FOLOTYN- pralatrexate injection
Folotyn by
Drug Labeling and Warnings
Folotyn by is a Prescription medication manufactured, distributed, or labeled by Acrotech Biopharma Inc, Baxter Oncology, GmbH, (Baxter). Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use FOLOTYN® safely and effectively. See full prescribing information for FOLOTYN.
FOLOTYN (pralatrexate injection), for intravenous use
Initial U.S. Approval: 2009INDICATIONS AND USAGE
FOLOTYN is a folate analog metabolic inhibitor indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL). This indication is based on overall response rate. Clinical benefit such as improvement in progression-free survival or overall survival has not been demonstrated. (1)
DOSAGE AND ADMINISTRATION
- The recommended dose of FOLOTYN is 30 mg/m2 administered as an intravenous push over 3 to 5 minutes once weekly for 6 weeks in 7-week cycles. (2.1)
- For patients with severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2), the recommended dose of FOLOTYN is 15 mg/m2 (2.1).
- Prior to initiating FOLOTYN, supplement patients with vitamin B12 1 mg intramuscularly every 8-10 weeks and folic acid 1.0-1.25 mg orally on a daily basis. (2.1)
- Dose omissions and/or dose reductions may be needed to manage adverse drug reactions. (2.2)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
- None. (4)
WARNINGS AND PRECAUTIONS
- Thrombocytopenia, neutropenia, and anemia: Monitor blood counts and omit and/or reduce dose for hematologic toxicities. (2.2, 5.1)
- Mucositis: Monitor at least weekly. If ≥ Grade 2 mucositis is observed, omit and/or reduce dose. (2.2, 5.2)
- Dermatologic reactions: Reactions, including fatal reactions, have occurred and may be progressive and increase in severity with further treatment. Monitor closely, and omit and/or reduce dose or discontinue FOLOTYN. (2.2, 5.3)
- Tumor lysis syndrome: Anticipate, monitor, and treat promptly. (5.4)
- Hepatic toxicity: Monitor for toxicity. For liver function test abnormalities Grade 3 or greater, omit until recovery then reduce dose or discontinue therapy as required. (2.2, 5.5)
- Risk of increased toxicity with renal impairment: Patients with moderate to severe renal function impairment may be at greater risk for increased exposure and toxicity. Monitor patients for renal function and systemic toxicity and adjust dosing accordingly. Avoid FOLOTYN use in patients with end stage renal disease including those undergoing dialysis unless the potential benefit justifies the potential risk. (2.1, 2.2, 5.6, 6.2)
- Embryo-Fetal toxicity: Women should avoid becoming pregnant while being treated with FOLOTYN. Inform pregnant women of the potential harm to the fetus. (5.7, 8.1)
ADVERSE REACTIONS
Most common adverse reactions (>35%) are mucositis, thrombocytopenia, nausea, and fatigue. Most common serious adverse reactions are pyrexia, mucositis, sepsis, febrile neutropenia, dehydration, dyspnea, and thrombocytopenia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Acrotech Biopharma LLC at 1-888-255-6788 or www.FOLOTYN.com or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
- Co-administration with probenecid or other drugs that may affect relevant transporter systems (eg, NSAIDs) requires close monitoring for signs of systemic toxicity. (7)
USE IN SPECIFIC POPULATIONS
- Women should be advised against breastfeeding while being treated with FOLOTYN. (8.3)
- Dose is reduced for patients with severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2) (2.1, 8.7)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 9/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing and Administration
2.2 Monitoring and Dose Modifications
2.3 Special Handling Precautions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Bone Marrow Suppression
5.2 Mucositis
5.3 Dermatologic Reactions
5.4 Tumor Lysis Syndrome
5.5 Hepatic Toxicity
5.6 Risk of Increased Toxicity in the Presence of Impaired Renal Function
5.7 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post Marketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
SPL UNCLASSIFIED SECTION
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 Need for Folic Acid and Vitamin B12
17.2 Low Blood Cell Counts
17.3 Mucositis
17.4 Fatal Dermatologic Reactions
17.5 Tumor Lysis Syndrome
17.6 Concomitant Medications
17.7 Pregnancy/Nursing
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing and Administration
Folic Acid: Patients should take folic acid 1.0-1.25 mg orally once daily beginning 10 days before the first dose of FOLOTYN. Continue folic acid during the full course of therapy and for 30 days after the last dose of FOLOTYN [see Warnings and Precautions (5.1, 5.2)].
Vitamin B12: Administer vitamin B12 1 mg intramuscularly within 10 weeks prior to the first dose of FOLOTYN and every 8-10 weeks thereafter. Subsequent vitamin B12 injections may be given the same day as treatment with FOLOTYN [see Warnings and Precautions (5.1, 5.2)].
Dosing and Administration
The recommended dose of FOLOTYN is 30 mg/m2 administered as an intravenous push over 3-5 minutes via the side port of a free-flowing 0.9% Sodium Chloride Injection, intravenous line once weekly for 6 weeks in 7-week cycles until progressive disease or unacceptable toxicity. The calculated dose of FOLOTYN should be aseptically withdrawn into a syringe for immediate use. Do not dilute FOLOTYN.
For patients with severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2), the recommended dose of FOLOTYN is 15 mg/m2.
FOLOTYN is a clear, yellow solution. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use any vials exhibiting particulate matter or discoloration.
2.2 Monitoring and Dose Modifications
Management of severe or intolerable adverse reactions may require dose omission, reduction, or discontinuation of FOLOTYN therapy.
Monitoring
Monitor complete blood cell counts and severity of mucositis at baseline and weekly. Perform serum chemistry tests, including renal and hepatic function, prior to the start of the first and fourth dose of each cycle.
Dose Modification Recommendations
Prior to administering any dose of FOLOTYN:
- Mucositis should be ≤ Grade 1.
- Platelet count should be ≥ 100,000/mcL for first dose and ≥ 50,000/mcL for all subsequent doses.
- Absolute neutrophil count (ANC) should be ≥ 1,000/mcL.
Doses may be omitted or reduced based on patient tolerance. Omitted doses will not be made up at the end of the cycle; once a dose reduction occurs for toxicity, do not re-escalate. For dose modifications and omissions, use the guidelines in Tables 1, 2, and 3.
For patients with severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2), the recommended starting dose of FOLOTYN is 15 mg/m2 with dose modification to 10 mg/m2 for the toxicities specified in Tables 1, 2, and 3.
Table 1 FOLOTYN Dose Modifications for Mucositis Mucositis Gradea on Day of Treatment Action
Dose upon Recovery to ≤ Grade 1
Dose Upon Recovery in Patients with Severe Renal Impairment
Grade 2 Omit dose
Continue prior dose
Continue prior dose
Grade 2 recurrence Omit dose
20 mg/m2
10 mg/m2
Grade 3 Omit dose
20 mg/m2
10 mg/m2
Grade 4 Stop therapy
a Per National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI CTCAE, Version 3.0)
Table 2 FOLOTYN Dose Modifications for Hematologic Toxicities Blood Count on Day of Treatment Duration of Toxicity
Action
Dose upon Restart
Dose Upon Recovery in Patients with Severe Renal Impairment
Platelet < 50,000/mcL 1 week
Omit dose
Continue prior dose
Continue prior dose
2 weeks
Omit dose
20 mg/m2
10 mg/m2
3 weeks
Stop therapy
ANC 500-1,000/mcL and no fever 1 week
Omit dose
Continue prior dose
Continue prior dose
ANC 500-1,000/mcL with fever or ANC < 500/mcL 1 week
Omit dose, give G‑CSF or GM‑CSF support
Continue prior dose with G-CSF or GM‑CSF support
Continue prior dose with G-CSF or GM‑CSF support
2 weeks or recurrence
Omit dose, give G‑CSF or GM‑CSF support
20 mg/m2 with G-CSF or GM-CSF support
10 mg/m2 with G-CSF or GM-CSF support
3 weeks or 2nd recurrence
Stop therapy
G-CSF=granulocyte colony-stimulating factor; GM-CSF=granulocyte macrophage colony-stimulating factor
Table 3 FOLOTYN Dose Modifications for All Other Treatment-related Toxicities Toxicity Grade a on Day of Treatment Action
Dose upon Recovery to ≤ Grade 2
Dose Upon Recovery in Patients with Severe Renal Impairment
Grade 3 Omit dose
20 mg/m2
10 mg/m2
Grade 4 Stop therapy
a Per National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI CTCAE, Version 3.0)
2.3 Special Handling Precautions
FOLOTYN is a cytotoxic anticancer agent. Caution should be exercised in handling, preparing, and administering of the solution. The use of gloves and other protective clothing is recommended. If FOLOTYN comes in contact with the skin, immediately and thoroughly wash with soap and water. If FOLOTYN comes in contact with mucous membranes, flush thoroughly with water.
Several published guidelines for handling and disposal of anticancer agents are available1-4.
- FOLOTYN vials should be refrigerated at 2-8°C (36-46°F) until use.
- FOLOTYN vials should be stored in original carton to protect from light until use.
- FOLOTYN vials contain no preservatives and are intended for single use only. After withdrawal of dose, discard vial including any unused portion.
- Unopened vial(s) of FOLOTYN are stable if stored in the original carton at room temperature for 72 hours. Any vials left at room temperature for greater than 72 hours should be discarded.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Bone Marrow Suppression
FOLOTYN can cause bone marrow suppression, manifested by thrombocytopenia, neutropenia, and/or anemia. Monitor complete blood counts and omit and/or reduce the dose based on ANC and platelet count prior to each dose as outlined in Section 2.2 Table 2. Administer vitamin B12 and instruct patients to take folic acid to reduce the risk of treatment-related hematological toxicity [see Dosage and Administration (2.1, 2.2) and Adverse Reactions (6.1)].
5.2 Mucositis
FOLOTYN can cause mucositis. Monitor for mucositis weekly and if ≥ Grade 2 mucositis is observed, omit and/or reduce the dose as outlined in Section 2.2 Table 1. Administer vitamin B12 and instruct patients to take folic acid to reduce the risk of mucositis [see Dosage and Administration (2.1, 2.2) and Adverse Reactions (6.1)].
5.3 Dermatologic Reactions
FOLOTYN can cause severe dermatologic reactions, which may result in death. These dermatologic reactions have been reported in clinical studies (14/663 patients [2.1%]) and post marketing experience, and have included skin exfoliation, ulceration, and toxic epidermal necrolysis (TEN). They may be progressive and increase in severity with further treatment, and may involve skin and subcutaneous sites of known lymphoma. Monitor patients with dermatologic reactions closely, and if severe, withhold or discontinue FOLOTYN [see Adverse Reactions (6.2) and Use in Specific Populations (8.7)].
5.4 Tumor Lysis Syndrome
FOLOTYN can cause tumor lysis syndrome (TLS). Monitor patients who are at increased risk of TLS and treat promptly.
5.5 Hepatic Toxicity
FOLOTYN can cause hepatic toxicity and liver function test abnormalities. Persistent liver function test abnormalities may be indicators of hepatic toxicity and require dose modification or discontinuation. Monitor liver function tests. Omit dose until recovery, adjust or discontinue therapy based on the severity of the hepatic toxicity [see Dosage and Administration (2.2) and Use in Specific Populations (8.6)].
5.6 Risk of Increased Toxicity in the Presence of Impaired Renal Function
Patients with moderate to severe renal function impairment may be at greater risk for increased exposure and toxicity. Monitor patients for renal function and systemic toxicity and adjust dosing accordingly.
Serious adverse drug reactions including toxic epidermal necrolysis and mucositis were reported in patients with end stage renal disease (ESRD) undergoing dialysis who were administered FOLOTYN therapy. Avoid FOLOTYN use in patients with end stage renal disease including those undergoing dialysis unless the potential benefit justifies the potential risk [see Dosage and Administration (2.2), Adverse Reactions (6.2), Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
5.7 Embryo-Fetal Toxicity
FOLOTYN can cause fetal harm when administered to a pregnant woman. FOLOTYN was embryotoxic and fetotoxic in rats and rabbits. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus [see Use in Specific Populations (8.1)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described below and elsewhere in the labeling:
- Bone Marrow Suppression [see Warnings and Precautions (5.1)]
- Mucositis [see Warnings and Precautions (5.2)]
- Dermatologic Reactions [see Warnings and Precautions (5.3)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.4)]
- Hepatic Toxicity [see Warnings and Precautions (5.5)]
The most common adverse reactions observed in patients with peripheral T-cell lymphoma (PTCL) treated with FOLOTYN were mucositis, thrombocytopenia, nausea, and fatigue.
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
The safety of FOLOTYN was evaluated in 111 PTCL patients in a single-arm clinical study in which patients received a starting dose of 30 mg/m2 once weekly for 6 weeks in 7-week cycles. The median duration of treatment was 70 days (range 1-540 days).
Most Frequent Adverse Reactions
Table 4 summarizes the most frequent adverse reactions, regardless of causality, using the National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI CTCAE, version 3.0).
Table 4 Adverse Reactions Occurring in PTCL Patients (Incidence ≥ 10% of patients) N=111
Total
Grade 3
Grade 4
Preferred Term
N
%
N
%
N
%
Any Adverse Event
111
100
48
43
34
31
Mucositisa
78
70
19
17
4
4
Thrombocytopeniab
45
41
15
14
21
19b
Nausea
44
40
4
4
0
0
Fatigue
40
36
5
5
2
2
Anemia
38
34
17
15
2
2
Constipation
37
33
0
0
0
0
Pyrexia
36
32
1
1
1
1
Edema
33
30
1
1
0
0
Cough
31
28
1
1
0
0
Epistaxis
29
26
0
0
0
0
Vomiting
28
25
2
2
0
0
Neutropenia
27
24
14
13
8
7
Diarrhea
23
21
2
2
0
0
Dyspnea
21
19
8
7
0
0
Anorexia
17
15
3
3
0
0
Hypokalemia
17
15
4
4
1
1
Rash
17
15
0
0
0
0
Pruritus
16
14
2
2
0
0
Pharyngolaryngeal pain
15
14
1
1
0
0
Liver function test abnormalc
14
13
6
5
0
0
Abdominal pain
13
12
4
4
0
0
Pain in extremity
13
12
0
0
0
0
Back pain
12
11
3
3
0
0
Leukopenia
12
11
3
3
4
4
Night sweats
12
11
0
0
0
0
Asthenia
11
10
1
1
0
0
Tachycardia
11
10
0
0
0
0
Upper respiratory tract infection
11
10
1
1
0
0
aStomatitis or mucosal inflammation of the gastrointestinal and genitourinary tracts.
bFive patients with platelets < 10,000/mcL
cAlanine aminotransferase, aspartate aminotransferase, and transaminases increasedSerious Adverse Events
Forty-four percent of patients (n = 49) experienced a serious adverse event while on study or within 30 days after their last dose of FOLOTYN. The most common serious adverse events (> 3%), regardless of causality, were pyrexia, mucositis, sepsis, febrile neutropenia, dehydration, dyspnea, and thrombocytopenia. One death from cardiopulmonary arrest in a patient with mucositis and febrile neutropenia was reported in this trial. Deaths from mucositis, febrile neutropenia, sepsis, and pancytopenia occurred in 1.2% of patients treated on all FOLOTYN trials at doses ranging from 30 to 325 mg/m2.
6.2 Post Marketing Experience
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Dermatologic Reactions
Toxic epidermal necrolysis, sometimes fatal, has been reported during post-marketing use of FOLOTYN. Fatal cases have been reported following the first dose of FOLOTYN, including when a reduced dose is given, and have been reported in patients with end-stage renal disease undergoing dialysis [see Warnings and Precautions (5.3), Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
-
7 DRUG INTERACTIONS
No formal clinical assessments of pharmacokinetic drug-drug interactions between FOLOTYN and other drugs have been conducted. The effect of co-administration of the uricosuric drug probenecid (an inhibitor of multiple transporter systems including the multidrug resistance-associated protein 2 (MRP2) efflux transporter) on pralatrexate pharmacokinetics was investigated in a Phase 1 clinical study. Co-administration of increasing doses of probenecid resulted in delayed clearance of pralatrexate and a commensurate increase in exposure [see Clinical Pharmacology (12.3)].
When administering FOLOTYN to patients receiving probenecid or other drugs that may affect relevant transporter systems (eg, NSAIDs), monitor patients closely for signs of systemic toxicity due to increased drug exposure.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category D [see Warnings and Precautions (5.7)]
Embryo-Fetal Toxicity
FOLOTYN can cause fetal harm when administered to a pregnant woman. Pralatrexate was embryotoxic and fetotoxic in rats at IV doses of 0.06 mg/kg/day (0.36 mg/m2/day or about 1.2% of the clinical dose on a mg/m2 basis) given on gestation days 7 through 20. Treatment with pralatrexate caused a dose-dependent decrease in fetal viability manifested as an increase in late, early, and total resorptions. There was also a dose-dependent increase in post-implantation loss. In rabbits, IV doses of 0.03 mg/kg/day (0.36 mg/m2/day) or greater given on gestation days 8 through 21 also caused abortion and fetal lethality. This toxicity manifested as early and total resorptions, post-implantation loss, and a decrease in the total number of live fetuses. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.
8.3 Nursing Mothers
It is not known whether pralatrexate is excreted in human milk. Because many drugs are excreted in human milk, and because of the potential for serious adverse reactions in nursing infants from this drug, a decision should be made whether to discontinue nursing or to discontinue FOLOTYN, taking into account the importance of FOLOTYN to the mother.
8.4 Pediatric Use
Pediatric patients were not included in clinical studies with FOLOTYN. The safety and effectiveness of FOLOTYN in pediatric patients have not been established.
8.5 Geriatric Use
In the PTCL efficacy study, 36% of patients (n = 40) were 65 years of age and over. No overall differences in efficacy and safety were observed in patients based on age (< 65 years compared with ≥ 65 years). Due to the contribution of renal excretion to overall clearance of pralatrexate (approximately 34%), age-related decline in renal function may lead to a reduction in clearance and a commensurate increase in plasma exposure. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. Since elderly patients may be at higher risk, monitor more closely. Omit dose and subsequently adjust or discontinue therapy for exposure related toxicity [see Dosage and Administration (2.2), Warnings and Precautions (5.5, 5.6), Use in Specific Populations (8.6, 8.7), and Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
The safety, efficacy and pharmacokinetics of FOLOTYN have not been evaluated in patients with hepatic impairment. Patients with the following laboratory values were excluded from the pralatrexate lymphoma clinical trials: total bilirubin > 1.5 mg/dL; aspartate aminotransferase (AST) or alanine aminotransferase (ALT) > 2.5 × upper limit of normal (ULN); and AST or ALT > 5 × ULN if documented hepatic involvement with lymphoma. Treatment with FOLOTYN can cause hepatic toxicity and liver function test abnormalities [see Dosage and Administration (2.2) and Warnings and Precautions (5.6)].
8.7 Renal Impairment
For patients with severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2), the recommended dose of FOLOTYN is 15 mg/m2. For patients with mild to moderate renal impairment, dose reduction is not necessary.
Serious adverse drug reactions, including TEN and mucositis have been reported in patients with ESRD undergoing dialysis. Monitor patients for renal function and for systemic toxicity due to increased drug exposure and adjust dosing accordingly. Avoid the use of FOLOTYN in patients with end stage renal disease undergoing dialysis unless the potential benefit justifies the potential risk [see Dosage and Administration (2.1, 2.2), Warnings and Precautions (5.3, 5.6), Adverse Reactions (6.2), and Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
FOLOTYN (pralatrexate injection) contains pralatrexate, which is an antineoplastic folate analog. Pralatrexate has the chemical name (2S)-2-[[4-[(1RS)-1-[(2, 4-diaminopteridin-6-yl)methyl]but-3-ynyl]benzoyl]amino]pentanedioic acid. The structural formula is as follows:
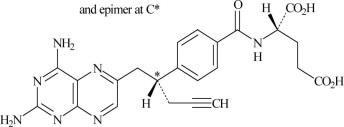
Pralatrexate is a 1:1 racemic mixture of S- and R- diastereomers at the C10 position (indicated with *).
The molecular formula is C23H23N7O5 and the molecular weight is 477.48 g/mol.
Pralatrexate is an off-white to yellow solid. It is soluble in aqueous solutions at pH 6.5 or higher. Pralatrexate is practically insoluble in chloroform and ethanol. The pKa values are 3.25, 4.76, and 6.17.
FOLOTYN is supplied as a preservative-free, sterile, isotonic, non-pyrogenic clear yellow aqueous parenteral solution contained in a single-dose clear glass vial (Type I) for intravenous administration. Each 1 mL of solution contains 20 mg of pralatrexate, sufficient sodium chloride to achieve an isotonic (280-300 mOsm) solution, and sufficient sodium hydroxide, and hydrochloric acid if needed, to adjust and maintain the pH at 7.5-8.5. FOLOTYN is supplied as either 20 mg (1 mL) or 40 mg (2 mL) single-dose vials at a concentration of 20 mg/mL.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Pralatrexate is a folate analog metabolic inhibitor that competitively inhibits dihydrofolate reductase. It is also a competitive inhibitor for polyglutamylation by the enzyme folylpolyglutamyl synthetase. This inhibition results in the depletion of thymidine and other biological molecules the synthesis of which depends on single carbon transfer.
12.3 Pharmacokinetics
Absorption
The pharmacokinetics of pralatrexate administered as a single agent at a dose of 30 mg/m2 administered as an intravenous push over 3-5 minutes once weekly for 6 weeks in 7-week cycles have been evaluated in 10 patients with PTCL. The total systemic clearance of pralatrexate diastereomers was 417 mL/min (S-diastereomer) and 191 mL/min (R-diastereomer). The terminal elimination half-life of pralatrexate was 12-18 hours (coefficient of variance [CV] = 62-120%). Pralatrexate total systemic exposure (AUC) and maximum plasma concentration (Cmax) increased proportionally with dose (dose range 30-325 mg/m2, including pharmacokinetics data from high-dose solid tumor clinical studies). The pharmacokinetics of pralatrexate did not change significantly over multiple treatment cycles, and no accumulation of pralatrexate was observed.
Distribution
Pralatrexate diastereomers showed a steady-state volume of distribution of 105 L (S-diastereomer) and 37 L (R-diastereomer). In vitro studies indicate that pralatrexate is approximately 67% bound to plasma proteins.
Elimination
Metabolism
In vitro studies using human hepatocytes, liver microsomes and S9 fractions, and recombinant human CYP450 isozymes showed that pralatrexate is not significantly metabolized by the phase I hepatic CYP450 isozymes or phase II hepatic glucuronidases.
Excretion
The mean fraction of unchanged pralatrexate diastereomers excreted in urine following a pralatrexate dose of 30 mg/m2 administered as an intravenous push over 3‑5 minutes was 31% (S‑diastereomer) (CV = 47%) and 38% (R-diastereomer) (CV = 45%), respectively. In a mass balance study conducted in patients with advanced cancer, an average of 39% (CV = 28%) of the administered radiolabeled pralatrexate dose was excreted in urine as parent, racemic pralatrexate (fe). An average of 34% (CV = 88%) of the administered dose was recovered in feces as total radiation (feTR) which included both parent pralatrexate and/or any metabolites. An average of 10% (CV = 95%) of total dose was exhaled as total radioactivity over 24 hours.
Pharmacokinetics in Specific Populations
Renal Impairment
In patients with cancer without renal impairment, approximately 34% of pralatrexate was excreted unchanged into urine following a single dose of 30 mg/m2 administered as an intravenous push over 3-5 minutes. The pharmacokinetics of FOLOTYN was studied in patients with varying degrees of renal impairment. In patients with severe renal impairment (eGFR 15 to <30 mL/min/1.73 m2), the FOLOTYN dose was 15 mg/m2. Patients with normal renal clearance, mild renal impairment, and moderate renal impairment were all dosed with 30 mg/m2. Mean exposures of the pralatrexate S-diastereomer and R-diastereomer were comparable across cohorts. The mean fraction of the administered dose excreted as unchanged diastereomers in urine (fe) decreased with declining renal function. The non-renal clearance and volume of distribution of pralatrexate were unaffected by renal impairment [see Use in Specific Populations (8.7)].
Hepatic Impairment
Pralatrexate has not been studied in patients with hepatic impairment.
Gender
There was no significant effect of gender on pharmacokinetics.
Drug Interactions
In vitro studies indicated that pralatrexate does not induce or inhibit the activity of CYP450 isozymes at concentrations of pralatrexate that can be reasonably expected clinically.
In vitro, pralatrexate is a substrate for the breast cancer resistance protein (BCRP), MRP2, multidrug resistance-associated protein 3 (MRP3), and organic anion transport protein 1B3 (OATP1B3) transporter systems at concentrations of pralatrexate that can be reasonably expected clinically. Pralatrexate is not a substrate of the P glycoprotein (P-gp), organic anion transport protein 1B1 (OATP1B1), organic cation transporter 2 (OCT2), organic anion transporter 1 (OAT1), and organic anion transporter 3 (OAT3) transporter systems.
In vitro, pralatrexate inhibits MRP2 and MRP3 transporter systems ([I]/IC50 > 0.1) at concentrations of pralatrexate that can be reasonably expected clinically. MRP3 is a transporter that may affect the transport of etoposide and teniposide.
In vitro, pralatrexate did not significantly inhibit the P-gp, BCRP, OCT2, OAT1, OAT3, OATP1B1, and OATP1B3 transporter systems at concentrations of pralatrexate that can be reasonably expected clinically.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
-
14 CLINICAL STUDIES
SPL UNCLASSIFIED SECTION
Peripheral T-cell Lymphoma (PTCL)
The safety and efficacy of FOLOTYN was evaluated in an open-label, single-arm, multi-center, international trial that enrolled 115 patients with relapsed or refractory PTCL. One hundred and eleven patients were treated with FOLOTYN at 30 mg/m2 once weekly by IV push over 3-5 minutes for 6 weeks in 7-week cycles until disease progression or unacceptable toxicity. Of the 111 patients treated, 109 patients were evaluable for efficacy. Evaluable patients had histologically confirmed PTCL by independent central review using the Revised European American Lymphoma (REAL) World Health Organization (WHO) disease classification, and relapsed or refractory disease after at least one prior treatment.
The primary efficacy endpoint was overall response rate (complete response, complete response unconfirmed, and partial response) as assessed by International Workshop Criteria (IWC). The key secondary efficacy endpoint was duration of response. Response assessments were scheduled at the end of cycle 1 and then every other cycle (every 14 weeks). Duration of response was measured from the first day of documented response to disease progression or death. Response and disease progression were evaluated by independent central review using the IWC.
The median age of treated patients was 59.0 years (range 21-85); 68% were male and 32% were female. Most patients were White (72%) and other racial origins included: Black (13%), Hispanic (8%), Asian (5%), other and unknown (<1% each). Patients had an Eastern Cooperative Oncology Group (ECOG) performance status at study entry of 0 (39%), 1 (44%), or 2 (17%). The median time from initial diagnosis to study entry was 15.6 months (range 0.8 – 322.3).
The median number of prior systemic therapies was 3 (range 1-12). Approximately one-fourth of patients (24%, n = 27) did not have evidence of response to any previous therapy. Approximately two-thirds of patients (63%, n = 70) did not have evidence of response to their most recent prior therapy before entering the study.
In all evaluable patients (n = 109) treated with FOLOTYN, the response rate, as determined by independent central review by IWC, was 27% (n = 29) (Table 5).
Table 5 Response Analysis per Independent Central Review (IWC) Evaluable Patients
(N=109)
N (%)
95% CI
Median Duration of Response
Range of Duration of Response
Overall Response
CR+CRu+PR
29 (27)
19, 36
287 days
(9.4 months)
1-503 days
CR/CRu
9 (8)
PR
20 (18)
Responses ≥ 14 weeks
CR+CRu+PR
13 (12)
7, 20
Not Reached
98-503 days
CR/CRu
7 (6)
PR
6 (6)
Fourteen patients went off treatment in cycle 1; 2 patients were unevaluable for response by IWC due to insufficient materials provided to central review.
CR = Complete Response, CRu = Complete Response unconfirmed, PR = Partial ResponseThe initial response assessment was scheduled at the end of cycle 1. Of the responders, 66% responded within cycle 1. The median time to first response was 45 days (range 37-349 days).
-
15 REFERENCES
- 1. Preventing Occupational Exposures to Antineoplastic and Other Hazardous Drugs in Health Care Settings. NIOSH Alert 2004-165.
- 2. OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling Occupational Exposure to Hazardous Drugs. OSHA, 1999. http://www.osha.gov/dts/osta/o tm/otm_vi/otm_vi_2.html
- 3. American Society of Health-System Pharmacists. ASHP guidelines on handling hazardous drugs. Am J Health-Syst Pharm. 2006;63:1172-1193.
- 4. Polovich, M., White, J. M., & Kelleher, L. O. (eds.) 2005. Chemotherapy and biotherapy guidelines and recommendations for practice (2nd. ed.) Pittsburgh, PA: Oncology Nursing Society.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
FOLOTYN is available in single-dose clear glass vials containing pralatrexate at a concentration of 20 mg/mL as a preservative-free, sterile, clear yellow solution individually packaged for intravenous use in the following presentations:
NDC: 72893-003-01: 20 mg of pralatrexate in 1 mL solution in a vial (20 mg / 1 mL)
NDC: 72893-005-01: 40 mg of pralatrexate in 2 mL solution in a vial (40 mg / 2 mL)Vials must be stored refrigerated at 2-8°C (36-46°F) (see USP Controlled Cold Temperature) in original carton to protect from light.
Handle and dispose of FOLOTYN according to guidelines issued for cytotoxic drugs, including the use of gloves and other protective clothing to prevent skin contact [see References (15)].
Each vial of FOLOTYN is intended for single use only. Any unused drug remaining after injection must be discarded.
-
17 PATIENT COUNSELING INFORMATION
See FDA-approved labeling (Patient Information).
Patients should be instructed to read the Patient Information carefully.
17.1 Need for Folic Acid and Vitamin B12
Advise patients treated with FOLOTYN to take folic acid and vitamin B12 as a prophylactic measure to reduce the risk of possible side effects [see Dosage and Administration (2.1)].
17.2 Low Blood Cell Counts
Inform patients of the risk of low blood cell counts and to immediately contact their physician should any signs of infection develop, including fever. Inform patients to contact their physician if bleeding or symptoms of anemia occur.
17.3 Mucositis
Inform patients of the signs and symptoms of mucositis. Instruct patients on ways to reduce the risk of its development, and on ways to maintain nutrition and control discomfort from mucositis if it occurs.
17.4 Fatal Dermatologic Reactions
Advise patients about the risks for and the signs and symptoms of dermatologic reactions. Instruct patients to immediately notify their physician if any skin reactions occur [see Warnings and Precautions (5.3)].
17.5 Tumor Lysis Syndrome
Inform patients about the risk of and the signs and symptoms of tumor lysis syndrome. Patients should be instructed to notify their physician if they experience these symptoms [see Warnings and Precautions (5.4)].
17.6 Concomitant Medications
Patients should be instructed to inform their physician if they are taking any concomitant medications including prescription drugs (such as trimethoprim/sulfamethoxazole) and nonprescription drugs (such as nonsteroidal anti-inflammatory drugs) [see Drug Interactions (7)].
17.7 Pregnancy/Nursing
Patients should be instructed to tell their physician if they are pregnant or plan to become pregnant due to the risk of fetal harm. Patients should be instructed to tell their physician if they are nursing.
Manufactured for:
Acrotech Biopharma LLC
East Windsor, NJ 08520
1-888-255-6788
FOLOTYN is a registered trademark of Acrotech Biopharma LLC.
U.S. Patents: 6,028,071, 7,622,470 and 8,299,078
Rev. September 2019© Acrotech Biopharma LLC. All rights reserved.
-
SPL PATIENT PACKAGE INSERT SECTION
Patient Information
FOLOTYN®(FOH-loh-tin)
(pralatrexate injection)Read the Patient Information that comes with FOLOTYN before you start treatment and each time you get treated with FOLOTYN. There may be new information. This leaflet does not take the place of talking to your doctor about your medical condition or treatment. Talk to your doctor if you have any questions about FOLOTYN.
What is FOLOTYN?
FOLOTYN is a prescription anti-cancer (chemotherapy) medicine. FOLOTYN is used to treat people with a type of cancer called Peripheral T-cell Lymphoma (PTCL) that does not go away, gets worse, or comes back after use of another cancer treatment.
What should I tell my doctor before receiving FOLOTYN?
Before you receive FOLOTYN, tell your doctor if you:
- have liver problems.
- have kidney problems.
- have any other medical conditions.
- are pregnant or plan to become pregnant. FOLOTYN can harm your unborn baby. Talk to your doctor about the best way to prevent pregnancy while taking FOLOTYN. Tell your doctor right away if you become pregnant while taking FOLOTYN.
- are breast-feeding or plan to breast-feed. It is not known if FOLOTYN passes into breast milk. You and your doctor should decide if you will take FOLOTYN or breast-feed. You should not do both. Talk to your doctor about the best way to feed your baby while you are being treated with FOLOTYN.
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. Some medicines may affect how FOLOTYN works, and FOLOTYN may affect how other medicines work. Especially tell your doctor if you take:
- sulfamethoxazole trimethoprim (Bactrim®, Septra®, Septra DS, Sulfatrim Pediatric, Sulfamethoprim, Sulfamethoprim-DS)
- non-steroidal anti-inflammatory drugs (NSAIDs)
- probenecid (Probalan, Col-Probenecid)
Ask your doctor or pharmacist if you are not sure if your medicine is listed above.
Know the medicines you take. Keep a list of them and show it to your doctor or pharmacist each time you start a new medicine.
How will I receive FOLOTYN?
- FOLOTYN will be given to you as directed by your doctor, as an intravenous (IV) injection into your vein over 3 to 5 minutes.
- FOLOTYN is usually given in cycles, one time each week for 6 weeks, with no treatment on the 7th week. Treatment with FOLOTYN may be continued as long as it is helpful to you.
To lower your chances of harmful side effects, it is important that you take folic acid and vitamin B12 during your treatment with FOLOTYN. Your doctor will give you specific instructions for vitamin supplementation.
- You will take folic acid by mouth for 10 days before your first dose of FOLOTYN. Do not take more or less folic acid than your doctor tells you to take. Continue taking folic acid every day until your doctor tells you to stop.
- Your doctor will give you a vitamin B12 injection into your muscle (intramuscular) before your first dose of FOLOTYN and about every 8 to 10 weeks during treatment with FOLOTYN.
You should have regular blood tests before and during your treatment with FOLOTYN. Your doctor may change your dose of FOLOTYN or delay treatment based on the results of your blood tests and on your general condition.
What are the possible side effects of FOLOTYN?
FOLOTYN may cause serious side effects, including:
- Low Blood Cell Counts: FOLOTYN can affect your bone marrow and cause you to have low blood cell counts. Your doctor will do blood tests as needed to check your blood cell counts.
- Low Platelet Count (thrombocytopenia): Tell your doctor right away if you have any unusual bleeding, such as nosebleeds, or bruising under your skin.
- Low White Blood Cell Count (neutropenia): A low white blood cell count can cause you to get infections, which may be serious. Serious illness or death can happen if an infection is not treated right away when white blood cell counts are very low. Tell your doctor right away if you have any of the following signs or symptoms of an infection:
- fever
- chills
- cough
- shortness of breath
- pain or burning on urination
- Low Red Blood Cell Count (anemia): Tell your doctor if you have any of these symptoms of anemia during treatment with FOLOTYN:
- feeling weak, tired, or you get tired easily
- you look pale
- you feel short of breath
- Redness and sores of the mucous membrane lining of the mouth, lips, throat, digestive tract, and genitals (mucositis). Discomfort or pain due to mucositis may happen as early as a few days after treatment with FOLOTYN. Your doctor should tell you about ways to reduce your risk of getting mucositis, and how to maintain nutrition and control the discomfort from mucositis.
- Severe skin reactions. Severe skin reactions may happen after treatment with FOLOTYN, especially if you have lymphoma in or under your skin. If your skin reactions are severe, they may lead to serious illness or death. Tell your doctor right away if you have any of the following skin reactions:
- rash
- peeling and loss of skin
- sores
- blisters
- Tumor Lysis Syndrome (TLS). FOLOTYN can cause the fast breakdown of certain types of cancer cells. This can lead to TLS. Your doctor may do blood tests to check you for TLS and treat you for TLS if needed.
- Harm to an unborn baby. Females should avoid becoming pregnant while being treated with FOLOTYN. Talk to your doctor about how to avoid pregnancy while taking FOLOTYN.
- Fever. Fever is often one of the most common and earliest signs of infection. Follow your doctor's instructions about how often to take your temperature, especially during the days after treatment with FOLOTYN. If you have a fever, tell your doctor or nurse right away.
- Loss of too much fluid from the body (dehydration). If you feel tired and weak this could be a sign of dehydration. Follow your doctor's instructions for what to do to help prevent or treat dehydration.
- Shortness of breath. Tell your doctor if this is a problem for you.
Common side effects of FOLOTYN include:
- nausea
- vomiting
- tiredness
- constipation
- swelling
- cough
- nosebleed
- diarrhea
These are not all the possible side effects of FOLOTYN. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You can report side effects to FDA at 1-800-FDA-1088.
General information about FOLOTYN
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. This patient information leaflet summarizes the most important information about FOLOTYN. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about FOLOTYN that is written for health professionals.
For more information, go to www.FOLOTYN.com or call 1-888-255-6788.
What are the ingredients in FOLOTYN?
Active ingredient: pralatrexate
Inactive ingredients: sodium chloride, sodium hydroxide, and hydrochloric acid
What is PTCL?
PTCL is a rare type of non-Hodgkin's lymphoma, a cancer of the lymphatic system. It happens when a type of T-cell (a kind of white blood cell) grows too much. PTCL may be found in different parts of the body, such as the lymph nodes, skin, bone marrow, liver, or spleen.
Acrotech Biopharma LLC
East Windsor, NJ 08520
Issued: May 2010
Revised: September 2019
FOLOTYN and the flower symbol are all registered trademarks of Acrotech Biopharma LLC.
© Acrotech Biopharma LLC. All rights reserved.
-
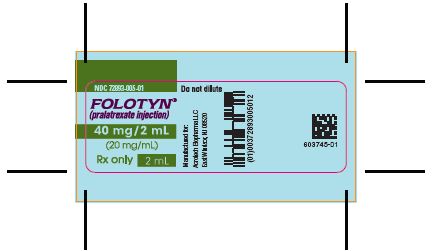
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
PACKAGE CARTON - FOLOTYN 20 mg/1 mL Vial
NDC: 72893-003-01
FOLOTYN®(pralatrexate injection)
20 mg/mL
For intravenous use
Rx only
1 mL
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
FOLOTYN
pralatrexate injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72893-003 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength pralatrexate (UNII: A8Q8I19Q20) (pralatrexate - UNII:A8Q8I19Q20) pralatrexate 20 mg in 1 mL Inactive Ingredients Ingredient Name Strength sodium chloride (UNII: 451W47IQ8X) sodium hydroxide (UNII: 55X04QC32I) hydrochloric acid (UNII: QTT17582CB) water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72893-003-01 1 in 1 CARTON 09/24/2009 1 1 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022468 09/24/2009 FOLOTYN
pralatrexate injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72893-005 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength pralatrexate (UNII: A8Q8I19Q20) (pralatrexate - UNII:A8Q8I19Q20) pralatrexate 40 mg in 2 mL Inactive Ingredients Ingredient Name Strength sodium chloride (UNII: 451W47IQ8X) sodium hydroxide (UNII: 55X04QC32I) hydrochloric acid (UNII: QTT17582CB) water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72893-005-01 1 in 1 CARTON 09/24/2009 1 2 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022468 09/24/2009 Labeler - Acrotech Biopharma LLC (116965616) Registrant - Acrotech Biopharma LLC (116965616) Establishment Name Address ID/FEI Business Operations Baxter Oncology, GmbH, (Baxter) 344276063 MANUFACTURE(72893-003, 72893-005)
Trademark Results [Folotyn]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 FOLOTYN 78881722 3743409 Live/Registered |
Allos Therapeutics, Inc. 2006-05-11 |
 FOLOTYN 77939090 3887969 Dead/Cancelled |
Allos Therapeutics, Inc. 2010-02-18 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.