PRALUENT- alirocumab injection, solution
Praluent by
Drug Labeling and Warnings
Praluent by is a Prescription medication manufactured, distributed, or labeled by Sanofi US Corporation, Sanofi-Aventis U.S. LLC, Regeneron Pharmaceuticals, Inc., Sanofi Chimie, Regeneron Ireland Unlimited Company (Raheen), Sanofi-Aventis Deutschland GmbH, Genzyme Ireland Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PRALUENT safely and effectively. See full prescribing information for PRALUENT.
PRALUENT® (alirocumab) injection, for subcutaneous use
Initial U.S. Approval: 2015RECENT MAJOR CHANGES
Warnings and Precautions (5.1) 3/2020 INDICATIONS AND USAGE
PRALUENT is a PCSK9 (proprotein convertase subtilisin kexin type 9) inhibitor antibody indicated:
- to reduce the risk of myocardial infarction, stroke, and unstable angina requiring hospitalization in adults with established cardiovascular disease. (1.1)
- as adjunct to diet, alone or in combination with other lipid-lowering therapies (e.g., statins, ezetimibe), for the treatment of adults with primary hyperlipidemia (including heterozygous familial hypercholesterolemia) to reduce low-density lipoprotein cholesterol LDL-C. (1.2)
DOSAGE AND ADMINISTRATION
The recommended starting dose of PRALUENT is 75 mg once every 2 weeks administered subcutaneously, since the majority of patients achieve sufficient LDL-C reduction with this dosage. An alternative starting dosage for patients who prefer less frequent dosing is 300 mg once every 4 weeks (monthly). (2.1)
If the LDL-C response is inadequate, the dosage may be adjusted to the maximum dosage of 150 mg administered every 2 weeks. (2.1)
The recommended dose of PRALUENT in patients with HeFH undergoing LDL apheresis is 150 mg once every 2 weeks. PRALUENT can be administered without regard to timing of apheresis. (2.1)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
History of a serious hypersensitivity reaction to PRALUENT. (4)
WARNINGS AND PRECAUTIONS
Allergic Reactions: Hypersensitivity reactions (e.g., pruritus, rash, urticaria), including some serious events (e.g., hypersensitivity vasculitis, angioedema, and hypersensitivity reactions requiring hospitalization), have been reported with PRALUENT treatment. If signs or symptoms of serious allergic reactions occur, discontinue treatment with PRALUENT, treat according to the standard of care, and monitor until signs and symptoms resolve. (5.1)
ADVERSE REACTIONS
The most commonly occurring adverse reactions (≥5% of patients treated with PRALUENT and occurring more frequently than with placebo) are nasopharyngitis, injection site reactions, and influenza. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Regeneron at 1-844-734-6643 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Prevention of Cardiovascular Events
1.2 Primary Hyperlipidemia (including heterozygous familial hypercholesterolemia)
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
2.2 Important Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Allergic Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Prevention of Cardiovascular Events
14.2 Primary Hyperlipidemia (including heterozygous familial hypercholesterolemia)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Prevention of Cardiovascular Events
PRALUENT® is indicated to reduce the risk of myocardial infarction, stroke, and unstable angina requiring hospitalization in adults with established cardiovascular disease.
1.2 Primary Hyperlipidemia (including heterozygous familial hypercholesterolemia)
PRALUENT is indicated as an adjunct to diet, alone or in combination with other lipid-lowering therapies (e.g., statins, ezetimibe), for the treatment of adults with primary hyperlipidemia to reduce low-density lipoprotein cholesterol (LDL-C).
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
The recommended starting dose of PRALUENT is 75 mg once every 2 weeks administered subcutaneously, since the majority of patients achieve sufficient LDL-C reduction with this dosage. An alternative starting dosage for patients who prefer less frequent dosing is 300 mg once every 4 weeks (monthly).
For patients receiving PRALUENT 75 mg every 2 weeks, measure LDL-C levels within 4 to 8 weeks of initiating PRALUENT. If the LDL-C response is inadequate, the dosage may be adjusted to the maximum dosage of 150 mg administered every 2 weeks. Reassess LDL-C within 4 to 8 weeks.
For patients receiving PRALUENT 300 mg every 4 weeks, measure LDL-C just prior to the next scheduled dose, since in some patients LDL-C can vary considerably between doses with this regimen [see Clinical Studies (14)]. If LDL-C reduction is inadequate, the dosage may be adjusted to 150 mg every 2 weeks, starting the new dose on the next scheduled dosing date. Reassess LDL-C within 4 to 8 weeks.
If an every-2-week dose is missed, instruct the patient to administer the injection within 7 days from the missed dose and then resume the patient's original schedule. If the missed dose is not administered within 7 days, instruct the patient to wait until the next dose on the original schedule.
If an every-4-week dose is missed, instruct the patient to administer the injection within 7 days from the missed dose and then resume the patient's original schedule. If the missed dose is not administered within 7 days, instruct the patient to administer the dose, starting a new schedule based on this date.
The recommended dose of PRALUENT in patients with HeFH undergoing LDL apheresis is 150 mg once every 2 weeks. PRALUENT can be administered without regard to the timing of apheresis.
2.2 Important Administration Instructions
- Provide proper training to patients and/or caregivers on the preparation and administration of PRALUENT prior to use according to the Instructions for Use. Instruct patients and/or caregivers to read and follow the Instructions for Use each time they use PRALUENT.
- Store PRALUENT in the refrigerator. Allow PRALUENT to warm to room temperature for 30 to 40 minutes prior to use. If needed, PRALUENT may be kept at room temperature up to 77°F (25°C) for a maximum of 30 days in original carton to protect from light. Do not store above 77°F (25°C). After removal from the refrigerator, PRALUENT must be used within 30 days or discarded [see How Supplied/Storage and Handling (16)].
- PRALUENT should be inspected visually for particulate matter and discoloration prior to administration. If the solution is discolored or contains visible particulate matter, the solution should not be used.
- Follow aseptic injection technique every time PRALUENT is administered.
- Administer PRALUENT by subcutaneous injection into the thigh, abdomen, or upper arm using a single-dose pre-filled pen or single-dose pre-filled syringe.
- Rotate the injection site with each injection.
- To administer the 300 mg dose, give two 150 mg PRALUENT injections consecutively at two different injection sites.
- Do NOT inject PRALUENT into areas of active skin disease or injury such as sunburns, skin rashes, inflammation, or skin infections.
- Do NOT co-administer PRALUENT with other injectable drugs at the same injection site.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
PRALUENT is contraindicated in patients with a history of a serious hypersensitivity reaction to PRALUENT. Reactions have included hypersensitivity vasculitis and hypersensitivity reactions requiring hospitalization. [See Warnings and Precautions (5.1).]
-
5 WARNINGS AND PRECAUTIONS
5.1 Allergic Reactions
Hypersensitivity reactions (e.g., pruritus, rash, urticaria), including some serious events (e.g., hypersensitivity vasculitis, angioedema, and hypersensitivity reactions requiring hospitalization), have been reported with PRALUENT treatment. If signs or symptoms of serious allergic reactions occur, discontinue treatment with PRALUENT, treat according to the standard of care, and monitor until signs and symptoms resolve [see Contraindications (4)].
-
6 ADVERSE REACTIONS
The following adverse reactions are also discussed in the other sections of the labeling:
- Allergic Reactions [See Warnings and Precautions (5.1).]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Common Adverse Reactions
The data in Table 1 are derived from 9 primary hyperlipidemia placebo-controlled trials that included 2476 patients treated with PRALUENT 75 mg and/or 150 mg every 2 weeks, including 2135 exposed for 6 months and 1999 exposed for more than 1 year (median treatment duration of 65 weeks). The mean age of the population was 59 years, 40% of the population were women, 90% were Caucasian, 4% were Black or African American, and 3% were Asian.
Adverse reactions reported in at least 2% of PRALUENT-treated patients, and more frequently than in placebo-treated patients, are shown in Table 1.
Table 1: Adverse Reactions Occurring in Greater Than or Equal to 2% of PRALUENT-Treated Patients and More Frequently Than with Placebo Adverse Reactions Placebo
(N=1276)PRALUENT*
(N=2476)- * 75 mg every 2 weeks and 150 mg every 2 weeks combined
- † Includes erythema/redness, itching, swelling, pain/tenderness
Nasopharyngitis 11.1% 11.3% Injection site reactions† 5.1% 7.2% Influenza 4.6% 5.7% Urinary tract infection 4.6% 4.8% Diarrhea 4.4% 4.7% Bronchitis 3.8% 4.3% Myalgia 3.4% 4.2% Muscle spasms 2.4% 3.1% Sinusitis 2.7% 3.0% Cough 2.3% 2.5% Contusion 1.3% 2.1% Musculoskeletal pain 1.6% 2.1% Adverse reactions led to discontinuation of treatment in 5.3% of patients treated with PRALUENT and 5.1% of patients treated with placebo. The most common adverse reactions leading to treatment discontinuation in patients treated with PRALUENT were allergic reactions (0.6% versus 0.2% for PRALUENT and placebo, respectively) and elevated liver enzymes (0.3% versus <0.1%).
In an analysis of ezetimibe-controlled trials in which 864 patients were exposed to PRALUENT for a median of 27 weeks and 618 patients were exposed to ezetimibe for a median of 24 weeks, the types and frequencies of common adverse reactions were similar to those listed above.
In a cardiovascular outcomes trial in which 9451 patients were exposed to PRALUENT for a median of 31 months and 9443 patients were exposed to placebo for a median of 32 months, common adverse reactions (greater than 5% of patients treated with PRALUENT and occurring more frequently than placebo) included non-cardiac chest pain (7.0% PRALUENT, 6.8% placebo), nasopharyngitis (6.0% PRALUENT, 5.6% placebo), and myalgia (5.6% PRALUENT, 5.3% placebo).
Local Injection Site Reactions
In a pool of placebo-controlled trials evaluating PRALUENT 75 mg and/or 150 mg administered every 2 weeks (Q2W), local injection site reactions including erythema/redness, itching, swelling, and pain/tenderness were reported more frequently in patients treated with PRALUENT (7.2% versus 5.1% for PRALUENT and placebo, respectively). Few patients discontinued treatment because of these reactions (0.2% versus 0.4% for PRALUENT and placebo, respectively), but patients receiving PRALUENT had a greater number of injection site reactions, had more reports of associated symptoms, and had reactions of longer average duration than patients receiving placebo.
In a 48-week placebo-controlled trial evaluating PRALUENT 300 mg every 4 weeks (Q4W) and 75 mg Q2W, in which all patients received an injection of drug or placebo every 2 weeks to maintain the blind, local injection site reactions were reported more frequently in patients treated with PRALUENT 300 mg Q4W as compared to those receiving PRALUENT 75 mg Q2W or placebo (16.6%, 9.6%, and 7.9%, respectively). Three patients (0.7%) treated with PRALUENT 300 mg Q4W discontinued treatment due to local injection site reactions versus no patients (0%) in the other 2 treatment groups.
In a cardiovascular outcomes trial, local injection site reactions were reported in 3.8% of patients treated with PRALUENT versus 2.1% patients treated with placebo, and led to permanent discontinuation in 26 patients (0.3%) versus 3 patients (<0.1%), respectively.
Allergic Reactions
Allergic reactions were reported more frequently in patients treated with PRALUENT than in those treated with placebo (8.6% versus 7.8%). The proportion of patients who discontinued treatment due to allergic reactions was higher among those treated with PRALUENT (0.6% versus 0.2%). Serious allergic reactions, such as hypersensitivity, nummular eczema, and hypersensitivity vasculitis were reported in patients using PRALUENT in controlled clinical trials [see Warnings and Precautions (5.1)].
Liver Enzyme Abnormalities
In the primary hyperlipidemia trials, liver-related disorders (primarily related to abnormalities in liver enzymes) were reported in 2.5% of patients treated with PRALUENT and 1.8% of patients treated with placebo, leading to treatment discontinuation in 0.4% and 0.2% of patients, respectively. Increases in serum transaminases to greater than 3 times the upper limit of normal occurred in 1.7% of patients treated with PRALUENT and 1.4% of patients treated with placebo.
Low LDL-C Values
In the placebo-controlled and active-controlled primary hyperlipidemia trials using an every 2 week or every 4 week dosing interval, 914 PRALUENT-treated patients had two consecutive calculated LDL-C values <25 mg/dL, and 335 had two consecutive calculated LDL-C values <15 mg/dL. LDL-C values <25 mg/dL and <15 mg/dL were observed more frequently in patients treated with the PRALUENT 150 mg Q2W or 300 mg Q4W dosing regimens. Changes to background lipid-altering therapy (e.g., maximally tolerated statins) were not made in response to low LDL-C values in these trials, and PRALUENT dosing was not modified or interrupted on this basis.
In a cardiovascular outcomes trial, 4305 PRALUENT-treated patients had two consecutive calculated LDL-C values <25 mg/dL, and 782 had two consecutive calculated LDL-C values <15 mg/dL. Because PRALUENT dosing was decreased or discontinued in the event of two consecutive LDL-C values <15 mg/dL in this trial, the effects of prolonged very low LDL-C with PRALUENT are unknown.
In published genetic studies as well as clinical and observational trials with lipid lowering therapies, an increased risk of new onset of diabetes has been associated with lower levels of LDL-C.
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity with PRALUENT. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to PRALUENT in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
In a cardiovascular outcomes trial, 5.5% (504/9091) of patients treated with PRALUENT 75 mg and/or 150 mg every 2 weeks (Q2W) had anti-drug antibodies (ADA) detected after initiating treatment compared with 1.6% (149/9097) of patients treated with placebo. Persistent ADA responses, defined as at least 2 consecutive post-baseline samples with positive ADA separated by at least a 16-week period, were observed in 0.7% of patients treated with PRALUENT and 0.4% of patients treated with placebo. Neutralizing antibody (NAb) responses were observed in 0.5% of patients treated with PRALUENT and in <0.1% of patients treated with placebo. Efficacy based on reductions in LDL-C was mostly similar in patients with or without ADA. However, some patients treated with PRALUENT with persistent or neutralizing antibodies experienced attenuation in LDL-C efficacy.
A higher incidence of injection site reactions were observed in patients with treatment-emergent ADA compared to patients who were ADA negative (7.5% vs 3.6%). In a pool of ten placebo-controlled and active-controlled trials of patients treated with PRALUENT 75 mg and/or 150 mg Q2W as well as in a separate clinical study of patients treated with PRALUENT 75 mg Q2W or 300 mg every 4 weeks (including some patients with dose adjustment to 150 mg Q2W), the incidence of detecting ADA and NAb was similar to the results from the trial described above.
The long-term consequences of continuing PRALUENT treatment in the presence of ADA are unknown.
6.3 Postmarketing Experience
The following adverse reactions have been reported during postapproval use of PRALUENT. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
-
General disorders and administration site conditions
- Flu-like illness
-
Allergic reactions
- Angioedema
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to PRALUENT during pregnancy.
Please contact 1-877-311-8972 or go to https://mothertobaby.org/ongoing-study/praluent/ to enroll in or to obtain information about the registry.
Risk Summary
There are no available data on use of PRALUENT in pregnant women to inform a drug-associated risk. In animal reproduction studies, there were no effects on embryo-fetal development when rats were subcutaneously administered alirocumab during organogenesis at dose exposures up to 12-fold the exposure at the maximum recommended human dose of 150 mg every two weeks. In monkeys, suppression of the humoral immune response was observed in infant monkeys when alirocumab was dosed during organogenesis to parturition at dose exposures 13-fold the exposure at the maximum recommended human dose of 150 mg every two weeks. No additional effects on pregnancy or neonatal/infant development were observed at dose exposures up to 81-fold the maximum recommended human dose of 150 mg every two weeks. Measurable alirocumab serum concentrations were observed in the infant monkeys at birth at comparable levels to maternal serum, indicating that alirocumab, like other IgG antibodies, crosses the placental barrier. FDA's experience with monoclonal antibodies in humans indicates that they are unlikely to cross the placenta in the first trimester; however, they are likely to cross the placenta in increasing amounts in the second and third trimester. Consider the benefits and risks of PRALUENT and possible risks to the fetus before prescribing PRALUENT to pregnant women.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%–4% and 15%–20%, respectively.
Data
Animal data
In Sprague Dawley rats, no effects on embryo-fetal development were observed when alirocumab was dosed at up to 75 mg/kg/dose by the subcutaneous route on gestation days 6 and 12 at exposures 12-fold the maximum recommended human dose of 150 mg every two weeks, based on serum AUC.
In cynomolgus monkeys, suppression of the humoral immune response to keyhole limpet hemocyanin (KLH) antigen was observed in infant monkeys at 4 to 6 months of age when alirocumab was dosed during organogenesis to parturition at 15 mg/kg/week and 75 mg/kg/week by the subcutaneous route, corresponding to 13-fold and 81-fold the human exposure at the maximum recommended human dose of 150 mg every two weeks, based on serum AUC. The lowest dose tested in the monkey resulted in humoral immune suppression; therefore, it is unknown if this effect would be observed at clinical exposure. No study designed to challenge the immune system of infant monkeys was conducted. No additional embryo-fetal, prenatal or postnatal effects were observed in infant monkeys, and no maternal effects were observed, when alirocumab was dosed at up to 75 mg/kg/week by the subcutaneous route, corresponding to maternal exposure of 81-fold the exposure at the maximum recommended human dose of 150 mg every two weeks, based on serum AUC.
8.2 Lactation
Risk Summary
There is no information regarding the presence of alirocumab in human milk, the effects on the breastfed infant, or the effects on milk production. The development and health benefits of breastfeeding should be considered along with the mother's clinical need for PRALUENT and any potential adverse effects on the breastfed infant from PRALUENT or from the underlying maternal condition. Human IgG is present in human milk, but published data suggest that breastmilk IgG antibodies do not enter the neonatal and infant circulation in substantial amounts.
8.5 Geriatric Use
In primary hypercholesterolemia controlled trials, 1158 patients treated with PRALUENT were ≥65 years of age and 241 patients treated with PRALUENT were ≥75 years of age. In a cardiovascular outcomes trial, 2505 patients treated with PRALUENT were ≥65 years of age and 493 patients treated with PRALUENT were ≥75 years of age. No overall differences in safety or effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
No dose adjustment is needed for patients with mild or moderately impaired renal function. No data are available in patients with severe renal impairment. [See Clinical Pharmacology (12.3).]
8.7 Hepatic Impairment
No dose adjustment is needed for patients with mild or moderate hepatic impairment. No data are available in patients with severe hepatic impairment. [See Clinical Pharmacology (12.3).]
-
11 DESCRIPTION
Alirocumab is a human monoclonal antibody (IgG1 isotype) that targets proprotein convertase subtilisin kexin type 9 (PCSK9). Alirocumab is a PCSK9 inhibitor produced by recombinant DNA technology in Chinese Hamster Ovary cell suspension culture. Alirocumab consists of two disulfide-linked human heavy chains, each covalently linked through a disulfide bond to a human kappa light chain. A single N-linked glycosylation site is located in each heavy chain within the CH2 domain of the Fc constant region of the molecule. The variable domains of the heavy and light chains combine to form the PCSK9 binding site within the antibody. Alirocumab has an approximate molecular weight of 146 kDa.
PRALUENT is a sterile, preservative-free, clear, colorless to pale yellow solution for subcutaneous injection. PRALUENT 75 mg/mL or 150 mg/mL solution for subcutaneous injection in a single-dose pre-filled pen or single-dose pre-filled syringe is supplied in a siliconized 1 mL Type-1 clear glass syringe. The needle shield is not made with natural rubber latex.
Each 75 mg/mL pre-filled pen or pre-filled syringe contains 75 mg alirocumab, histidine (8 mM), polysorbate 20 (0.1 mg), sucrose (100 mg), and Water for Injection USP, to pH 6.0.
Each 150 mg/mL pre-filled pen or pre-filled syringe contains 150 mg alirocumab, histidine (6 mM), polysorbate 20 (0.1 mg), sucrose (100 mg), and Water for Injection USP, to pH 6.0.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Alirocumab is a human monoclonal antibody that binds to proprotein convertase subtilisin kexin type 9 (PCSK9). PCSK9 binds to the low-density lipoprotein receptors (LDLR) on the surface of hepatocytes to promote LDLR degradation within the liver. LDLR is the primary receptor that clears circulating LDL, therefore the decrease in LDLR levels by PCSK9 results in higher blood levels of LDL-C. By inhibiting the binding of PCSK9 to LDLR, alirocumab increases the number of LDLRs available to clear LDL, thereby lowering LDL-C levels.
12.2 Pharmacodynamics
Alirocumab reduced free PCSK9 in a concentration-dependent manner. Following a single subcutaneous administration of alirocumab 75 or 150 mg, maximal suppression of free PCSK9 occurred within 4 to 8 hours. Free PCSK9 concentrations returned to baseline when alirocumab concentrations decreased below the limit of quantitation.
12.3 Pharmacokinetics
Absorption
After subcutaneous (SC) administration of 75 mg to 300 mg alirocumab, median times to maximum serum concentrations (tmax) were 3–7 days. The pharmacokinetics of alirocumab after single SC administration of 75 mg into the abdomen, upper arm, or thigh were similar. The absolute bioavailability of alirocumab after SC administration was about 85% as determined by population pharmacokinetics analysis. A slightly greater than dose proportional increase was observed, with a 2.1-fold to 2.7-fold increase in total alirocumab concentrations for a 2-fold increase in dose from 75 mg every 2 weeks to 150 mg every 2 weeks. Monthly dose normalized exposure with 300 mg every 4 weeks treatment was similar to that of 150 mg every 2 weeks. Steady state was reached after 2 to 3 doses with an accumulation ratio up to a maximum of about 2-fold.
Distribution
Following IV administration, the volume of distribution was about 0.04 to 0.05 L/kg indicating that alirocumab is distributed primarily in the circulatory system.
Metabolism and Elimination
Specific metabolism studies were not conducted, because alirocumab is a protein. Alirocumab is expected to degrade to small peptides and individual amino acids. In clinical studies where alirocumab was administered in combination with atorvastatin or rosuvastatin, no relevant changes in statin concentrations were observed in the presence of repeated administration of alirocumab, indicating that cytochrome P450 enzymes (mainly CYP3A4 and CYP2C9) and transporter proteins such as P-gp and OATP were not affected by alirocumab.
Two elimination phases were observed for alirocumab. At low concentrations, the elimination is predominately through saturable binding to target (PCSK9), while at higher concentrations the elimination of alirocumab is largely through a non-saturable proteolytic pathway.
Based on a population pharmacokinetic analysis, the median apparent half-life of alirocumab at steady state was 17 to 20 days in patients receiving alirocumab at subcutaneous doses of 75 mg Q2W or 150 mg Q2W.
Specific Populations
A population pharmacokinetic analysis was conducted on data from 2799 patients. Age, body weight, gender, race, and creatinine clearance were found not to significantly influence alirocumab pharmacokinetics. No dose adjustments are recommended for these demographics.
Pediatric
PRALUENT has not been studied in pediatric patients [see Use in Specific Populations (8.4)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with alirocumab. The mutagenic potential of alirocumab has not been evaluated; however, monoclonal antibodies are not expected to alter DNA or chromosomes.
There were no adverse effects on surrogate markers of fertility (e.g., estrous cyclicity, testicular volume, ejaculate volume, sperm motility, or total sperm count per ejaculate) in a 6-month chronic toxicology study in sexually-mature monkeys subcutaneously administered at 5, 15, and 75 mg/kg/week at systemic exposures up to 103-fold the 150 mg every two weeks subcutaneous clinical dose based on serum AUC. In addition, there were no adverse alirocumab-related anatomic pathology or histopathology findings in reproductive tissues in rat or monkey toxicology studies at systemic exposures up to 11-fold and 103-fold respectively, in the 6-month studies, compared to clinical systemic exposure following a 150 mg every two weeks dose, based on serum AUC.
13.2 Animal Toxicology and/or Pharmacology
During a 13-week toxicology study of 75 mg/kg once weekly alirocumab in combination with 40 mg/kg once daily atorvastatin in adult monkeys, there were no effects of PRALUENT on the humoral immune response to keyhole limpet hemocyanin (KLH) after one to two months at exposures 100-fold greater than the exposure at the maximum recommended human dose of 150 mg every two weeks, based on AUC.
-
14 CLINICAL STUDIES
14.1 Prevention of Cardiovascular Events
Study 1 (ODYSSEY OUTCOMES, NTC01663402) was a multicenter, double-blind, placebo-controlled trial in 18,924 adult patients (9462 PRALUENT; 9462 placebo) followed for up to 5 years. Patients had an acute coronary syndrome (ACS) event 4 to 52 weeks prior to randomization and were treated with a lipid-modifying–therapy (LMT) regimen that was statin-intensive (defined as atorvastatin 40 or 80 mg, or rosuvastatin 20 or 40 mg) or at maximally tolerated dose of a statin, with or without other LMT. Patients were randomized to receive either PRALUENT 75 mg once every two weeks (Q2W) or placebo Q2W. The PRALUENT 300 mg Q4W dose [see Dosage and Administration (2.1)] was not evaluated in this study.
At month 2, if additional LDL-C lowering was required based on pre-specified LDL-C criteria (LDL-C ≥50 mg/dL), PRALUENT was adjusted to 150 mg Q2W. For patients who had their dose adjusted to 150 mg Q2W and who had two consecutive LDL-C values below 25 mg/dL, down-titration from 150 mg Q2W to 75 mg Q2W was performed. Patients on 75 mg Q2W who had two consecutive LDL-C values below 15 mg/dL were switched to placebo in a blinded fashion. Approximately 2615 (27.7%) of 9451 patients treated with PRALUENT required dose adjustment to 150 mg Q2W. Of these 2615 patients, 805 (30.8%) were down-titrated to 75 mg Q2W. Overall, 730 (7.7%) of 9451 patients switched to placebo.
A total of 99.5% of patients were followed for survival until the end of the trial. The median follow-up duration was 33 months.
The mean age at baseline was 59 years (range 39–92), with 25% women, and 27% at least 65 years old. The trial population was 79% Caucasian, 3% Black, and 13% Asian; 17% identified as Hispanic/Latino ethnicity. The index ACS event was a myocardial infarction in 83% of patients and unstable angina in 17% of patients. Prior to the index ACS event, 19% had prior myocardial infarction and 23% had coronary revascularization procedures (CABG/PCI). Selected additional baseline risk factors included hypertension (65%), diabetes mellitus (25%), New York Association class I or II congestive heart failure (15%), and eGFR <60 mL/min/1.73 m2 (13%). Most patients (89%) were receiving statin-intensive therapy with or without other LMT at randomization. The mean LDL-C value at baseline was 92.4 mg/dL.
PRALUENT significantly reduced the risk for the primary composite endpoint (time to first occurrence of coronary heart disease death, non-fatal myocardial infarction, fatal and non-fatal ischemic stroke, or unstable angina requiring hospitalization: p=0.0003). The results are presented in Table 2.
Table 2: Cardiovascular Outcomes in Patients with Established Cardiovascular Disease Endpoint PRALUENT
N=9462Placebo
N=9462Hazard Ratio
(95% CI)*n (%) Incidence Rate per 100 Patient Years
(95% CI)n (%) Incidence Rate per 100 Patient Years
(95% CI)- * Cox-proportional hazards model with treatment as a factor and stratified by geographic region
- † Primary composite endpoint defined as: time to first occurrence of coronary heart disease death, non-fatal myocardial infarction, fatal and non-fatal ischemic stroke, or unstable angina requiring hospitalization
- ‡ First occurrence of specified event at any time; patients may have experienced more than one adjudicated event
- § Statistical testing performed outside hierarchy; therefore not considered statistically significant
Primary composite endpoint† 903
(9.5%)3.5
(3.3 to 3.8)1052
(11.1%)4.2
(3.9 to 4.4)0.85
(0.78, 0.93)Components of the Primary Composite Endpoint‡ CHD death 205
(2.2%)0.8
(0.7 to 0.9)222
(2.3%)0.8
(0.7 to 0.9)0.92
(0.76, 1.11)Non-fatal MI§ 626
(6.6%)2.4
(2.2 to 2.6)722
(7.6%)2.8
(2.6 to 3.0)0.86
(0.77, 0.96)Fatal or non-fatal ischemic stroke§ 111
(1.2%)0.4
(0.3 to 0.5)152
(1.6%)0.6
(0.5 to 0.7)0.73
(0.57, 0.93)Unstable angina requiring hospitalization§ 37
(0.4%)0.1
(0.1 to 0.2)60
(0.6%)0.2
(0.2 to 0.3)0.61
(0.41, 0.92)Mortality Endpoint (not statistically significant per pre-specified method to control for type I error) All-cause mortality 334
(3.5%)1.2
(1.1 to 1.4)392
(4.1%)1.5
(1.3 to 1.6)0.85
(0.73, 0.98)The Kaplan-Meier estimates of the cumulative incidence of the primary endpoint over time is presented in Figure 1.
Figure 1: Primary Composite Endpoint Cumulative Incidence over 4 Years in ODYSSEY OUTCOMES
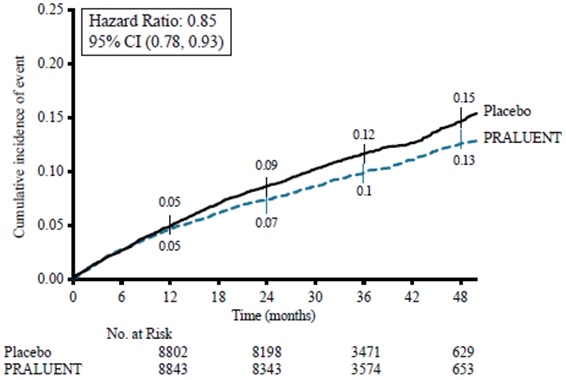
14.2 Primary Hyperlipidemia (including heterozygous familial hypercholesterolemia)
Study 2 (ODYSSEY LONG TERM, NCT01507831) was a multicenter, double-blind, placebo-controlled trial that randomly assigned 1553 patients to PRALUENT 150 mg Q2W and 788 patients to placebo. All patients were taking maximally tolerated doses of statins with or without other lipid-modifying therapy, and required additional LDL-C reduction. The mean age was 61 years (range 18–89), 38% were women, 93% were Caucasian, 3% were Black, and 5% were Hispanic/Latino. The average LDL-C at baseline was 122 mg/dL.
The proportion of patients who prematurely discontinued study drug prior to the 24-week primary endpoint was 8% among those treated with PRALUENT and 8% among those treated with placebo.
At week 24, the treatment difference between PRALUENT and placebo in mean LDL-C percent change was -58% (95% CI: -61%, -56%; p-value: <0.0001).
For additional results see Table 3 and Figure 2.
Table 3: Mean Percent Change from Baseline and Difference* from Placebo in Lipid Parameters at Week 24 in ODYSSEY LONG TERM† Treatment Group LDL-C Total-C Non-HDL-C Apo B - * Difference is PRALUENT minus Placebo
- † A pattern-mixture model approach was used with multiple imputation of missing post-treatment values based on a patient's own baseline value and multiple imputation of missing on-treatment values based on a model including available on-treatment values
Week 24 (Mean Percent Change from Baseline) Placebo 1 0 1 1 PRALUENT
(150 mg)-58 -36 -49 -50 Difference from placebo (LS Mean)
(95% CI)-58
(-61, -56)-36
(-37, -34)-50
(-52, -47)-51
(-53, -48)Figure 2: Mean Percent Change from Baseline in LDL-C Over 52 Weeks in Patients on Maximally Tolerated Statin Treated with PRALUENT 150 mg Q2W and Placebo Q2W (ODYSSEY LONG TERM)a
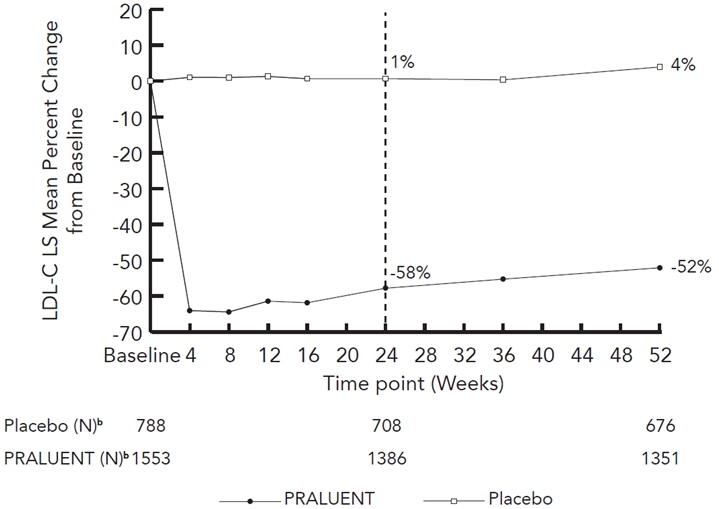
a The means were estimated based on all randomized patients, with multiple imputation of missing data taking into account treatment adherence.
b Number of patients with observed data.
Study 3 (ODYSSEY COMBO I, NCT01644175) was a multicenter, double-blind, placebo-controlled trial that randomly assigned 209 patients to PRALUENT and 107 patients to placebo. Patients were taking maximally tolerated doses of statins with or without other lipid-modifying therapy, and required additional LDL-C reduction.
The mean age was 63 years (range 39–87), 34% were women, 82% were Caucasian, 16% were Black, and 11% were Hispanic/Latino. Mean baseline LDL-C was 102 mg/dL.
The proportion of patients who prematurely discontinued study drug prior to the 24-week primary endpoint was 11% among those treated with PRALUENT and 12% among those treated with placebo.
At week 12, the mean percent change from baseline in LDL-C was -45% with PRALUENT compared to 1% with placebo, and the treatment difference between PRALUENT 75 mg Q2W and placebo in mean LDL-C percent change was -46% (95% CI: -53%, -39%).
At week 12, if additional LDL-C lowering was required based on pre-specified LDL-C criteria, PRALUENT was up-titrated to 150 mg Q2W for the remainder of the trial. The dose was up-titrated to 150 mg Q2W in 32 (17%) of 191 patients treated with PRALUENT for at least 12 weeks. At week 24, the mean percent change from baseline in LDL-C was -44% with PRALUENT and -2% with placebo, and the treatment difference between PRALUENT and placebo in mean LDL-C percent change was -43% (95% CI: -50%, -35%; p-value: <0.0001).
Studies 4 (ODYSSEY FH I, NCT01623115) and 5 (ODYSSEY FH II, NCT01709500) were multicenter, double-blind, placebo-controlled trials that, combined, randomly assigned 490 patients to PRALUENT and 245 patients to placebo. The trials were similar with regard to both design and eligibility criteria. All patients had HeFH, were taking a maximally tolerated dose of statin with or without other lipid-modifying therapy, and required additional LDL-C reduction. The diagnosis of HeFH was made either by genotyping or clinical criteria ("definite FH" using either the Simon Broome or WHO/Dutch Lipid Network criteria). The mean age was 52 years (range 20–87), 45% were women, 94% were Caucasian, 1% were Black, and 3% were Hispanic/Latino. The average LDL-C at baseline was 141 mg/dL.
Considering both trials together, the proportion of patients who prematurely discontinued study drug prior to the 24-week primary endpoint was 6% among those treated with PRALUENT and 4% among those treated with placebo.
At week 12, the treatment difference between PRALUENT 75 mg Q2W and placebo in mean LDL-C percent change was -48% (95% CI: -52%, -44%).
At week 12, if additional LDL-C lowering was required based on pre-specified LDL-C criteria, PRALUENT was up-titrated to 150 mg Q2W for the remainder of the trials. The dose was up-titrated to 150 mg Q2W in 196 (42%) of 469 patients treated with PRALUENT for at least 12 weeks. At week 24, the mean treatment difference between PRALUENT and placebo in mean LDL-C percent change from baseline was -54% (95% CI: -59%, -50%; p-value: ˂0.0001). The LDL-C-lowering effect was sustained to week 52.
For additional results see Table 4 and Figure 3.
Table 4: Mean Percent Change from Baseline and Difference* from Placebo in Lipid Parameters at Week 12 and Week 24 in Patients with HeFH (ODYSSEY FH I and FH II Pooled)† Treatment Group LDL-C Total-C Non-HDL-C Apo B - * Difference is PRALUENT minus Placebo
- † A pattern-mixture model approach was used with multiple imputation of missing post-treatment values based on a patient's own baseline value and multiple imputation of missing on-treatment values based on a model including available on-treatment values
- ‡ Dose was up-titrated to 150 mg Q2W in 196 (42%) patients treated for at least 12 weeks
Week 12 (Mean Percent Change from Baseline) Placebo 5 4 5 2 PRALUENT (75 mg) -43 -27 -38 -34 Difference from placebo (LS Mean) (95% CI) -48
(-52, -44)-31
(-34, -28)-42
(-46, -39)-36
(-39, -33)Week 24 (Mean Percent Change from Baseline) Placebo 7 5 7 2 PRALUENT (75/up 150 mg‡) -47 -30 -42 -40 Difference from placebo
(LS Mean) (95% CI)-54
(-59, -50)-36
(-39, -33)-49
(-53, -45)-42
(-45, -39)Figure 3: Mean Percent Change from Baseline in LDL-C Over 52 Weeks in Patients with HeFH on Maximally Tolerated Statin Treated with PRALUENT 75/150 mg Q2W and Placebo Q2W (ODYSSEY FH I and FH II Pooled)a
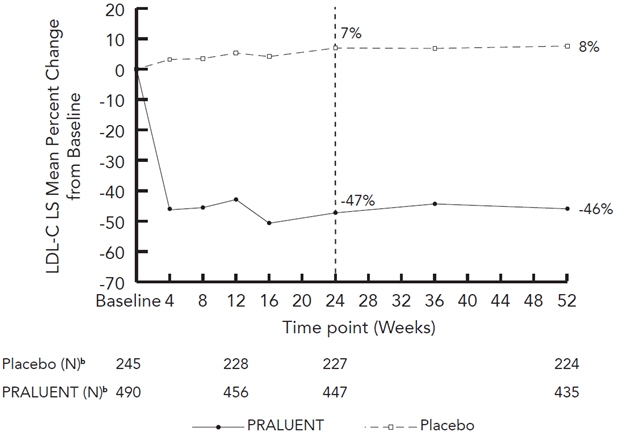
a The means were estimated based on all randomized patients, with multiple imputation of missing data taking into account treatment adherence.
b Number of patients with observed data.
Study 6 (ODYSSEY HIGH FH, NCT01617655) was a multicenter, double-blind, placebo-controlled trial that randomly assigned 72 patients to PRALUENT 150 mg Q2W and 35 patients to placebo. Patients had HeFH with a baseline LDL-C ≥160 mg/dL while taking a maximally tolerated dose of statin with or without other lipid-modifying therapy. The mean age was 51 years (range 18–80), 47% were women, 88% were Caucasian, 2% were Black, and 6% were Hispanic/Latino. The average LDL-C at baseline was 198 mg/dL.
The proportion of patients who discontinued study drug prior to the 24-week primary endpoint was 10% among those treated with PRALUENT and 0% among those treated with placebo.
At week 24, the mean percent change from baseline in LDL-C was -43% with PRALUENT and -7% with placebo, and the treatment difference between PRALUENT and placebo in mean LDL-C percent change was -36% (95% CI: -49%, -24%; p-value: <0.0001).
Study 7 (ODYSSEY CHOICE I, NCT01926782) was a multicenter, double-blind, placebo-controlled trial that randomly assigned 458 patients with primary hyperlipidemia to PRALUENT 300 mg Q4W, 115 patients to PRALUENT 75 mg Q2W, and 230 patients to placebo. Patients were stratified based on whether or not they were treated concomitantly with statin.
The mean age was 61 years (range 21–88), 42% were women, 87% were Caucasian, 11% were Black, and 3% were Hispanic/Latino.
The proportion of patients who discontinued study drug prior to the 24-week primary endpoint was 12% among those treated with PRALUENT 300 mg Q4W, 14% among those treated with PRALUENT 75 mg Q2W, and 15% among those treated with placebo.
In the cohort of patients on background statin, the mean LDL-C at baseline was 113 mg/dL. At week 12, the treatment difference between PRALUENT 300 mg Q4W and placebo in mean percent change in LDL-C from baseline was -54% (97.5% CI: -61%, -48%), and the treatment difference between PRALUENT 75 mg Q2W and placebo in mean percent change in LDL-C was -44% (97.5% CI: -53%, -35%) (Figure 4).
- * The means were estimated based on all randomized patients, with multiple imputation of missing data taking into account treatment adherence.
Figure 4: Mean Percent Change from Baseline in LDL-C up to Week 12 in Patients on Concomitant Statin Treated with PRALUENT 75 mg Q2W, PRALUENT 300 mg Q4W or Placebo* 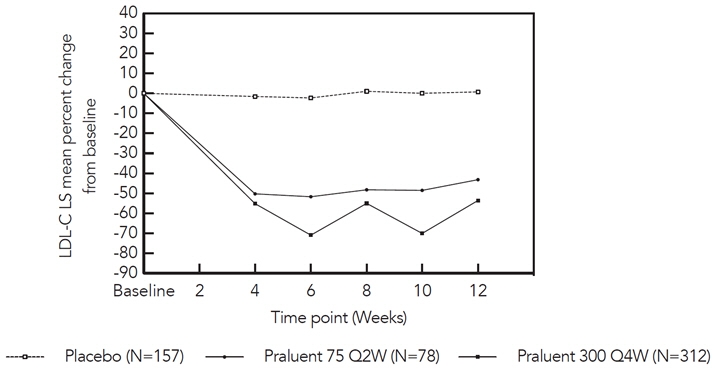
At week 12, if additional LDL-C lowering was required based on pre-specified LDL-C criteria, PRALUENT was adjusted to 150 mg Q2W for the remainder of the trial. The dose was adjusted to 150 mg Q2W in approximately 20% of patients treated with PRALUENT 75 mg Q2W or 300 mg Q4W for at least 12 weeks.
At week 24, the treatment difference between initial assignment to PRALUENT 300 mg Q4W and placebo in mean percent change in LDL-C from baseline was -56% (97.5% CI: -62%, -49%; p-value: <0.0001), and the treatment difference between initial assignment to PRALUENT 75 mg Q2W and placebo in mean percent change in LDL-C from baseline was -48% (97.5% CI: -57%, -39%).
In the cohort of patients not treated with a concomitant statin, the mean LDL-C at baseline was 142 mg/dL. The treatment difference between PRALUENT and placebo were similar to the cohort of patients treated with a concomitant statin.
Study 8 (ODYSSEY ESCAPE, NCT02326220) was a multicenter, double-blind, placebo-controlled trial that randomly assigned patients with HeFH who were undergoing LDL apheresis to PRALUENT 150 mg Q2W (N=41) or placebo (N=21). Patients were treated in combination with their usual LDL apheresis schedule for 6 weeks. The mean age was 59 years (range 27–79), 42% were women, 97% were Caucasian, 3% were Black, and 0% were Hispanic/Latino. The mean LDL-C at baseline, measured before the apheresis procedure, was 181 mg/dL. The proportion of patients who discontinued study drug prior to the 6-week endpoint was 2% among those treated with PRALUENT 150 mg Q2W and 5% among those treated with placebo. At week 6, the mean percent change from baseline in pre-apheresis LDL-C was -53% in patients in the PRALUENT group compared to 1% in patients who received placebo.
Study 9 (ODYSSEY COMBO II, NCT01644188) was a multicenter, double-blind, ezetimibe-controlled trial that randomly assigned 479 patients to PRALUENT 75 mg Q2W/150 mg Q2W and 241 patients to ezetimibe 10 mg/day. Patients were taking a maximally tolerated dose of a statin and required additional LDL-C reduction.
The mean age was 62 years (range 29–88), 26% were women, 85% were Caucasian, 4% were Black, and 3% were Hispanic/Latino. Mean baseline LDL-C was 107 mg/dL.
The proportion of patients who prematurely discontinued study drug prior to the 24-week primary endpoint was 9% among those treated with PRALUENT and 10% among those treated with ezetimibe.
At week 12, the mean percent change from baseline in LDL-C was -50% with PRALUENT compared to -22% with ezetimibe, and the treatment difference between PRALUENT and ezetimibe in mean LDL-C percent change was -28% (95% CI: -32%, -23%).
At week 12, if additional LDL-C lowering was required based on pre-specified LDL-C criteria, PRALUENT was up-titrated to 150 mg Q2W for the remainder of the trial. The dose was up-titrated to 150 mg Q2W in 82 (18%) of 446 patients treated with PRALUENT for at least 12 weeks. At week 24, the mean percent change from baseline in LDL-C was -48% with PRALUENT and -20% with ezetimibe, and the treatment difference between PRALUENT and ezetimibe in mean LDL-C percent change was -28% (95% CI: -33%, -23%; p-value: <0.0001).
Study 10 (ODYSSEY MONO, NCT01644474) was a multicenter, double-blind, ezetimibe-controlled trial in patients with a moderate CV risk, not taking statins or other lipid-modifying therapies, and a baseline LDL-C between 100 mg/dL to 190 mg/dL that randomly assigned 52 patients to PRALUENT 75 mg Q2W and 51 patients to ezetimibe 10 mg/day.
The mean age was 60 years (range 45–72), 47% were women, 90% were Caucasian and 10% were Black. Mean baseline LDL-C was 140 mg/dL.
The proportion of patients who prematurely discontinued study drug prior to the 24-week endpoint was 15% among those treated with PRALUENT and 14% among those treated with ezetimibe.
At week 12, the mean percent change from baseline in LDL-C was -48% with PRALUENT compared to -19% with ezetimibe, and the treatment difference between PRALUENT 75 mg Q2W and ezetimibe in mean LDL-C percent change was -29% (95% CI: -37%, -22%). At week 12, if additional LDL-C lowering was required based on pre-specified LDL-C criteria, PRALUENT was up-titrated to 150 mg Q2W for the remainder of the trial. The dose was up-titrated to 150 mg Q2W in 14 (30%) of 46 patients treated with PRALUENT for at least 12 weeks. At week 24, the mean percent change from baseline in LDL-C was -45% with PRALUENT and -14% with ezetimibe, and the treatment difference between PRALUENT and ezetimibe in mean LDL-C percent change was -31% (95% CI: -40%, -22%; p-value: <0.0001).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
PRALUENT is a clear, colorless to pale yellow solution, supplied in single-dose pre-filled pens and single-dose pre-filled glass syringes. Each pre-filled pen or pre-filled syringe of PRALUENT is designed to deliver 1 mL of 75 mg/mL or 150 mg/mL solution.
PRALUENT is available in cartons containing 1 or 2 pre-filled pens and 1 or 2 pre-filled syringes.
Pack Size 75 mg/mL Pre-filled Pen 150 mg/mL Pre-filled Pen Pack of 1 pen NDC: 0024-5901-01 NDC: 0024-5902-01 Pack of 2 pens NDC: 0024-5901-02
NDC: 72733-5901-2NDC: 0024-5902-02
NDC: 72733-5902-2Pack Size 75 mg/mL Pre-filled Syringe 150 mg/mL Pre-filled Syringe Pack of 1 syringe NDC: 0024-5903-01 NDC: 0024-5904-01 Pack of 2 syringes NDC: 0024-5903-02 NDC: 0024-5904-02 Storage
Pharmacy
Store in a refrigerator at 36°F to 46°F (2°C to 8°C) in the original carton in order to protect from light.
Patient/Caregiver
Store in a refrigerator at 36°F to 46°F (2°C to 8°C) in the original carton in order to protect from light. If needed, PRALUENT may be kept at room temperature up to 77°F (25°C) for a maximum of 30 days in original carton to protect from light. Do not store above 77°F (25°C). After removal from the refrigerator, PRALUENT must be used within 30 days or discarded.
Do NOT freeze. Do NOT expose to extreme heat. Do NOT shake.
-
17 PATIENT COUNSELING INFORMATION
See FDA-Approved Patient Labeling (Patient Information and Instructions for Use).
Pregnancy Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to PRALUENT during pregnancy. Encourage participation in the registry [see Use in Specific Populations (8.1)].
Allergic Reactions
- Advise patients to discontinue PRALUENT and seek prompt medical attention if any signs or symptoms of serious allergic reactions occur.
Instructions for Administration
- Instruct patients and caregivers to read the Patient Information and Instructions for Use (IFU) before the patient starts using PRALUENT, and each time the patient gets a refill as there may be new information they need to know.
- Provide guidance to patients and caregivers on proper subcutaneous injection technique, including aseptic technique, and how to use the pre-filled pen or pre-filled syringe correctly (see Instructions for Use leaflet). Inform patients that it may take up to 20 seconds to inject PRALUENT.
- The pre-filled pen or pre-filled syringe should be allowed to warm to room temperature for 30 to 40 minutes prior to use.
- Patients and caregivers should be cautioned that the pre-filled pen or pre-filled syringe must not be re-used and instructed in the technique of proper pen and syringe disposal in a puncture-resistant container. Do not recycle the container.
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: April 2020 Patient Information
PRALUENT® (PRAHL-u-ent)
(alirocumab)
injection, for subcutaneous useWhat is PRALUENT?
PRALUENT is an injectable prescription medicine used:- in adults with cardiovascular disease to reduce the risk of heart attack, stroke, and certain types of chest pain conditions (unstable angina) requiring hospitalization.
- along with diet, alone or together with other cholesterol-lowering medicines in adults with high blood cholesterol levels called primary hyperlipidemia (including a type of high cholesterol called heterozygous familial hypercholesterolemia), to reduce low-density lipoprotein cholesterol (LDL-C) or bad cholesterol.
Who should not use PRALUENT?
Do not use PRALUENT if you are allergic to alirocumab or to any of the ingredients in PRALUENT. See the end of this leaflet for a complete list of ingredients in PRALUENT.What should I tell my healthcare provider before using PRALUENT?
Before you start using PRALUENT, tell your healthcare provider about all of your medical conditions, including allergies, and if you:- are pregnant or plan to become pregnant. It is not known if PRALUENT will harm your unborn baby. Tell your healthcare provider if you become pregnant while taking PRALUENT.
Pregnancy Registry. There is a pregnancy registry for women who take PRALUENT during pregnancy. The purpose of this registry is to collect information about your health and your baby's health. You can talk to your healthcare provider or contact 1-877-311-8972 or go to https://mothertobaby.org/ongoing-study/praluent/ to enroll in this registry or get more information. - are breastfeeding or plan to breastfeed. You and your healthcare provider should decide if you will take PRALUENT or breastfeed. You should not do both without talking to your healthcare provider first.
How should I use PRALUENT? - See the detailed "Instructions for Use" that comes with this Patient Information about the right way to prepare and give your PRALUENT injections.
- Use PRALUENT exactly as your healthcare provider tells you to use it.
- PRALUENT comes as a single-dose (1 time) pre-filled pen (autoinjector), or as a single-dose pre-filled syringe. Your healthcare provider will prescribe the type and dosage that is best for you.
- If your healthcare provider decides that you or a caregiver can give the injections of PRALUENT, you or your caregiver should receive training on the right way to prepare and give PRALUENT. Do not try to inject PRALUENT until you have been shown the right way by your healthcare provider or nurse.
- PRALUENT is injected under the skin (subcutaneously) every 2 weeks or every 4 weeks (monthly).
- If your healthcare provider prescribes you the monthly dose, you will give yourself 2 separate injections in a row, using a different syringe or pen for each injection and 2 different injection sites.
- Do not inject PRALUENT together with other injectable medicines at the same injection site.
- Always check the label of your pen or syringe to make sure you have the correct medicine and the correct dose of PRALUENT before each injection.
- If you forget to use PRALUENT or are not able to take the dose at your regular time, inject your missed dose as soon as you remember, within 7 days. Then, if you inject every 2 weeks take your next dose in 2 weeks from the day you missed your dose or if you inject every 4 weeks take your next dose in 4 weeks from the day you missed your dose. This will put you back on your original schedule.
- If you missed a dose by more than 7 days and you inject every 2 weeks wait until your next scheduled dose to re-start PRALUENT or if you inject every 4 weeks start a new schedule from the time you remember to take your dose.
If you are not sure when to re-start PRALUENT, ask your healthcare provider or pharmacist. - If you use more PRALUENT than you should, talk to your healthcare provider or pharmacist.
- Do not stop using PRALUENT without talking with your healthcare provider. If you stop using PRALUENT, your cholesterol levels can increase.
What are the possible side effects of PRALUENT?
PRALUENT can cause serious side effects, including:- allergic reactions. PRALUENT may cause allergic reactions that can be severe and require treatment in a hospital. Stop using PRALUENT and call your healthcare provider or go to the nearest hospital emergency room right away if you have any symptoms of an allergic reaction including:
- a severe rash
- redness
- hives
- severe itching
- trouble breathing
- swelling of the face, lips, throat or tongue
The most common side effects of PRALUENT include: - redness, itching, swelling, pain, or tenderness at the injection site
- symptoms of the common cold
- flu or flu-like symptoms
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all of the possible side effects of PRALUENT. Ask your healthcare provider or pharmacist for more information.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of PRALUENT.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use PRALUENT for a condition for which it was not prescribed. Do not give PRALUENT to other people, even if they have the same symptoms that you have. It may harm them.
This Patient Information summarizes the most important information about PRALUENT. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about PRALUENT that is written for health professionals.
For more information about PRALUENT, go to www.PRALUENT.com or call 1-844-PRALUENT (1-844-772-5836).What are the ingredients in PRALUENT? - Active ingredient: alirocumab
- Inactive ingredients: histidine, polysorbate 20, sucrose, and Water for Injection, USP.
Manufactured by: Regeneron Pharmaceuticals, Inc. Tarrytown, NY 10591, U.S. License # 1760; PRALUENT is a registered trademark of Regeneron Pharmaceuticals, Inc. / ©2020 Regeneron Pharmaceuticals, Inc. All rights reserved. -
INSTRUCTIONS FOR USE
Instructions For Use
PRALUENT® (PRAHL-u-ent)
(alirocumab)
Injection, for Subcutaneous Injection
Single-Dose Pre-Filled Pen (75 mg/mL)Important Information - The device is a single-dose disposable pen. It contains 75 mg of PRALUENT (alirocumab) in 1 mL.
- The PRALUENT pen contains medicine prescribed by your healthcare provider.
- The medicine is injected under your skin and can be given by yourself or someone else (caregiver).
- It is important that you do not try to give yourself or someone else the injection unless you have received training from your healthcare provider.
- This pen can only be used for 1 single injection, and must be discarded after use.
- Read all of the instructions carefully before using the PRALUENT pen.
- Follow these instructions every time you use a PRALUENT pen.
- Do not touch the yellow safety cover.
- Do not use the pen if it has been dropped or damaged.
- Do not use the pen if the blue cap is missing or not securely attached.
- Do not re-use a pen.
Storage of PRALUENT - Store unused pens in the refrigerator at 36°F to 46°F (2°C to 8°C) in the original carton to protect from light.
- Do not freeze.
- Do not expose the pen to extreme heat or direct sunlight.
- Do not shake.
- PRALUENT should be allowed to warm to room temperature for 30 to 40 minutes before use.
- If needed, PRALUENT may be kept at room temperature up to 77°F (25°C) for 30 days in the original carton to protect from light. Do not store above 77°F (25°C).
- After PRALUENT is removed from the refrigerator it must be used within 30 days or thrown away.
- Keep the PRALUENT pens and all medicines out of the reach of children.
Keep this leaflet. If you have questions, ask your healthcare provider or call 1-844-PRALUENT (1-844-772-5836).
The parts of the PRALUENT pen are shown in this picture.
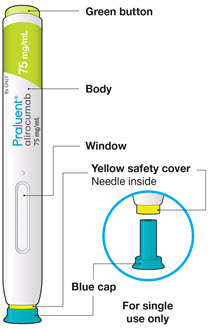
Step A: Getting ready for your injection
Before you start you will need:
- the PRALUENT pen
- 1 alcohol wipe
- 1 cotton ball or gauze
- a puncture-resistant container (see Step B8)
A1: Look at the label on the pen.
- Check that you have the correct product and the correct dose.
- Check the expiration date: do not use if this date has passed.
A2: Look at the window.
- Check the liquid is clear, colorless to pale yellow and free from particles (see Figure A).
- You may see an air bubble. This is normal.
- Do not use if the window appears solid yellow (see Figure B).
- Do not use this medicine if the solution is discolored or cloudy, or if it contains visible flakes or particles.
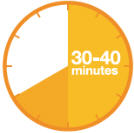
A3: Let the pen warm up at room temperature for 30 to 40 minutes.
- This is important for administering the entire dose and helps minimize discomfort.
- Take PRALUENT out of the refrigerator to warm up before using.
- Do not heat the pen, let it warm up on its own.
- Do not put the pen back in the refrigerator.
A4: Prepare the injection site.
- Wash your hands with soap and water and dry with a towel.
- Clean skin in the injection area with an alcohol wipe.
- You can inject into your (see below picture):
- thighs
- stomach (except for the 2 inch area around your navel)
- upper arms
- You can stand or sit to give yourself an injection.
Important:
- Change (rotate) your injection site each time you give yourself an injection. If you need to use the same injection site, make sure it is not the same spot on the site you used last time.
- Do not inject into areas where the skin is injured, tender, hard, red, or hot. Do not inject PRALUENT into areas with visible veins, scars or stretch marks.
Step B: How to give your injection
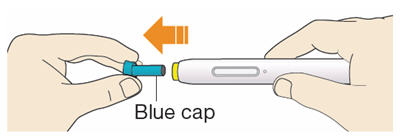
B1: After completing all steps in "Step A: Getting ready for your injection", pull off the blue cap.
- Do not pull off the cap until you are ready to inject.
- Do not put the blue cap back on.
B2: Hold the PRALUENT pen like this.
- Do not touch the yellow safety cover.
- Make sure you can see the window.
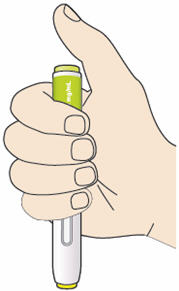
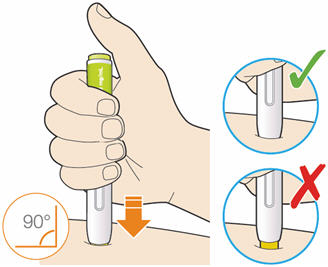
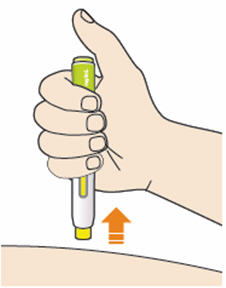
B3: Press the yellow safety cover on your skin at roughly a 90° angle.
- Press and firmly hold the pen against your body until the yellow safety cover is no longer visible. The pen will not work if the yellow safety cover is not depressed fully.
- If needed, pinch the skin to make sure the injection site is firm.
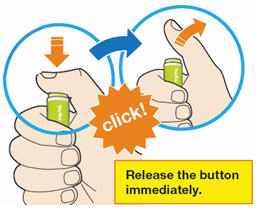
B4: Push and immediately release the green button with your thumb.
- You will hear a click. Your injection has now started.
- The window will start to turn yellow.
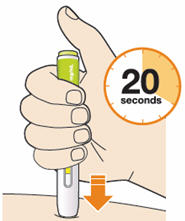
B5: Keep holding the pen against your skin after releasing the button.
- The injection may take up to 20 seconds.
- The time required for injection to give the entire dose may be longer than for other injectable medicines.
B6: Check the window has turned yellow, before removing the pen.
- Do not remove the pen until the entire window has turned yellow.
- Your injection is complete when the window has turned completely yellow, you may hear a second click.
- If the window does not turn completely yellow, call 1-844-772-5836 for help. Do not give yourself a second dose without speaking to your healthcare provider.

B7: Pull pen away from your skin.
- Do not rub the skin after the injection.
- If you see any blood, press a cotton ball or gauze on the site until the bleeding stops.
- Do not put the blue cap back on.
- Throw away pen and cap in a puncture-resistant container immediately after they have been used.
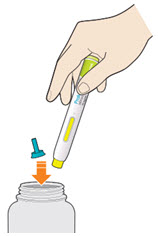
Disposing of used pens:
- Put your used pens in a FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) pens and caps in your household trash.
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
Keep PRALUENT and all medicines out of the reach of children.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Regeneron Pharmaceuticals, Inc.
Tarrytown, NY 10591
U.S. License # 1760PRALUENT is a registered trademark of Regeneron Pharmaceuticals, Inc.
©2020 Regeneron Pharmaceuticals, Inc. All rights reserved.
Revised: April 2020
-
INSTRUCTIONS FOR USE
Instructions For Use
PRALUENT® (PRAHL-u-ent)
(alirocumab)
Injection, for Subcutaneous Injection
Single-Dose Pre-Filled Pen (150 mg/mL)Important Information - The device is a single-dose disposable pen. It contains 150 mg of PRALUENT (alirocumab) in 1 mL.
- The PRALUENT pen contains medicine prescribed by your healthcare provider.
- The medicine is injected under your skin and can be given by yourself or someone else (caregiver).
- It is important that you do not try to give yourself or someone else the injection unless you have received training from your healthcare provider.
- This pen can only be used for 1 single injection, and must be discarded after use.
- Read all of the instructions carefully before using the PRALUENT pen.
- Follow these instructions every time you use a PRALUENT pen.
- Do not touch the yellow safety cover.
- Do not use the pen if it has been dropped or damaged.
- Do not use the pen if the blue cap is missing or not securely attached.
- Do not re-use a pen.
Storage of PRALUENT - Store unused pens in the refrigerator at 36°F to 46°F (2°C to 8°C) in the original carton to protect from light.
- Do not freeze.
- Do not expose the pen to extreme heat or direct sunlight.
- Do not shake.
- PRALUENT should be allowed to warm to room temperature for 30 to 40 minutes before use.
- If needed, PRALUENT may be kept at room temperature up to 77°F (25°C) for 30 days in the original carton to protect from light. Do not store above 77°F (25°C).
- After PRALUENT is removed from the refrigerator it must be used within 30 days or thrown away.
- Keep the PRALUENT pens and all medicines out of the reach of children.
Keep this leaflet. If you have questions, ask your healthcare provider or call 1-844-PRALUENT (1-844-772-5836).
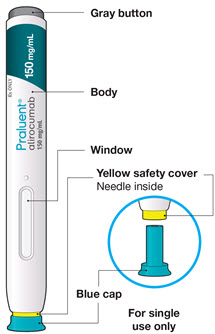
The parts of the PRALUENT pen are shown in this picture.
Step A: Getting ready for your injection
Before you start you will need:
- the PRALUENT pen
- 1 alcohol wipe
- 1 cotton ball or gauze
- a puncture-resistant container (see Step B8)
A1: Look at the label on the pen.
- Check that you have the correct product and the correct dose.
- Check the expiration date: do not use if this date has passed.
A2: Look at the window.
- Check the liquid is clear, colorless to pale yellow and free from particles (see Figure A).
- You may see an air bubble. This is normal.
- Do not use if the window appears solid yellow (see Figure B).
- Do not use this medicine if the solution is discolored or cloudy, or if it contains visible flakes or particles.
A3: Let the pen warm up at room temperature for 30 to 40 minutes.
- This is important for administering the entire dose and helps minimize discomfort.
- Take PRALUENT out of the refrigerator to warm up before using.
- Do not heat the pen, let it warm up on its own.
- Do not put the pen back in the refrigerator.
A4: Prepare the injection site.
- Wash your hands with soap and water and dry with a towel.
- Clean skin in the injection area with an alcohol wipe.
- You can inject into your (see below picture):
- thighs
- stomach (except for the 2 inch area around your navel)
- upper arms
- You can stand or sit to give yourself an injection.
Important:
- Change (rotate) your injection site each time you give yourself an injection. If you need to use the same injection site, make sure it is not the same spot on the site you used last time.
- Do not inject into areas where the skin is injured, tender, hard, red, or hot. Do not inject PRALUENT into areas with visible veins, scars or stretch marks.
Step B: How to give your injection
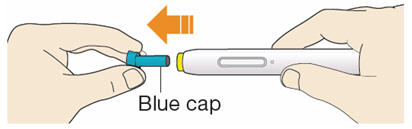
B1: After completing all steps in "Step A: Getting ready for your injection", pull off the blue cap.
- Do not pull off the cap until you are ready to inject.
- Do not put the blue cap back on.
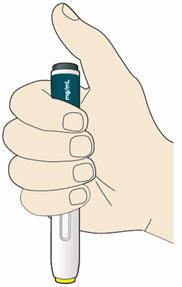
B2: Hold the PRALUENT pen like this.
- Do not touch the yellow safety cover.
- Make sure you can see the window.
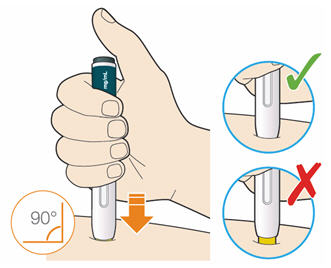
B3: Press the yellow safety cover on your skin at roughly a 90° angle.
- Press and firmly hold the pen against your body until the yellow safety cover is no longer visible. The pen will not work if the yellow safety cover is not depressed fully.
- If needed, pinch the skin to make sure the injection site is firm.
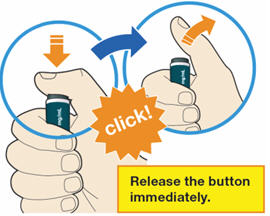
B4: Push and immediately release the gray button with your thumb.
- You will hear a click. Your injection has now started.
- The window will start to turn yellow.
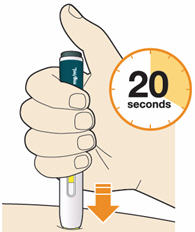
B5: Keep holding the pen against your skin after releasing the button.
- The injection may take up to 20 seconds.
- The time required for injection to give the entire dose may be longer than for other injectable medicines.
B6: Check the window has turned yellow, before removing the pen.
- Do not remove the pen until the entire window has turned yellow.
- Your injection is complete when the window has turned completely yellow, you may hear a second click.
- If the window does not turn completely yellow, call 1-844-772-5836 for help. Do not give yourself a second dose without speaking to your healthcare provider.

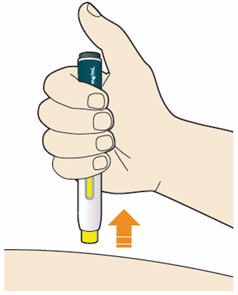
B7: Pull pen away from your skin.
- Do not rub the skin after the injection.
- If you see any blood, press a cotton ball or gauze on the site until the bleeding stops.
- Do not put the blue cap back on.
- Throw away pen and cap in a puncture-resistant container immediately after they have been used.
Disposing of used pens:
- Put your used pens in a FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) pens and caps in your household trash.
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
Keep PRALUENT and all medicines out of the reach of children.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Regeneron Pharmaceuticals, Inc.
Tarrytown, NY 10591
U.S. License # 1760PRALUENT is a registered trademark of Regeneron Pharmaceuticals, Inc.
©2020 Regeneron Pharmaceuticals, Inc. All rights reserved.
Revised: April 2020
-
INSTRUCTIONS FOR USE
Instructions For Use
PRALUENT® (PRAHL-u-ent)
(alirocumab)
Injection, for Subcutaneous Injection
Single-Dose Pre-Filled Syringe (75 mg/mL)Important Information - The device is a single-dose pre-filled syringe. It contains 75 mg of PRALUENT (alirocumab) in 1 mL.
- The PRALUENT syringe contains medicine prescribed by your healthcare provider.
- The medicine is injected under your skin and can be given by yourself or someone else (caregiver).
- It is important that you do not try to give yourself or someone else the injection unless you have received training from your healthcare provider.
- This syringe can only be used for 1 single injection, and must be discarded after use.
- Read all of the instructions carefully before using the PRALUENT syringe.
- Follow these instructions every time you use a PRALUENT syringe.
- Do not touch the needle.
- Do not use the syringe if it has been dropped or damaged.
- Do not use the syringe if the gray needle cap is missing or not securely attached.
- Do not re-use a syringe.
Storage of PRALUENT - Store unused syringes in the refrigerator at 36°F to 46°F (2°C to 8°C) in the original carton to protect from light.
- Do not freeze.
- Do not expose the syringe to extreme heat or direct sunlight.
- Do not shake.
- PRALUENT should be allowed to warm to room temperature for 30 to 40 minutes before use.
- If needed, PRALUENT may be kept at room temperature up to 77°F (25°C) for 30 days in the original carton to protect from light. Do not store above 77°F (25°C).
- After PRALUENT is removed from the refrigerator it must be used within 30 days or thrown away.
- Keep the PRALUENT syringes and all medicines out of the reach of children.
Keep this leaflet. If you have questions, ask your healthcare provider or call 1-844-PRALUENT (1-844-772-5836).
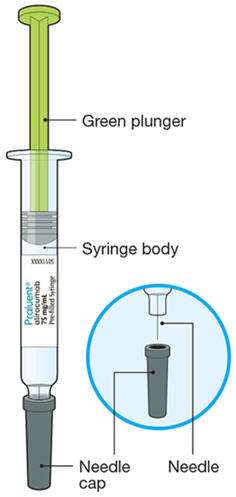
The parts of the PRALUENT syringe are shown in this picture.
Step A: Getting ready for your injection
Before you start you will need:
- the PRALUENT syringe
- 1 alcohol wipe
- 1 cotton ball or gauze
- a puncture-resistant container (see Step B6)
A1: Before you start.
- Take the syringe out of the packaging by holding the syringe body.
- Check the expiration date: do not use if this date has passed.
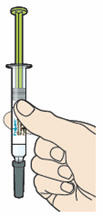
A2: Look at the label on the syringe.
- Check that you have the correct product and the correct dose (green plunger for 75 mg/mL).
- Check the liquid is clear, colorless to pale yellow and free from particles .
- Check that the syringe is not open or damaged.
- Do not use this medicine if the solution is discolored or cloudy, or if it contains visible flakes or particles.
A3: Let the syringe warm up at room temperature for 30 to 40 minutes.
- This is important for administering the entire dose and helps minimize discomfort.
- Take PRALUENT out of the refrigerator to warm up before using.
- Do not heat the syringe, let it warm up on its own.
- Do not put the syringe back in the refrigerator.
A4: Prepare the injection site.
- Wash your hands with soap and water and dry with a towel.
- Clean skin in the injection area with an alcohol wipe.
- You can inject into your (see below picture):
- thighs
- stomach (except for the 2 inch area around your navel)
- upper arms
- You can stand or sit to give yourself an injection.
Important:
- Change (rotate) your injection site each time you give yourself an injection. If you need to use the same injection site, make sure it is not the same spot on the site you used last time.
- Do not inject into areas where the skin is injured, tender, hard, red, or hot. Do not inject PRALUENT into areas with visible veins, scars or stretch marks.
Step B: How to give your injection
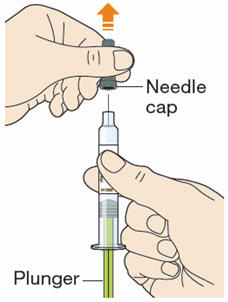
B1: After completing all steps in "Step A: Getting ready for your injection", pull off the needle cap.
- Do not pull off the cap until you are ready to inject.
- Hold the syringe in the middle of the syringe body with the needle pointing away from you.
- Keep your hand away from the plunger.
- Do not try to remove any air bubbles in the syringe before the injection.
- Do not put the gray cap back on the needle.
B2: If needed pinch the skin.
- Use your thumb and first finger to pinch a fold of skin at the injection site.
- Hold the skin like this during the entire injection.
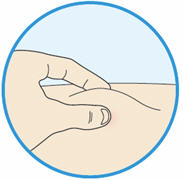
B3: Insert the needle into the fold of skin with a quick dart-like motion.
- Inject the medicine at a 90° angle if you can pinch 2 inches of skin.
- Inject the medicine at a 45° angle if you can only pinch 1 inch of skin.
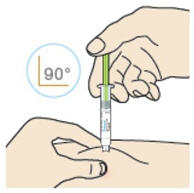
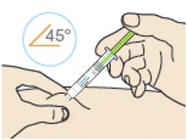
B4: Push the plunger down.
- Inject all of the medicine by slowly and steadily pushing down the plunger.
- You may have to push harder on the plunger than for other injectable medicines.
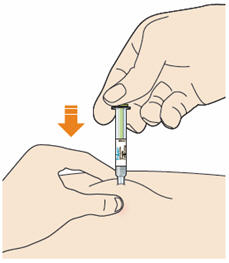
B5: Before you remove the needle check the syringe is empty.
- The time required for injection to give the entire dose may be longer than for other injectable medicines.
- Do not remove the syringe until it is completely empty.
- Pull the needle out of the skin at the same angle as it was inserted.
- Do not rub the skin after the injection.
- If you see any blood, press a cotton ball or gauze on the site until the bleeding stops.
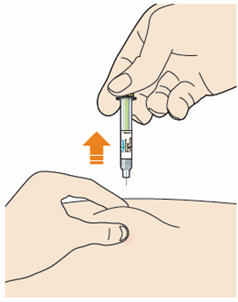
- Do not put the gray needle cap back on.
- Do not re-use the syringe.
- Throw away syringe and cap in a puncture-resistant container immediately after they have been used.
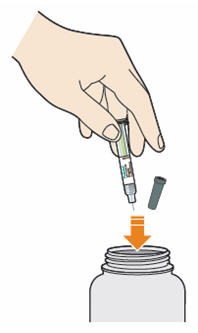
Disposing of used prefilled syringes and needles:
- Put your used needles and syringes in a FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles and syringes in your household trash.
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
Keep PRALUENT and all medicines out of the reach of children.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Regeneron Pharmaceuticals, Inc.
Tarrytown, NY 10591
U.S. License # 1760PRALUENT is a registered trademark of Regeneron Pharmaceuticals, Inc.
©2020 Regeneron Pharmaceuticals, Inc. All rights reserved.
Revised: April 2020
-
INSTRUCTIONS FOR USE
Instructions For Use
PRALUENT® (PRAHL-u-ent)
(alirocumab)
Injection, for Subcutaneous Injection
Single-Dose Pre-Filled Syringe (150 mg/mL)Important Information - The device is a single-dose pre-filled syringe. It contains 150 mg of PRALUENT (alirocumab) in 1 mL.
- The PRALUENT syringe contains medicine prescribed by your healthcare provider.
- The medicine is injected under your skin and can be given by yourself or someone else (caregiver).
- It is important that you do not try to give yourself or someone else the injection unless you have received training from your healthcare provider.
- This syringe can only be used for 1 single injection, and must be discarded after use.
- Read all of the instructions carefully before using the PRALUENT syringe.
- Follow these instructions every time you use a PRALUENT syringe.
- Do not touch the needle.
- Do not use the syringe if it has been dropped or damaged.
- Do not use the syringe if the gray needle cap is missing or not securely attached.
- Do not re-use a syringe.
Storage of PRALUENT - Store unused syringes in the refrigerator at 36°F to 46°F (2°C to 8°C) in the original carton to protect from light.
- Do not freeze.
- Do not expose the syringe to extreme heat or direct sunlight.
- Do not shake.
- PRALUENT should be allowed to warm to room temperature for 30 to 40 minutes before use.
- If needed, PRALUENT may be kept at room temperature up to 77°F (25°C) for 30 days in the original carton to protect from light. Do not store above 77°F (25°C).
- After PRALUENT is removed from the refrigerator it must be used within 30 days or thrown away.
- Keep the PRALUENT syringes and all medicines out of the reach of children.
Keep this leaflet. If you have questions, ask your healthcare provider or call 1-844-PRALUENT (1-844-772-5836).
The parts of the PRALUENT syringe are shown in this picture.
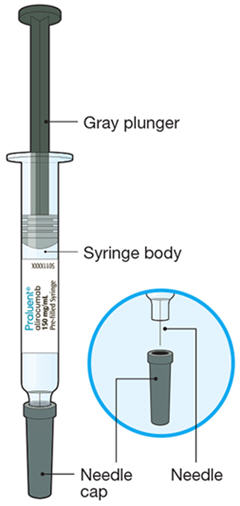
Step A: Getting ready for your injection
Before you start you will need:
- the PRALUENT syringe
- 1 alcohol wipe
- 1 cotton ball or gauze
- a puncture-resistant container (see Step B6)
A1: Before you start.
- Take the syringe out of the packaging by holding the syringe body.
- Check the expiration date: do not use if this date has passed.
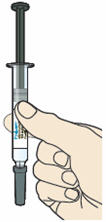
A2: Look at the label on the syringe.
- Check that you have the correct product and the correct dose (gray plunger for 150 mg/mL).
- Check the liquid is clear, colorless to pale yellow and free from particles.
- Check that the syringe is not open or damaged.
- Do not use this medicine if the solution is discolored or cloudy, or if it contains visible flakes or particles.
A3: Let the syringe warm up at room temperature for 30 to 40 minutes.
- This is important for administering the entire dose and helps minimize discomfort.
- Take PRALUENT out of the refrigerator to warm up before using.
- Do not heat the syringe, let it warm up on its own.
- Do not put the syringe back in the refrigerator.
A4: Prepare the injection site.
- Wash your hands with soap and water and dry with a towel.
- Clean skin in the injection area with an alcohol wipe.
- You can inject into your (see below picture):
- thighs
- stomach (except for the 2 inch area around your navel)
- upper arms
- You can stand or sit to give yourself an injection.
Important:
- Change (rotate) your injection site each time you give yourself an injection. If you need to use the same injection site, make sure it is not the same spot on the site you used last time.
- Do not inject into areas where the skin is injured, tender, hard, red, or hot. Do not inject PRALUENT into areas with visible veins, scars or stretch marks.
Step B: How to give your injection
B1: After completing all steps in "Step A: Getting ready for your injection", pull off the needle cap.
- Do not pull off the cap until you are ready to inject.
- Hold the syringe in the middle of the syringe body with the needle pointing away from you.
- Keep your hand away from the plunger.
- Do not try to remove any air bubbles in the syringe before the injection.
- Do not put the gray cap back on the needle.
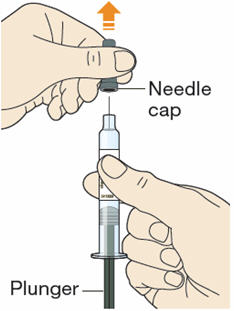
B2: If needed pinch the skin.
- Use your thumb and first finger to pinch a fold of skin at the injection site.
- Hold the skin like this during the entire injection.
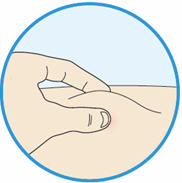
B3: Insert the needle into the fold of skin with a quick dart-like motion.
- Inject the medicine at a 90° angle if you can pinch 2 inches of skin.
- Inject the medicine at a 45° angle if you can only pinch 1 inch of skin.
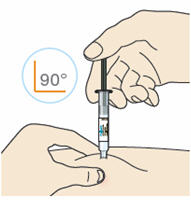
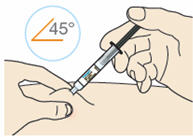
B4: Push the plunger down.
- Inject all of the medicine by slowly and steadily pushing down the plunger.
- You may have to push harder on the plunger than for other injectable medicines.
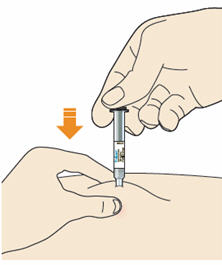
B5: Before you remove the needle check the syringe is empty.
- The time required for injection to give the entire dose may be longer than for other injectable medicines.
- Do not remove the syringe until it is completely empty.
- Pull the needle out of the skin at the same angle as it was inserted.
- Do not rub the skin after the injection.
- If you see any blood, press a cotton ball or gauze on the site until the bleeding stops.
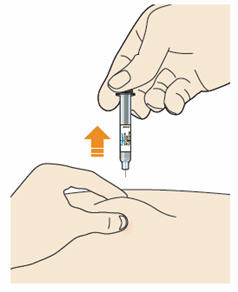
- Do not put the gray needle cap back on.
- Do not re-use the syringe.
- Throw away syringe and cap in a puncture-resistant container immediately after they have been used.
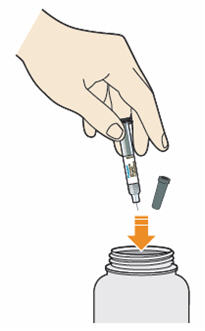
Disposing of used prefilled syringes and needles:
- Put your used needles and syringes in a FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles and syringes in your household trash.
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
Keep PRALUENT and all medicines out of the reach of children.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Regeneron Pharmaceuticals, Inc.
Tarrytown, NY 10591
U.S. License # 1760PRALUENT is a registered trademark of Regeneron Pharmaceuticals, Inc.
©2020 Regeneron Pharmaceuticals, Inc. All rights reserved.
Revised: April 2020
-
PRINCIPAL DISPLAY PANEL - 75 mg/mL Pen Carton
NDC: 72733-5901-2
Rx ONLYPraluent®
alirocumab
Injection
75 mg/mLTwo Pre-filled Pens
For subcutaneous injection only. Single-dose.
Carton contains: Two single-dose pre-filled pens, the Package Insert,
Patient Information, and Instructions for Use.75 mg/mL
OPEN
HEREREGENERON
SANOFI

-
PRINCIPAL DISPLAY PANEL - 150 mg/mL Pen Carton
NDC: 72733-5902-2
Rx ONLYPraluent®
alirocumab
Injection
150 mg/mLTwo Pre-filled Pens
For subcutaneous injection only. Single-dose.
Carton contains: Two single-dose pre-filled pens, the Package Insert,
Patient Information, and Instructions for Use.150 mg/mL
OPEN
HEREREGENERON
SANOFI

-
INGREDIENTS AND APPEARANCE
PRALUENT
alirocumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72733-5901 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ALIROCUMAB (UNII: PP0SHH6V16) (ALIROCUMAB - UNII:PP0SHH6V16) ALIROCUMAB 75 mg in 1 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) 8 mmol in 1 mL SUCROSE (UNII: C151H8M554) 100 mg in 1 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.1 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72733-5901-2 2 in 1 CARTON 02/15/2019 1 1 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125559 02/15/2019 PRALUENT
alirocumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72733-5902 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ALIROCUMAB (UNII: PP0SHH6V16) (ALIROCUMAB - UNII:PP0SHH6V16) ALIROCUMAB 150 mg in 1 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) 6 mmol in 1 mL SUCROSE (UNII: C151H8M554) 100 mg in 1 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.1 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72733-5902-2 2 in 1 CARTON 02/15/2019 1 1 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125559 02/15/2019 Labeler - Sanofi US Corporation (116902801) Registrant - Sanofi-Aventis U.S. LLC (824676584) Establishment Name Address ID/FEI Business Operations Regeneron Pharmaceuticals, Inc. 945589711 ANALYSIS(72733-5901, 72733-5902) , API MANUFACTURE(72733-5901, 72733-5902) Establishment Name Address ID/FEI Business Operations Sanofi Chimie 291592785 ANALYSIS(72733-5901, 72733-5902) , API MANUFACTURE(72733-5901, 72733-5902) Establishment Name Address ID/FEI Business Operations Regeneron Ireland Unlimited Company (Raheen) 985528196 ANALYSIS(72733-5901, 72733-5902) , API MANUFACTURE(72733-5901, 72733-5902) Establishment Name Address ID/FEI Business Operations Sanofi-Aventis Deutschland GmbH 313218430 ANALYSIS(72733-5901, 72733-5902) , LABEL(72733-5901, 72733-5902) , MANUFACTURE(72733-5901, 72733-5902) , PACK(72733-5901, 72733-5902) Establishment Name Address ID/FEI Business Operations Genzyme Ireland Limited 985127419 ANALYSIS(72733-5901, 72733-5902) , LABEL(72733-5901, 72733-5902) , MANUFACTURE(72733-5901, 72733-5902) , PACK(72733-5901, 72733-5902)
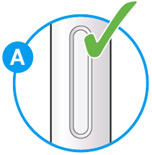
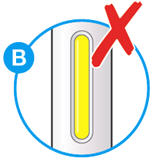
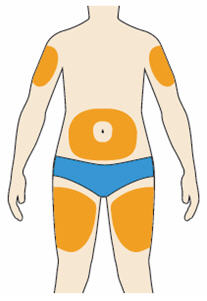
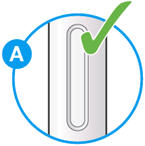
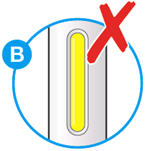
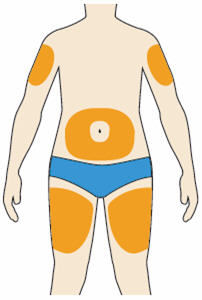
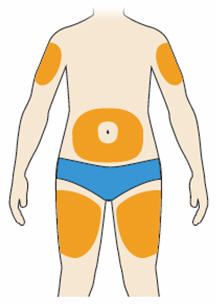
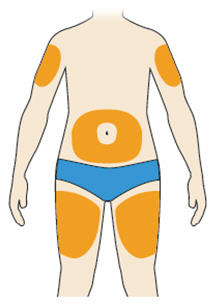
Trademark Results [Praluent]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 PRALUENT 86318389 4659691 Live/Registered |
SANOFI BIOTECHNOLOGY 2014-06-24 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.