XELODA- capecitabine tablet, film coated
Xeloda by
Drug Labeling and Warnings
Xeloda by is a Prescription medication manufactured, distributed, or labeled by Genentech, Inc., Shanghai Roche Pharmaceuticals Ltd, F. Hoffmann-La Roche Ltd, F. Hoffmann-La Roche AG. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use XELODA® safely and effectively. See full prescribing information for XELODA®.
XELODA® (capecitabine) tablets, for oral use
Initial U.S. Approval: 1998WARNING: XELODA-WARFARIN INTERACTION
See full prescribing information for complete boxed warning.
Patients receiving concomitant XELODA and oral coumarin-derivative anticoagulants such as warfarin and phenprocoumon should have their anticoagulant response (INR or prothrombin time) monitored frequently in order to adjust the anticoagulant dose accordingly. Altered coagulation parameters and/or bleeding, including death, have been reported during concomitant use.
- Occurrence: Within several days and up to several months after initiating XELODA therapy; may also be seen within 1 month after stopping XELODA
- Predisposing factors: age>60 and diagnosis of cancer
INDICATIONS AND USAGE
XELODA (capecitabine) is a nucleoside metabolic inhibitor with antineoplastic activity indicated for:
-
Adjuvant Colon Cancer (1.1)
- – Patients with Dukes' C colon cancer
-
Metastatic Colorectal Cancer (1.1)
- – First-line as monotherapy when treatment with fluoropyrimidine therapy alone is preferred
-
Metastatic Breast Cancer (1.2)
- – In combination with docetaxel after failure of prior anthracycline-containing therapy
- – As monotherapy in patients resistant to both paclitaxel and an anthracycline-containing regimen
DOSAGE AND ADMINISTRATION
- Take XELODA with water within 30 min after a meal (2.1)
- Monotherapy: 1250 mg/m2 twice daily orally for 2 weeks followed by a one week rest period in 3-week cycles (2.2)
- Adjuvant treatment is recommended for a total of 6 months (8 cycles) (2.2)
- In combination with docetaxel, the recommended dose of XELODA is 1250 mg/m2 twice daily for 2 weeks followed by a 7-day rest period, combined with docetaxel at 75 mg/m2 as a 1-hour IV infusion every 3 weeks (2.2)
- XELODA dosage may need to be individualized to optimize patient management (2.3)
- Reduce the dose of XELODA by 25% in patients with moderate renal impairment (2.4)
DOSAGE FORMS AND STRENGTHS
- Tablets: 150 mg and 500 mg (3)
WARNINGS AND PRECAUTIONS
- Coagulopathy: May result in bleeding, death. Monitor anticoagulant response (e.g., INR) and adjust anticoagulant dose accordingly. (5.1)
- Diarrhea: May be severe. Interrupt XELODA treatment immediately until diarrhea resolves or decreases to grade 1. Recommend standard antidiarrheal treatments. (5.2)
- Cardiotoxicity: Common in patients with a prior history of coronary artery disease. (5.3)
- Increased Risk of Severe or Fatal Adverse Reactions in Patients with Low or Absent Dihydropyrimidine Dehydrogenase (DPD) Activity: Withhold or permanently discontinue XELODA in patients with evidence of acute early-onset or unusually severe toxicity, which may indicate near complete or total absence of DPD activity. No XELODA dose has been proven safe in patients with absent DPD activity. (5.4)
- Dehydration and Renal Failure: Interrupt XELODA treatment until dehydration is corrected. Potential risk of acute renal failure secondary to dehydration. Monitor and correct dehydration. (5.5).
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.6, 8.1, 8.3)
- Mucocutaneous and Dermatologic Toxicity: Severe mucocutaneous reactions, Steven-Johnson Syndrome (SJS) and Toxic Epidermal Necrolysis (TEN), have been reported. XELODA should be permanently discontinued in patients who experience a severe mucocutaneous reaction during treatment. XELODA may induce hand-and-foot syndrome. Persistent or severe hand-and-foot syndrome can lead to loss of fingerprints which could impact patient identification. Interrupt XELODA treatment until the hand-and-foot syndrome event resolves or decreases in intensity. (5.7)
- Hyperbilirubinemia: Interrupt XELODA treatment immediately until the hyperbilirubinemia resolves or decreases in intensity. (5.8)
- Hematologic: Do not treat patients with neutrophil counts <1.5 × 109/L or thrombocyte counts <100 × 109/L. If grade 3-4 neutropenia or thrombocytopenia occurs, stop therapy until condition resolves. (5.9)
ADVERSE REACTIONS
Most common adverse reactions (≥30%) were diarrhea, hand-and-foot syndrome, nausea, vomiting, abdominal pain, fatigue/weakness, and hyperbilirubinemia. Other adverse reactions, including serious adverse reactions, have been reported. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Genentech at 1-888-835-2555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Anticoagulants: Monitor anticoagulant response (INR or prothrombin time) frequently in order to adjust the anticoagulant dose as needed. (5.2, 7.1)
- Phenytoin: Monitor phenytoin levels in patients taking XELODA concomitantly with phenytoin. The phenytoin dose may need to be reduced. (7.1)
- Leucovorin: The concentration of 5-fluorouracil is increased and its toxicity may be enhanced by leucovorin. (7.1)
- CYP2C9 substrates: Care should be exercised when XELODA is coadministered with CYP2C9 substrates. (7.1)
- Allopurinol: Avoid the use of allopurinol during treatment with XELODA.
- Food reduced both the rate and extent of absorption of capecitabine. (2, 7.2, 12.3)
USE IN SPECIFIC POPULATIONS
- Lactation: Advise women not to breastfeed. (8.2)
- Females and Males of Reproductive Potential: Verify pregnancy status of females prior to initiation of XELODA. Advise males with female partners of reproductive potential to use effective contraception. (8.3)
- Geriatric: Greater incidence of adverse reactions. Monitoring required. (8.5)
- Hepatic Impairment: Monitoring is recommended in patients with mild to moderate hepatic impairment. (8.6)
- Renal Impairment: Reduce XELODA starting dose in patients with moderate renal impairment (2.4, 8.7, 12.3)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: XELODA-WARFARIN INTERACTION
1 INDICATIONS AND USAGE
1.1 Colorectal Cancer
1.2 Breast Cancer
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
2.2 Standard Starting Dose
2.3 Dose Management Guidelines
2.4 Adjustment of Starting Dose in Special Populations
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Severe Renal Impairment
4.2 Hypersensitivity
5 WARNINGS AND PRECAUTIONS
5.1 Coagulopathy
5.2 Diarrhea
5.3 Cardiotoxicity
5.4 Dihydropyrimidine Dehydrogenase Deficiency
5.5 Dehydration and Renal Failure
5.6 Embryo-Fetal Toxicity
5.7 Mucocutaneous and Dermatologic Toxicity
5.8 Hyperbilirubinemia
5.9 Hematologic
5.10 Geriatric Patients
5.11 Hepatic Insufficiency
5.12 Combination With Other Drugs
6 ADVERSE REACTIONS
6.1 Adjuvant Colon Cancer
6.2 Metastatic Colorectal Cancer
6.3 Breast Cancer
6.4 Clinically Relevant Adverse Events in <5% of Patients
6.5 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Drug-Drug Interactions
7.2 Drug-Food Interaction
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Insufficiency
8.7 Renal Insufficiency
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Adjuvant Colon Cancer
14.2 Metastatic Colorectal Cancer
14.3 Breast Cancer
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: XELODA-WARFARIN INTERACTION
XELODA Warfarin Interaction: Patients receiving concomitant capecitabine and oral coumarin-derivative anticoagulant therapy should have their anticoagulant response (INR or prothrombin time) monitored frequently in order to adjust the anticoagulant dose accordingly. A clinically important XELODA-Warfarin drug interaction was demonstrated in a clinical pharmacology trial [see Warnings and Precautions (5.2) and Drug Interactions (7.1)]. Altered coagulation parameters and/or bleeding, including death, have been reported in patients taking XELODA concomitantly with coumarin-derivative anticoagulants such as warfarin and phenprocoumon. Postmarketing reports have shown clinically significant increases in prothrombin time (PT) and INR in patients who were stabilized on anticoagulants at the time XELODA was introduced. These events occurred within several days and up to several months after initiating XELODA therapy and, in a few cases, within 1 month after stopping XELODA. These events occurred in patients with and without liver metastases. Age greater than 60 and a diagnosis of cancer independently predispose patients to an increased risk of coagulopathy.
-
1 INDICATIONS AND USAGE
1.1 Colorectal Cancer
- XELODA is indicated as a single agent for adjuvant treatment in patients with Dukes' C colon cancer who have undergone complete resection of the primary tumor when treatment with fluoropyrimidine therapy alone is preferred. XELODA was non-inferior to 5-fluorouracil and leucovorin (5-FU/LV) for disease-free survival (DFS). Physicians should consider results of combination chemotherapy trials, which have shown improvement in DFS and OS, when prescribing single-agent XELODA in the adjuvant treatment of Dukes' C colon cancer.
- XELODA is indicated as first-line treatment of patients with metastatic colorectal carcinoma when treatment with fluoropyrimidine therapy alone is preferred. Combination chemotherapy has shown a survival benefit compared to 5-FU/LV alone. A survival benefit over 5-FU/LV has not been demonstrated with XELODA monotherapy. Use of XELODA instead of 5-FU/LV in combinations has not been adequately studied to assure safety or preservation of the survival advantage.
1.2 Breast Cancer
- XELODA in combination with docetaxel is indicated for the treatment of patients with metastatic breast cancer after failure of prior anthracycline-containing chemotherapy.
- XELODA monotherapy is also indicated for the treatment of patients with metastatic breast cancer resistant to both paclitaxel and an anthracycline-containing chemotherapy regimen or resistant to paclitaxel and for whom further anthracycline therapy is not indicated (e.g., patients who have received cumulative doses of 400 mg/m2 of doxorubicin or doxorubicin equivalents). Resistance is defined as progressive disease while on treatment, with or without an initial response, or relapse within 6 months of completing treatment with an anthracycline-containing adjuvant regimen.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
XELODA tablets should be swallowed whole with water within 30 minutes after a meal. XELODA is a cytotoxic drug. Follow applicable special handling and disposal procedures.1 If XELODA tablets must be cut or crushed, this should be done by a professional trained in safe handling of cytotoxic drugs using appropriate equipment and safety procedures. XELODA dose is calculated according to body surface area.
2.2 Standard Starting Dose
Monotherapy (Metastatic Colorectal Cancer, Adjuvant Colorectal Cancer, Metastatic Breast Cancer)
The recommended dose of XELODA is 1250 mg/m2 administered orally twice daily (morning and evening; equivalent to 2500 mg/m2 total daily dose) for 2 weeks followed by a 1-week rest period given as 3-week cycles (see Table 1).
Adjuvant treatment in patients with Dukes' C colon cancer is recommended for a total of 6 months [ie, XELODA 1250 mg/m2 orally twice daily for 2 weeks followed by a 1-week rest period, given as 3-week cycles for a total of 8 cycles (24 weeks)].
Table 1 XELODA Dose Calculation According to Body Surface Area Dose Level 1250 mg/m2
Twice a DayNumber of Tablets to be Taken at Each Dose (Morning and Evening) Surface Area
(m2)Total Daily Dose* (mg) 150 mg 500 mg - * Total Daily Dose divided by 2 to allow equal morning and evening doses
≤ 1.25 3000 0 3 1.26-1.37 3300 1 3 1.38-1.51 3600 2 3 1.52-1.65 4000 0 4 1.66-1.77 4300 1 4 1.78-1.91 4600 2 4 1.92-2.05 5000 0 5 2.06-2.17 5300 1 5 ≥ 2.18 5600 2 5 In Combination With Docetaxel (Metastatic Breast Cancer)
In combination with docetaxel, the recommended dose of XELODA is 1250 mg/m2 twice daily for 2 weeks followed by a 1-week rest period, combined with docetaxel at 75 mg/m2 as a 1-hour intravenous infusion every 3 weeks. Pre-medication, according to the docetaxel labeling, should be started prior to docetaxel administration for patients receiving the XELODA plus docetaxel combination. Table 1 displays the total daily dose of XELODA by body surface area and the number of tablets to be taken at each dose.
2.3 Dose Management Guidelines
General
XELODA dosage may need to be individualized to optimize patient management. Patients should be carefully monitored for toxicity and doses of XELODA should be modified as necessary to accommodate individual patient tolerance to treatment [see Clinical Studies (14)]. Toxicity due to XELODA administration may be managed by symptomatic treatment, dose interruptions and adjustment of XELODA dose. Once the dose has been reduced, it should not be increased at a later time. Doses of XELODA omitted for toxicity are not replaced or restored; instead the patient should resume the planned treatment cycles.
The dose of phenytoin and the dose of coumarin-derivative anticoagulants may need to be reduced when either drug is administered concomitantly with XELODA [see Drug Interactions (7.1)].
Monotherapy (Metastatic Colorectal Cancer, Adjuvant Colorectal Cancer, Metastatic Breast Cancer)
XELODA dose modification scheme as described below (see Table 2) is recommended for the management of adverse reactions.
Table 2 Recommended Dose Modifications of XELODA Toxicity NCIC Grades* During a Course of Therapy Dose Adjustment for Next Treatment (% of starting dose) - * National Cancer Institute of Canada Common Toxicity Criteria were used except for the hand-and-foot syndrome [see Warnings and Precautions (5)].
Grade 1 Maintain dose level Maintain dose level Grade 2 -1st appearance Interrupt until resolved to grade 0-1 100% -2nd appearance 75% -3rd appearance 50% -4th appearance Discontinue treatment permanently - Grade 3 -1st appearance Interrupt until resolved to grade 0-1 75% -2nd appearance 50% -3rd appearance Discontinue treatment permanently - Grade 4 -1st appearance Discontinue permanently
OR
If physician deems it to be in the patient's best interest to continue, interrupt until resolved to grade 0-150% In Combination With Docetaxel (Metastatic Breast Cancer)
Dose modifications of XELODA for toxicity should be made according to Table 2 above for XELODA. At the beginning of a treatment cycle, if a treatment delay is indicated for either XELODA or docetaxel, then administration of both agents should be delayed until the requirements for restarting both drugs are met.
The dose reduction schedule for docetaxel when used in combination with XELODA for the treatment of metastatic breast cancer is shown in Table 3.
Table 3 Docetaxel Dose Reduction Schedule in Combination with XELODA Toxicity NCIC Grades* Grade 2 Grade 3 Grade 4 - * National Cancer Institute of Canada Common Toxicity Criteria were used except for hand-and-foot syndrome [see Warnings and Precautions (5)].
1st appearance Delay treatment until resolved to grade 0-1; Resume treatment with original dose of 75 mg/m2 docetaxel Delay treatment until resolved to grade 0-1;
Resume treatment at 55 mg/m2 of docetaxel.Discontinue treatment with docetaxel 2nd appearance Delay treatment until resolved to grade 0-1; Resume treatment at 55 mg/m2 of docetaxel. Discontinue treatment with docetaxel - 3rd appearance Discontinue treatment with docetaxel - - 2.4 Adjustment of Starting Dose in Special Populations
Renal Impairment
No adjustment to the starting dose of XELODA is recommended in patients with mild renal impairment (creatinine clearance = 51 to 80 mL/min [Cockroft and Gault, as shown below]). In patients with moderate renal impairment (baseline creatinine clearance = 30 to 50 mL/min), a dose reduction to 75% of the XELODA starting dose when used as monotherapy or in combination with docetaxel (from 1250 mg/m2 to 950 mg/m2 twice daily) is recommended [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)]. Subsequent dose adjustment is recommended as outlined in Table 2 and Table 3 (depending on the regimen) if a patient develops a grade 2 to 4 adverse event [see Warnings and Precautions (5.5)]. The starting dose adjustment recommendations for patients with moderate renal impairment apply to both XELODA monotherapy and XELODA in combination use with docetaxel.
Cockroft and Gault Equation:
Creatinine clearance for males = (140 - age [yrs]) (body wt [kg]) (72) (serum creatinine [mg/dL]) Creatinine clearance for females = 0.85 × male value
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
4.1 Severe Renal Impairment
XELODA is contraindicated in patients with severe renal impairment (creatinine clearance below 30 mL/min [Cockroft and Gault]) [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Coagulopathy
Patients receiving concomitant capecitabine and oral coumarin-derivative anticoagulant therapy should have their anticoagulant response (INR or prothrombin time) monitored closely with great frequency and the anticoagulant dose should be adjusted accordingly [see Boxed Warning and Drug Interactions (7.1)].
5.2 Diarrhea
XELODA can induce diarrhea, sometimes severe. Patients with severe diarrhea should be carefully monitored and given fluid and electrolyte replacement if they become dehydrated. In 875 patients with either metastatic breast or colorectal cancer who received XELODA monotherapy, the median time to first occurrence of grade 2 to 4 diarrhea was 34 days (range from 1 to 369 days). The median duration of grade 3 to 4 diarrhea was 5 days. National Cancer Institute of Canada (NCIC) grade 2 diarrhea is defined as an increase of 4 to 6 stools/day or nocturnal stools, grade 3 diarrhea as an increase of 7 to 9 stools/day or incontinence and malabsorption, and grade 4 diarrhea as an increase of ≥10 stools/day or grossly bloody diarrhea or the need for parenteral support. If grade 2, 3 or 4 diarrhea occurs, administration of XELODA should be immediately interrupted until the diarrhea resolves or decreases in intensity to grade 1 [see Dosage and Administration (2.3)]. Standard antidiarrheal treatments (e.g., loperamide) are recommended.
Necrotizing enterocolitis (typhlitis) has been reported.
5.3 Cardiotoxicity
The cardiotoxicity observed with XELODA includes myocardial infarction/ischemia, angina, dysrhythmias, cardiac arrest, cardiac failure, sudden death, electrocardiographic changes, and cardiomyopathy. These adverse reactions may be more common in patients with a prior history of coronary artery disease.
5.4 Dihydropyrimidine Dehydrogenase Deficiency
Based on postmarketing reports, patients with certain homozygous or certain compound heterozygous mutations in the DPD gene that result in complete or near complete absence of DPD activity are at increased risk for acute early-onset of toxicity and severe, life-threatening, or fatal adverse reactions caused by XELODA (e.g., mucositis, diarrhea, neutropenia, and neurotoxicity). Patients with partial DPD activity may also have increased risk of severe, life-threatening, or fatal adverse reactions caused by XELODA.
Withhold or permanently discontinue XELODA based on clinical assessment of the onset, duration and severity of the observed toxicities in patients with evidence of acute early-onset or unusually severe toxicity, which may indicate near complete or total absence of DPD activity. No XELODA dose has been proven safe for patients with complete absence of DPD activity. There is insufficient data to recommend a specific dose in patients with partial DPD activity as measured by any specific test.
5.5 Dehydration and Renal Failure
Dehydration has been observed and may cause acute renal failure which can be fatal. Patients with pre-existing compromised renal function or who are receiving concomitant XELODA with known nephrotoxic agents are at higher risk. Patients with anorexia, asthenia, nausea, vomiting or diarrhea may rapidly become dehydrated. Monitor patients when XELODA is administered to prevent and correct dehydration at the onset. If grade 2 (or higher) dehydration occurs, XELODA treatment should be immediately interrupted and the dehydration corrected. Treatment should not be restarted until the patient is rehydrated and any precipitating causes have been corrected or controlled. Dose modifications should be applied for the precipitating adverse event as necessary [see Dosage and Administration (2.3)].
Patients with moderate renal impairment at baseline require dose reduction [see Dosage and Administration (2.4)]. Patients with mild and moderate renal impairment at baseline should be carefully monitored for adverse reactions. Prompt interruption of therapy with subsequent dose adjustments is recommended if a patient develops a grade 2 to 4 adverse event as outlined in Table 2 [see Dosage and Administration (2.3), Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
5.6 Embryo-Fetal Toxicity
Based on findings from animal reproduction studies and its mechanism of action, XELODA may cause fetal harm when given to a pregnant woman [see Clinical Pharmacology (12.1)]. Limited available data are not sufficient to inform use of XELODA in pregnant women. In animal reproduction studies, administration of capecitabine to pregnant animals during the period of organogenesis caused embryolethality and teratogenicity in mice and embryolethality in monkeys at 0.2 and 0.6 times the exposure (AUC) in patients receiving the recommended dose respectively [see Use in Specific Populations (8.1)]. Apprise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment and for 6 months following the last dose of XELODA [see Use in Specific Populations (8.3)].
5.7 Mucocutaneous and Dermatologic Toxicity
Severe mucocutaneous reactions, some with fatal outcome, such as Stevens-Johnson syndrome and Toxic Epidermal Necrolysis (TEN) can occur in patients treated with XELODA [see Adverse Reactions (6.4)]. XELODA should be permanently discontinued in patients who experience a severe mucocutaneous reaction possibly attributable to XELODA treatment.
Hand-and-foot syndrome (palmar-plantar erythrodysesthesia or chemotherapy-induced acral erythema) is a cutaneous toxicity. Median time to onset was 79 days (range from 11 to 360 days) with a severity range of grades 1 to 3 for patients receiving XELODA monotherapy in the metastatic setting. Grade 1 is characterized by any of the following: numbness, dysesthesia/paresthesia, tingling, painless swelling or erythema of the hands and/or feet and/or discomfort which does not disrupt normal activities. Grade 2 hand-and-foot syndrome is defined as painful erythema and swelling of the hands and/or feet and/or discomfort affecting the patient's activities of daily living. Grade 3 hand-and-foot syndrome is defined as moist desquamation, ulceration, blistering or severe pain of the hands and/or feet and/or severe discomfort that causes the patient to be unable to work or perform activities of daily living. Persistent or severe hand-and-foot syndrome (grade 2 and above) can eventually lead to loss of fingerprints which could impact patient identification. If grade 2 or 3 hand-and-foot syndrome occurs, administration of XELODA should be interrupted until the event resolves or decreases in intensity to grade 1. Following grade 3 hand-and-foot syndrome, subsequent doses of XELODA should be decreased [see Dosage and Administration (2.3)].
5.8 Hyperbilirubinemia
In 875 patients with either metastatic breast or colorectal cancer who received at least one dose of XELODA 1250 mg/m2 twice daily as monotherapy for 2 weeks followed by a 1-week rest period, grade 3 (1.5-3 × ULN) hyperbilirubinemia occurred in 15.2% (n=133) of patients and grade 4 (>3 × ULN) hyperbilirubinemia occurred in 3.9% (n=34) of patients. Of 566 patients who had hepatic metastases at baseline and 309 patients without hepatic metastases at baseline, grade 3 or 4 hyperbilirubinemia occurred in 22.8% and 12.3%, respectively. Of the 167 patients with grade 3 or 4 hyperbilirubinemia, 18.6% (n=31) also had postbaseline elevations (grades 1 to 4, without elevations at baseline) in alkaline phosphatase and 27.5% (n=46) had postbaseline elevations in transaminases at any time (not necessarily concurrent). The majority of these patients, 64.5% (n=20) and 71.7% (n=33), had liver metastases at baseline. In addition, 57.5% (n=96) and 35.3% (n=59) of the 167 patients had elevations (grades 1 to 4) at both prebaseline and postbaseline in alkaline phosphatase or transaminases, respectively. Only 7.8% (n=13) and 3.0% (n=5) had grade 3 or 4 elevations in alkaline phosphatase or transaminases.
In the 596 patients treated with XELODA as first-line therapy for metastatic colorectal cancer, the incidence of grade 3 or 4 hyperbilirubinemia was similar to the overall clinical trial safety database of XELODA monotherapy. The median time to onset for grade 3 or 4 hyperbilirubinemia in the colorectal cancer population was 64 days and median total bilirubin increased from 8 µm/L at baseline to 13 µm/L during treatment with XELODA. Of the 136 colorectal cancer patients with grade 3 or 4 hyperbilirubinemia, 49 patients had grade 3 or 4 hyperbilirubinemia as their last measured value, of which 46 had liver metastases at baseline.
In 251 patients with metastatic breast cancer who received a combination of XELODA and docetaxel, grade 3 (1.5 to 3 × ULN) hyperbilirubinemia occurred in 7% (n=17) and grade 4 (>3 × ULN) hyperbilirubinemia occurred in 2% (n=5).
If drug-related grade 3 to 4 elevations in bilirubin occur, administration of XELODA should be immediately interrupted until the hyperbilirubinemia decreases to ≤3.0 × ULN [see recommended dose modifications under Dosage and Administration (2.3)].
5.9 Hematologic
In 875 patients with either metastatic breast or colorectal cancer who received a dose of 1250 mg/m2 administered twice daily as monotherapy for 2 weeks followed by a 1-week rest period, 3.2%, 1.7%, and 2.4% of patients had grade 3 or 4 neutropenia, thrombocytopenia or decreases in hemoglobin, respectively. In 251 patients with metastatic breast cancer who received a dose of XELODA in combination with docetaxel, 68% had grade 3 or 4 neutropenia, 2.8% had grade 3 or 4 thrombocytopenia, and 9.6% had grade 3 or 4 anemia.
Patients with baseline neutrophil counts of <1.5 × 109/L and/or thrombocyte counts of <100 × 109/L should not be treated with XELODA. If unscheduled laboratory assessments during a treatment cycle show grade 3 or 4 hematologic toxicity, treatment with XELODA should be interrupted.
5.10 Geriatric Patients
Patients ≥80 years old may experience a greater incidence of grade 3 or 4 adverse reactions. In 875 patients with either metastatic breast or colorectal cancer who received XELODA monotherapy, 62% of the 21 patients ≥80 years of age treated with XELODA experienced a treatment-related grade 3 or 4 adverse event: diarrhea in 6 (28.6%), nausea in 3 (14.3%), hand-and-foot syndrome in 3 (14.3%), and vomiting in 2 (9.5%) patients. Among the 10 patients 70 years of age and greater (no patients were >80 years of age) treated with XELODA in combination with docetaxel, 30% (3 out of 10) of patients experienced grade 3 or 4 diarrhea and stomatitis, and 40% (4 out of 10) experienced grade 3 hand-and-foot syndrome.
Among the 67 patients ≥60 years of age receiving XELODA in combination with docetaxel, the incidence of grade 3 or 4 treatment-related adverse reactions, treatment-related serious adverse reactions, withdrawals due to adverse reactions, treatment discontinuations due to adverse reactions and treatment discontinuations within the first two treatment cycles was higher than in the <60 years of age patient group.
In 995 patients receiving XELODA as adjuvant therapy for Dukes' C colon cancer after resection of the primary tumor, 41% of the 398 patients ≥65 years of age treated with XELODA experienced a treatment-related grade 3 or 4 adverse event: hand-and-foot syndrome in 75 (18.8%), diarrhea in 52 (13.1%), stomatitis in 12 (3.0%), neutropenia/granulocytopenia in 11 (2.8%), vomiting in 6 (1.5%), and nausea in 5 (1.3%) patients. In patients ≥65 years of age (all randomized population; capecitabine 188 patients, 5-FU/LV 208 patients) treated for Dukes' C colon cancer after resection of the primary tumor, the hazard ratios for disease-free survival and overall survival for XELODA compared to 5-FU/LV were 1.01 (95% C.I. 0.80 – 1.27) and 1.04 (95% C.I. 0.79 – 1.37), respectively.
5.11 Hepatic Insufficiency
Patients with mild to moderate hepatic dysfunction due to liver metastases should be carefully monitored when XELODA is administered. The effect of severe hepatic dysfunction on the disposition of XELODA is not known [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
-
6 ADVERSE REACTIONS
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
6.1 Adjuvant Colon Cancer
Table 4 shows the adverse reactions occurring in ≥5% of patients from one phase 3 trial in patients with Dukes' C colon cancer who received at least one dose of study medication and had at least one safety assessment. A total of 995 patients were treated with 1250 mg/m2 twice a day of XELODA administered for 2 weeks followed by a 1-week rest period, and 974 patients were administered 5-FU and leucovorin (20 mg/m2 leucovorin IV followed by 425 mg/m2 IV bolus 5-FU on days 1-5 every 28 days). The median duration of treatment was 164 days for capecitabine-treated patients and 145 days for 5-FU/LV-treated patients. A total of 112 (11%) and 73 (7%) capecitabine and 5-FU/LV-treated patients, respectively, discontinued treatment because of adverse reactions. A total of 18 deaths due to all causes occurred either on study or within 28 days of receiving study drug: 8 (0.8%) patients randomized to XELODA and 10 (1.0%) randomized to 5-FU/LV.
Table 5 shows grade 3/4 laboratory abnormalities occurring in ≥1% of patients from one phase 3 trial in patients with Dukes' C colon cancer who received at least one dose of study medication and had at least one safety assessment.
Table 4 Percent Incidence of Adverse Reactions Reported in ≥5% of Patients Treated With XELODA or 5-FU/LV for Colon Cancer in the Adjuvant Setting (Safety Population) Adjuvant Treatment for Colon Cancer (N=1969) XELODA
(N=995)5-FU/LV
(N=974)Body System/
Adverse EventAll Grades Grade 3/4 All Grades Grade 3/4 Gastrointestinal Disorders Diarrhea 47 12 65 14 Nausea 34 2 47 2 Stomatitis 22 2 60 14 Vomiting 15 2 21 2 Abdominal Pain 14 3 16 2 Constipation 9 - 11 <1 Upper Abdominal Pain 7 <1 7 <1 Dyspepsia 6 <1 5 - Skin and Subcutaneous Tissue Disorders Hand-and-Foot Syndrome 60 17 9 <1 Alopecia 6 - 22 <1 Rash 7 - 8 - Erythema 6 1 5 <1 General Disorders and Administration Site Conditions Fatigue 16 <1 16 1 Pyrexia 7 <1 9 <1 Asthenia 10 <1 10 1 Lethargy 10 <1 9 <1 Nervous System Disorders Dizziness 6 <1 6 - Headache 5 <1 6 <1 Dysgeusia 6 - 9 - Metabolism and Nutrition Disorders Anorexia 9 <1 11 <1 Eye Disorders Conjunctivitis 5 <1 6 <1 Blood and Lymphatic System Disorders Neutropenia 2 <1 8 5 Respiratory Thoracic and Mediastinal Disorders Epistaxis 2 - 5 - Table 5 Percent Incidence of Grade 3/4 Laboratory Abnormalities Reported in ≥1% of Patients Receiving XELODA Monotherapy for Adjuvant Treatment of Colon Cancer (Safety Population) Adverse Event XELODA
(n=995)
Grade 3/4 %IV 5-FU/LV
(n=974)
Grade 3/4 %- * The incidence of grade 3/4 white blood cell abnormalities was 1.3% in the XELODA arm and 4.9% in the IV 5-FU/LV arm.
- † It should be noted that grading was according to NCIC CTC Version 1 (May, 1994). In the NCIC-CTC Version 1, hyperbilirubinemia grade 3 indicates a bilirubin value of 1.5 to 3.0 × upper limit of normal (ULN) range, and grade 4 a value of > 3.0 × ULN. The NCI CTC Version 2 and above define a grade 3 bilirubin value of >3.0 to 10.0 × ULN, and grade 4 values >10.0 × ULN.
Increased ALAT (SGPT) 1.6 0.6 Increased calcium 1.1 0.7 Decreased calcium 2.3 2.2 Decreased hemoglobin 1.0 1.2 Decreased lymphocytes 13.0 13.0 Decreased neutrophils* 2.2 26.2 Decreased neutrophils/granulocytes 2.4 26.4 Decreased platelets 1.0 0.7 Increased bilirubin† 20 6.3 6.2 Metastatic Colorectal Cancer
Monotherapy
Table 6 shows the adverse reactions occurring in ≥5% of patients from pooling the two phase 3 trials in first line metastatic colorectal cancer. A total of 596 patients with metastatic colorectal cancer were treated with 1250 mg/m2 twice a day of XELODA administered for 2 weeks followed by a 1-week rest period, and 593 patients were administered 5-FU and leucovorin in the Mayo regimen (20 mg/m2 leucovorin IV followed by 425 mg/m2 IV bolus 5-FU, on days 1-5, every 28 days). In the pooled colorectal database the median duration of treatment was 139 days for capecitabine-treated patients and 140 days for 5-FU/LV-treated patients. A total of 78 (13%) and 63 (11%) capecitabine and 5-FU/LV-treated patients, respectively, discontinued treatment because of adverse reactions/intercurrent illness. A total of 82 deaths due to all causes occurred either on study or within 28 days of receiving study drug: 50 (8.4%) patients randomized to XELODA and 32 (5.4%) randomized to 5-FU/LV.
Table 6 Pooled Phase 3 Colorectal Trials: Percent Incidence of Adverse Reactions in ≥5% of Patients Adverse Event XELODA
(n=596)5-FU/LV
(n=593)Total
%Grade 3
%Grade 4
%Total
%Grade 3
%Grade 4
%– Not observed
NA = Not Applicable- * Excluding vertigo
Number of Patients With > One Adverse Event 96 52 9 94 45 9 Body System/Adverse Event GI Diarrhea 55 13 2 61 10 2 Nausea 43 4 – 51 3 <1 Vomiting 27 4 <1 30 4 <1 Stomatitis 25 2 <1 62 14 1 Abdominal Pain 35 9 <1 31 5 – Gastrointestinal Motility Disorder 10 <1 – 7 <1 – Constipation 14 1 <1 17 1 – Oral Discomfort 10 – – 10 – – Upper GI Inflammatory Disorders 8 <1 – 10 1 – Gastrointestinal Hemorrhage 6 1 <1 3 1 – Ileus 6 4 1 5 2 1 Skin and Subcutaneous Hand-and-Foot Syndrome 54 17 NA 6 1 NA Dermatitis 27 1 – 26 1 – Skin Discoloration 7 <1 – 5 – – Alopecia 6 – – 21 <1 – General Fatigue/Weakness 42 4 – 46 4 – Pyrexia 18 1 – 21 2 – Edema 15 1 – 9 1 – Pain 12 1 – 10 1 – Chest Pain 6 1 – 6 1 <1 Neurological Peripheral Sensory Neuropathy 10 – – 4 – – Headache 10 1 – 7 – – Dizziness* 8 <1 – 8 <1 – Insomnia 7 – – 7 – – Taste Disturbance 6 1 – 11 <1 1 Metabolism Appetite Decreased 26 3 <1 31 2 <1 Dehydration 7 2 <1 8 3 1 Eye Eye Irritation 13 – – 10 <1 – Vision Abnormal 5 – – 2 – – Respiratory Dyspnea 14 1 – 10 <1 1 Cough 7 <1 1 8 – – Pharyngeal Disorder 5 – – 5 – – Epistaxis 3 <1 – 6 – – Sore Throat 2 – – 6 – – Musculoskeletal Back Pain 10 2 – 9 <1 – Arthralgia 8 1 – 6 1 – Vascular Venous Thrombosis 8 3 <1 6 2 – Psychiatric Mood Alteration 5 – – 6 <1 – Depression 5 – – 4 <1 – Infections Viral 5 <1 – 5 <1 – Blood and Lymphatic Anemia 80 2 <1 79 1 <1 Neutropenia 13 1 2 46 8 13 Hepatobiliary Hyperbilirubinemia 48 18 5 17 3 3 6.3 Breast Cancer
In Combination with Docetaxel
The following data are shown for the combination study with XELODA and docetaxel in patients with metastatic breast cancer in Table 7 and Table 8. In the XELODA and docetaxel combination arm the treatment was XELODA administered orally 1250 mg/m2 twice daily as intermittent therapy (2 weeks of treatment followed by 1 week without treatment) for at least 6 weeks and docetaxel administered as a 1-hour intravenous infusion at a dose of 75 mg/m2 on the first day of each 3-week cycle for at least 6 weeks. In the monotherapy arm docetaxel was administered as a 1-hour intravenous infusion at a dose of 100 mg/m2 on the first day of each 3-week cycle for at least 6 weeks. The mean duration of treatment was 129 days in the combination arm and 98 days in the monotherapy arm. A total of 66 patients (26%) in the combination arm and 49 (19%) in the monotherapy arm withdrew from the study because of adverse reactions. The percentage of patients requiring dose reductions due to adverse reactions was 65% in the combination arm and 36% in the monotherapy arm. The percentage of patients requiring treatment interruptions due to adverse reactions in the combination arm was 79%. Treatment interruptions were part of the dose modification scheme for the combination therapy arm but not for the docetaxel monotherapy-treated patients.
Table 7 Percent Incidence of Adverse Events Considered Related or Unrelated to Treatment in ≥5% of Patients Participating in the XELODA and Docetaxel Combination vs Docetaxel Monotherapy Study Adverse Event XELODA 1250 mg/m2/bid With Docetaxel
75 mg/m2/3 weeksDocetaxel
100 mg/m2/3 weeks(n=251) (n=255) Total
%Grade 3
%Grade 4
%Total
%Grade 3
%Grade 4
%– Not observed
NA = Not ApplicableNumber of Patients With at Least One Adverse Event 99 76.5 29.1 97 57.6 31.8 Body System/Adverse Event GI Diarrhea 67 14 <1 48 5 <1 Stomatitis 67 17 <1 43 5 – Nausea 45 7 – 36 2 – Vomiting 35 4 1 24 2 – Constipation 20 2 – 18 – – Abdominal Pain 30 <3 <1 24 2 – Dyspepsia 14 – – 8 1 – Dry Mouth 6 <1 – 5 – – Skin and Subcutaneous Hand-and-Foot Syndrome 63 24 NA 8 1 NA Alopecia 41 6 – 42 7 – Nail Disorder 14 2 – 15 – – Dermatitis 8 – – 11 1 – Rash Erythematous 9 <1 – 5 – – Nail Discoloration 6 – – 4 <1 – Onycholysis 5 1 – 5 1 – Pruritus 4 – – 5 – – General Pyrexia 28 2 – 34 2 – Asthenia 26 4 <1 25 6 – Fatigue 22 4 – 27 6 – Weakness 16 2 – 11 2 – Pain in Limb 13 <1 – 13 2 – Lethargy 7 – – 6 2 – Pain 7 <1 – 5 1 – Chest Pain (non-cardiac) 4 <1 – 6 2 – Influenza-like Illness 5 – – 5 – – Neurological Taste Disturbance 16 <1 – 14 <1 – Headache 15 3 – 15 2 – Paresthesia 12 <1 – 16 1 – Dizziness 12 – – 8 <1 – Insomnia 8 – – 10 <1 – Peripheral Neuropathy 6 – – 10 1 – Hypoaesthesia 4 <1 – 8 <1 – Metabolism Anorexia 13 1 – 11 <1 – Appetite Decreased 10 – – 5 – – Weight Decreased 7 – – 5 – – Dehydration 10 2 – 7 <1 <1 Eye Lacrimation Increased 12 – – 7 <1 – Conjunctivitis 5 – – 4 – – Eye Irritation 5 – – 1 – – Musculoskeletal Arthralgia 15 2 – 24 3 – Myalgia 15 2 – 25 2 – Back Pain 12 <1 – 11 3 – Bone Pain 8 <1 – 10 2 – Cardiac Edema 33 <2 – 34 <3 1 Blood Neutropenic Fever 16 3 13 21 5 16 Respiratory Dyspnea 14 2 <1 16 2 – Cough 13 1 – 22 <1 – Sore Throat 12 2 – 11 <1 – Epistaxis 7 <1 – 6 – – Rhinorrhea 5 – – 3 – – Pleural Effusion 2 1 – 7 4 – Infection Oral Candidiasis 7 <1 – 8 <1 – Urinary Tract Infection 6 <1 – 4 – – Upper Respiratory Tract 4 – – 5 1 – Vascular Flushing 5 – – 5 – – Lymphoedema 3 <1 – 5 1 – Psychiatric Depression 5 – – 5 1 – Table 8 Percent of Patients With Laboratory Abnormalities Participating in the XELODA and Docetaxel Combination vs Docetaxel Monotherapy Study Adverse Event XELODA 1250 mg/m2/bid With Docetaxel
75 mg/m2/3 weeksDocetaxel
100 mg/m2/3 weeks(n=251) (n=255) Body System/Adverse Event Total
%Grade 3
%Grade 4
%Total
%Grade 3
%Grade 4
%Hematologic Leukopenia 91 37 24 88 42 33 Neutropenia/Granulocytopenia 86 20 49 87 10 66 Thrombocytopenia 41 2 1 23 1 2 Anemia 80 7 3 83 5 <1 Lymphocytopenia 99 48 41 98 44 40 Hepatobiliary Hyperbilirubinemia 20 7 2 6 2 2 Monotherapy
The following data are shown for the study in stage IV breast cancer patients who received a dose of 1250 mg/m2 administered twice daily for 2 weeks followed by a 1-week rest period. The mean duration of treatment was 114 days. A total of 13 out of 162 patients (8%) discontinued treatment because of adverse reactions/intercurrent illness.
Table 9 Percent Incidence of Adverse Reactions Considered Remotely, Possibly or Probably Related to Treatment in ≥5% of Patients Participating in the Single Arm Trial in Stage IV Breast Cancer Adverse Event Phase 2 Trial in Stage IV Breast Cancer
(n=162)Body System/Adverse Event Total
%Grade 3
%Grade 4
%– Not observed
NA = Not ApplicableGI Diarrhea 57 12 3 Nausea 53 4 – Vomiting 37 4 – Stomatitis 24 7 – Abdominal Pain 20 4 – Constipation 15 1 – Dyspepsia 8 – – Skin and Subcutaneous Hand-and-Foot Syndrome 57 11 NA Dermatitis 37 1 – Nail Disorder 7 – – General Fatigue 41 8 – Pyrexia 12 1 – Pain in Limb 6 1 – Neurological Paresthesia 21 1 – Headache 9 1 – Dizziness 8 – – Insomnia 8 – – Metabolism Anorexia 23 3 – Dehydration 7 4 1 Eye Eye Irritation 15 – – Musculoskeletal Myalgia 9 – – Cardiac Edema 9 1 – Blood Neutropenia 26 2 2 Thrombocytopenia 24 3 1 Anemia 72 3 1 Lymphopenia 94 44 15 Hepatobiliary Hyperbilirubinemia 22 9 2 6.4 Clinically Relevant Adverse Events in <5% of Patients
Clinically relevant adverse events reported in <5% of patients treated with XELODA either as monotherapy or in combination with docetaxel that were considered at least remotely related to treatment are shown below; occurrences of each grade 3 and 4 adverse event are provided in parentheses.
Monotherapy (Metastatic Colorectal Cancer, Adjuvant Colorectal Cancer, Metastatic Breast Cancer)
Gastrointestinal: abdominal distension, dysphagia, proctalgia, ascites (0.1%), gastric ulcer (0.1%), ileus (0.3%), toxic dilation of intestine, gastroenteritis (0.1%) Skin & Subcutan.: nail disorder (0.1%), sweating increased (0.1%), photosensitivity reaction (0.1%), skin ulceration, pruritus, radiation recall syndrome (0.2%) General: chest pain (0.2%), influenza-like illness, hot flushes, pain (0.1%), hoarseness, irritability, difficulty in walking, thirst, chest mass, collapse, fibrosis (0.1%), hemorrhage, edema, sedation Neurological: insomnia, ataxia (0.5%), tremor, dysphasia, encephalopathy (0.1%), abnormal coordination, dysarthria, loss of consciousness (0.2%), impaired balance Metabolism: increased weight, cachexia (0.4%), hypertriglyceridemia (0.1%), hypokalemia, hypomagnesemia Eye: conjunctivitis Respiratory: cough (0.1%), epistaxis (0.1%), asthma (0.2%), hemoptysis, respiratory distress (0.1%), dyspnea Cardiac: tachycardia (0.1%), bradycardia, atrial fibrillation, ventricular extrasystoles, extrasystoles, myocarditis (0.1%), pericardial effusion Infections: laryngitis (1.0%), bronchitis (0.2%), pneumonia (0.2%), bronchopneumonia (0.2%), keratoconjunctivitis, sepsis (0.3%), fungal infections (including candidiasis) (0.2%) Musculoskeletal: myalgia, bone pain (0.1%), arthritis (0.1%), muscle weakness Blood & Lymphatic: leukopenia (0.2%), coagulation disorder (0.1%), bone marrow depression (0.1%), idiopathic thrombocytopenia purpura (1.0%), pancytopenia (0.1%) Vascular: hypotension (0.2%), hypertension (0.1%), lymphoedema (0.1%), pulmonary embolism (0.2%), cerebrovascular accident (0.1%) Psychiatric: depression, confusion (0.1%) Renal: renal impairment (0.6%) Ear: vertigo Hepatobiliary: hepatic fibrosis (0.1%), hepatitis (0.1%), cholestatic hepatitis (0.1%), abnormal liver function tests Immune System: drug hypersensitivity (0.1%) XELODA In Combination With Docetaxel (Metastatic Breast Cancer)
Gastrointestinal: ileus (0.4%), necrotizing enterocolitis (0.4%), esophageal ulcer (0.4%), hemorrhagic diarrhea (0.8%) Neurological: ataxia (0.4%), syncope (1.2%), taste loss (0.8%), polyneuropathy (0.4%), migraine (0.4%) Cardiac: supraventricular tachycardia (0.4%) Infection: neutropenic sepsis (2.4%), sepsis (0.4%), bronchopneumonia (0.4%) Blood & Lymphatic: agranulocytosis (0.4%), prothrombin decreased (0.4%) Vascular: hypotension (1.2%), venous phlebitis and thrombophlebitis (0.4%), postural hypotension (0.8%) Renal: renal failure (0.4%) Hepatobiliary: jaundice (0.4%), abnormal liver function tests (0.4%), hepatic failure (0.4%), hepatic coma (0.4%), hepatotoxicity (0.4%) Immune System: hypersensitivity (1.2%) 6.5 Postmarketing Experience
The following adverse reactions have been observed in the postmarketing setting: hepatic failure, lacrimal duct stenosis, acute renal failure secondary to dehydration including fatal outcome [see Warnings and Precautions (5.5)], cutaneous lupus erythematosus, corneal disorders including keratitis, toxic leukoencephalopathy, severe skin reactions such as Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis (TEN) [see Warnings and Precautions (5.7)], persistent or severe hand-and-foot syndrome can eventually lead to loss of fingerprints [see Warnings and Precautions (5.7)]
In instances of exposure to crushed XELODA tablets, the following adverse reactions have been reported: eye irritation and swelling, skin rash, diarrhea, paresthesia, headache, gastric irritation, vomiting, and nausea.
-
7 DRUG INTERACTIONS
7.1 Drug-Drug Interactions
Anticoagulants
Altered coagulation parameters and/or bleeding have been reported in patients taking XELODA concomitantly with coumarin-derivative anticoagulants such as warfarin and phenprocoumon [see Boxed Warning]. These events occurred within several days and up to several months after initiating XELODA therapy and, in a few cases, within 1 month after stopping XELODA. These events occurred in patients with and without liver metastases. In a drug interaction study with single-dose warfarin administration, there was a significant increase in the mean AUC of S-warfarin [see Clinical Pharmacology (12.3)]. The maximum observed INR value increased by 91%. This interaction is probably due to an inhibition of cytochrome P450 2C9 by capecitabine and/or its metabolites.
Phenytoin
The level of phenytoin should be carefully monitored in patients taking XELODA and phenytoin dose may need to be reduced [see Dosage and Administration (2.3)]. Postmarketing reports indicate that some patients receiving XELODA and phenytoin had toxicity associated with elevated phenytoin levels. Formal drug-drug interaction studies with phenytoin have not been conducted, but the mechanism of interaction is presumed to be inhibition of the CYP2C9 isoenzyme by capecitabine and/or its metabolites.
Leucovorin
The concentration of 5-fluorouracil is increased and its toxicity may be enhanced by leucovorin. Deaths from severe enterocolitis, diarrhea, and dehydration have been reported in elderly patients receiving weekly leucovorin and fluorouracil.
CYP2C9 substrates
Other than warfarin, no formal drug-drug interaction studies between XELODA and other CYP2C9 substrates have been conducted. Care should be exercised when XELODA is coadministered with CYP2C9 substrates.
Allopurinol
Concomitant use with allopurinol may decrease concentration of capecitabine's active metabolites [see Clinical Pharmacology (12.3)], which may decrease XELODA efficacy. Avoid the use of allopurinol during treatment with XELODA.
7.2 Drug-Food Interaction
Food was shown to reduce both the rate and extent of absorption of capecitabine [see Clinical Pharmacology (12.3)]. In all clinical trials, patients were instructed to administer XELODA within 30 minutes after a meal. It is recommended that XELODA be administered with food [see Dosage and Administration (2)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animal reproduction studies and its mechanism of action, XELODA can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. Limited available human data are not sufficient to inform the drug-associated risk during pregnancy. In animal reproduction studies, administration of capecitabine to pregnant animals during the period of organogenesis caused embryo lethality and teratogenicity in mice and embryo lethality in monkeys at 0.2 and 0.6 times the exposure (AUC) in patients receiving the recommended dose respectively [see Data]. Apprise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
Oral administration of capecitabine to pregnant mice during the period of organogenesis at a dose of 198 mg/kg/day caused malformations and embryo lethality. In separate pharmacokinetic studies, this dose in mice produced 5'-DFUR AUC values that were approximately 0.2 times the AUC values in patients administered the recommended daily dose. Malformations in mice included cleft palate, anophthalmia, microphthalmia, oligodactyly, polydactyly, syndactyly, kinky tail and dilation of cerebral ventricles. Oral administration of capecitabine to pregnant monkeys during the period of organogenesis at a dose of 90 mg/kg/day, caused fetal lethality. This dose produced 5'-DFUR AUC values that were approximately 0.6 times the AUC values in patients administered the recommended daily dose.
8.2 Lactation
Risk Summary
There is no information regarding the presence of capecitabine in human milk, or on its effects on milk production or the breast-fed infant. Capecitabine metabolites were present in the milk of lactating mice [see Data]. Because of the potential for serious adverse reactions from capecitabine exposure in breast-fed infants, advise women not to breastfeed during treatment with XELODA and for 2 weeks after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential prior to initiating XELODA.
Contraception
Females
XELODA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment and for 6 months following the final dose of XELODA.
Males
Based on genetic toxicity findings, advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months following the last dose of XELODA [see Nonclinical Toxicology (13.1)].
Infertility
Based on animal studies, XELODA may impair fertility in females and males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of XELODA in pediatric patients have not been established. No clinical benefit was demonstrated in two single arm trials in pediatric patients with newly diagnosed brainstem gliomas and high grade gliomas. In both trials, pediatric patients received an investigational pediatric formulation of capecitabine concomitantly with and following completion of radiation therapy (total dose of 5580 cGy in 180 cGy fractions). The relative bioavailability of the investigational formulation to XELODA was similar.
The first trial was conducted in 22 pediatric patients (median age 8 years, range 5-17 years) with newly diagnosed non-disseminated intrinsic diffuse brainstem gliomas and high grade gliomas. In the dose-finding portion of the trial, patients received capecitabine with concomitant radiation therapy at doses ranging from 500 mg/m2 to 850 mg/m2 every 12 hours for up to 9 weeks. After a 2 week break, patients received 1250 mg/m2 capecitabine every 12 hours on Days 1-14 of a 21-day cycle for up to 3 cycles. The maximum tolerated dose (MTD) of capecitabine administered concomitantly with radiation therapy was 650 mg/m2 every 12 hours. The major dose limiting toxicities were palmar-plantar erythrodysesthesia and alanine aminotransferase (ALT) elevation.
The second trial was conducted in 34 additional pediatric patients with newly diagnosed non-disseminated intrinsic diffuse brainstem gliomas (median age 7 years, range 3-16 years) and 10 pediatric patients who received the MTD of capecitabine in the dose-finding trial and met the eligibility criteria for this trial. All patients received 650 mg/m2 capecitabine every 12 hours with concomitant radiation therapy for up to 9 weeks. After a 2 week break, patients received 1250 mg/m2 capecitabine every 12 hours on Days 1-14 of a 21-day cycle for up to 3 cycles.
There was no improvement in one-year progression-free survival rate and one-year overall survival rate in pediatric patients with newly diagnosed intrinsic brainstem gliomas who received capecitabine relative to a similar population of pediatric patients who participated in other clinical trials.
The adverse reaction profile of capecitabine was consistent with the known adverse reaction profile in adults, with the exception of laboratory abnormalities which occurred more commonly in pediatric patients. The most frequently reported laboratory abnormalities (per-patient incidence ≥40%) were increased ALT (75%), lymphocytopenia (73%), leukopenia (73%), hypokalemia (68%), thrombocytopenia (57%), hypoalbuminemia (55%), neutropenia (50%), low hematocrit (50%), hypocalcemia (48%), hypophosphatemia (45%) and hyponatremia (45%).
8.5 Geriatric Use
Physicians should pay particular attention to monitoring the adverse effects of XELODA in the elderly [see Warnings and Precautions (5.10)].
8.6 Hepatic Insufficiency
Exercise caution when patients with mild to moderate hepatic dysfunction due to liver metastases are treated with XELODA. The effect of severe hepatic dysfunction on XELODA is not known [see Warnings and Precautions (5.11) and Clinical Pharmacology (12.3)].
8.7 Renal Insufficiency
Patients with moderate (creatinine clearance = 30 to 50 mL/min) and severe (creatinine clearance <30 mL/min) renal impairment showed higher exposure for capecitabine, 5-DFUR, and FBAL than in those with normal renal function [see Contraindications (4.2), Warnings and Precautions (5.5), Dosage and Administration (2.4), and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
The manifestations of acute overdose would include nausea, vomiting, diarrhea, gastrointestinal irritation and bleeding, and bone marrow depression. Medical management of overdose should include customary supportive medical interventions aimed at correcting the presenting clinical manifestations. Although no clinical experience using dialysis as a treatment for XELODA overdose has been reported, dialysis may be of benefit in reducing circulating concentrations of 5'-DFUR, a low–molecular-weight metabolite of the parent compound.
Single doses of XELODA were not lethal to mice, rats, and monkeys at doses up to 2000 mg/kg (2.4, 4.8, and 9.6 times the recommended human daily dose on a mg/m2 basis).
-
11 DESCRIPTION
XELODA (capecitabine) is a fluoropyrimidine carbamate with antineoplastic activity. It is an orally administered systemic prodrug of 5'-deoxy-5-fluorouridine (5'-DFUR) which is converted to 5-fluorouracil.
The chemical name for capecitabine is 5'-deoxy-5-fluoro-N-[(pentyloxy) carbonyl]-cytidine and has a molecular weight of 359.35. Capecitabine has the following structural formula:
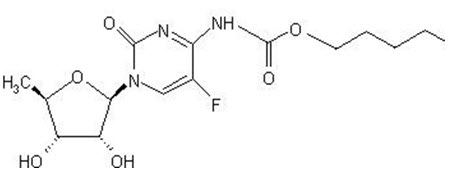
Capecitabine is a white to off-white crystalline powder with an aqueous solubility of 26 mg/mL at 20ºC.
XELODA is supplied as biconvex, oblong film-coated tablets for oral administration. Each light peach-colored tablet contains 150 mg capecitabine and each peach-colored tablet contains 500 mg capecitabine. The inactive ingredients in XELODA include: anhydrous lactose, croscarmellose sodium, hydroxypropyl methylcellulose, microcrystalline cellulose, magnesium stearate and purified water. The peach or light peach film coating contains hydroxypropyl methylcellulose, talc, titanium dioxide, and synthetic yellow and red iron oxides.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Enzymes convert capecitabine to 5-fluorouracil (5-FU) in vivo. Both normal and tumor cells metabolize 5-FU to 5-fluoro-2'-deoxyuridine monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). These metabolites cause cell injury by two different mechanisms. First, FdUMP and the folate cofactor, N5-10-methylenetetrahydrofolate, bind to thymidylate synthase (TS) to form a covalently bound ternary complex. This binding inhibits the formation of thymidylate from 2'-deoxyuridylate. Thymidylate is the necessary precursor of thymidine triphosphate, which is essential for the synthesis of DNA, so that a deficiency of this compound can inhibit cell division. Second, nuclear transcriptional enzymes can mistakenly incorporate FUTP in place of uridine triphosphate (UTP) during the synthesis of RNA. This metabolic error can interfere with RNA processing and protein synthesis.
12.3 Pharmacokinetics
Absorption
Following oral administration of 1255 mg/m2 BID to cancer patients, capecitabine reached peak blood levels in about 1.5 hours (Tmax) with peak 5-FU levels occurring slightly later, at 2 hours. Food reduced both the rate and extent of absorption of capecitabine with mean Cmax and AUC0-∞ decreased by 60% and 35%, respectively. The Cmax and AUC0-∞ of 5-FU were also reduced by food by 43% and 21%, respectively. Food delayed Tmax of both parent and 5-FU by 1.5 hours [see Warnings and Precautions (5), Dosage and Administration (2), and Drug-Food Interaction (7.2)].
The pharmacokinetics of XELODA and its metabolites have been evaluated in about 200 cancer patients over a dosage range of 500 to 3500 mg/m2/day. Over this range, the pharmacokinetics of XELODA and its metabolite, 5'-DFCR were dose proportional and did not change over time. The increases in the AUCs of 5'-DFUR and 5-FU, however, were greater than proportional to the increase in dose and the AUC of 5-FU was 34% higher on day 14 than on day 1. The interpatient variability in the Cmax and AUC of 5-FU was greater than 85%.
Distribution
Plasma protein binding of capecitabine and its metabolites is less than 60% and is not concentration-dependent. Capecitabine was primarily bound to human albumin (approximately 35%). XELODA has a low potential for pharmacokinetic interactions related to plasma protein binding.
Bioactivation and Metabolism
Capecitabine is extensively metabolized enzymatically to 5-FU. In the liver, a 60 kDa carboxylesterase hydrolyzes much of the compound to 5'-deoxy-5-fluorocytidine (5'-DFCR). Cytidine deaminase, an enzyme found in most tissues, including tumors, subsequently converts 5'-DFCR to 5'-DFUR. The enzyme, thymidine phosphorylase (dThdPase), then hydrolyzes 5'-DFUR to the active drug 5-FU. Many tissues throughout the body express thymidine phosphorylase. Some human carcinomas express this enzyme in higher concentrations than surrounding normal tissues. Following oral administration of XELODA 7 days before surgery in patients with colorectal cancer, the median ratio of 5-FU concentration in colorectal tumors to adjacent tissues was 2.9 (range from 0.9 to 8.0). These ratios have not been evaluated in breast cancer patients or compared to 5-FU infusion.
Metabolic Pathway of capecitabine to 5-FU 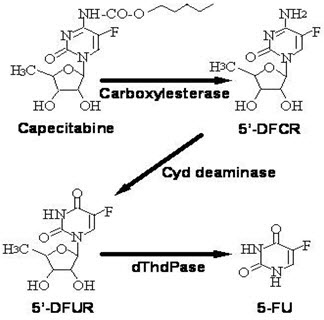
The enzyme dihydropyrimidine dehydrogenase hydrogenates 5-FU, the product of capecitabine metabolism, to the much less toxic 5-fluoro-5, 6-dihydro-fluorouracil (FUH2). Dihydropyrimidinase cleaves the pyrimidine ring to yield 5-fluoro-ureido-propionic acid (FUPA). Finally, β-ureido-propionase cleaves FUPA to α-fluoro-β-alanine (FBAL) which is cleared in the urine.
In vitro enzymatic studies with human liver microsomes indicated that capecitabine and its metabolites (5'-DFUR, 5'-DFCR, 5-FU, and FBAL) did not inhibit the metabolism of test substrates by cytochrome P450 isoenzymes 1A2, 2A6, 3A4, 2C19, 2D6, and 2E1.
Excretion
Capecitabine and its metabolites are predominantly excreted in urine; 95.5% of administered capecitabine dose is recovered in urine. Fecal excretion is minimal (2.6%). The major metabolite excreted in urine is FBAL which represents 57% of the administered dose. About 3% of the administered dose is excreted in urine as unchanged drug. The elimination half-life of both parent capecitabine and 5-FU was about 0.75 hour.
Effect of Age, Gender, and Race on the Pharmacokinetics of Capecitabine
A population analysis of pooled data from the two large controlled studies in patients with metastatic colorectal cancer (n=505) who were administered XELODA at 1250 mg/m2 twice a day indicated that gender (202 females and 303 males) and race (455 white/Caucasian patients, 22 black patients, and 28 patients of other race) have no influence on the pharmacokinetics of 5'-DFUR, 5-FU and FBAL. Age has no significant influence on the pharmacokinetics of 5'-DFUR and 5-FU over the range of 27 to 86 years. A 20% increase in age results in a 15% increase in AUC of FBAL [see Warnings and Precautions (5.11) and Dosage and Administration (2.4)].
Following oral administration of 825 mg/m2 capecitabine twice daily for 14 days, Japanese patients (n=18) had about 36% lower Cmax and 24% lower AUC for capecitabine than the Caucasian patients (n=22). Japanese patients had also about 25% lower Cmax and 34% lower AUC for FBAL than the Caucasian patients. The clinical significance of these differences is unknown. No significant differences occurred in the exposure to other metabolites (5'-DFCR, 5'-DFUR, and 5-FU).
Effect of Hepatic Insufficiency
XELODA has been evaluated in 13 patients with mild to moderate hepatic dysfunction due to liver metastases defined by a composite score including bilirubin, AST/ALT and alkaline phosphatase following a single 1255 mg/m2 dose of XELODA. Both AUC0-∞ and Cmax of capecitabine increased by 60% in patients with hepatic dysfunction compared to patients with normal hepatic function (n=14). The AUC0-∞ and Cmax of 5-FU were not affected. In patients with mild to moderate hepatic dysfunction due to liver metastases, caution should be exercised when XELODA is administered. The effect of severe hepatic dysfunction on XELODA is not known [see Warnings and Precautions (5.11) and Use in Special Populations (8.6)].
Effect of Renal Insufficiency
Following oral administration of 1250 mg/m2 capecitabine twice a day to cancer patients with varying degrees of renal impairment, patients with moderate (creatinine clearance = 30 to 50 mL/min) and severe (creatinine clearance <30 mL/min) renal impairment showed 85% and 258% higher systemic exposure to FBAL on day 1 compared to normal renal function patients (creatinine clearance >80 mL/min). Systemic exposure to 5'-DFUR was 42% and 71% greater in moderately and severely renal impaired patients, respectively, than in normal patients. Systemic exposure to capecitabine was about 25% greater in both moderately and severely renal impaired patients [see Dosage and Administration (2.4), Contraindications (4.2), Warnings and Precautions (5.5), and Use in Special Populations (8.7)].
Effect of Capecitabine on the Pharmacokinetics of Warfarin
In four patients with cancer, chronic administration of capecitabine (1250 mg/m2 bid) with a single 20 mg dose of warfarin increased the mean AUC of S-warfarin by 57% and decreased its clearance by 37%. Baseline corrected AUC of INR in these 4 patients increased by 2.8-fold, and the maximum observed mean INR value was increased by 91% [see Boxed Warning and Drug Interactions (7.1)].
Effect of Antacids on the Pharmacokinetics of Capecitabine
When Maalox® (20 mL), an aluminum hydroxide- and magnesium hydroxide-containing antacid, was administered immediately after XELODA (1250 mg/m2, n=12 cancer patients), AUC and Cmax increased by 16% and 35%, respectively, for capecitabine and by 18% and 22%, respectively, for 5'-DFCR. No effect was observed on the other three major metabolites (5'-DFUR, 5-FU, FBAL) of XELODA.
Effect of Allopurinol on Capecitabine
Published literature reported that concomitant use with allopurinol may decrease conversion of capecitabine to the active metabolites, FdUMP and FUTP; however, the clinical significance was not fully characterized.
Effect of Capecitabine on the Pharmacokinetics of Docetaxel and Vice Versa
A Phase 1 study evaluated the effect of XELODA on the pharmacokinetics of docetaxel (Taxotere®) and the effect of docetaxel on the pharmacokinetics of XELODA was conducted in 26 patients with solid tumors. XELODA was found to have no effect on the pharmacokinetics of docetaxel (Cmax and AUC) and docetaxel has no effect on the pharmacokinetics of capecitabine and the 5-FU precursor 5'-DFUR.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Adequate studies investigating the carcinogenic potential of capecitabine have not been conducted. Capecitabine was not mutagenic in vitro to bacteria (Ames test) or mammalian cells (Chinese hamster V79/HPRT gene mutation assay). Capecitabine was clastogenic in vitro to human peripheral blood lymphocytes but not clastogenic in vivo to mouse bone marrow (micronucleus test). Fluorouracil causes mutations in bacteria and yeast. Fluorouracil also causes chromosomal abnormalities in the mouse micronucleus test in vivo.
In studies of fertility and general reproductive performance in female mice, oral capecitabine doses of 760 mg/kg/day (about 2300 mg/m2/day) disturbed estrus and consequently caused a decrease in fertility. In mice that became pregnant, no fetuses survived this dose. The disturbance in estrus was reversible. In males, this dose caused degenerative changes in the testes, including decreases in the number of spermatocytes and spermatids. In separate pharmacokinetic studies, this dose in mice produced 5'-DFUR AUC values about 0.7 times the corresponding values in patients administered the recommended daily dose.
-
14 CLINICAL STUDIES
14.1 Adjuvant Colon Cancer
A multicenter randomized, controlled phase 3 clinical trial in patients with Dukes' C colon cancer (X-ACT) provided data concerning the use of XELODA for the adjuvant treatment of patients with colon cancer. The primary objective of the study was to compare disease-free survival (DFS) in patients receiving XELODA to those receiving IV 5-FU/LV alone. In this trial, 1987 patients were randomized either to treatment with XELODA 1250 mg/m2 orally twice daily for 2 weeks followed by a 1-week rest period, given as 3-week cycles for a total of 8 cycles (24 weeks) or IV bolus 5-FU 425 mg/m2 and 20 mg/m2 IV leucovorin on days 1 to 5, given as 4-week cycles for a total of 6 cycles (24 weeks). Patients in the study were required to be between 18 and 75 years of age with histologically-confirmed Dukes' stage C colon cancer with at least one positive lymph node and to have undergone (within 8 weeks prior to randomization) complete resection of the primary tumor without macroscopic or microscopic evidence of remaining tumor. Patients were also required to have no prior cytotoxic chemotherapy or immunotherapy (except steroids), and have an ECOG performance status of 0 or 1 (KPS ≥ 70%), ANC ≥ 1.5×109/L, platelets ≥ 100×109/L, serum creatinine ≤ 1.5 ULN, total bilirubin ≤ 1.5 ULN, AST/ALT ≤ 2.5 ULN and CEA within normal limits at time of randomization.
The baseline demographics for XELODA and 5-FU/LV patients are shown in Table 10. The baseline characteristics were well-balanced between arms.
Table 10 Baseline Demographics XELODA
(n=1004)5-FU/LV
(n=983)Age (median, years) 62 63 Range (25-80) (22-82) Gender Male (n, %) 542 (54) 532 (54) Female (n, %) 461 (46) 451 (46) ECOG PS 0 (n, %) 849 (85) 830 (85) 1 (n, %) 152 (15) 147 (15) Staging – Primary Tumor PT1 (n, %) 12 (1) 6 (0.6) PT2 (n, %) 90 (9) 92 (9) PT3 (n, %) 763 (76) 746 (76) PT4 (n, %) 138 (14) 139 (14) Other (n, %) 1 (0.1) 0 (0) Staging – Lymph Node pN1 (n, %) 695 (69) 694 (71) pN2 (n, %) 305 (30) 288 (29) Other (n, %) 4 (0.4) 1 (0.1) All patients with normal renal function or mild renal impairment began treatment at the full starting dose of 1250 mg/m2 orally twice daily. The starting dose was reduced in patients with moderate renal impairment (calculated creatinine clearance 30 to 50 mL/min) at baseline [see Dosage and Administration (2.4)]. Subsequently, for all patients, doses were adjusted when needed according to toxicity. Dose management for XELODA included dose reductions, cycle delays and treatment interruptions (see Table 11).
Table 11 Summary of Dose Modifications in X-ACT Study XELODA
N = 9955-FU/LV
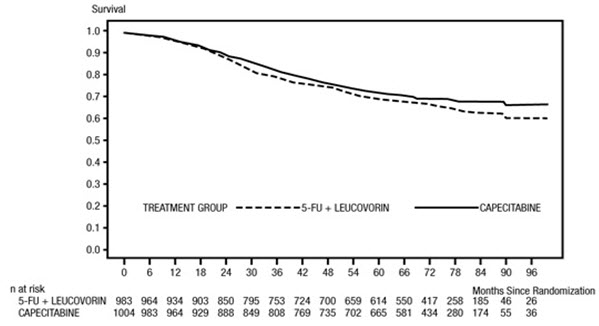
N = 974Median relative dose intensity (%) 93 92 Patients completing full course of treatment (%) 83 87 Patients with treatment interruption (%) 15 5 Patients with cycle delay (%) 46 29 Patients with dose reduction (%) 42 44 Patients with treatment interruption, cycle delay, or dose reduction (%) 57 52 The median follow-up at the time of the analysis was 83 months (6.9 years). The hazard ratio for DFS for XELODA compared to 5-FU/LV was 0.88 (95% C.I. 0.77 – 1.01) (see Table 12 and Figure 1). Because the upper 2-sided 95% confidence limit of hazard ratio was less than 1.20, XELODA was non-inferior to 5-FU/LV. The choice of the non-inferiority margin of 1.20 corresponds to the retention of approximately 75% of the 5-FU/LV effect on DFS. The hazard ratio for XELODA compared to 5-FU/LV with respect to overall survival was 0.86 (95% C.I. 0.74 – 1.01). The 5-year overall survival rates were 71.4% for XELODA and 68.4% for 5-FU/LV (see Figure 2).
Table 12 Efficacy of XELODA vs 5-FU/LV in Adjuvant Treatment of Colon Cancer* All Randomized Population XELODA
(n=1004)5-FU/LV
(n=983)- * Approximately 93.4% had 5-year DFS information
- † Based on Kaplan-Meier estimates
- ‡ Test of superiority of XELODA vs 5-FU/LV (Wald chi-square test)
Median follow-up (months) 83 83 5-year Disease-free Survival Rates (%)† 59.1 54.6 Hazard Ratio
(XELODA/5-FU/LV)
(95% C.I. for Hazard Ratio)0.88
(0.77 - 1.01)p-value‡ p = 0.068 - * XELODA has been demonstrated to be non-inferior to 5-FU/LV.
Figure 1 Kaplan-Meier Estimates of Disease-Free Survival (All Randomized Population)* 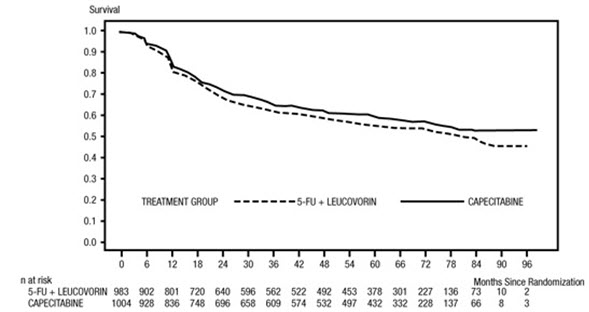
Figure 2 Kaplan-Meier Estimates of Overall Survival (All Randomized Population)
14.2 Metastatic Colorectal Cancer
General
The recommended dose of XELODA was determined in an open-label, randomized clinical study, exploring the efficacy and safety of continuous therapy with capecitabine (1331 mg/m2/day in two divided doses, n=39), intermittent therapy with capecitabine (2510 mg/m2/day in two divided doses, n=34), and intermittent therapy with capecitabine in combination with oral leucovorin (LV) (capecitabine 1657 mg/m2/day in two divided doses, n=35; leucovorin 60 mg/day) in patients with advanced and/or metastatic colorectal carcinoma in the first-line metastatic setting. There was no apparent advantage in response rate to adding leucovorin to XELODA; however, toxicity was increased. XELODA, 1250 mg/m2 twice daily for 14 days followed by a 1-week rest, was selected for further clinical development based on the overall safety and efficacy profile of the three schedules studied.
Monotherapy
Data from two open-label, multicenter, randomized, controlled clinical trials involving 1207 patients support the use of XELODA in the first-line treatment of patients with metastatic colorectal carcinoma. The two clinical studies were identical in design and were conducted in 120 centers in different countries. Study 1 was conducted in the US, Canada, Mexico, and Brazil; Study 2 was conducted in Europe, Israel, Australia, New Zealand, and Taiwan. Altogether, in both trials, 603 patients were randomized to treatment with XELODA at a dose of 1250 mg/m2 twice daily for 2 weeks followed by a 1-week rest period and given as 3-week cycles; 604 patients were randomized to treatment with 5-FU and leucovorin (20 mg/m2 leucovorin IV followed by 425 mg/m2 IV bolus 5-FU, on days 1 to 5, every 28 days).
In both trials, overall survival, time to progression and response rate (complete plus partial responses) were assessed. Responses were defined by the World Health Organization criteria and submitted to a blinded independent review committee (IRC). Differences in assessments between the investigator and IRC were reconciled by the sponsor, blinded to treatment arm, according to a specified algorithm. Survival was assessed based on a non-inferiority analysis.
The baseline demographics for XELODA and 5-FU/LV patients are shown in Table 13.
Table 13 Baseline Demographics of Controlled Colorectal Trials Study 1 Study 2 XELODA
(n=302)5-FU/LV
(n=303)XELODA
(n=301)5-FU/LV
(n=301)Age (median, years) 64 63 64 64 Range (23-86) (24-87) (29-84) (36-86) Gender Male (%) 181 (60) 197 (65) 172 (57) 173 (57) Female (%) 121 (40) 106 (35) 129 (43) 128 (43) Karnofsky PS (median) 90 90 90 90 Range (70-100) (70-100) (70-100) (70-100) Colon (%) 222 (74) 232 (77) 199 (66) 196 (65) Rectum (%) 79 (26) 70 (23) 101 (34) 105 (35) Prior radiation therapy (%) 52 (17) 62 (21) 42 (14) 42 (14) Prior adjuvant 5-FU (%) 84 (28) 110 (36) 56 (19) 41 (14) The efficacy endpoints for the two phase 3 trials are shown in Table 14 and Table 15.
Table 14 Efficacy of XELODA vs 5-FU/LV in Colorectal Cancer (Study 1) XELODA
(n=302)5-FU/LV
(n=303)Overall Response Rate (%, 95% C.I.) 21 (16-26) 11 (8-15) (p-value) 0.0014 Time to Progression (Median, days, 95% C.I.) 128 (120-136) 131 (105-153) Hazard Ratio (XELODA/5-FU/LV)
95% C.I. for Hazard Ratio0.99
(0.84-1.17)Survival (Median, days, 95% C.I.) 380 (321-434) 407 (366-446) Hazard Ratio (XELODA/5-FU/LV)
95% C.I. for Hazard Ratio1.00
(0.84-1.18)Table 15 Efficacy of XELODA vs 5-FU/LV in Colorectal Cancer (Study 2) XELODA
(n=301)5-FU/LV
(n=301)Overall Response Rate (%, 95% C.I.) 21 (16-26) 14 (10-18) (p-value) 0.027 Time to Progression (Median, days, 95% C.I.) 137 (128-165) 131 (102-156) Hazard Ratio (XELODA/5-FU/LV)
95% C.I. for Hazard Ratio0.97
(0.82-1.14)Survival (Median, days, 95% C.I.) 404 (367-452) 369 (338-430) Hazard Ratio (XELODA/5-FU/LV)
95% C.I. for Hazard Ratio0.92
(0.78-1.09)Figure 3 Kaplan-Meier Curve for Overall Survival of Pooled Data (Studies 1 and 2)
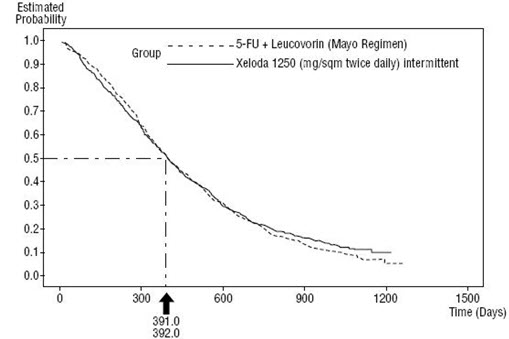
XELODA was superior to 5-FU/LV for objective response rate in Study 1 and Study 2. The similarity of XELODA and 5-FU/LV in these studies was assessed by examining the potential difference between the two treatments. In order to assure that XELODA has a clinically meaningful survival effect, statistical analyses were performed to determine the percent of the survival effect of 5-FU/LV that was retained by XELODA. The estimate of the survival effect of 5-FU/LV was derived from a meta-analysis of ten randomized studies from the published literature comparing 5-FU to regimens of 5-FU/LV that were similar to the control arms used in these Studies 1 and 2. The method for comparing the treatments was to examine the worst case (95% confidence upper bound) for the difference between 5-FU/LV and XELODA, and to show that loss of more than 50% of the 5-FU/LV survival effect was ruled out. It was demonstrated that the percent of the survival effect of 5-FU/LV maintained was at least 61% for Study 2 and 10% for Study 1. The pooled result is consistent with a retention of at least 50% of the effect of 5-FU/LV. It should be noted that these values for preserved effect are based on the upper bound of the 5-FU/LV vs XELODA difference. These results do not exclude the possibility of true equivalence of XELODA to 5-FU/LV (see Table 14, Table 15, and Figure 3).
14.3 Breast Cancer
XELODA has been evaluated in clinical trials in combination with docetaxel (Taxotere®) and as monotherapy.
In Combination With Docetaxel
The dose of XELODA used in the phase 3 clinical trial in combination with docetaxel was based on the results of a phase 1 study, where a range of doses of docetaxel administered in 3-week cycles in combination with an intermittent regimen of XELODA (14 days of treatment, followed by a 7-day rest period) were evaluated. The combination dose regimen was selected based on the tolerability profile of the 75 mg/m2 administered in 3-week cycles of docetaxel in combination with 1250 mg/m2 twice daily for 14 days of XELODA administered in 3-week cycles. The approved dose of 100 mg/m2 of docetaxel administered in 3-week cycles was the control arm of the phase 3 study.
XELODA in combination with docetaxel was assessed in an open-label, multicenter, randomized trial in 75 centers in Europe, North America, South America, Asia, and Australia. A total of 511 patients with metastatic breast cancer resistant to, or recurring during or after an anthracycline-containing therapy, or relapsing during or recurring within 2 years of completing an anthracycline-containing adjuvant therapy were enrolled. Two hundred and fifty-five (255) patients were randomized to receive XELODA 1250 mg/m2 twice daily for 14 days followed by 1 week without treatment and docetaxel 75 mg/m2 as a 1-hour intravenous infusion administered in 3-week cycles. In the monotherapy arm, 256 patients received docetaxel 100 mg/m2 as a 1-hour intravenous infusion administered in 3-week cycles. Patient demographics are provided in Table 16.
Table 16 Baseline Demographics and Clinical Characteristics XELODA and Docetaxel Combination vs Docetaxel in Breast Cancer Trial XELODA + Docetaxel
(n=255)Docetaxel
(n=256)- * Includes 10 patients in combination and 18 patients in monotherapy arms treated with an anthracenedione
Age (median, years) 52 51 Karnofsky PS (median) 90 90 Site of Disease Lymph nodes 121 (47%) 125 (49%) Liver 116 (45%) 122 (48%) Bone 107 (42%) 119 (46%) Lung 95 (37%) 99 (39%) Skin 73 (29%) 73 (29%) Prior Chemotherapy Anthracycline* 255 (100%) 256 (100%) 5-FU 196 (77%) 189 (74%) Paclitaxel 25 (10%) 22 (9%) Resistance to an Anthracycline No resistance 19 (7%) 19 (7%) Progression on anthracycline therapy 65 (26%) 73 (29%) Stable disease after 4 cycles of anthracycline therapy 41 (16%) 40 (16%) Relapsed within 2 years of completion of anthracycline-adjuvant therapy 78 (31%) 74 (29%) Experienced a brief response to anthracycline therapy, with subsequent progression while on therapy or within 12 months after last dose 51 (20%) 50 (20%) No. of Prior Chemotherapy Regimens for Treatment of Metastatic Disease 0 89 (35%) 80 (31%) 1 123 (48%) 135 (53%) 2 43 (17%) 39 (15%) 3 0 (0%) 2 (1%) XELODA in combination with docetaxel resulted in statistically significant improvement in time to disease progression, overall survival and objective response rate compared to monotherapy with docetaxel as shown in Table 17, Figure 4, and Figure 5.
Table 17 Efficacy of XELODA and Docetaxel Combination vs Docetaxel Monotherapy Efficacy Parameter Combination Therapy Monotherapy p-value Hazard Ratio - * The response rate reported represents a reconciliation of the investigator and IRC assessments performed by the sponsor according to a predefined algorithm.
- † NA = Not Applicable
Time to Disease Progression Median Days 186 128 0.0001 0.643 95% C.I. (165-198) (105-136) Overall Survival Median Days 442 352 0.0126 0.775 95% C.I. (375-497) (298-387) Response Rate* 32% 22% 0.009 NA† Figure 4 Kaplan-Meier Estimates for Time to Disease Progression XELODA and Docetaxel vs Docetaxel
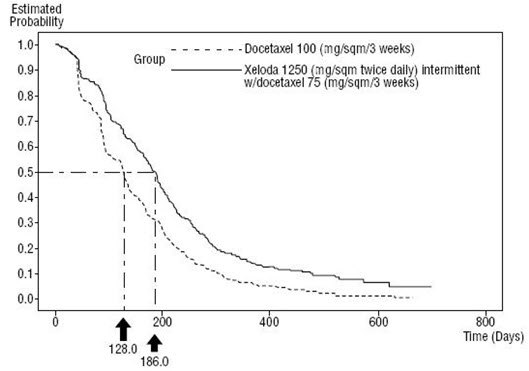
Figure 5 Kaplan-Meier Estimates of Survival XELODA and Docetaxel vs Docetaxel
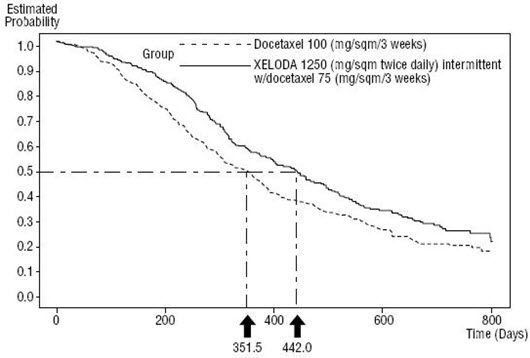
Monotherapy
The antitumor activity of XELODA as a monotherapy was evaluated in an open-label single-arm trial conducted in 24 centers in the US and Canada. A total of 162 patients with stage IV breast cancer were enrolled. The primary endpoint was tumor response rate in patients with measurable disease, with response defined as a ≥50% decrease in sum of the products of the perpendicular diameters of bidimensionally measurable disease for at least 1 month. XELODA was administered at a dose of 1255 mg/m2 twice daily for 2 weeks followed by a 1-week rest period and given as 3-week cycles. The baseline demographics and clinical characteristics for all patients (n=162) and those with measurable disease (n=135) are shown in Table 18. Resistance was defined as progressive disease while on treatment, with or without an initial response, or relapse within 6 months of completing treatment with an anthracycline-containing adjuvant chemotherapy regimen.
Table 18 Baseline Demographics and Clinical Characteristics Single-Arm Breast Cancer Trial Patients With Measurable Disease
(n=135)All Patients
(n=162)- * Lung, pleura, liver, peritoneum
- † Includes 2 patients treated with an anthracenedione
Age (median, years) 55 56 Karnofsky PS 90 90 No. Disease Sites 1-2 43 (32%) 60 (37%) 3-4 63 (46%) 69 (43%) >5 29 (22%) 34 (21%) Dominant Site of Disease Visceral* 101 (75%) 110 (68%) Soft Tissue 30 (22%) 35 (22%) Bone 4 (3%) 17 (10%) Prior Chemotherapy Paclitaxel 135 (100%) 162 (100%) Anthracycline† 122 (90%) 147 (91%) 5-FU 110 (81%) 133 (82%) Resistance to Paclitaxel 103 (76%) 124 (77%) Resistance to an Anthracycline† 55 (41%) 67 (41%) Resistance to both Paclitaxel and an Anthracycline† 43 (32%) 51 (31%) Antitumor responses for patients with disease resistant to both paclitaxel and an anthracycline are shown in Table 19.
Table 19 Response Rates in Doubly-Resistant Patients Single-Arm Breast Cancer Trial Resistance to Both Paclitaxel and an Anthracycline
(n=43)- * Includes 2 patients treated with an anthracenedione
- † From date of first response
CR 0 PR* 11 CR + PR* 11 Response Rate*
(95% C.I.)25.6%
(13.5, 41.2)Duration of Response,* Median in days†
(Range)154
(63-233)For the subgroup of 43 patients who were doubly resistant, the median time to progression was 102 days and the median survival was 255 days. The objective response rate in this population was supported by a response rate of 18.5% (1 CR, 24 PRs) in the overall population of 135 patients with measurable disease, who were less resistant to chemotherapy (see Table 18). The median time to progression was 90 days and the median survival was 306 days.
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
150 mg
Color: Light peach Engraving: XELODA on one side and 150 on the other 150 mg tablets are packaged in bottles of 60 (NDC: 0004-1100-20), individually packaged in a carton. 500 mg
Color: Peach Engraving: XELODA on one side and 500 on the other 500 mg tablets are packaged in bottles of 120 (NDC: 0004-1101-50), individually packaged in a carton. Storage and Handling
Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F). [See USP Controlled Room Temperature]. KEEP TIGHTLY CLOSED.
XELODA is a cytotoxic drug. Follow applicable special handling and disposal procedures.1 Any unused product should be disposed of in accordance with local requirements, or drug take back programs.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Diarrhea
Inform patients experiencing grade 2 diarrhea (an increase of 4 to 6 stools/day or nocturnal stools) or greater or experiencing severe bloody diarrhea with severe abdominal pain and fever to stop taking XELODA. Advise patients on the use of antidiarrheal treatments (e.g., loperamide) to manage diarrhea [see Warnings and Precautions (5.2)].
Cardiotoxicity
Advise patients of the risk of cardiotoxicity and to immediately contact their healthcare provider or to go to an emergency room for new onset of chest pain, shortness of breath, dizziness, or lightheadedness [see Warnings and Precautions (5.3)].
Dihydropyrimidine Dehydrogenase Deficiency
Advise patients to notify their healthcare provider if they have a known DPD deficiency. Advise patients if they have complete or near complete absence of DPD activity they are at an increased risk of acute early-onset of toxicity and severe, life-threatening, or fatal adverse reactions caused by XELODA (e.g., mucositis, diarrhea, neutropenia, and neurotoxicity) [see Warnings and Precautions (5.4)].
Dehydration and Renal Failure
Instruct patients experiencing grade 2 or higher dehydration (IV fluids indicated < 24 hours) to stop taking XELODA immediately and to call their healthcare provider to correct the dehydration. Advise patients to not restart XELODA until rehydrated and any precipitating causes have been corrected or controlled [see Warnings and Precautions (5.5)].
Important Administration Instructions
Advise patients to swallow XELODA tablets whole with water within 30 minutes of a meal. Advise patients and caregivers not to crush or cut XELODA tablets. Advise patients if they cannot swallow XELODA tablets whole, to inform their healthcare provider [see Dosage and Administration (2.1)].
Nausea
Instruct patients experiencing grade 2 nausea (food intake significantly decreased but able to eat intermittently) or greater to stop taking XELODA immediately and to contact their healthcare provider for management of nausea [see Adverse Reactions (6.1)].
Vomiting
Instruct patients experiencing grade 2 vomiting (2 to 5 episodes in a 24-hour period) or greater to stop taking XELODA immediately and to contact their healthcare provider for management of vomiting [see Adverse Reactions (6.1)].
Hand-and-Foot Syndrome
Instruct patients experiencing grade 2 hand-and-foot syndrome (painful erythema and swelling of the hands and/or feet and/or discomfort affecting the patients' activities of daily living) or greater to stop taking XELODA immediately and to contact their healthcare provider. Inform patients that initiation of symptomatic treatment is recommended and hand-and-foot syndrome can lead to loss of fingerprints which could impact personal identification [see Adverse Reactions (6.1)].
Stomatitis
Inform patients experiencing grade 2 stomatitis (painful erythema, edema or ulcers of the mouth or tongue, but able to eat) or greater to stop taking XELODA immediately and to contact their healthcare provider [see Adverse Reactions (6.1)].
Fever and Neutropenia
Inform patients who develop a fever of 100.5°F or greater or other evidence of potential infection to contact their healthcare provider [see Adverse Reactions (6.1)].
Embryo-Fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment with XELODA and for 6 months after the last dose. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.6), Use in Specific Populations (8.1 and 8.3)].
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with XELODA and for 3 months after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise females not to breastfeed during treatment with XELODA and for 2 weeks after the last dose [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 02/2019 Patient Information
XELODA® (zeh-LOE-duh)
(capecitabine)
tabletsWhat is the most important information I should know about XELODA?
XELODA can cause serious side effects, including:
- XELODA can interact with blood thinner medicines, such as warfarin (COUMADIN®). Taking XELODA with these medicines can cause changes in how fast your blood clots and can cause bleeding that can lead to death. This can happen as soon as a few days after you start taking XELODA, or later during treatment, and possibly even within 1 month after you stop taking XELODA. Your risk may be higher because you have cancer, and if you are over 60 years of age.
- Before taking XELODA, tell your healthcare provider if you are taking warfarin (COUMADIN) or another blood thinner medicine.
- If you take warfarin (COUMADIN) or another blood thinner that is like warfarin (COUMADIN) during treatment with XELODA, your healthcare provider should do blood tests often, to check how fast your blood clots during and after you stop treatment with XELODA. Your healthcare provider may change your dose of the blood thinner medicine if needed.
See "What are the possible side effects of XELODA?" for more information about side effects.
What is XELODA?
XELODA is a prescription medicine used to treat people with:
- cancer of the colon that has spread to lymph nodes in the area close to the colon (Dukes' C stage), after they have surgery.
- cancer of the colon or rectum (colorectal) that has spread to other parts of the body (metastatic).
- breast cancer that has spread to other parts of the body (metastatic) together with another medicine called docetaxel after treatment with certain other anti-cancer medicines have not worked.
- breast cancer that has spread to other parts of the body and has not improved after treatment with paclitaxel and certain other anti-cancer medicines, or who cannot receive any more treatment with certain anti-cancer medicines.
It is not known if XELODA is safe and effective in children.
Do not take XELODA if you:
- have severe kidney problems.
- are allergic to capecitabine, 5-fluorouracil, or any of the ingredients in XELODA. See the end of this leaflet for a complete list of ingredients in XELODA.
Talk to your healthcare provider before taking XELODA if you are not sure if you have any of the conditions listed above.
Before taking XELODA, tell your healthcare provider about all your medical conditions, including if you:
See "What is the most important information I should know about XELODA?"
- have had heart problems.
- have kidney or liver problems.
- have been told that you lack the enzyme DPD (dihydropyrimidine dehydrogenase).
- are pregnant or plan to become pregnant. XELODA can harm your unborn baby. Your healthcare provider should do a pregnancy test before you start treatment with XELODA. Tell your healthcare provider right away if you become pregnant or think you might be pregnant during treatment with XELODA.
- Females who are able to become pregnant should use effective birth control during treatment and for 6 months after the final dose. Talk to your healthcare provider about birth control choices that may be right for you during treatment with XELODA.
- Males who have female partners who are able to become pregnant should use effective birth control during treatment and for 3 months after the final dose.
- are breastfeeding or plan to breastfeed. It is not known if XELODA passes into your breast milk. Do not breastfeed during treatment with XELODA and for 2 weeks after the final dose.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. XELODA may affect the way other medicines work, and other medicines may affect the way XELODA works.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I take XELODA?
- Take XELODA exactly as your healthcare provider tells you to take it.
- Your healthcare provider will tell you how much XELODA to take and when to take it.
- Take XELODA 2 times a day, 1 time in the morning and 1 time in the evening.
- Take XELODA within 30 minutes after finishing a meal.
- Swallow XELODA tablets whole with water. Do not crush or cut XELODA tablets. If you cannot swallow XELODA tablets whole, tell your healthcare provider.
- Your healthcare provider may change your dose, temporarily stop, or permanently stop treatment with XELODA if you develop side effects.
- If you take too much XELODA, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of XELODA?
XELODA may cause serious side effects including:
See "What is the most important information I should know about XELODA?".
- Diarrhea. Diarrhea is common with XELODA and can sometimes be severe. Stop taking XELODA and call your healthcare provider right away if the number of bowel movements you have in a day increases by 4 or more than is usual for you. Ask your healthcare provider about what medicines you can take to treat your diarrhea. If you have severe bloody diarrhea with severe abdominal pain and fever, call your healthcare provider or go to the nearest hospital emergency room right away.
- Heart problems. XELODA can cause heart problems including: heart attack and decreased blood flow to the heart, chest pain, irregular heartbeats, changes in the electrical activity of your heart seen on an electrocardiogram (ECG), problems with your heart muscle, heart failure, and sudden death. Stop taking XELODA and call your healthcare provider right away if you get any of the following symptoms:
- chest pain
- feeling faint
- sudden weight gain
- shortness of breath
- irregular heartbeats or skipping beats
- swollen ankles or legs
-
Loss of too much body fluid (dehydration) and kidney failure. Dehydration can happen with XELODA and may cause sudden kidney failure that can lead to death. You are at higher risk if you have kidney problems before taking XELODA and also take other medicines that can cause kidney problems.
Nausea, and vomiting are common with XELODA. If you lose your appetite, feel weak, and have nausea, vomiting, or diarrhea, you can quickly become dehydrated.
Stop taking XELODA and call your doctor right away if you:- vomit 2 or more times in a day.
- are only able to eat or drink a little now and then, or not at all due to nausea.
- have diarrhea. See "diarrhea" above.
-
Serious skin and mouth reactions.
- XELODA can cause serious skin reactions that may lead to death. Tell your Healthcare provider right away if you develop a skin rash, blisters and peeling of your skin. Your healthcare provider may tell you to stop taking XELODA if you have a serious skin reaction. Do not take XELODA again if this happens.
- XELODA can also cause "hand and foot syndrome." Hand and foot syndrome is common with XELODA and can cause you to have numbness and changes in sensation in your hands and feet, or cause redness, pain, swelling of your hands and feet. Stop taking XELODA and call your healthcare provider right away if you have any of these symptoms and you are not able to do your usual activities.
Hand and foot syndrome can lead to loss of fingerprints which could impact your identification. - you may get sores in your mouth or on your tongue when taking XELODA. Stop taking XELODA and call your healthcare provider if you get painful redness, swelling, or ulcers in your mouth and tongue, or if you are having problems eating.
- Increased level of bilirubin in your blood and liver problems. Increased bilirubin in your blood is common with XELODA. Your healthcare provider will check you for these problems during treatment with XELODA.
-
Decreased white blood cells, platelets, and red blood cell counts. Your healthcare provider will do blood tests during treatment with XELODA to check your blood cell counts.
If your white blood cell count is very low, you are at increased risk for infection. Call your healthcare provider right away if you develop a fever of 100.5°F or greater or have other signs and symptoms of infection.
People 80 years of age or older may be more likely to develop severe or serious side effects with XELODA.
The most common side effects of XELODA include:
- diarrhea
- hand and foot syndrome
- nausea
- vomiting
- stomach-area (abdominal) pain
- weakness and tiredness
- increased amounts of red blood cell breakdown products (bilirubin) in your blood
XELODA may cause fertility problems in females and males. This may affect the ability to have a child. Talk to your healthcare provider if you have concerns about fertility.
These are not all the possible side effects of XELODA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store XELODA?
- Store XELODA at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep XELODA in a tightly closed container.
- Ask your healthcare provider or pharmacist how to safely throw away any unused XELODA.
Keep XELODA and all medicines out of the reach of children.
General information about the safe and effective use of XELODA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use XELODA for a condition for which it was not prescribed. Do not give XELODA to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about XELODA that is written for health professionals.
What are the ingredients in XELODA?
Active ingredient: capecitabine
Inactive ingredients: anhydrous lactose, croscarmellose sodium, hydroxypropyl methylcellulose, microcrystalline cellulose, magnesium stearate and purified water. The peach or light peach film coating contains hydroxypropyl methylcellulose, talc, titanium dioxide, and synthetic yellow and red iron oxides.
For more information, go to http://www.gene.com/patients/medicines/xeloda or call 1-877-436-3683.
Distributed by:
Genentech USA, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA 94080-4990XELODA® is a registered trademark of Hoffmann-La Roche, Inc.© 2019 Genentech, Inc. All rights reserved.
- XELODA can interact with blood thinner medicines, such as warfarin (COUMADIN®). Taking XELODA with these medicines can cause changes in how fast your blood clots and can cause bleeding that can lead to death. This can happen as soon as a few days after you start taking XELODA, or later during treatment, and possibly even within 1 month after you stop taking XELODA. Your risk may be higher because you have cancer, and if you are over 60 years of age.
-
SPL UNCLASSIFIED SECTION
Representative sample of labeling (see the HOW SUPPLIED section for complete listing):
-
PRINCIPAL DISPLAY PANEL - 150 mg Tablet Bottle Carton
NDC: 0004-1100-20
Xeloda®
(capecitabine)
Tablets150 mg
Each tablet contains 150 mg
capecitabine.Rx only
60 tablets
Genentech10210268

-
PRINCIPAL DISPLAY PANEL - 500 mg Tablet Bottle Carton
NDC: 0004-1101-50
Xeloda®
(capecitabine)
Tablets500 mg
Each tablet contains 500 mg
capecitabine.Rx only
120 tablets
Genentech10210269

-
INGREDIENTS AND APPEARANCE
XELODA
capecitabine tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0004-1100 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength capecitabine (UNII: 6804DJ8Z9U) (capecitabine - UNII:6804DJ8Z9U) capecitabine 150 mg Inactive Ingredients Ingredient Name Strength croscarmellose sodium (UNII: M28OL1HH48) anhydrous lactose (UNII: 3SY5LH9PMK) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) HYPROMELLOSE 2208 (3 MPA.S) (UNII: 9H4L916OBU) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) magnesium stearate (UNII: 70097M6I30) talc (UNII: 7SEV7J4R1U) titanium dioxide (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color PINK (light peach) Score no score Shape OVAL Size 11mm Flavor Imprint Code XELODA;150 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0004-1100-20 60 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 04/30/1998 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020896 04/30/1998 XELODA
capecitabine tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0004-1101 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength capecitabine (UNII: 6804DJ8Z9U) (capecitabine - UNII:6804DJ8Z9U) capecitabine 500 mg Inactive Ingredients Ingredient Name Strength croscarmellose sodium (UNII: M28OL1HH48) anhydrous lactose (UNII: 3SY5LH9PMK) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) HYPROMELLOSE 2208 (3 MPA.S) (UNII: 9H4L916OBU) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) magnesium stearate (UNII: 70097M6I30) talc (UNII: 7SEV7J4R1U) titanium dioxide (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color PINK (peach) Score no score Shape OVAL Size 11mm Flavor Imprint Code XELODA;500 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0004-1101-50 120 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 04/30/1998 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020896 04/30/1998 Labeler - Genentech, Inc. (080129000) Establishment Name Address ID/FEI Business Operations Shanghai Roche Pharmaceuticals Ltd 654520048 MANUFACTURE(0004-1101) , ANALYSIS(0004-1101) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche Ltd 485244961 ANALYSIS(0004-1100, 0004-1101) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche Ltd 482242971 ANALYSIS(0004-1100, 0004-1101)
Trademark Results [Xeloda]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 XELODA 75241696 2200434 Live/Registered |
Hoffmann-La Roche Inc. 1997-02-13 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.