dialysis, or kidney transplant) for each visit and treatment group.
Values reported in the figure were expressed as percent change from baseline and 95% CI, estimated from the regression model in Table 2.
Abbreviations: CI, confidence interval; MMRM, mixed model repeated measures; UPCR, urine protein-to-creatinine ratio.The treatment effect on UPCR at Week 36 was consistent across subgroups including age, sex, race, baseline disease characteristics (such as baseline eGFR and proteinuria levels) within the main cohort. The treatment effect on UPCR at Week 36 was also consistent in the exploratory SGLT2i cohort.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
VANRAFIA is supplied as follows:
- 0.75 mg tablets are film-coated, round, biconvex, white to off-white tablets debossed with “7” on one side and unmarked on the other side, packaged in a high-density polyethylene (HDPE) bottle containing a desiccant, with induction seal and child-resistant cap. Each bottle contains 30 tablets (NDC: 0078-1420-15).
Storage
Store at 20°C to 25°C (68°F to 77°F). Excursions are permitted between 15°C and 30°C (59°F and 86°F). [See USP Controlled Room Temperature]. Store and dispense VANRAFIA in its original container.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide)
Embryo-Fetal Toxicity
Educate and counsel female patients of reproductive potential to use effective contraception prior to starting treatment with VANRAFIA, during treatment and for two weeks after treatment discontinuation. Patients who can become pregnant should have a negative pregnancy test prior to treatment with VANRAFIA [see Dosage and Administration (2.1), Contraindications (4.1), Warnings and Precautions (5.1), Use in Specific Populations (8.1, 8.3)].
Patients should be instructed to immediately contact their physician if they suspect they may be pregnant. Patients should seek additional contraceptive advice as needed.
Educate and counsel patients who can become pregnant on the use of emergency contraception in the event of unprotected sex or contraceptive failure.
Advise patients to contact their healthcare provider if they want to change the form of birth control which is used to ensure that another acceptable form of birth control is selected.
Hepatotoxicity
Some members of this pharmacological class are hepatotoxic. Educate patients on signs of hepatotoxicity. Advise patients that they should contact their doctor if they have unexplained nausea, vomiting, right upper quadrant pain, fatigue, anorexia, jaundice, dark urine, fever, or itching [see Warnings and Precautions (5.2)].
Fluid Retention
Educate patients on signs of fluid retention. Advise patients that they should contact their doctor if they have unusual weight increase or swelling of the ankles or legs [see Warnings and Precautions (5.3)].
Lactation
Advise patients not to breastfeed during treatment with VANRAFIA [see Use in Specific Populations (8.2)].
Other Risks Associated with VANRAFIA
Instruct patients that the risks associated with VANRAFIA also include the following:
- Decreases in sperm count [see Warnings and Precautions (5.4)]
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936© 2025 Novartis
T2025-18
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Issued: April 2025 MEDICATION GUIDE
VANRAFIA™ (van-rah-fee-ah)
(atrasentan)
tabletsWhat is the most important information I should know about VANRAFIA?
VANRAFIA can cause serious birth defects if taken during pregnancy.- Females should not be pregnant when they start taking VANRAFIA or become pregnant during treatment with VANRAFIA or for two weeks after stopping treatment with VANRAFIA.
- Females who can become pregnant should have a negative pregnancy test before starting VANRAFIA.
- Females who can become pregnant are those who:
- have entered puberty, even if they have not started their menstrual period, and
- have a uterus, and
- have not gone through menopause. Menopause means that you have not had a menstrual period for at least 12 months for natural reasons, or that you have had your ovaries removed.
- Females who cannot become pregnant are those who:
- have not yet entered puberty, or
- do not have a uterus, or
- have gone through menopause. Menopause means that you have not had a menstrual period for at least 12 months for natural reasons, or that you have had your ovaries removed, or
- are infertile for any other medical reason and this infertility is permanent and cannot be reversed.
- Females who can become pregnant are those who:
-
Females who can become pregnant should use effective birth control before starting treatment with VANRAFIA, during treatment with VANRAFIA and for 2 weeks after stopping VANRAFIA because the medicine may still be in your body.
- Talk with your healthcare provider or gynecologist (a healthcare provider who specializes in reproduction) to find out about options for effective forms of birth control that you may use to prevent pregnancy during treatment with VANRAFIA.
- If you decide that you want to change the form of birth control that you use, talk with your healthcare provider or gynecologist to be sure that you choose another effective form of birth control.
- Do not have unprotected sex. Talk to your healthcare provider or pharmacist right away if you have unprotected sex or if you think your birth control has failed. Your healthcare provider may talk with you about using emergency birth control.
- Tell your healthcare provider right away if you miss a menstrual period or think you may be pregnant.
What is VANRAFIA?
VANRAFIA is a prescription medicine used to reduce levels of protein in the urine (proteinuria) in adults with a kidney disease called primary immunoglobulin A nephropathy (IgAN) who are at risk of their disease getting worse quickly.
It is not known if VANRAFIA is safe and effective in children.Who should not take VANRAFIA?
Do not take VANRAFIA if you are- pregnant, plan to become pregnant, or become pregnant during treatment with VANRAFIA. VANRAFIA can cause serious birth defects. See “What is the most important information I should know about VANRAFIA?”
- allergic to atrasentan or any of the ingredients in VANRAFIA. See the end of this Medication Guide for a complete list of ingredients in VANRAFIA.
Before taking VANRAFIA, tell your healthcare provider about all your medical conditions including if you: - have liver problems
- are pregnant or plan to become pregnant during VANRAFIA treatment. VANRAFIA can cause serious birth defects. See "What is the most important information I should know about VANRAFIA?”
- are breastfeeding or plan to breastfeed. It is not known if VANRAFIA passes into your breast milk. Do not breastfeed during treatment with VANRAFIA. Talk with your healthcare provider about the best way to feed your baby if you take VANRAFIA.
How should I take VANRAFIA? - Take VANRAFIA exactly as your healthcare provider tells you. Do not change your dose or stop taking unless your healthcare provider tells you to.
- Take 1 VANRAFIA tablet 1 time each day with or without food.
- Swallow VANRAFIA tablets whole. Do not cut, crush, or chew VANRAFIA tablets.
- If you miss a dose or more doses of VANRAFIA, skip the missed dose and take your next dose at your regularly scheduled time. Do not take 2 doses at the same time to make up for a missed dose.
- If you take too much VANRAFIA, call your healthcare provider right away or go to the nearest hospital or emergency room.
What are the possible side effects of VANRAFIA?
VANRAFIA can cause serious side effects, including:- Serious birth defects. See "What is the most important information I should know about VANRAFIA?”
- Liver problems. Medicines like VANRAFIA can cause liver problems, including liver failure. VANRAFIA can increase liver enzymes in your blood. Your healthcare provider will do blood tests to check your liver enzymes before starting treatment and if needed during treatment. Your healthcare provider may temporarily stop or permanently stop treatment with VANRAFIA if your liver enzymes increase or if you develop symptoms of liver problems. Tell your healthcare provider if you develop any of the following symptoms of liver problems during treatment with VANRAFIA.
- nausea or vomiting
- pain in the upper right stomach
- tiredness
- loss of appetite
- yellowing of your skin or whites of your eyes
- dark urine
- fever
- itching
- Fluid retention. VANRAFIA can cause your body to hold too much water. Tell your healthcare provider if you develop any unusual weight gain, trouble breathing, or swelling of your ankles or legs during treatment. Your healthcare provider may prescribe other medicines (diuretics) and may temporarily stop VANRAFIA if you develop fluid retention.
- Decreased sperm count. VANRAFIA may cause decreased sperm counts in males and may affect the ability to father a child. Tell your healthcare provider if being able to have children is important to you.
- swelling of hands, legs, ankles, and feet (peripheral edema)
- low red blood cells (anemia)
How should I store VANRAFIA? - Store VANRAFIA at room temperature between 68°F to 77°F (20°C to 25°C).
- Store VANRAFIA in the original container.
- The bottle has a child-resistant cap and contains a desiccant to help keep the tablets dry.
General information about the safe and effective use of VANRAFIA
Medicines are sometimes prescribed for purposes other than those listed in the Medication Guide. Do not use VANRAFIA for a condition for which it was not prescribed. Do not give VANRAFIA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about VANRAFIA that is written for health professionals.What are the ingredients in VANRAFIA?
Active ingredient: atrasentan
Inactive ingredients: crospovidone, glyceryl dibehenate, hypromellose, lactose monohydrate, L-cysteine hydrochloride monohydrate, polyethylene glycol, and silicon dioxide.Distributed by: Novartis Pharmaceuticals Corporation, East Hanover, New Jersey 07936
© 2025 Novartis
For more information, go to www.VANRAFIA.com or call 1-888-669-6682.T2025-19
-
PRINCIPAL DISPLAY PANEL
NDC: 0078-1420-15
Rx only
VANRAFIA™
(atrasentan) tablets
0.75 mg
Dispense with accompanying
Medication Guide.Swallow tablets whole.
Do not cut, crush, or chew.30 tablets
NOVARTIS

-
INGREDIENTS AND APPEARANCE
VANRAFIA
atrasentan tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-1420 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ATRASENTAN HYDROCHLORIDE (UNII: E4G31X93ZA) (ATRASENTAN - UNII:V6D7VK2215) ATRASENTAN 0.75 mg Inactive Ingredients Ingredient Name Strength CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) GLYCERYL DIBEHENATE (UNII: R8WTH25YS2) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) CYSTEINE HYDROCHLORIDE (UNII: ZT934N0X4W) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) Product Characteristics Color WHITE (white to off-white) Score no score Shape ROUND (biconvex) Size 7mm Flavor Imprint Code 7;unmarked Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-1420-15 30 in 1 BOTTLE; Type 0: Not a Combination Product 04/02/2025 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219208 04/02/2025 Labeler - Novartis Pharmaceuticals Corporation (002147023)
Trademark Results [VANRAFIA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 VANRAFIA 97626972 not registered Live/Pending |
Novartis AG 2022-10-11 |
 VANRAFIA 79238110 5640110 Live/Registered |
NOVARTIS AG 2018-05-30 |
 VANRAFIA 79157838 4741479 Dead/Cancelled |
NOVARTIS AG 2014-11-11 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.