TECARTUS- brexucabtagene autoleucel suspension
TECARTUS by
Drug Labeling and Warnings
TECARTUS by is a Other medication manufactured, distributed, or labeled by Kite Pharma, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TECARTUS safely and effectively. See full prescribing information for TECARTUS.
TECARTUS® (brexucabtagene autoleucel) suspension for intravenous infusion
Initial U.S. Approval: 2020WARNING: CYTOKINE RELEASE SYNDROME, NEUROLOGIC TOXICITIES, AND SECONDARY HEMATOLOGICAL MALIGNANCIES
See full prescribing information for complete boxed warning.
- Cytokine Release Syndrome (CRS), including life-threatening reactions, occurred in patients receiving TECARTUS. Do not administer TECARTUS to patients with active infection or inflammatory disorders. Treat severe or life-threatening CRS with tocilizumab or tocilizumab and corticosteroids (2.2, 2.3, 5.1).
- Neurologic toxicities, including life-threatening reactions, occurred in patients receiving TECARTUS, including concurrently with CRS or after CRS resolution. Monitor for neurologic toxicities after treatment with TECARTUS. Provide supportive care and/or corticosteroids, as needed (2.2, 2.3, 5.2).
- T cell malignancies have occurred following treatment of hematologic malignancies with BCMA- and CD19-directed genetically modified autologous T cell immunotherapies (5.8).
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
TECARTUS is a CD19-directed genetically modified autologous T cell immunotherapy indicated for the treatment of: (1)
- Adult patients with relapsed or refractory mantle cell lymphoma (MCL).
- Adult patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL).
DOSAGE AND ADMINISTRATION
For autologous use only. For intravenous use only.
- Do NOT use a leukodepleting filter.
- Administer a lymphodepleting regimen of cyclophosphamide and fludarabine before infusion of TECARTUS. (2.2)
- Verify the patient's identity prior to infusion. (2.2)
- Premedicate with acetaminophen and diphenhydramine. (2.2)
- Confirm availability of tocilizumab prior to infusion. (2.2, 5.1)
- Dosing of TECARTUS is based on the number of chimeric antigen receptor (CAR)-positive viable T cells. (2.1)
- MCL: dose is 2 × 106 CAR-positive viable T cells per kg body weight, with a maximum of 2 × 108 CAR-positive viable T cells. (2.1)
- ALL: dose is 1 × 106 CAR-positive viable T cells per kg body weight, with a maximum of 1 × 108 CAR-positive viable T cells. (2.1)
DOSAGE FORMS AND STRENGTHS
- TECARTUS is available as a cell suspension for infusion.
- MCL: Comprises a suspension of 2 × 106 CAR-positive viable T cells per kg of body weight, with a maximum of 2 × 108 CAR-positive viable T cells in approximately 68 mL. (3)
- ALL: Comprises a suspension of 1 × 106 CAR-positive viable T cells per kg of body weight, with a maximum of 1 × 108 CAR-positive viable T cells in approximately 68 mL. (3)
CONTRAINDICATIONS
- None. (4)
WARNINGS AND PRECAUTIONS
- Hemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome: Administer treatment per institutional standards. (5.3)
- Hypersensitivity Reactions: Monitor for hypersensitivity reactions during infusion. (5.4)
- Severe Infections: Monitor patients for signs and symptoms of infection; treat appropriately. (5.5)
- Prolonged Cytopenias: Patients may exhibit Grade 3 or higher cytopenias for several weeks following TECARTUS infusion. Monitor complete blood counts. (5.6)
- Hypogammaglobulinemia: Monitor and provide replacement therapy. (5.7)
- Secondary Malignancies: T cell malignancies have occurred following treatment of hematologic malignancies with BCMA- and CD19-directed genetically modified autologous T cell immunotherapies. In the event that a secondary malignancy occurs after treatment with TECARTUS, contact Kite at 1-844-454-KITE (5483). (5.8)
ADVERSE REACTIONS
The most common non-laboratory adverse reactions (incidence greater than or equal to 20%) are:
- MCL: CRS, fever, encephalopathy, hypotension, infection with pathogen unspecified, viral infections, fatigue, tachycardias, chills, hypoxia, tremor, cough, musculoskeletal pain, nausea, edema, headache, constipation, diarrhea, decreased appetite, dyspnea, rash, insomnia, pleural effusion, aphasia, motor dysfunction.
- ALL: fever, CRS, hypotension, encephalopathy, tachycardia, nausea, chills, headache, fatigue, febrile neutropenia, diarrhea, musculoskeletal pain, hypoxia, rash, edema, tremor, infection with pathogen unspecified, constipation, decreased appetite, and vomiting. (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Kite at 1-844-454-KITE (5483) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 4/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: CYTOKINE RELEASE SYNDROME, NEUROLOGIC TOXICITIES, and SECONDARY HEMATOLOGICAL MALIGNANCIES
1 INDICATIONS AND USAGE
1.1 Mantle Cell Lymphoma
1.2 Acute Lymphoblastic Leukemia
2 DOSAGE AND ADMINISTRATION
2.1 Dose
2.2 Administration
2.3 Management of Severe Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Cytokine Release Syndrome
5.2 Neurologic Toxicities
5.3 Hemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome
5.4 Hypersensitivity Reactions
5.5 Severe Infections
5.6 Prolonged Cytopenias
5.7 Hypogammaglobulinemia
5.8 Secondary Malignancies
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Relapsed or Refractory Mantle Cell Lymphoma
14.2 Relapsed or Refractory B-cell precursor Acute Lymphoblastic Leukemia
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: CYTOKINE RELEASE SYNDROME, NEUROLOGIC TOXICITIES, and SECONDARY HEMATOLOGICAL MALIGNANCIES
- Cytokine Release Syndrome (CRS), including life-threatening reactions, occurred in patients receiving TECARTUS. Do not administer TECARTUS to patients with active infection or inflammatory disorders. Treat severe or life-threatening CRS with tocilizumab or tocilizumab and corticosteroids [see Dosage and Administration (2.2, 2.3), Warnings and Precautions (5.1)].
- Neurologic toxicities, including life-threatening reactions, occurred in patients receiving TECARTUS, including concurrently with CRS or after CRS resolution. Monitor for neurologic toxicities after treatment with TECARTUS. Provide supportive care and/or corticosteroids, as needed [see Dosage and Administration (2.2, 2.3), Warnings and Precautions (5.2)].
- T cell malignancies have occurred following treatment of hematologic malignancies with BCMA- and CD19-directed genetically modified autologous T cell immunotherapies [see Warnings and Precautions (5.8)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dose
For autologous use only. For intravenous use only.
2.2 Administration
TECARTUS is for autologous use only. The patient's identity must match the patient identifiers on the TECARTUS cassette and infusion bag. Do not infuse TECARTUS if the information on the patient-specific label does not match the intended patient.
Preparing Patient for TECARTUS Infusion
Confirm availability of TECARTUS prior to starting the lymphodepleting chemotherapy regimen.
Pre-treatment
- MCL: Administer a lymphodepleting chemotherapy regimen of cyclophosphamide 500 mg/m2 intravenously and fludarabine 30 mg/m2 intravenously on each of the fifth, fourth, and third day before infusion of TECARTUS.
- ALL: Administer a lymphodepleting chemotherapy regimen of fludarabine 25 mg/m2 intravenously over 30 minutes on the fourth, third, and second day and administer cyclophosphamide 900 mg/m2 over 60 minutes on the second day before infusion of TECARTUS.
Preparation of TECARTUS for infusion
Coordinate the timing of TECARTUS thaw and infusion. Confirm the infusion time in advance, and adjust the start time of TECARTUS thaw such that TECARTUS will be available for infusion when the patient is ready.
- Confirm patient identity: Prior to TECARTUS preparation, match the patient's identity with the patient identifiers on the TECARTUS cassette.
- Do not remove the TECARTUS infusion bag from the cassette if the patient information on the cassette label does not match the intended patient.
- Once patient identity is confirmed, remove the TECARTUS infusion bag from the cassette and check that the patient information on the cassette label matches the patient information on the bag label.
- Inspect the infusion bag for any breaches of container integrity such as breaks or cracks before thawing. If the bag is compromised, follow the local guidelines (or call Kite at 1-844-454-KITE).
- Place the infusion bag inside a second sterile bag per local guidelines.
- Thaw the infusion bag at approximately 37°C using either a water bath or dry-thaw method until there is no visible ice in the infusion bag.
- Gently mix the contents of the bag to disperse clumps of cellular material. If visible cell clumps remain, continue to gently mix the contents of the bag. Small clumps of cellular material should disperse with gentle manual mixing. Do not wash, spin down, and/or re-suspend TECARTUS in new media prior to infusion.
- Once thawed, TECARTUS should be administered within 30 minutes but may be stored at room temperature (20°C to 25°C) for up to 3 hours.
Administration
- For autologous use only.
- Confirm that tocilizumab and emergency equipment are available prior to infusion and during the recovery period.
- Do NOT use a leukodepleting filter.
- Central venous access is recommended for the administration of TECARTUS.
- Confirm that the patient's identity matches the patient identifiers on the TECARTUS infusion bag.
- Prime the tubing with normal saline prior to infusion.
- Infuse the entire contents of the TECARTUS bag within 30 minutes by either gravity or a peristaltic pump. TECARTUS is stable at room temperature for up to 3 hours after thaw.
- Gently agitate the TECARTUS bag during infusion to prevent cell clumping.
- After the entire contents of the TECARTUS bag are infused, rinse the tubing with normal saline at the same infusion rate to ensure all product is delivered.
TECARTUS contains human blood cells that are genetically modified with replication-incompetent retroviral vector. Follow universal precautions and local biosafety guidelines for handling and disposal of TECARTUS to avoid potential transmission of infectious diseases.
Monitoring
- Monitor patients daily for at least 7 days following infusion for signs and symptoms of Cytokine Release Syndrome (CRS) and neurologic events.
- Instruct patients to remain within proximity of a healthcare facility for at least 2 weeks following infusion.
- Advise patients to avoid driving for at least 2 weeks following infusion.
2.3 Management of Severe Adverse Reactions
Cytokine Release Syndrome (CRS)
Identify CRS based on clinical presentation [see Warnings and Precautions (5.1)]. Evaluate for and treat other causes of fever, hypoxia, and hypotension. If CRS is suspected, manage according to the recommendations in Table 1. Patients who experience Grade 2 or higher CRS (e.g., hypotension, not responsive to fluids, or hypoxia requiring supplemental oxygenation) should be monitored with continuous cardiac telemetry and pulse oximetry. For patients experiencing severe CRS, consider performing an echocardiogram to assess cardiac function. For severe or life-threatening CRS, consider intensive care supportive therapy. Physicians may also consider management per current practice guidelines.
Table 1. CRS Grading and Management Guidance CRS Grade* Supportive Care Tocilizumab Corticosteroids Abbreviations: ICU, intensive care unit; IV, intravenous/intravenously - * Lee et al. 2014. /
- † Refer to tocilizumab Prescribing Information for details.
- ‡ Refer to Table 2 for management of neurologic toxicity.
- § Refer to current practice guidelines for alternative immunosuppressant agents.
Grade 1
Symptoms require symptomatic treatment only (e.g., fever, nausea, fatigue, headache, myalgia, malaise).Supportive care with antipyretics, IV hydration, and symptomatic management of organ toxicities and constitutional symptoms If not improving after 24 hours, administer tocilizumab† 8 mg/kg IV over 1 hour (not to exceed 800 mg). Not applicable. Grade 2
Symptoms require and respond to moderate intervention.
Oxygen requirement less than 40% FiO2 or hypotension responsive to fluids or low dose of one vasopressor or Grade 2 organ toxicity.‡Continue supportive care as per Grade 1 and include IV fluid bolus and/or supplemental oxygen as needed Administer tocilizumab 8 mg/kg IV over 1 hour (not to exceed 800 mg).
Repeat tocilizumab every 8 hours as needed if not responsive to IV fluids or increasing supplemental oxygen. Limit to a maximum of 3 doses in a 24-hour period; maximum total of 4 doses if no clinical improvement in the signs and symptoms of CRS.
If improving, discontinue tocilizumab.In patients with hypotension that persists after 2 fluid boluses and after 1 to 2 doses of tocilizumab, may consider dexamethasone 10 mg IV (or equivalent) every 12 hours for 1 to 2 doses and then reassess.
Manage per Grade 3 if no improvement within 24 hours after starting tocilizumab.
If improving, taper corticosteroids.Grade 3
Symptoms require and respond to aggressive intervention.
Oxygen requirement greater than or equal to 40% FiO2 or hypotension requiring high-dose or multiple vasopressors or Grade 3 organ toxicity or Grade 4 transaminitis.Transfer to ICU
Continue supportive care per Grade 2.
If echocardiogram was not already performed, obtain ECHO to assess cardiac function and conduct hemodynamic monitoring.Per Grade 2
If improving, discontinue tocilizumab.Administer dexamethasone 10 mg IV every 6-8 hours or methylprednisolone 1 mg/kg IV twice daily until Grade 1, then taper corticosteroids.
If improving, manage as Grade 2.
If not improving, manage as Grade 4.Grade 4
Life-threatening symptoms.
Requirements for ventilator support or continuous veno-venous hemodialysis (CVVHD), or Grade 4 organ toxicity (excluding transaminitis).Transfer to ICU
Continue supportive care as per Grade 2.Per Grade 2
If improving, discontinue tocilizumab.
If not improving, consider alternate immunosuppressants§Administer methylprednisolone 1000 mg IV per day for 3 days.
If improving, taper corticosteroids, and manage as Grade 3.
If not improving during first 24 hours, consider adding alternate immunosuppressants.Neurologic Toxicity
Monitor patients daily for signs and symptoms of neurologic toxicities/immune effector cell-associated neurotoxicity syndrome (ICANS) (Table 2). Rule out other causes of neurologic symptoms. Patients who experience Grade 2 or higher neurologic toxicities/ICANS should be monitored with continuous cardiac telemetry and pulse oximetry and provide neuroimaging of the brain (brain MRI or CT scan); for persisting Grade 2 or higher neurologic toxicities/ICANS consider repeat neuroimaging every 2 – 3 days. Provide intensive care supportive therapy for severe or life-threatening neurologic toxicities/ICANS. Consider non-sedating anti-seizure medicines (e.g., levetiracetam) for seizure prophylaxis for any Grade 2 or higher neurologic toxicities. Physicians may also consider management per current practice guidelines.
Table 2. Neurologic Toxicity/ICANS Grading and Management Guidance Neurologic Event* Concurrent CRS No Concurrent CRS Abbreviation: ADLs, activities of daily living; ICE, immune effector cell–associated encephalopathy; ICU, intensive care unit; IV, intravenous/intravenously - * Severity based on Common Terminology Criteria for Adverse Events.
- † Severity based on American Society of Transplantation and Cellular Therapy (ASTCT) consensus grading
- ‡ Refer to current practice guidelines for alternative immunosuppressant agents.
Grade 1
ICANS† Grade 1: ICE score 7-9
Examples include:
Somnolence – mild drowsiness or sleepiness
Confusion – mild disorientation
Encephalopathy – mild limiting of ADLs
Dysphasia – not impairing ability to communicateAdminister tocilizumab 8 mg/kg IV over 1 hour (not to exceed 800 mg).
Repeat tocilizumab every 8 hours as needed. Limit to a maximum of 3 doses in a 24-hour period; maximum total of 4 doses. Consider adding corticosteroids (dexamethasone 10mg IV) to tocilizumab past the first dose.Supportive care. If persisting, consider corticosteroids (dexamethasone 10 mg IV). Grade 2
ICANS† Grade 2: ICE score 3-6
Examples include:
Somnolence – awakening to voice moderate limiting instrumental ADLs
Confusion – moderate disorientation
Encephalopathy – limiting instrumental ADLs
Dysphasia moderate impairing ability to communicate spontaneously
Seizure(s)Administer tocilizumab per Table 1 for management of Grade 2 CRS. Consider adding corticosteroids (dexamethasone 10 mg IV or methylprednisolone 1 mg/kg IV every 12 hours) to tocilizumab past the first dose, if symptoms are refractory to tocilizumab. Consider transfer in ICU, if ICANS associated with ≥ Grade 2 CRS.
If not improving within 24 hours after starting tocilizumab, administer dexamethasone 10 mg IV every 6 hours or methylprednisolone 1 mg/kg IV every 12 hours until the event is Grade 1 or less, then rapidly taper corticosteroids.
If improving, discontinue tocilizumab.
If still not improving, manage as Grade 3.Administer dexamethasone 10 mg IV every 6 hours until the event is Grade 1 or less.
If improving, taper corticosteroids.Consider non-sedating, anti-seizure medicines (e.g., levetiracetam) for seizure prophylaxis. Grade 3
ICANS† Grade 3: ICE score 0-2
Examples include:
Somnolence – obtundation or stupor; awakening only to tactile stimulus
Confusion – severe disorientation
Encephalopathy – limiting self-care ADLs
Dysphasia – severe receptive or expressive characteristics, impairing ability to read, write, or communicate intelligibly
Seizure(s) focal / non-convulsive seizures on EEG,
Focal or local edema on neuroimagingTransfer to ICU
Administer tocilizumab per Table 1 for management of Grade 2 CRS.
In addition, administer dexamethasone 10 mg IV every 6 hours or methylprednisolone 1 mg/kg IV every 8 to 12 hours starting with the first dose of tocilizumab and repeat dose every 6 hours. Continue dexamethasone or methylprednisolone use until the event is Grade 1 or less, then taper corticosteroids.
If improving, discontinue tocilizumab and manage as Grade 2.
If still not improving, manage as Grade 4.Transfer to ICU
Administer dexamethasone 10 mg IV every 6 hours or methylprednisolone 1 mg/kg IV every 8 to 12 hours.
Continue dexamethasone/methylprednisolone use until the event is Grade 1 or less, then taper corticosteroids.
If not improving, manage as Grade 4.Consider non-sedating anti-seizure medicines (e.g., levetiracetam) for seizure prophylaxis. Grade 4
ICANS† Grade 4: ICE score 0
Life-threatening consequences
Urgent intervention required
Requirement for mechanical ventilation
Life-threatening / prolonged seizure
Diffuse cerebral edema on neuro-imagingTransfer to ICU
Administer tocilizumab per Table 1 for management of Grade 2 CRS.
Administer methylprednisolone 1000 mg IV 1-2 times per day with first dose of tocilizumab and continue methylprednisolone 1000 mg IV per day for 2 more days.
If improving, then manage as Grade 3.
If not improving, consider alternate immunosuppressants.‡Transfer to ICU
Administer methylprednisolone 1000 mg IV 1-2 times per day for 3 days.
If improving, then manage as Grade 3.
If not improving, consider alternate immunosuppressants.‡Consider non-sedating anti-seizure medicines (e.g., levetiracetam) for seizure prophylaxis. -
3 DOSAGE FORMS AND STRENGTHS
TECARTUS is available as a cell suspension for infusion.
- MCL: A single dose of TECARTUS contains 2 × 106 CAR-positive viable T cells per kg of body weight [maximum of 2 × 108 CAR-positive viable T cells (for patients 100 kg and above)] in approximately 68 mL suspension in an infusion bag [see How Supplied/Storage and Handling (16)].
- ALL: A single dose of TECARTUS contains 1 × 106 CAR-positive viable T cells per kg of body weight [maximum of 1 × 108 CAR-positive viable T cells (for patients 100 kg and above)] in approximately 68 mL suspension in an infusion bag [see How Supplied/Storage and Handling (16)].
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Cytokine Release Syndrome
Cytokine Release Syndrome (CRS), including fatal or life-threatening reactions, occurred following treatment with TECARTUS. CRS occurred in 93% (157/168) of patients with MCL, including ≥ Grade 3 (Lee 2014 grading system1) CRS in 12% of patients in Study 1. Among the patients with MCL who died after receiving TECARTUS, 1 patient had a fatal CRS event. The median time to onset of CRS was 4 days (range: 1 to 13 days). The median duration of CRS was 7 days (range: 1 to 50 days). CRS occurred in 92% (72/78) of patients with ALL, including ≥ Grade 3 (Lee grading system1) CRS in 26% of patients. Three patients with ALL had ongoing CRS events at the time of death. The median time to onset of CRS was 5 days (range: 1 to 12 days) and the median duration of CRS was 8 days (range: 2 to 63 days) for patients with ALL.
Confirm that a minimum of 2 doses of tocilizumab are available for each patient prior to infusion of TECARTUS. Monitor patients daily for at least 7 days following infusion for signs and symptoms of CRS. Monitor patients for signs or symptoms of CRS for 2 weeks after infusion. Counsel patients to seek immediate medical attention should signs or symptoms of CRS occur at any time. At the first sign of CRS, institute treatment with supportive care, tocilizumab, or tocilizumab and corticosteroids as indicated [see Dosage and Administration (2.3)].
5.2 Neurologic Toxicities
Neurologic toxicities (including ICANS), which may be severe, life-threatening or fatal, occurred following treatment with TECARTUS. Neurologic events occurred in 80% (135/168) of patients with MCL, including ≥ Grade 3 in 33% of patients in Study 1. The median time to onset for neurologic events was 6 days (range: 1 to 32 days). The median duration of neurologic events was 19 days (range: 1 to 828 days). Neurologic events occurred in 87% (68/78) of patients with ALL, including ≥ Grade 3 in 35% of patients. The median time to onset for neurologic events was 7 days (range: 1 to 51 days) with a median duration of 15 days (range: 1 to 397 days) in patients with ALL. For patients with MCL 105 (63%) patients experienced CRS before the onset of neurological events. Six (4%) patients did not experience CRS with neurologic events and 25 patients (15%) developed neurological events after the resolution of CRS. Neurologic events resolved for 167 out of 203 (82%) patients treated with TECARTUS. Fourteen patients (8 patients with MCL and 6 patients with ALL) had ongoing neurologic events at the time of death. For patients with ALL, neurologic events occurred before, during, and after CRS in 4 (5%), 57 (73%), and 8 (10%) of patients; respectively. Three patients (4%) had neurologic events without CRS. The onset of neurologic events can be concurrent with CRS, following resolution of CRS or in the absence of CRS.
Monitor patients daily for at least 7 days following infusion for signs and symptoms of neurologic toxicity/ICANS. Monitor patients for signs or symptoms of neurologic toxicities for 2 weeks after infusion and treat promptly [see Dosage and Administration (2.3)]. Advise patients to avoid driving for at least 2 weeks following infusion.
5.3 Hemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome
Hemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome (HLH/MAS), including life-threatening reactions, occurred following treatment with TECARTUS. HLH/MAS occurred in 4% (3/78) of patients with ALL. Two patients experienced Grade 3 events and 1 patient experienced a Grade 4 event. The median time to onset for HLH/MAS was 8 days (range: 6 to 9 days) with a median duration of 5 days (range: 2 to 8 days). All 3 patients with HLH/MAS had concurrent CRS symptoms and neurologic events after TECARTUS infusion. Treatment of HLH/MAS should be administered per institutional standards.
5.4 Hypersensitivity Reactions
Serious hypersensitivity reactions, including anaphylaxis, may occur due to dimethyl sulfoxide (DMSO) or residual gentamicin in TECARTUS.
5.5 Severe Infections
Severe or life-threatening infections occurred in patients after TECARTUS infusion. Infections (all grades) occurred in 63% (105/168) of patients with MCL and 44% (34/78) of patients with ALL. Grade 3 or higher infections, including bacterial, viral, and fungal infections, occurred in 33% of patients with MCL and 29% of patients with ALL. TECARTUS should not be administered to patients with clinically significant active systemic infections. Monitor patients for signs and symptoms of infection before and after TECARTUS infusion and treat appropriately. Administer prophylactic antimicrobials according to local guidelines.
Febrile neutropenia was observed in 4% of patients with MCL and 35% of patients with ALL after TECARTUS infusion and may be concurrent with CRS. The febrile neutropenia in 27 (35%) of patients with ALL includes events of "febrile neutropenia" (11 (14%)) plus the concurrent events of "fever" and "neutropenia" (16 (21%)). In the event of febrile neutropenia, evaluate for infection and manage with broad spectrum antibiotics, fluids, and other supportive care as medically indicated.
In immunosuppressed patients, life-threatening and fatal opportunistic infections have been reported. The possibility of rare infectious etiologies (e.g., fungal and viral infections such as HHV-6 and progressive multifocal leukoencephalopathy) should be considered in patients with neurologic events and appropriate diagnostic evaluations should be performed.
Hepatitis B Reactivation
Hepatitis B virus (HBV) reactivation, in some cases resulting in fulminant hepatitis, hepatic failure, and death, can occur in patients treated with drugs directed against B cells. Perform screening for HBV, hepatitis C virus (HCV), and human immunodeficiency virus (HIV) in accordance with clinical guidelines before collection of cells for manufacturing.
5.6 Prolonged Cytopenias
Patients may exhibit cytopenias for several weeks following lymphodepleting chemotherapy and TECARTUS infusion. In patients with MCL, Grade 3 or higher cytopenias not resolved by Day 30 following TECARTUS infusion occurred in 55% (92/168) of patients and included thrombocytopenia (32%), neutropenia (42%), and anemia (14%). In patients with ALL who were responders to TECARTUS treatment, Grade 3 or higher cytopenias not resolved by Day 30 following TECARTUS infusion occurred in 20% (7/35) of the patients and included neutropenia (12%) and thrombocytopenia (12%); Grade 3 or higher cytopenias not resolved by Day 60 following TECARTUS infusion occurred in 11% (4/35) of the patients and included neutropenia (9%) and thrombocytopenia (6%). Monitor blood counts after TECARTUS infusion.
5.7 Hypogammaglobulinemia
B cell aplasia and hypogammaglobulinemia can occur in patients receiving treatment with TECARTUS. Hypogammaglobulinemia was reported in 14% (23/168) of patients with MCL and 9% (7/78) of patients with ALL. Monitor immunoglobulin levels after treatment with TECARTUS and manage using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement.
The safety of immunization with live viral vaccines during or following TECARTUS treatment has not been studied. Vaccination with live virus vaccines is not recommended for at least 6 weeks prior to the start of lymphodepleting chemotherapy, during TECARTUS treatment, and until immune recovery following treatment with TECARTUS.
5.8 Secondary Malignancies
Patients treated with TECARTUS may develop secondary malignancies. T cell malignancies have occurred following treatment of hematologic malignancies with BCMA- and CD19-directed genetically modified autologous T cell immunotherapies. Mature T cell malignancies, including CAR-positive tumors, may present as soon as weeks following infusion, and may include fatal outcomes. [see Boxed Warning, Adverse Reactions (6.2),].
Monitor life-long for secondary malignancies. In the event that a secondary malignancy occurs, contact Kite at 1-844-454-KITE (5483) to obtain instructions on patient samples to collect for testing.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Study 1 (Relapsed/Refractory Mantle Cell Lymphoma)
Cohort 1 and 2
The safety of TECARTUS in patients with relapsed/refractory Mantle cell lymphoma (MCL) was evaluated in a single-arm study (Study 1; Cohorts 1 and 2) in which a total of 82 patients received a single dose of TECARTUS (2 × 106 or 0.5 × 106 anti-CD19 CAR T cells/kg) [see Clinical Studies (14.1)].
The most common serious adverse reactions (> 2%) were encephalopathy, fever, infection with pathogen unspecified, CRS, hypoxia, aphasia, renal insufficiency, pleural effusion, respiratory failure, bacterial infections, dyspnea, fatigue, arrhythmia, tachycardia, and viral infections. Table 3 summarizes the adverse reactions that occurred in at least 10% of patients in Study 1 Cohorts 1 and 2 and Table 4 lists the laboratory abnormalities of Grade 3 or 4 that occurred in at least 10% of patients in Study 1 Cohorts 1 and 2.
Table 3. Adverse Reactions Observed in at Least 10% of Patients in Study 1 (Cohorts 1 and 2) (N=82) Adverse Reaction Any Grade (%) Grade ≥3 (%) - * Includes multiple related terms.
- † Motor dysfunction includes asthenia, intensive care acquired weakness, mobility decreased, muscle twitching, muscular weakness, myopathy.
- ‡ Encephalopathy includes encephalopathy, altered state of consciousness, amnesia, balance disorder, cognitive disorder, confusional state, disturbance in attention, dysgraphia, dyskinesia, memory impairment, mental status changes, neurotoxicity, somnolence.
- § Dizziness includes dizziness, presyncope, syncope.
- ¶ Neuropathy includes hyperaesthesia, neuropathy peripheral, paraesthesia, paraesthesia oral.
- # Delirium includes delirium, agitation, disorientation, hallucination, hypomania, irritability, nervousness, personality change.
- Þ Thrombosis includes thrombosis, deep vein thrombosis, embolism, pulmonary embolism.
Blood and Lymphatic System Disorders Coagulopathy * 10 2 Cardiac Disorders Tachycardias * 45 0 Bradycardias * 10 0 Non-ventricular Arrhythmias * 10 4 Gastrointestinal Disorders Nausea 35 1 Constipation 29 0 Diarrhea 28 5 Abdominal pain * 17 0 Oral pain * 16 0 Vomiting * 13 0 Dysphagia 10 2 General Disorders and Administration Site Conditions Fever 94 15 Fatigue * 48 1 Chills 41 0 Edema * 35 2 Pain * 17 2 Immune System Disorders Cytokine release syndrome 91 18 Hypogammaglobulinemia * 16 1 Infections and Infestations Infection with pathogen unspecified 43 24 Viral infections 18 4 Bacterial infections 13 6 Metabolism and Nutrition Disorders Decreased appetite 26 0 Musculoskeletal and Connective Tissue Disorders Musculoskeletal pain * 37 2 Motor dysfunction † 17 4 Nervous System Disorders Encephalopathy ‡ 51 24 Tremor 38 2 Headache * 35 1 Aphasia * 20 7 Dizziness § 18 6 Neuropathy ¶ 13 2 Psychiatric Disorders Insomnia 21 0 Delirium # 18 5 Anxiety 16 0 Renal and Urinary Disorders Renal insufficiency * 18 9 Urine output decreased * 11 1 Respiratory, Thoracic and Mediastinal Disorders Hypoxia 40 20 Cough * 38 0 Dyspnea * 24 6 Pleural effusion 21 5 Skin and Subcutaneous Tissue Disorders Rash * 22 4 Vascular Disorders Hypotension * 57 27 Hypertension 18 11 Thrombosis Þ 17 4 Other clinically important adverse reactions that occurred in less than 10% of patients include the following:
- Gastrointestinal disorders: dry mouth (7%)
- Infections and infestations disorders: fungal infections (9%)
- Metabolism and nutrition disorders: dehydration (6%)
- Nervous system disorders: ataxia (7%), seizure (5%), increased intracranial pressure (2%)
- Respiratory, thoracic and mediastinal disorders: respiratory failure (6%), pulmonary edema (4%)
- Skin and subcutaneous tissue disorders: rash (9%)
- Vascular disorders: hemorrhage (7%)
Table 4. Grade 3 or 4 Laboratory Abnormalities Occurring in ≥ 10% of Patients in Study 1 (Cohorts 1 and 2) Following TECARTUS Infusion (N = 82) Grades 3 or 4 (%) Leukopenia 95 Neutropenia 95 Lymphopenia 86 Thrombocytopenia 63 Anemia 55 Hypophosphatemia 30 Hypocalcemia 21 Blood uric acid increased 17 Hyponatremia 16 Aspartate Aminotransferase increased 15 Alanine Aminotransferase increased 15 Hypokalemia 10 Cohort 3
The safety of TECARTUS in patients with relapsed/refractory MCL who had been treated with up to 5 prior treatment regimens but had not received prior therapy with a Bruton tyrosine kinase inhibitor (BTKi) was evaluated in Study 1 Cohort 3. A total of 86 patients were treated with a single dose of TECARTUS (2 × 106 or 0.5 × 106 anti-CD19 CAR T cells/kg) [see Clinical Studies (14.1)].
Serious adverse reactions occurred in 65% of patients. The most common serious adverse reactions (>2%) were non-ventricular arrhythmias, tachycardias, pyrexia, cytokine release syndrome, unspecified pathogen infections, viral infections, bacterial infections, fungal infections, musculoskeletal pain, motor dysfunction, encephalopathy, aphasia, tremor, seizure, delirium, hypoxia, hypotension, hemorrhage, and thrombosis.
Table 5 summarizes the adverse reactions that occurred in at least 10% of patients in Study 1 Cohort 3 and Table 6 lists the laboratory abnormalities of Grade 3 or 4 that occurred in at least 10% of patients in Study 1 Cohort 3.
Table 5. Adverse Reactions Observed in at Least 10% of Patients in Study 1 (Cohort 3) (N=86) Adverse Reaction Any Grade (%) Grade ≥3 - * Includes multiple related terms
- † Motor dysfunction includes Asthenia, Facial paresis, Fine motor skill dysfunction, Monoparesis, Motor dysfunction, Muscular weakness, Muscle spasms, Myoclonus, Myopathy, Paraparesis, Peroneal nerve palsy
- ‡ Encephalopathy includes Agnosia, Agraphia, Altered state of consciousness, Amnesia, Anal incontinence, Aphasia, Balance disorder, Bradyphrenia, Cerebellar syndrome, Cognitive disorder, Confusional state, Depressed level of consciousness, Disturbance in attention, Dysarthria, Dysgeusia, Dysgraphia, Dysmetria, Dysphagia, Encephalopathy, Lethargy, Loss of consciousness, Memory impairment, Mental status changes, Neurological decompensation, Somnolence, Speech disorder, Toxic encephalopathy
- § Tremor includes Action tremor, Postural tremor, Tremor
- ¶ Dizziness includes Dizziness, Syncope
- # Peripheral Neuropathy includes Hypoesthesia, Neuropathy peripheral, Paresthesia, Paresthesia oral, Peripheral motor neuropathy, Peripheral sensory neuropathy, Polyneuropathy
- Þ Delirium includes Agitation, Delirium, Disorientation, Hallucination, Hallucinations, mixed, Nervousness
- ß Thrombosis includes Deep vein thrombosis, Embolism, Jugular vein thrombosis, Peripheral vein thrombosis, Pulmonary embolism, Venous thrombosis
Cardiac Disorders Tachycardias * 20 1 Non-ventricular Arrhythmias * 13 3 Gastrointestinal Disorders Nausea 33 1 Constipation 28 0 Diarrhea 24 0 Abdominal pain * 19 1 Vomiting 15 0 Oral pain * 14 1 General Disorders and Administration Site Conditions Fever 94 17 Fatigue * 43 2 Edema * 21 0 Chills 19 0 Immune System Disorders Cytokine release syndrome 95 6 Infections and Infestations Infection with pathogen unspecified * 49 23 Viral infections * 49 16 Bacterial infections * 14 7 Fungal infections * 14 2 Metabolism and Nutrition Disorders Decreased appetite 24 9 Weight decreased 12 1 Musculoskeletal and Connective Tissue Disorders Musculoskeletal pain * 24 0 Motor dysfunction † 20 3 Nervous System Disorders Encephalopathy ‡ 66 24 Tremor § 30 2 Headache 29 0 Dizziness ¶ 16 5 Peripheral neuropathy # 12 1 Psychiatric Disorders Delirium Þ 17 1 Insomnia * 12 0 Renal and Urinary Disorders Renal insufficiency * 19 2 Respiratory, Thoracic and Mediastinal Disorders Hypoxia * 24 10 Cough * 14 0 Dyspnea * 14 5 Skin and Subcutaneous Tissue Disorders Rash * 16 0 Vascular Disorders Hypotension * 52 8 Hemorrhage * 16 5 Thrombosis ß 13 3 Hypertension 10 2 Other clinically important adverse reactions that occurred in less than 10% of patients include the following:
- Blood and lymphatic system disorders: coagulopathy (5%), febrile neutropenia (1%)
- Cardiac disorders: Bradycardia (1%)
- Eye disorders: Visual impairment (6%)
- Gastrointestinal disorders: dry mouth (7%), dysphagia (1%)
- General disorders and administration site conditions: Pain (9%)
- Immune system disorders: hypersensitivity (1%), hypogammaglobulinemia (9%)
- Metabolism and nutrition disorders: dehydration (6%)
- Nervous system disorders: ataxia (9%), increased intracranial pressure (1%), seizure (5%)
- Psychiatric disorders: anxiety (8%)
- Renal and urinary disorders: urine output decreased (1%)
- Respiratory, thoracic and mediastinal disorders: pleural effusion (1%), pulmonary edema (3%), respiratory failure (6%)
Table 6. Grade 3 or 4 Laboratory Abnormalities Occurring in ≥ 10% of Patients in Study 1 (Cohort 3) Following TECARTUS Infusion (N=86) Grades 3 or 4 (%) Leukopenia 97 Lymphopenia 96 Neutropenia 91 Thrombocytopenia 44 Hypophosphatemia 40 Anemia 31 Hyperglycemia 29 Blood uric acid increased 28 Alanine Aminotransferase increased 27 Hyponatremia 22 Calcium increased 12 Creatinine increased 12 Direct bilirubin increased 12 Magnesium increased 12 Aspartate Aminotransferase increased 10 Hypocalcemia 10 Study 2 (Relapsed/Refractory B-cell precursor Acute Lymphoblastic Leukemia)
The safety of TECARTUS in patients with relapsed/refractory ALL was evaluated in an open-label, multicenter study in which a total of 78 patients received a single dose of CAR-positive T cells (1 x 106 anti-CD19 CAR T cells/kg) [see Clinical Studies (14.2)].
The most common serious adverse reactions (≥ 2%) were cytokine release syndrome, febrile neutropenia, hypotension, encephalopathy, fever, infection with pathogen unspecified, hypoxia, tachycardia, bacterial infections, respiratory failure, seizure, diarrhea, dyspnea, fungal infections, viral infections, coagulopathy, delirium, fatigue, hemophagocytic lymphohistiocytosis, musculoskeletal pain, edema, and paraparesis.
Fatal adverse reactions occurred in 5% (4/78) of patients including cerebral edema, sepsis, and fungal pneumonia. Of the 4 patients who had fatal adverse reactions: 1 patient with fatal pneumonia had pre-existing pneumonia prior to study enrollment, and 1 patient with fatal sepsis had prolonged cytopenia and immunosuppression from prior therapies and underlying disease.
Table 7 summarizes the adverse reactions that occurred in at least 10% of patients in Study 2 and Table 8 describes the laboratory abnormalities of Grade 3 or 4 that occurred in at least 10% of patients in Study 2.
Table 7. Adverse Reactions Observed in at Least 10% of Patients in Study 2 (N=78) Adverse Reaction Any Grade (%) Grade ≥3 (%) - * Febrile neutropenia includes febrile neutropenia (11 (14%)) and fever occurring concurrently with neutropenia events (16 (21%))
- † Represents a composite of multiple, related preferred terms
- ‡ Arrhythmia includes cardiac arrest, .
- § Encephalopathy includes altered state of consciousness, aphasia, cognitive disorder, confusional state, depressed level of consciousness, disturbance in attention, dysarthria, dysgraphia, encephalopathy, immune effector cell-associated neurotoxicity syndrome, memory impairment, mental status changes, slow response to stimuli, slow speech, somnolence, speech disorder.
- ¶ Dizziness includes dizziness, syncope.
- # Delirium includes agitation, delirium, delusion, disorientation, hallucination.
- Þ Hemorrhage includes conjunctival hemorrhage, contusion, epistaxis, gastric hemorrhage, hematoma, hematoma muscle, hemorrhage intracranial, hemorrhoidal hemorrhage, menorrhagia, petechiae, pulmonary alveolar hemorrhage, retinal hemorrhage, vaginal hemorrhage, vitreous hemorrhage.
Blood and Lymphatic System Disorders Febrile Neutropenia* 35 35 Coagulopathy† 17 5 Cardiac Disorders Tachycardias† 63 6 Arrhythmia†‡ 15 1 Gastrointestinal Disorders Nausea 41 1 Diarrhea 32 6 Abdominal pain† 19 0 Constipation 24 0 Vomiting 21 3 General Disorders and Administration Site Conditions Fever 96 38 Chills 40 0 Edema† 29 5 Fatigue† 37 1 Pain 13 1 Immune System Disorders Cytokine release syndrome 92 26 Infections and Infestations Infection with pathogen unspecified† 28 22 Bacterial infections† 15 8 Fungal infections† 13 5 Metabolism and Nutrition Disorders Decreased appetite 22 1 Musculoskeletal and Connective Tissue Disorders Musculoskeletal pain† 32 5 Muscular weakness 14 1 Nervous System Disorders Encephalopathy§ 63 27 Headache 38 1 Tremor 29 1 Dizziness¶ 13 1 Psychiatric Disorders Delirium# 18 5 Anxiety 12 0 Insomnia 13 0 Respiratory, Thoracic and Mediastinal Disorders Hypoxia 31 23 Cough† 12 0 Dyspnea 12 1 Skin and Subcutaneous Tissue Disorders Rash† 31 0 Vascular Disorders Hypotension† 69 33 HemorrhageÞ 13 4 Hypertension 13 6 Other clinically important adverse reactions that occurred in less than 10% of patients include the following:
- Cardiac disorder: cardiac failure (4%), palpitations (3%)
- Eye disorders: visual impairment (9%)
- Gastrointestinal disorders: dry mouth (6%), dysphagia (4%), oral pain (1%)
- Immune system disorders: hypogammaglobulinemia (9%), hemophagocytic lymphohistiocytosis (4%), drug hypersensitivity (1%)
- Infections and infestations: viral infections (6%)
- Metabolism and nutrition disorders: dehydration (5%), tumor lysis syndrome (1%)
- Musculoskeletal and connective tissue disorders: muscle spasms (4%), musculoskeletal stiffness (3%)
- Nervous system disorders: seizure (8%), ataxia (5%), peripheral neuropathy (4%), myoclonus (3%), paraparesis (3%), brain edema (1%), brain herniation (1%), cauda equina syndrome (1%), monoplegia (1%)
- Renal and urinary disorders: renal impairment (6%)
- Respiratory, thoracic and mediastinal disorders: respiratory failure (9%), pulmonary edema (6%), pleural effusion (4%), pneumonitis (4%)
- Skin and subcutaneous tissue disorders: skin lesion (4%), decubitus ulcer (3%), dry skin (3%), skin ulcer (3%), alopecia (1%), hyperhidrosis (1%), skin hyperpigmentation (1%)
- Vascular disorders: thrombosis (4%)
Table 8. Grade 3 or 4 Laboratory Abnormalities Occurring in ≥ 10% of Patients in Study 2 Following TECARTUS Infusion (N = 78) Grades 3 or 4 (%) Leukopenia 99 Neutropenia 97 Lymphopenia 96 Thrombocytopenia 87 Anemia 77 Hypophosphatemia 47 Alanine aminotransferase increased 31 Aspartate aminotransferase increased 23 Hyperglycemia 22 Hypocalcemia 22 Blood uric acid increased 19 Direct bilirubin increased 19 Hyponatremia 19 Hypokalemia 13 Hyperbilirubinemia 10 6.2 Postmarketing Experience
Because adverse events to marketed products are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to product exposure.
The following adverse event has been identified during postmarketing use of TECARTUS:
Immune System Disorders: Infusion related reaction
The following adverse event has been identified during postmarketing use of BCMA- or CD19-directed genetically modified autologous T cell immunotherapies:
Neoplasms: T cell malignancies
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data with TECARTUS use in pregnant women. No animal reproductive and developmental toxicity studies have been conducted with TECARTUS to assess whether TECARTUS can cause fetal harm when administered to a pregnant woman. It is not known if TECARTUS has the potential to be transferred to the fetus. Based on the mechanism of action of TECARTUS, if the transduced cells cross the placenta, they may cause fetal toxicity, including B cell lymphocytopenia. Therefore, TECARTUS is not recommended for women who are pregnant. Pregnancy after TECARTUS infusion should be discussed with the treating physician.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% – 4% and 15% – 20%, respectively.
8.2 Lactation
Risk Summary
There is no information regarding the presence of TECARTUS in human milk, the effect on the breastfed infant, and the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for TECARTUS and any potential adverse effects on the breastfed infant from TECARTUS or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Pregnancy status of females with reproductive potential should be verified. Sexually active females of reproductive potential should have a negative pregnancy test prior to starting treatment with TECARTUS.
Contraception
See the Prescribing Information for fludarabine and cyclophosphamide for information on the need for effective contraception in patients who receive the lymphodepleting chemotherapy.
There are insufficient exposure data to provide a recommendation concerning duration of contraception following treatment with TECARTUS.
8.4 Pediatric Use
The safety and efficacy of TECARTUS have not been established in pediatric patients.
8.5 Geriatric Use
Of the 82 patients treated with TECARTUS for MCL, 42 (51%) were 65 years of age and over. Of the 78 patients treated with TECARTUS for ALL, 12 (15%) were 65 years of age and over. No overall differences in safety or effectiveness were observed between these patients and younger patients.
-
11 DESCRIPTION
TECARTUS (brexucabtagene autoleucel) is a CD19-directed genetically modified autologous T cell immunotherapy. To prepare TECARTUS, a patient's own T cells are harvested and genetically modified ex vivo by retroviral transduction to express a chimeric antigen receptor (CAR) comprising a murine anti-CD19 single-chain variable fragment (scFv) linked to CD28 and CD3-zeta co-stimulatory domains. The anti-CD19 CAR T cells are expanded and infused back into the patient, where they can recognize and eliminate CD19-expressing target cells.
TECARTUS is prepared from the patient's peripheral blood mononuclear cells, which are obtained via a standard leukapheresis procedure. The mononuclear cells are enriched for T cells and activated with anti-CD3 and anti-CD28 antibodies in the presence of IL-2, then transduced with a replication-incompetent retroviral vector containing the anti-CD19 CAR transgene. The transduced T cells are expanded in cell culture, washed, formulated into a suspension, and cryopreserved. The manufacture of TECARTUS includes a T cell enrichment step that may reduce the likelihood of circulating CD19-expressing tumor cells in patients' leukapheresis material driving the activation, expansion, and exhaustion of the anti-CD19 CAR T cells during the ex vivo manufacturing process. The product must pass a sterility test before release for shipping as a frozen suspension in a patient-specific infusion bag. The product is thawed prior to infusion [see Dosage and Administration (2.2), How Supplied/Storage and Handling (16)].
In addition to T cells, TECARTUS may contain natural killer (NK) cells. The formulation contains CryoStor (dimethyl sulfoxide [DMSO], final concentration, 5%), sodium chloride (NaCl), and Human Serum Albumin (HSA).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
TECARTUS, a CD19-directed genetically modified autologous T cell immunotherapy, binds to CD19-expressing cancer cells and normal B cells. Studies demonstrated that following anti-CD19 CAR T cell engagement with CD19-expressing target cells, the CD28 and CD3-zeta co-stimulatory domains activate downstream signaling cascades that lead to T cell activation, proliferation, acquisition of effector functions, and secretion of inflammatory cytokines and chemokines. This sequence of events leads to killing of CD19-expressing cells.
12.2 Pharmacodynamics
After TECARTUS infusion, pharmacodynamic responses were evaluated over a four-week interval by measuring transient elevation of cytokines, chemokines, and other molecules in blood. Levels of cytokines and chemokines such as IL-6, IL-8, IL-10, IL-15, TNF-α, IFN-γ, and sIL2Rα were analyzed. Peak elevation was generally observed within 8 days after infusion. The majority of serum analytes returned to baseline levels by Week 4; however, ferritin, IFN-γ, IL-6, and IL-15 remained elevated ≥ 2-fold relative to baseline at Week 4.
Due to the on-target effect of TECARTUS, a period of B cell aplasia is expected.
12.3 Pharmacokinetics
Following infusion (target dose of 2 × 106 anti-CD19 CAR T cells/kg) of TECARTUS in Study 1, anti-CD19 CAR T cells exhibited an initial rapid expansion followed by a decline to near baseline levels by 3 months. Peak levels of anti-CD19 CAR T cells occurred within the first 15 days after TECARTUS infusion. Following infusion (target dose of 1 × 106 anti-CD19 CAR T cells/kg) of TECARTUS in Study 2 (Phase 2), anti-CD19 CAR T cells exhibited an initial rapid expansion followed by a decline to near baseline levels by 6 months. Median anti- CD19 CAR T cell time to peak was 15 days after TECARTUS infusion.
Description of Pharmacokinetics in Adult r/r MCL
In Study 1 Cohort 1, the number of anti-CD19 CAR T cells in blood was associated with objective response [complete remission (CR) or partial remission (PR)]. Median peak anti-CD19 CAR T cell level in responders was 102.4 cells/µL (range: 0.2 to 2589.5 cells/µL; n = 51), and in nonresponders was 12.0 cells/µL (range: 0.2 to 1364.0 cells/µL, n = 8). The median AUC0-28 in patients with an objective response was 1487.0 cells/µLdays (range: 3.8 to 2.77E+04 cells/µLdays; n = 51) versus 169.5 cells/µLdays in nonresponders (range: 1.8 to 1.17E+04 cells/µLdays; n = 8). In Study 1 Cohort 3 , median peak anti-CD19 CAR T cell level in responders [patients who achieved complete remission (CR) or partial remission (PR)] was 72.9 cells/μL (range: 0.5 to 1874.5 cells/μL; n = 77), and in nonresponders was 288.8 cells/μL (range: 1.32 to 912.6 cells/μL, n = 7). The median AUC0-28 in patients with an objective response was 941.53 cells/μLdays (range: 8.8 to 1.63E+04 cells/μLdays; n = 77) versus 2141.4 cells/μLdays in nonresponders (range: 14.4 to 1.02E+04 cells/μLdays; n = 7).
In Study 1 Cohort 1, median peak anti-CD19 CAR T cell and AUC0-28 levels in patients who received neither corticosteroids nor tocilizumab (peak: 24.7 cells/μL; AUC0-28: 360.4 cells/μLdays, n = 18) was similar to patients who received corticosteroids alone (peak: 24.2 cells/μL; AUC0-28: 367.8 cells/μLdays, n = 2); both groups were lower than patients who received tocilizumab alone (peak: 86.5 cells/μL; AUC0-28: 1188.9 cells/μLdays, n = 10); the highest exposure was in patients who received both corticosteroids and tocilizumab (peak: 167.2 cells/μL; AUC0-28: 1996.0 cells/μLdays, n = 37). In Study 1 Cohort 3, median peak anti-CD19 CAR T cell and AUC0-28 levels were lowest in patients who received neither corticosteroids nor tocilizumab (peak: 16.9 cells/μL; AUC0-28: 131.4 cells/μLdays, n = 13). Median peak anti-CD19 CAR T cell and AUC0-28 levels in patients who received tocilizumab alone (peak 61.7 cells/ µL; AUC0-28: 594.3 cells/μLdays, n=8) were higher than patients who received neither corticosteroids nor tocilizumab. Two patients who received corticosteroids alone (peak: 122.5 cells/μL; AUC0-28: 1189.1 cells/μLdays, n = 2) and patients who received both corticosteroids and tocilizumab (peak: 108.4 cells/μL; AUC0-28: 1249.9 cells/μLdays, n = 61) had highest median peak anti-CD19 CAR T cell and AUC0-28 levels.
In Study 1 Cohort 1, median peak anti-CD19 CAR T cell values were 74.1 cells/μL in patients ≥ 65 years of age (n = 39) and 112.5 cells/μL in patients < 65 years of age (n = 28). Median anti-CD19 CAR T cell AUC0-28 values were 876.5 cells/μLdays in patients ≥ 65 years of age and 1640.2 cells/μLdays in patients < 65 years of age. In Study 1 Cohort 3, median peak anti-CD19 CAR T cell values were 105.5 cells/μL in patients ≥ 65 years of age (n = 40) and 58.5 cells/μL in patients < 65 years of age (n = 44). Median anti-CD19 CAR T cell AUC0-28 values were 1080.7 cells/μLdays in patients ≥ 65 years of age and 711 cells/μLdays in patients < 65 years of age.
Gender had no significant impact on AUC Day 0-28 and Cmax of TECARTUS.
Description of Pharmacokinetics in Adult r/r B-cell precursor ALL
Median peak anti-CD19 CAR T cell levels was 38.4 cells/μL (range: 1.31 to 1533.4 cells/μL; n = 32) in patients who had overall complete remission (CR+CRi), and 0.9 cells/μL (range: 0.00 to 183.5 cells/μL, n = 17) in patients who had non-complete remission. The median AUC0-28 in patients who had overall complete remission (CR+CRi) was 424.0 cells/μLdays (range: 14.12 to 19,390.4 cells/μLdays; n = 32) vs 7.9 cells/μLdays in patients who had non-complete remission (range: 0.00 to 889.0 cells/μLdays; n=17).
Median peak anti-CD19 CAR T cell and AUC0-28 levels were lowest in patients who received neither corticosteroids nor tocilizumab (peak 5.7 cells/µL; AUC0-28: 60.7 cells/µLdays, n=11). Median peak anti-CD19 CAR T cell and AUC0-28 levels in patients who received tocilizumab alone (peak: 11.2 cells/µL; AUC0-28: 137.4 cells/μLdays, n=8) were lower than in the one patient who received corticosteroids alone (peak: 36.2 cells/ µL; AUC0-28: 423.1 cells/μLdays, n= 1); the highest exposure was observed in evaluable patients who received both corticosteroids and tocilizumab (peak: 49.2 cells/µL; AUC0-28: 454.1 cells/ μLdays, n=30).
Hepatic and renal impairment studies of TECARTUS were not conducted.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-product antibodies in the studies described below with the incidence of anti-product antibodies in other studies, including those of TECARTUS or of other similar products.
The potential for TECARTUS to induce anti-product antibodies was evaluated in Study 1 and Study 2 using an enzyme-linked immunosorbent assay (ELISA) for the detection of binding antibodies against FMC63, the originating antibody of the anti-CD19 CAR. To date, no anti-CAR T cell antibody immunogenicity has been observed in patients with MCL in Study 1 (Cohorts 1, 2, and 3). No patient in Study 1 had a confirmed positive antibody result post treatment. Two patients in Study 2 had confirmed positive antibody results post treatment: 1 patient had a confirmed positive antibody result at Month 6 and 1 patient had a confirmed antibody result at retreatment Day 28 and Month 3. There is no evidence that the kinetics of initial expansion and persistence of TECARTUS, or the safety or effectiveness of TECARTUS, were altered in these patients.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
14.1 Relapsed or Refractory Mantle Cell Lymphoma
A single-arm, open-label, multicenter trial (Study 1; NCT02601313) evaluated the efficacy of a single infusion of TECARTUS in adult patients with relapsed or refractory mantle cell lymphoma (MCL) who had previously received anthracycline- or bendamustine-containing chemotherapy, an anti-CD20 antibody, and a Bruton tyrosine kinase inhibitor (BTKi; ibrutinib or acalabrutinib). Eligible patients also had disease progression after their last regimen or refractory disease to their most recent therapy. The study excluded patients with active or serious infections, prior allogeneic hematopoietic stem cell transplant (HSCT), detectable cerebrospinal fluid malignant cells or brain metastases, and any history of central nervous system (CNS) lymphoma or CNS disorders.
Seventy-four patients were leukapheresed, 5 (7%) of whom did not begin conditioning chemotherapy or receive TECARTUS: 3 (4%) experienced manufacturing failure, 1 (1%) died of progressive disease, and 1 (1%) withdrew from the study. One patient (1%) received lymphodepleting chemotherapy but did not receive TECARTUS due to ongoing active atrial fibrillation. Sixty-eight of the patients who were leukapheresed received a single infusion of TECARTUS, and 60 of these patients were followed for at least 6 months after their first objective disease response, qualifying them as efficacy-evaluable. Among the 60 efficacy-evaluable patients, 2 × 106 CAR-positive viable T cells/kg were administered to 54 (90%). The remaining 6 (10%) patients received doses of 1.0, 1.6, 1.8, 1.8, 1.9, and 1.9 × 106 CAR-positive viable T cells/kg.
Of the 60 efficacy-evaluable patients, the median age was 65 years (range: 38 to 79 years), 51 patients (85%) were male, and 56 patients (93%) were white, 1 patient (2%) was Black or African American, 1 patient (2%) was Pacific Islander and 2 patient (3%) were of “Other” race. Fifty patients (83%) had stage IV disease, 8 patients (13%) had stage III disease and 2 patients (3%) had stage II disease. Twenty patients (33% of 60) had baseline bone marrow examinations performed per protocol; of these, 10 (50%) were negative, 8 (40%) were positive, and 2 (10%) were indeterminate. The median number of prior therapies was 3 (range: 2 to 5). Twenty-six (43%) of the patients had relapsed after or were refractory to autologous HSCT. Twenty-one (35%) had relapsed after their last therapy for MCL, while 36 (60%) were refractory to their last therapy for MCL. Fourteen patients (23%) had blastoid MCL, 35 patients (58%) had Classical MCL, 1 patient (2%) had Other and 10 patients (17%) had Unknown MCL type. Following leukapheresis and prior to administration of TECARTUS, 21 (35%) of the 60 patients received bridging therapy. Sixteen (27%) were treated with a BTKi, 9 (15%) with a corticosteroid, and 4 (7%) with both a BTKi and a corticosteroid.
Among the 60 efficacy-evaluable patients, the median time from leukapheresis to product delivery was 15 days (range: 11 to 28 days), and the median time from leukapheresis to product infusion was 27 days (range: 19 to 63 days). The protocol-defined lymphodepleting chemotherapy regimen of cyclophosphamide 500 mg/m2 intravenously and fludarabine 30 mg/m2 intravenously, both given on each of the fifth, fourth, and third days before TECARTUS infusion, was administered to 53 (88%) of the 60 efficacy-evaluable patients. The remaining 7 patients (12%) either received lymphodepletion over 4 or more days or received TECARTUS 4 or more days after completing lymphodepletion. All treated patients received TECARTUS infusion on Day 0 and were hospitalized until at least Day 7.
The primary endpoint of objective response rate (ORR) per the Lugano Classification (2014) in patients treated with TECARTUS in Study 1 Cohorts 1 as determined by an independent review committee is provided in Table 9. The median time to response was 28 days (range: 24 to 92 days) with a median follow-up time for DOR of 8.6 months.
Table 9. Efficacy Results in Adult Patients with Relapsed/Refractory MCL (Study 1 Cohort 1) Efficacy-Evaluable Patients
N = 60All Leukapheresed Patients (ITT)
N = 74CI, confidence interval; CR, complete remission; DOR, duration of response; NE, not estimable; NR, not reached; PR, partial remission. - * Among all responders. DOR is measured from the date of first objective response to the date of progression or death.
- † A + sign indicates a censored value.
- ‡ At the time of primary analysis.
Response Rate Objective Response Rate*, n (%) [95% CI] 52 (87%) [75, 94] 59 (80%) [69, 88] Complete Remission Rate, n (%) [95% CI] 37 (62%) [48, 74] 41 (55%) [43, 67] Partial Remission Rate, n (%) [95% CI] 15 (25%) [15, 38] 18 (24%) [15, 36] Duration of Response (DOR) Median in months [95% CI]
Range† in monthsNR [8.6, NE]
0.0+, 29.2+NR [8.6, NE]
0.0+, 29.2+DOR, if best response is CR, median in months [95% CI]
Range† in monthsNR [13.6, NE]
1.9+, 29.2+NR [13.6, NE]
0.0+, 29.2+DOR, if best response is PR, median in months [95% CI]
Range† in months2.2 [1.5, 5.1]
0.0+, 22.1+2.2 [1.5, 5.1]
0.0+, 22.1+Median Follow-up for DOR in months‡ 8.6 8.1 A subsequent, open label cohort in Study 1 (Cohort 3) evaluated the efficacy of TECARTUS in patients with relapsed or refractory MCL who had been treated with up to 5 prior treatment regimens but had not received prior therapy with a BTKi. A total of 95 patients were enrolled and leukapheresed, 8 (8%) of whom did not begin conditioning chemotherapy or receive TECARTUS: 3 (3%) experienced an AE, 1 (1%) died due to an AE, 1 (1%) withdrew from the study as the product did not meet specifications, 1 (1%) withdrew consent, 1 (1%) experienced deterioration of their general condition, and 1 (1%) experienced progressive disease. One patient (1%) received lymphodepleting chemotherapy but did not receive TECARTUS due to rapid and uncontrolled disease progression. Eighty-six patients who were leukapheresed received a single infusion of TECARTUS, and were followed for at least 18 months after their first objective disease response, qualifying them as efficacy-evaluable.
Of the 86 efficacy-evaluable patients, the median age was 64 years (range: 40 to 82 years), 67 (78%) were male, and 19 (22%) were females, 78 (91%) were white, 2 (2%) other, 1(1%) Asian and 5 (6%) were not reported. Most (56 patients; 65%) had stage IV disease, 3 (3%) had stage I, 17 (20%) had stage II, and 10 (12%) had stage III disease. All patients had baseline bone marrow examinations performed per protocol; of these, 52 (60%) were negative and 34 (40%) were positive. The median number of prior therapies was 1 (range: 1 to 5). Seventy-four patients (86%) had relapsed after their last therapy for MCL, while 12 (14%) were refractory to their last therapy for MCL. Sixty-five patients (76%) had classical MCL, 6 (7%) had blastoid MCL, 6 (7%) had pleomorphic MCL, and 9 (10%) were other. Following leukapheresis and prior to administration of TECARTUS, 31 (36%) of the 86 patients received bridging therapy.
The median time from leukapheresis to product delivery was 24 days (range: 14 to 43 days), and the median time from leukapheresis to product infusion was 33.5 days (range: 19 to 85 days). The protocol-defined lymphodepleting chemotherapy regimen of cyclophosphamide 500 mg/m2 intravenously and fludarabine 30 mg/m2 intravenously, both given concurrently for 3 days (Day 5 to Day 3) before TECARTUS infusion to all 86 efficacy-evaluable patients. All treated patients received TECARTUS infusion on Day 0 and were hospitalized until at least Day 7.
The primary endpoint was objective response rate (ORR) per the Lugano Classification (2014) as determined by an independent review committee.
The efficacy results are summarized in Table 10 below.
Table 10. Efficacy Results in (Study 1 Cohort 3) Efficacy evaluable patients*
N = 86All Leukapheresed Patients (ITT)
N = 95CI, confidence interval; CR, complete remission; DOR, duration of response; NE, not estimable; NR, not reached; PR, partial remission. - * Of the 95 patients that were enrolled (and leukapheresed), 87 patients received lymphodepleting chemotherapy, and 86 patients received TECARTUS.
- † Per the International Working Group Lugano Classification (Cheson 2014), as assessed by the Independent Radiology Review Committee.
- ‡ Among all responders. DOR is measured from the date of first objective response to the date of progression or death prior to new anticancer therapy (including SCT).
- § A + sign indicates a censored value.
- ¶ At the time of primary analysis.
Response Rate Objective Response Rate†, n (%) [95% CI] 78 (91%) [82.5, 95.9] 78 (82%) [72.9, 89.2] Complete Remission Rate, n (%) [95% CI] 68 (79%) [69.0, 87.1] 68 (72%) [61.4, 80.4] Partial Remission Rate, n (%) [95% CI] 10 (12%) [5.7, 20.3] 10 (11%) [5.2, 18.5] Duration of Response (DOR)‡ Median in months [95% CI]
Range§ in monthsNR [26.2, NE]
0.0+, 35.3+DOR, if best response is CR, median in months [95% CI]
Range§ in monthsNR [26.2, NE]
0.5, 35.3+DOR, if best response is PR, median in months [95% CI]
Range§ in months13.3 [2.1, NE]
0.0+, 16.5+Median Follow-up for DOR in months¶ 23.0 The median time to response was 1 month (range: 0.8 to 1.9 months)
14.2 Relapsed or Refractory B-cell precursor Acute Lymphoblastic Leukemia
The efficacy of TECARTUS was evaluated in Study 2 (NCT02614066), an open-label, single-arm, multicenter trial in adult patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL). Eligible patients were adults with primary refractory ALL, first relapse following a remission lasting ≤ 12 months, relapsed or refractory ALL after second-line or higher therapy, or relapsed or refractory ALL at least 100 days after allogeneic stem cell transplantation (HSCT). The study excluded patients with active or serious infections, active graft-vs-host disease or taking immunosuppressive medications within 4 weeks prior to enrollment, and any history of CNS disorders, including CNS-2 disease with neurologic changes and CNS-3 disease irrespective of neurological changes. Treatment consisted of lymphodepleting chemotherapy (fludarabine 25 mg/m2 IV daily on Days -4, -3 and -2; cyclophosphamide 900 mg/m2 iv on Day -2) followed by a single intravenous infusion of TECARTUS at a target dose of 1 × 106 anti-CD19 CAR T cells/kg (maximum 1 × 108 cells) on Day 0. All treated patients were hospitalized until at least Day 7.
Seventy-one patients were enrolled and leukapheresed; 6 of these patients did not receive TECARTUS due to manufacturing failure, 8 patients were not treated primarily due to adverse events following leukapheresis, 2 patients underwent leukapheresis and received lymphodepleting chemotherapy but were not treated with TECARTUS, and 1 patient treated with TECARTUS was inevaluable for efficacy. Among the remaining 54 efficacy-evaluable patients, the median time from leukapheresis to product delivery was 16 days (range: 11 to 39 days) and the median time from leukapheresis to TECARTUS infusion was 29 days (range: 20 to 60 days).
Of the 54 patients who were efficacy-evaluable, the median age was 40 years (range: 19 to 84 years), 61% were male, and 67% were White, 6% were Asian, 2% were Black or African American, and 2% were American Indian or Alaska Native. At enrollment, 46% had refractory relapse, 26% had primary refractory disease, 20% had untreated second or later relapse, and 7% had first untreated relapse. Among prior therapies, 43% of patients were previously treated with allo-SCT, 46% with blinatumomab, and 22% with inotuzumab. Twenty-six percent of patients were Philadelphia chromosome positive (Ph+). Fifty (93%) patients had received bridging therapy between leukapheresis and lymphodepleting chemotherapy to control disease burden.
The efficacy of TECARTUS was established on the basis of complete remission (CR) within 3 months after infusion and the duration of CR (DOCR). Twenty-eight (51.9%) of the 54 evaluable patients achieved CR, and with a median follow-up for responders of 7.1 months, the median DOCR was not reached (Table 11). The median time to CR was 56 days (range: 25 to 86 days). All efficacy-evaluable patients had potential follow-up for ≥ 10 months with a median actual follow-up time of 12.3 months (range: 0.3 to 22.1 months).
Table 11. Efficacy Results in Study 2 Efficacy Evaluable Patients*
N= 54All Leukapheresed Patients
N = 71CI, confidence interval; CR, complete remission; CRi, complete remission with incomplete blood count recovery; DOR, duration of remission; NE, not estimable; NR, not reached, OCR, overall complete remission; NE, not estimable - * Of the 71 patients that were enrolled (and leukapheresed), 57 patients received lymphodepleting chemotherapy, and 55 patients received TECARTUS. 54 patients were included in the efficacy-evaluable population.
- † A + sign indicates a censored value.
OCR rate (CR + CRi), n (%) [95% CI] 35 (64.8) [51, 77] 36 (50.7) [39, 63] CR rate, n (%) [95% CI] 28 (51.9) [37.8, 65.7] 29 (40.9) [29.3, 53.2] Duration of Remission, Median in months [95% CI]
(Range† in months)13.6 [9.4, NE]
(0.03+, 16.07+)13.6 [8.7, NE]
(0.03+, 16.07+)DOR, if best response is CR, median in months [95% CI]
(Range in months)NR [9.6, NE]
(0.03+, 16.07+)13.6 [9.4, NE]
(0.03+, 16.07+)DOR, if best response is CRi, median in months [95% CI]
(Range in months)8.7 [1.0, NE]
(0.03+, 10.15+)8.7 [1.0, NE]
(0.03+, 10.15+)Median Follow-up for CR in months 7.1 (0.03+, 16.1+) 5.0 (0.03+, 16.1+) - 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
TECARTUS is supplied in an infusion bag containing approximately 68 mL of frozen suspension of genetically modified autologous T cells in 5% DMSO and human serum albumin.
Each TECARTUS infusion bag is individually packed in a metal cassette. TECARTUS is supplied in a liquid nitrogen dry shipper.
Indication Infusion Bag NDC number Metal Cassette NDC number MCL 71287-219-01 71287-219-02 ALL 71287-220-01 71287-220-02 - Match the identity of the patient with the patient identifiers on the cassette and infusion bag upon receipt.
- Store TECARTUS frozen in the vapor phase of liquid nitrogen (less than or equal to -150°C). TECARTUS may be stored for a single time at -80°C (+/- 10°C), for up to 90 days, and not exceeding the labeled expiration date. Do not return TECARTUS to storage in the vapor phase of liquid nitrogen after storage at -80°C.
- Thaw before using [see Dosage and Administration (2)].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Ensure that patients understand the risk of manufacturing failure (4% in clinical trial). In case of a manufacturing failure, a second manufacturing of TECARTUS may be attempted. In addition, while the patient awaits the product, additional chemotherapy (not the lymphodepletion) may be necessary and may increase the risk of adverse events during the pre-infusion period.
Advise patients to seek immediate attention for any of the following:
- Cytokine Release Syndrome (CRS) - Signs or symptoms associated with CRS, including fever, chills, fatigue, tachycardia, nausea, hypoxia, and hypotension [see Warnings and Precautions (5.1) and Adverse Reactions (6)].
- Neurologic Toxicities - Signs or symptoms associated with neurologic events, including encephalopathy, seizures, changes in level of consciousness, speech disorders, tremors, and confusion [see Warnings and Precautions (5.2) and Adverse Reactions (6)].
- Severe Infections - Signs or symptoms associated with infection [see Warnings and Precautions (5.5) and Adverse Reactions (6)].
- Prolonged Cytopenias - Signs or symptoms associated with bone marrow suppression, including neutropenia, anemia, thrombocytopenia, or febrile neutropenia [see Warnings and Precautions (5.6) and Adverse Reactions (6)].
- Secondary Malignancies - Secondary malignancies, including T cell malignancies, have occurred following treatment with BCMA- and CD19-directed genetically modified autologous T cell immunotherapies [see Boxed Warning, Warnings and Precautions (5.8), Adverse Reactions (6.2)].
Advise patients of the need to:
- Avoid driving for at least 2 weeks.
- Contact Kite at 1-844-454-KITE (5483) if they are diagnosed with a secondary malignancy [see Warnings and Precautions (5.8)].
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: April 2026 MEDICATION GUIDE
TECARTUS (pronounced tek-ahr-tuhs)
(brexucabtagene autoleucel)Read this Medication Guide before you start your TECARTUS treatment. The more you know about your treatment, the more active you can be in your care. Talk with your healthcare provider if you have questions about your health condition or treatment. Reading this Medication Guide does not take the place of talking with your healthcare provider about your treatment. What is the most important information I should know about TECARTUS?
TECARTUS may cause side effects that are life-threatening and can lead to death. Call or see your healthcare provider or get emergency help right away if you get any of the following:- Fever (100.4°F/38°C or higher)
- Difficulty breathing
- Chills or shaking chills
- Confusion
- Dizziness or lightheadedness
- Severe nausea, vomiting, or diarrhea
- Fast or irregular heartbeat
- Severe fatigue or weakness
What is TECARTUS?
TECARTUS is a treatment for adults with mantle cell lymphoma or acute lymphoblastic leukemia. It is used following disease progression while on or after other treatment. TECARTUS is different than other cancer medicines because it is made from your own white blood cells, which have been modified to recognize and attack your lymphoma cells.Before getting TECARTUS, tell your healthcare provider about all your medical problems, including if you have or have had: - Neurologic problems (such as seizures, stroke, or memory loss)
- Lung or breathing problems
- Heart problems
- Liver problems
- Kidney problems
- A recent or active infection
How will I receive TECARTUS? - Since TECARTUS is made from your own white blood cells, your blood will be collected by a process called "leukapheresis" (loo-kah-fur-ee-sis), which will concentrate your white blood cells.
- Your blood cells will be sent to a manufacturing center to make your TECARTUS.
- Before you get TECARTUS, you will get 3 days of chemotherapy to prepare your body.
- When your TECARTUS is ready, your healthcare provider will give it to you through a catheter placed into your vein (intravenous infusion). The infusion usually takes less than 30 minutes.
- You will be monitored daily for at least 7 days after the infusion.
- You should plan to stay close to a healthcare facility for at least 2 weeks after getting TECARTUS. Your healthcare provider will help you with any side effects that may occur.
- You may be hospitalized for side effects. Your healthcare provider will discharge you if your side effects are under control and it is safe for you to leave the hospital.
- Your healthcare provider will want to do blood tests to follow your progress. It is important that you do have your blood tested. If you miss an appointment, call your healthcare provider as soon as possible to reschedule.
- Avoid driving for at least 2 weeks after you get TECARTUS.
- Do not donate blood, organs, tissues, or cells for transplantation.
What are the possible or reasonably likely side effects of TECARTUS?
The most common side effects of TECARTUS include:- Fever (100.4°F/38°C or higher)
- Low white blood cells (can occur with a fever)
- Low red blood cells
- Low blood pressure (dizziness or lightheadedness, headache, feeling tired, short of breath)
- Fast heartbeat
- Confusion
- Difficulty speaking or slurred speech
- Nausea
- Diarrhea
These are not all the possible side effects of TECARTUS. Call your healthcare provider about any side effects that concern you. You may report side effects to the FDA at 1-800-FDA-1088.General information about the safe and effective use of TECARTUS
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. If you would like more information about TECARTUS, talk with your healthcare provider. You can ask your healthcare provider for information about TECARTUS that is written for health professionals. You can get additional information by contacting Kite at 1-844-454-KITE (5483) or at www.Tecartus.com.What are the ingredients in TECARTUS?
Active ingredients: brexucabtagene autoleucel.
Inactive ingredients: albumin (human); DMSO.
TECARTUS is a trademark of Kite Pharma, Inc. All other trademarks referenced herein are the property of their respective owners.
©2026 Kite Pharma, Inc. All Rights Reserved. -
PRINCIPAL DISPLAY PANEL - 68 mL Bag Label - Product - MK-00379
NDC: 71287-219-01
brexucabtagene autoleucel
TECARTUS®RX ONLY
FOR AUTOLOGOUS & INTRAVENOUS USE ONLY
No U.S. standard of potencyDose: One sterile bag for infusion.
Contents: Maximum of 2 x 108 autologous anti-CD19 CAR T cells in
approximately 68 mL suspension containing 5% DMSO USP.Gently mix the contents of the bag
while thawingSee package insert for full prescribing
information and instructions for
administrationShip and store in vapor phase of liquid
nitrogen ≤ -150°C. Optional one time
storage at -80°C as indicated in the
package insert.DO NOT USE A
LEUKODEPLETING FILTERDO NOT IRRADIATE
Manufactured with gentamicin
Not evaluated for infectious
substancesPreservative free
Manufacturer: Kite Pharma, Inc., 2400 Broadway, Santa Monica, CA 90404
Phone: 1-844-454-KITE U.S. Lic. #2064MK-00379

-
PRINCIPAL DISPLAY PANEL - 68 mL Cassette Label - Product - MK-00376
NDC: 71287-219-02
brexucabtagene autoleucel
TECARTUS®RX ONLY
FOR AUTOLOGOUS & INTRAVENOUS USE ONLY
No U.S. standard of potencyDose: One sterile bag for infusion.
Contents: Maximum of 2 x 108 autologous anti-CD19 CAR T cells in
approximately 68 mL suspension containing 5% DMSO USP.Gently mix the contents of
the bag while thawingSee package insert for full
prescribing information and
instructions for administrationShip and store in vapor phase of
liquid nitrogen ≤ -150°C. Optional
one time storage at -80°C as
indicated in the package insert.DO NOT USE A
LEUKODEPLETING FILTERDO NOT IRRADIATE
Manufactured with gentamicin
Not evaluated for infectious
substancesPreservative free
Manufacturer: Kite Pharma, Inc., 2400 Broadway, Santa Monica, CA 90404
Phone: 1-844-454-KITE U.S. Lic. #2064Kite
A GILEAD CompanyMK-00376

- PRINCIPAL DISPLAY PANEL - 68 mL Bag Label - Patient - AS-02442
-
PRINCIPAL DISPLAY PANEL - 68 mL Cassette Label - Patient - AS-02444
STOP
Confirm patient
ID prior to
infusionVERIFY
PATIENT IDbrexucabtagene autoleucel
TECARTUS®Lot: 123456789-0X
Kite Patient ID: 987654321
Expiration Date: YYYY-Mmm-DD
First Name M.I.: FIRST NAME W
Last Name: LAST NAME
DOB: YYYY-Mmm-DD
Hospital Patient ID: 1234567890123456
DIN:
W0123 45 678900
00
9AS-02444
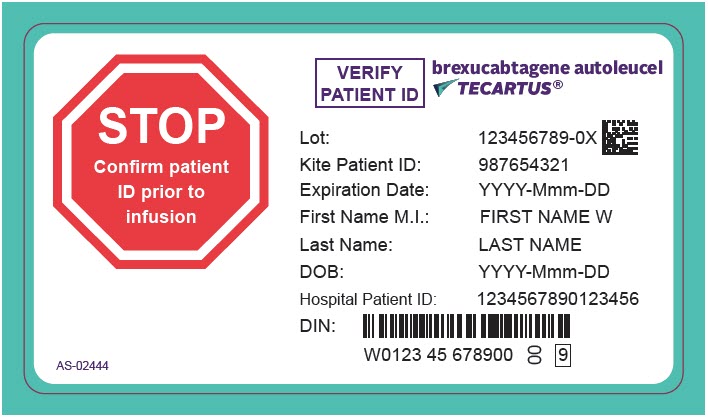
- PRINCIPAL DISPLAY PANEL - 68 mL LN2 Label - AS-02447
-
PRINCIPAL DISPLAY PANEL - 68 mL Bag Label - Product - MK-00380
NDC: 71287-220-01
brexucabtagene autoleucel
TECARTUS®RX ONLY
FOR AUTOLOGOUS & INTRAVENOUS USE ONLY
No U.S. standard of potencyDose: One sterile bag for infusion.
Contents: Maximum of 1 x 108 autologous anti-CD19 CAR T cells in
approximately 68 mL suspension containing 5% DMSO USP.Gently mix the contents of the bag
while thawingSee package insert for full prescribing
information and instructions for
administrationShip and store in vapor phase of liquid
nitrogen ≤ -150°C. Optional one time
storage at -80°C as indicated in the
package insert.DO NOT USE A
LEUKODEPLETING FILTERDO NOT IRRADIATE
Manufactured with gentamicin
Not evaluated for infectious
substancesPreservative free
Manufacturer: Kite Pharma, Inc., 2400 Broadway, Santa Monica, CA 90404
Phone: 1-844-454-KITE U.S. Lic. #2064MK-00380

-
PRINCIPAL DISPLAY PANEL - 68 mL Cassette Label - Product - MK-00377
NDC: 71287-220-02
brexucabtagene autoleucel
TECARTUS®RX ONLY
FOR AUTOLOGOUS & INTRAVENOUS USE ONLY
No U.S. standard of potencyDose: One sterile bag for infusion.
Contents: Maximum of 1 x 108 autologous anti-CD19 CAR T cells in
approximately 68 mL suspension containing 5% DMSO USP.Gently mix the contents of
the bag while thawingSee package insert for full
prescribing information and
instructions for administrationShip and store in vapor phase of
liquid nitrogen ≤ -150°C. Optional
one time storage at -80°C as
indicated in the package insert.DO NOT USE A
LEUKODEPLETING FILTERDO NOT IRRADIATE
Manufactured with gentamicin
Not evaluated for infectious
substancesPreservative free
Manufacturer: Kite Pharma, Inc., 2400 Broadway, Santa Monica, CA 90404
Phone: 1-844-454-KITE U.S. Lic. #2064Kite
A GILEAD CompanyMK-00377

- PRINCIPAL DISPLAY PANEL - 68 mL Bag Label - Patient - AS-02532
-
PRINCIPAL DISPLAY PANEL - 68 mL Cassette Label - Patient - AS-02534
STOP
Confirm patient
ID prior to
infusionVERIFY
PATIENT IDbrexucabtagene autoleucel
TECARTUS®Lot: 123456789-0X
Kite Patient ID: 987654321
Expiration Date: YYYY-Mmm-DD
First Name M.I.: FIRST NAME W
Last Name: LAST NAME
DOB: YYYY-Mmm-DD
Hospital Patient ID: 1234567890123456
DIN:
W0123 45 678900
00
9AS-02534

- PRINCIPAL DISPLAY PANEL - 68 mL LN2 Label - AS-02536
-
INGREDIENTS AND APPEARANCE
TECARTUS
brexucabtagene autoleucel suspensionProduct Information Product Type CELLULAR THERAPY Item Code (Source) NDC: 71287-219 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BREXUCABTAGENE AUTOLEUCEL (UNII: 4MD2J2T8SJ) (BREXUCABTAGENE AUTOLEUCEL - UNII:4MD2J2T8SJ) BREXUCABTAGENE AUTOLEUCEL 2000000 in 68 mL Inactive Ingredients Ingredient Name Strength DIMETHYL SULFOXIDE (UNII: YOW8V9698H) ALBUMIN HUMAN (UNII: ZIF514RVZR) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71287-219-02 1 in 1 PACKAGE 1 NDC: 71287-219-01 68 mL in 1 BAG; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125703 07/24/2020 TECARTUS
brexucabtagene autoleucel suspensionProduct Information Product Type CELLULAR THERAPY Item Code (Source) NDC: 71287-220 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BREXUCABTAGENE AUTOLEUCEL (UNII: 4MD2J2T8SJ) (BREXUCABTAGENE AUTOLEUCEL - UNII:4MD2J2T8SJ) BREXUCABTAGENE AUTOLEUCEL 1000000 in 68 mL Inactive Ingredients Ingredient Name Strength DIMETHYL SULFOXIDE (UNII: YOW8V9698H) ALBUMIN HUMAN (UNII: ZIF514RVZR) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71287-220-02 1 in 1 PACKAGE 1 NDC: 71287-220-01 68 mL in 1 BAG; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125703 10/01/2021 Labeler - Kite Pharma, Inc. (963353359) Establishment Name Address ID/FEI Business Operations Gilead Sciences, Inc. 117649816 API MANUFACTURE (71287-219) , ANALYSIS(71287-219) Establishment Name Address ID/FEI Business Operations Kite Pharma, Inc. 116931311 ANALYSIS(71287-219)




Trademark Results [TECARTUS]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TECARTUS 88560305 not registered Live/Pending |
Kite Pharma, Inc. 2019-08-01 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.