NESINA- alogliptin tablet, film coated
Nesina by
Drug Labeling and Warnings
Nesina by is a Prescription medication manufactured, distributed, or labeled by Takeda Pharmaceuticals America, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NESINA safely and effectively. See full prescribing information for NESINA.
NESINA (alogliptin) tablets, for oral use
Initial U.S. Approval: 2013INDICATIONS AND USAGE
NESINA is a dipeptidyl peptidase-4 (DPP-4) inhibitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. (1.1, 14)
Important Limitations of Use: Not for treatment of type 1 diabetes or diabetic ketoacidosis. (1.1)
DOSAGE AND ADMINISTRATION
- The recommended dose in patients with normal renal function or mild renal impairment is 25 mg once daily. (2.1)
- Can be taken with or without food. (2.1)
- Adjust dose if moderate or severe renal impairment or end-stage renal disease (ESRD). (2.2)
Degree of Renal Impairment Creatinine Clearance (mL/min) Recommended Dosing Moderate ≥30 to <60 12.5 mg once daily Severe/ESRD <30 6.25 mg once daily DOSAGE FORMS AND STRENGTHS
Tablets: 25 mg, 12.5 mg and 6.25 mg (3)
CONTRAINDICATIONS
History of a serious hypersensitivity reaction to alogliptin-containing products, such as anaphylaxis, angioedema or severe cutaneous adverse reactions. (4)
WARNINGS AND PRECAUTIONS
- Acute pancreatitis: There have been postmarketing reports of acute pancreatitis. If pancreatitis is suspected, promptly discontinue NESINA. (5.1)
- Heart failure: Consider the risks and benefits of NESINA prior to initiating treatment in patients at risk for heart failure. If heart failure develops, evaluate and manage according to current standards of care and consider discontinuation of NESINA (5.2).
- Hypersensitivity: There have been postmarketing reports of serious hypersensitivity reactions in patients treated with NESINA such as anaphylaxis, angioedema and severe cutaneous adverse reactions, including Stevens-Johnson syndrome. In such cases, promptly discontinue NESINA, assess for other potential causes, institute appropriate monitoring and treatment and initiate alternative treatment for diabetes. (5.3)
- Hepatic effects: Postmarketing reports of hepatic failure, sometimes fatal. Causality cannot be excluded. If liver injury is detected, promptly interrupt NESINA and assess patient for probable cause, then treat cause if possible, to resolution or stabilization. Do not restart NESINA if liver injury is confirmed and no alternative etiology can be found. (5.4)
- Hypoglycemia: When an insulin secretagogue (e.g., sulfonylurea) or insulin is used in combination with NESINA, a lower dose of the insulin secretagogue or insulin may be required to minimize the risk of hypoglycemia. (5.5)
- Arthralgia: Severe and disabling arthralgia has been reported in patients taking DPP-4 inhibitors. Consider as a possible cause for severe joint pain and discontinue drug if appropriate. (5.6)
- Bullous pemphigoid: There have been postmarketing reports of bullous pemphigoid requiring hospitalization in patients taking DPP-4 inhibitors. Tell patients to report development of blisters or erosions. If bullous pemphigoid is suspected, discontinue NESINA. (5.7)
- Macrovascular outcomes: There have been no clinical studies establishing conclusive evidence of macrovascular risk reduction with NESINA or any other antidiabetic drug. (5.8)
ADVERSE REACTIONS
The most common adverse reactions (4% or greater incidence) are nasopharyngitis, headache and upper respiratory tract infection. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Takeda Pharmaceuticals at 1-877-TAKEDA-7 (1-877-825-3327) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Monotherapy and Combination Therapy
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
2.2 Patients with Renal Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Pancreatitis
5.2 Heart Failure
5.3 Hypersensitivity Reactions
5.4 Hepatic Effects
5.5 Use with Medications Known to Cause Hypoglycemia
5.6 Severe and Disabling Arthralgia
5.7 Bullous Pemphigoid
5.8 Macrovascular Outcomes
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Patients with Inadequate Glycemic Control on Diet and Exercise
14.2 Combination Therapy
14.3 Cardiovascular Safety Trial
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Monotherapy and Combination Therapy
NESINA is indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus [see Clinical Studies (14)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
The recommended dose of NESINA is 25 mg once daily. NESINA may be taken with or without food.
2.2 Patients with Renal Impairment
No dose adjustment of NESINA is necessary for patients with mild renal impairment (creatinine clearance [CrCl] ≥60 mL/min).
The dose of NESINA is 12.5 mg once daily for patients with moderate renal impairment (CrCl ≥30 to <60 mL/min).
The dose of NESINA is 6.25 mg once daily for patients with severe renal impairment (CrCl ≥15 to <30 mL/min) or with end-stage renal disease (ESRD) (CrCl <15 mL/min or requiring hemodialysis). NESINA may be administered without regard to the timing of dialysis. NESINA has not been studied in patients undergoing peritoneal dialysis [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
Because there is a need for dose adjustment based upon renal function, assessment of renal function is recommended prior to initiation of NESINA therapy and periodically thereafter.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Pancreatitis
Acute pancreatitis has been reported in the postmarketing setting and in randomized clinical trials. In glycemic control trials in patients with type 2 diabetes, acute pancreatitis was reported in 6 (0.2%) patients treated with NESINA 25 mg and 2 (<0.1%) patients treated with active comparators or placebo. In the EXAMINE trial (a cardiovascular outcomes trial of patients with type 2 diabetes and high cardiovascular (CV) risk), acute pancreatitis was reported in 10 (0.4%) of patients treated with NESINA and in 7 (0.3%) of patients treated with placebo.
It is unknown whether patients with a history of pancreatitis are at increased risk for pancreatitis while using NESINA.
After initiation of NESINA, patients should be observed for signs and symptoms of pancreatitis. If pancreatitis is suspected, NESINA should promptly be discontinued and appropriate management should be initiated.
5.2 Heart Failure
In the EXAMINE trial which enrolled patients with type 2 diabetes and recent acute coronary syndrome, 106 (3.9%) of patients treated with NESINA and 89 (3.3%) of patients treated with placebo were hospitalized for congestive heart failure.
Consider the risks and benefits of NESINA prior to initiating treatment in patients at risk for heart failure, such as those with a prior history of heart failure and a history of renal impairment, and observe these patients for signs and symptoms of heart failure during therapy. Patients should be advised of the characteristic symptoms of heart failure and should be instructed to immediately report such symptoms. If heart failure develops, evaluate and manage according to current standards of care and consider discontinuation of NESINA.
5.3 Hypersensitivity Reactions
There have been postmarketing reports of serious hypersensitivity reactions in patients treated with NESINA. These reactions include anaphylaxis, angioedema and severe cutaneous adverse reactions, including Stevens-Johnson syndrome. If a serious hypersensitivity reaction is suspected, discontinue NESINA, assess for other potential causes for the event and institute alternative treatment for diabetes [see Adverse Reactions (6.2)]. Use caution in patients with a history of angioedema with another dipeptidyl peptidase-4 (DPP-4) inhibitor because it is unknown whether such patients will be predisposed to angioedema with NESINA.
5.4 Hepatic Effects
There have been postmarketing reports of fatal and nonfatal hepatic failure in patients taking NESINA, although some of the reports contain insufficient information necessary to establish the probable cause [see Adverse Reactions (6.2)].
In glycemic control trials in patients with type 2 diabetes, serum alanine aminotransferase (ALT) elevations greater than three times the upper limit of normal (ULN) were reported in 1.3% of patients treated with NESINA 25 mg and 1.7% of patients treated with active comparators or placebo. In the EXAMINE trial (a cardiovascular outcomes trial of patients with type 2 diabetes and high cardiovascular (CV) risk), increases in serum alanine aminotransferase three times the upper limit of the reference range occurred in 2.4% of patients treated with NESINA and in 1.8% of patients treated with placebo.
Measure liver tests promptly in patients who report symptoms that may indicate liver injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine or jaundice. In this clinical context, if the patient is found to have clinically significant liver enzyme elevations and if abnormal liver tests persist or worsen, NESINA should be interrupted and investigation done to establish the probable cause. NESINA should not be restarted in these patients without another explanation for the liver test abnormalities.
5.5 Use with Medications Known to Cause Hypoglycemia
Insulin and insulin secretagogues, such as sulfonylureas, are known to cause hypoglycemia. Therefore, a lower dose of insulin or insulin secretagogue may be required to minimize the risk of hypoglycemia when used in combination with NESINA.
5.6 Severe and Disabling Arthralgia
There have been postmarketing reports of severe and disabling arthralgia in patients taking DPP-4 inhibitors. The time to onset of symptoms following initiation of drug therapy varied from one day to years. Patients experienced relief of symptoms upon discontinuation of the medication. A subset of patients experienced a recurrence of symptoms when restarting the same drug or a different DPP-4 inhibitor. Consider DPP-4 inhibitors as a possible cause for severe joint pain and discontinue drug if appropriate.
5.7 Bullous Pemphigoid
Postmarketing cases of bullous pemphigoid requiring hospitalization have been reported with DPP-4 inhibitor use. In reported cases, patients typically recovered with topical or systemic immunosuppressive treatment and discontinuation of the DPP-4 inhibitor. Tell patients to report development of blisters or erosions while receiving NESINA. If bullous pemphigoid is suspected, NESINA should be discontinued and referral to a dermatologist should be considered for diagnosis and appropriate treatment.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described below or elsewhere in the prescribing information:
- Pancreatitis [see Warnings and Precautions (5.1)]
- Heart Failure [see Warnings and Precautions (5.2)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.3)]
- Hepatic Effects [see Warnings and Precautions (5.4)]
- Severe and Disabling Arthralgia [see Warnings and Precautions (5.6)]
- Bullous Pemphigoid [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 14,778 patients with type 2 diabetes participated in 14 randomized, double-blind, controlled clinical trials of whom 9052 subjects were treated with NESINA, 3469 subjects were treated with placebo and 2257 were treated with an active comparator. The mean duration of diabetes was seven years, the mean body mass index (BMI) was 31 kg/m2 (49% of patients had a BMI ≥30 kg/m2), and the mean age was 58 years (26% of patients ≥65 years of age). The mean exposure to NESINA was 49 weeks with 3348 subjects treated for more than one year.
In a pooled analysis of these 14 controlled clinical trials, the overall incidence of adverse reactions was 73% in patients treated with NESINA 25 mg compared to 75% with placebo and 70% with active comparator. Overall discontinuation of therapy due to adverse reactions was 6.8% with NESINA 25 mg compared to 8.4% with placebo or 6.2% with active comparator.
Adverse reactions reported in ≥4% of patients treated with NESINA 25 mg and more frequently than in patients who received placebo are summarized in Table 1.
Table 1. Adverse Reactions Reported in ≥4% Patients Treated with NESINA 25 mg and More Frequently Than in Patients Given Placebo in Pooled Studies Number of Patients (%) NESINA 25 mg Placebo Active Comparator N=6447 N=3469 N=2257 Nasopharyngitis 309 (4.8) 152 (4.4) 113 (5.0) Upper Respiratory Tract Infection 287 (4.5) 121 (3.5) 113 (5.0) Headache 278 (4.3) 101 (2.9) 121 (5.4) Hypoglycemia
Hypoglycemic events were documented based upon a blood glucose value and/or clinical signs and symptoms of hypoglycemia.
In the monotherapy study, the incidence of hypoglycemia was 1.5% in patients treated with NESINA compared to 1.6% with placebo. The use of NESINA as add-on therapy to glyburide or insulin did not increase the incidence of hypoglycemia compared to placebo. In a monotherapy study comparing NESINA to a sulfonylurea in elderly patients, the incidence of hypoglycemia was 5.4% with NESINA compared to 26% with glipizide (Table 2).
Table 2. Incidence and Rate of Hypoglycemia* in Placebo and Active-Controlled Studies when NESINA Was Used as Add-On Therapy to Glyburide, Insulin, Metformin, Pioglitazone or Compared to Glipizide or Metformin - * Adverse reactions of hypoglycemia were based on all reports of symptomatic and asymptomatic hypoglycemia; a concurrent glucose measurement was not required; intent-to-treat population.
- † Severe events of hypoglycemia were defined as those events requiring medical assistance or exhibiting depressed level or loss of consciousness or seizure.
Add-On to Glyburide
(26 Weeks)NESINA 25 mg Placebo N=198 N=99 Overall (%) 19 (9.6) 11 (11.1) Severe (%)† 0 1 (1) Add-On to Insulin (± Metformin)
(26 Weeks)NESINA 25 mg Placebo N=129 N=129 Overall (%) 35 (27) 31 (24) Severe (%)† 1 (0.8) 2 (1.6) Add-On to Metformin
(26 Weeks)NESINA 25 mg Placebo N=207 N=104 Overall (%) 0 3 (2.9) Severe (%)† 0 0 Add-On to Pioglitazone (± Metformin or Sulfonylurea)
(26 Weeks)NESINA 25 mg Placebo N=199 N=97 Overall (%) 14 (7.0) 5 (5.2) Severe (%)† 0 1 (1) Compared to Glipizide
(52 Weeks)NESINA 25 mg Glipizide N=222 N=219 Overall (%) 12 (5.4) 57 (26) Severe (%)† 0 3 (1.4) Compared to Metformin
(26 Weeks)NESINA 25 mg Metformin 500 mg twice daily N=112 N=109 Overall (%) 2 (1.8) 2 (1.8) Severe (%)† 0 0 Add-On to Metformin Compared to Glipizide
(52 Weeks)NESINA 25 mg Glipizide N=877 N=869 Overall (%) 12 (1.4) 207 (23.8) Severe (%)† 0 4 (0.5) In the EXAMINE trial, the incidence of investigator reported hypoglycemia was 6.7% in patients receiving NESINA and 6.5% in patients receiving placebo. Serious adverse reactions of hypoglycemia were reported in 0.8% of patients treated with NESINA and in 0.6% of patients treated with placebo.
Renal Impairment
In glycemic control trials in patients with type 2 diabetes, 3.4% of patients treated with NESINA and 1.3% of patients treated with placebo had renal function adverse reactions. The most commonly reported adverse reactions were renal impairment (0.5% for NESINA and 0.1% for active comparators or placebo), decreased creatinine clearance (1.6% for NESINA and 0.5% for active comparators or placebo) and increased blood creatinine (0.5% for NESINA and 0.3% for active comparators or placebo) [see Use in Specific Populations (8.6)].
In the EXAMINE trial of high CV risk type 2 diabetes patients, 23% of patients treated with NESINA and 21% of patients treated with placebo had an investigator reported renal impairment adverse reaction. The most commonly reported adverse reactions were renal impairment (7.7% for NESINA and 6.7% for placebo), decreased glomerular filtration rate (4.9% for NESINA and 4.3% for placebo) and decreased renal clearance (2.2% for NESINA and 1.8% for placebo). Laboratory measures of renal function were also assessed. Estimated glomerular filtration rate decreased by 25% or more in 21.1% of patients treated with NESINA and 18.7% of patients treated with placebo. Worsening of chronic kidney disease stage was seen in 16.8% of patients treated with NESINA and in 15.5% of patients treated with placebo.
6.2 Postmarketing Experience
The following adverse reactions have been identified during the postmarketing use of NESINA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Acute pancreatitis, hypersensitivity reactions including anaphylaxis, angioedema, rash, urticaria and severe cutaneous adverse reactions, including Stevens-Johnson syndrome, hepatic enzyme elevations, fulminant hepatic failure, severe and disabling arthralgia, bullous pemphigoid, rhabdomyolysis, and diarrhea, constipation, nausea, and ileus [see Warnings and Precautions (5.1, 5.3, 5.4, 5.6, 5.7)].
-
7 DRUG INTERACTIONS
NESINA is primarily renally excreted. Cytochrome (CYP) P450-related metabolism is negligible. No significant drug-drug interactions were observed with the CYP-substrates or inhibitors tested or with renally excreted drugs [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Limited data with NESINA in pregnant women are not sufficient to determine a drug-associated risk for major birth defects or miscarriage. There are risks to the mother and fetus associated with poorly controlled diabetes in pregnancy [see Clinical Considerations].
No adverse developmental effects were observed when alogliptin was administered to pregnant rats and rabbits during organogenesis at exposures 180- and 149-times the 25 mg clinical dose, respectively, based on plasma drug exposure (AUC) [see Data].
The estimated background risk of major birth defects is 6-10% in women with pre-gestational diabetes with a HbA1c >7 and has been reported to be as high as 20-25% in women with HbA1c >10. The estimated background risk of miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Poorly controlled diabetes in pregnancy increases the maternal risk for diabetic ketoacidosis, pre-eclampsia, spontaneous abortions, preterm delivery, still birth and delivery complications. Poorly controlled diabetes increases the fetal risk for major birth defects, still birth, and macrosomia related morbidity.
Data
Animal Data
Alogliptin administered to pregnant rabbits and rats during the period of organogenesis did not cause adverse developmental effects at doses of up to 200 mg/kg and 500 mg/kg, or 149 times and 180 times, the 25 mg clinical dose, respectively, based on plasma drug exposure (AUC). Placental transfer of alogliptin into the fetus was observed following oral dosing to pregnant rats.
No adverse developmental outcomes were observed in offspring when alogliptin was administered to pregnant rats during gestation and lactation at doses up to 250 mg/kg (~ 95 times the 25 mg clinical dose, based on AUC).
8.2 Lactation
Risk Summary
There is no information regarding the presence of alogliptin in human milk, the effects on the breastfed infant, or the effects on milk production. Alogliptin is present in rat milk: however, due to species specific differences in lactation physiology, animal lactation data may not reliably predict levels in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for NESINA and any potential adverse effects on the breastfed infant from NESINA or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of NESINA in pediatric patients have not been established.
8.5 Geriatric Use
Of the total number of patients (N=9052) in clinical safety and efficacy studies treated with NESINA, 2257 (24.9%) patients were 65 years and older and 386 (4.3%) patients were 75 years and older. No overall differences in safety or effectiveness were observed between patients 65 years and over and younger patients. While this clinical experience has not identified differences in responses between the elderly and younger patients, greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
A total of 602 patients with moderate renal impairment (eGFR ≥30 and <60 mL/min/1.73 m2) and 4 patients with severe renal impairment/end-stage renal disease (eGFR <30 mL/min/1.73 m2 or <15 mL/min/1.73 m2, respectively) at baseline were treated with NESINA in clinical trials in patients with type 2 diabetes. Reductions in HbA1c were generally similar in this subgroup of patients. The overall incidence of adverse reactions was generally balanced between NESINA and placebo treatments in this subgroup of patients.
In the EXAMINE trial of high CV risk type 2 diabetes patients, 694 patients had moderate renal impairment and 78 patients had severe renal impairment or end-stage renal disease at baseline. The overall incidences of adverse reactions, serious adverse reactions and adverse reactions leading to study drug discontinuation were generally similar between the treatment groups.
8.7 Hepatic Impairment
No dose adjustments are required in patients with mild to moderate hepatic impairment (Child-Pugh Grade A and B) based on insignificant change in systemic exposures (e.g., AUC) compared to subjects with normal hepatic function in a pharmacokinetic study. NESINA has not been studied in patients with severe hepatic impairment (Child-Pugh Grade C). Use caution when administering NESINA to patients with liver disease [see Warnings and Precautions (5.3)].
-
10 OVERDOSAGE
The highest doses of NESINA administered in clinical trials were single doses of 800 mg to healthy subjects and doses of 400 mg once daily for 14 days to patients with type 2 diabetes (equivalent to 32 times and 16 times the maximum recommended clinical dose of 25 mg, respectively). No serious adverse reactions were observed at these doses.
In the event of an overdose, it is reasonable to institute the necessary clinical monitoring and supportive therapy as dictated by the patient's clinical status. Per clinical judgment, it may be reasonable to initiate removal of unabsorbed material from the gastrointestinal tract.
Alogliptin is minimally dialyzable; over a three-hour hemodialysis session, approximately 7% of the drug was removed. Therefore, hemodialysis is unlikely to be beneficial in an overdose situation. It is not known if NESINA is dialyzable by peritoneal dialysis.
-
11 DESCRIPTION
NESINA tablets contain the active ingredient alogliptin, which is a selective, orally bioavailable inhibitor of the enzymatic activity of dipeptidyl peptidase-4 (DPP-4).
Chemically, alogliptin is prepared as a benzoate salt, which is identified as 2-({6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl}methyl)benzonitrile monobenzoate. It has a molecular formula of C18H21N5O2C7H6O2 and a molecular weight of 461.51 daltons. The structural formula is:
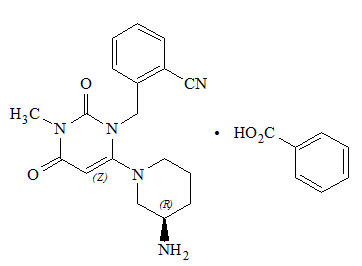
Alogliptin benzoate is a white to off-white crystalline powder containing one asymmetric carbon in the aminopiperidine moiety. It is soluble in dimethylsulfoxide, sparingly soluble in water and methanol, slightly soluble in ethanol and very slightly soluble in octanol and isopropyl acetate.
Each NESINA tablet contains 34 mg, 17 mg or 8.5 mg alogliptin benzoate, which is equivalent to 25 mg, 12.5 mg or 6.25 mg, respectively, of alogliptin and the following inactive ingredients: mannitol, microcrystalline cellulose, hydroxypropyl cellulose, croscarmellose sodium and magnesium stearate. In addition, the film coating contains the following inactive ingredients: hypromellose, titanium dioxide, ferric oxide (red or yellow) and polyethylene glycol, and is marked with printing ink (Gray F1).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Increased concentrations of the incretin hormones such as glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are released into the bloodstream from the small intestine in response to meals. These hormones cause insulin release from the pancreatic beta cells in a glucose-dependent manner but are inactivated by the dipeptidyl peptidase-4 (DPP-4) enzyme within minutes. GLP-1 also lowers glucagon secretion from pancreatic alpha cells, reducing hepatic glucose production. In patients with type 2 diabetes, concentrations of GLP-1 are reduced but the insulin response to GLP-1 is preserved. Alogliptin is a DPP-4 inhibitor that slows the inactivation of the incretin hormones, thereby increasing their bloodstream concentrations and reducing fasting and postprandial glucose concentrations in a glucose-dependent manner in patients with type 2 diabetes mellitus. Alogliptin selectively binds to and inhibits DPP-4 but not DPP-8 or DPP-9 activity in vitro at concentrations approximating therapeutic exposures.
12.2 Pharmacodynamics
Single-dose administration of NESINA to healthy subjects resulted in a peak inhibition of DPP-4 within two to three hours after dosing. The peak inhibition of DPP-4 exceeded 93% across doses of 12.5 mg to 800 mg. Inhibition of DPP-4 remained above 80% at 24 hours for doses greater than or equal to 25 mg. Peak and total exposure over 24 hours to active GLP-1 were three- to four-fold greater with NESINA (at doses of 25 to 200 mg) than placebo. In a 16-week, double-blind, placebo-controlled study, NESINA 25 mg demonstrated decreases in postprandial glucagon while increasing postprandial active GLP-1 levels compared to placebo over an eight-hour period following a standardized meal. It is unclear how these findings relate to changes in overall glycemic control in patients with type 2 diabetes mellitus. In this study, NESINA 25 mg demonstrated decreases in two-hour postprandial glucose compared to placebo (-30 mg/dL versus 17 mg/dL, respectively).
Multiple-dose administration of alogliptin to patients with type 2 diabetes also resulted in a peak inhibition of DPP-4 within one to two hours and exceeded 93% across all doses (25 mg, 100 mg and 400 mg) after a single dose and after 14 days of once-daily dosing. At these doses of NESINA, inhibition of DPP-4 remained above 81% at 24 hours after 14 days of dosing.
Cardiac Electrophysiology
In a randomized, placebo-controlled, four-arm, parallel-group study, 257 subjects were administered either alogliptin 50 mg, alogliptin 400 mg, moxifloxacin 400 mg or placebo once daily for a total of seven days. No increase in corrected QT (QTc) was observed with either dose of alogliptin. At the 400 mg dose, peak alogliptin plasma concentrations were 19-fold higher than the peak concentrations following the maximum recommended clinical dose of 25 mg.
12.3 Pharmacokinetics
The pharmacokinetics of NESINA has been studied in healthy subjects and in patients with type 2 diabetes. After administration of single, oral doses up to 800 mg in healthy subjects, the peak plasma alogliptin concentration (median Tmax) occurred one to two hours after dosing. At the maximum recommended clinical dose of 25 mg, NESINA was eliminated with a mean terminal half-life (T1/2) of approximately 21 hours.
After multiple-dose administration up to 400 mg for 14 days in patients with type 2 diabetes, accumulation of alogliptin was minimal with an increase in total [e.g., area under the plasma concentration curve (AUC)] and peak (i.e., Cmax) alogliptin exposures of 34% and 9%, respectively. Total and peak exposure to alogliptin increased proportionally across single doses and multiple doses of alogliptin ranging from 25 mg to 400 mg. The intersubject coefficient of variation for alogliptin AUC was 17%. The pharmacokinetics of NESINA was also shown to be similar in healthy subjects and in patients with type 2 diabetes.
Absorption
The absolute bioavailability of NESINA is approximately 100%. Administration of NESINA with a high-fat meal results in no significant change in total and peak exposure to alogliptin. NESINA may therefore be administered with or without food.
Distribution
Following a single, 12.5 mg intravenous infusion of alogliptin to healthy subjects, the volume of distribution during the terminal phase was 417 L, indicating that the drug is well distributed into tissues.
Alogliptin is 20% bound to plasma proteins.
Metabolism
Alogliptin does not undergo extensive metabolism and 60% to 71% of the dose is excreted as unchanged drug in the urine.
Two minor metabolites were detected following administration of an oral dose of [14C] alogliptin, N-demethylated, M-I (less than 1% of the parent compound), and N-acetylated alogliptin, M-II (less than 6% of the parent compound). M-I is an active metabolite and is an inhibitor of DPP-4 similar to the parent molecule; M-II does not display any inhibitory activity toward DPP-4 or other DPP-related enzymes. In vitro data indicate that CYP2D6 and CYP3A4 contribute to the limited metabolism of alogliptin.
Alogliptin exists predominantly as the (R)-enantiomer (more than 99%) and undergoes little or no chiral conversion in vivo to the (S)-enantiomer. The (S)-enantiomer is not detectable at the 25 mg dose.
Excretion
The primary route of elimination of [14C] alogliptin-derived radioactivity occurs via renal excretion (76%) with 13% recovered in the feces, achieving a total recovery of 89% of the administered radioactive dose. The renal clearance of alogliptin (9.6 L/hr) indicates some active renal tubular secretion and systemic clearance was 14.0 L/hr.
Special Populations
Renal Impairment
A single-dose, open-label study was conducted to evaluate the pharmacokinetics of alogliptin 50 mg in patients with chronic renal impairment compared with healthy subjects.
In patients with mild renal impairment (creatinine clearance [CrCl] ≥60 to <90 mL/min), an approximate 1.2-fold increase in plasma AUC of alogliptin was observed. Because increases of this magnitude are not considered clinically relevant, dose adjustment for patients with mild renal impairment is not recommended.
In patients with moderate renal impairment (CrCl ≥30 to <60 mL/min), an approximate two-fold increase in plasma AUC of alogliptin was observed. To maintain similar systemic exposures of NESINA to those with normal renal function, the recommended dose is 12.5 mg once daily in patients with moderate renal impairment.
In patients with severe renal impairment (CrCl ≥15 to <30 mL/min) and end-stage renal disease (ESRD) (CrCl <15 mL/min or requiring dialysis), an approximate three- and four-fold increase in plasma AUC of alogliptin were observed, respectively. Dialysis removed approximately 7% of the drug during a three-hour dialysis session. NESINA may be administered without regard to the timing of the dialysis. To maintain similar systemic exposures of NESINA to those with normal renal function, the recommended dose is 6.25 mg once daily in patients with severe renal impairment, as well as in patients with ESRD requiring dialysis.
Hepatic Impairment
Total exposure to alogliptin was approximately 10% lower and peak exposure was approximately 8% lower in patients with moderate hepatic impairment (Child-Pugh Grade B) compared to healthy subjects. The magnitude of these reductions is not considered to be clinically meaningful. Patients with severe hepatic impairment (Child-Pugh Grade C) have not been studied. Use caution when administering NESINA to patients with liver disease [see Use in Specific Populations (8.6) and Warnings and Precautions (5.3)].
Gender
No dose adjustment of NESINA is necessary based on gender. Gender did not have any clinically meaningful effect on the pharmacokinetics of alogliptin.
Geriatric
No dose adjustment of NESINA is necessary based on age. Age did not have any clinically meaningful effect on the pharmacokinetics of alogliptin.
Drug Interactions
In Vitro Assessment of Drug Interactions
In vitro studies indicate that alogliptin is neither an inducer of CYP1A2, CYP2B6, CYP2C9, CYP2C19 and CYP3A4, nor an inhibitor of CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP3A4 and CYP2D6 at clinically relevant concentrations.
In Vivo Assessment of Drug Interactions
Effects of Alogliptin on the Pharmacokinetics of Other Drugs
In clinical studies, alogliptin did not meaningfully increase the systemic exposure to the following drugs that are metabolized by CYP isozymes or excreted unchanged in urine (Figure 1). No dose adjustment of NESINA is recommended based on results of the described pharmacokinetic studies.
Figure 1. Effect of Alogliptin on the Pharmacokinetic Exposure to Other Drugs
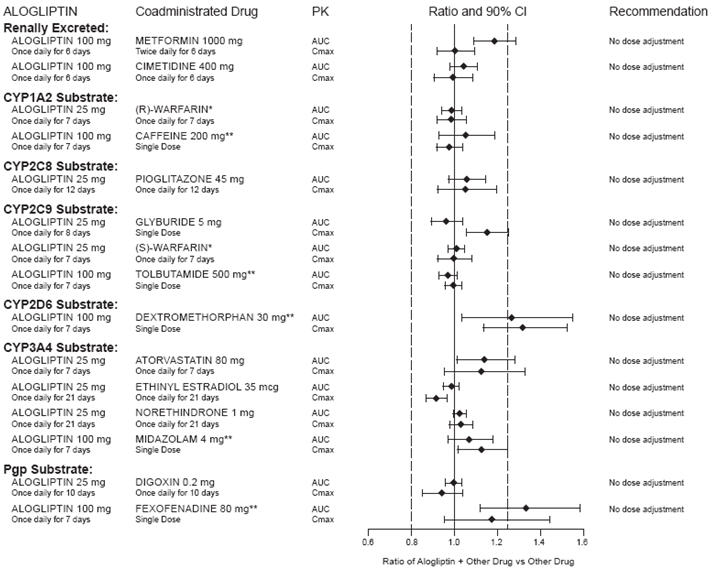
*Warfarin was given once daily at a stable dose in the range of 1 mg to 10 mg. Alogliptin had no significant effect on the prothrombin time (PT) or International Normalized Ratio (INR).
**Caffeine (1A2 substrate), tolbutamide (2C9 substrate), dextromethorphan (2D6 substrate), midazolam (3A4 substrate) and fexofenadine (P-gp substrate) were administered as a cocktail.
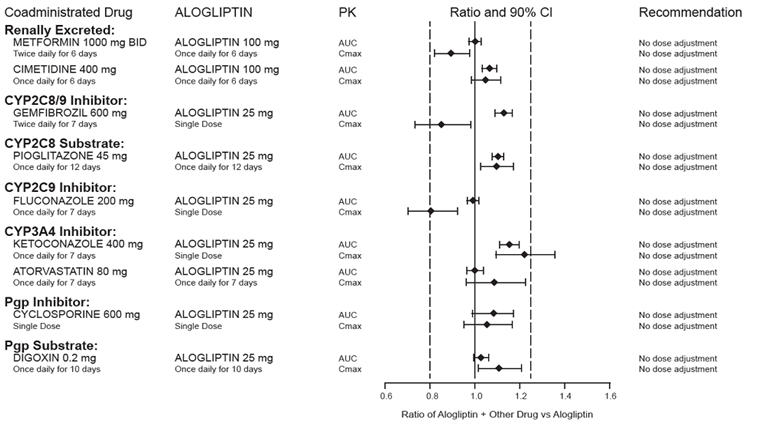
Effects of Other Drugs on the Pharmacokinetics of Alogliptin
There are no clinically meaningful changes in the pharmacokinetics of alogliptin when NESINA is administered concomitantly with the drugs described below (Figure 2).
Figure 2. Effect of Other Drugs on the Pharmacokinetic Exposure of Alogliptin
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Rats were administered oral doses of 75, 400 and 800 mg/kg alogliptin for two years. No drug-related tumors were observed up to 75 mg/kg or approximately 32 times the maximum recommended clinical dose of 25 mg, based on area under the plasma concentration curve (AUC) exposure. At higher doses (approximately 308 times the maximum recommended clinical dose of 25 mg), a combination of thyroid C-cell adenomas and carcinomas increased in male but not female rats. No drug-related tumors were observed in mice after administration of 50, 150 or 300 mg/kg alogliptin for two years, or up to approximately 51 times the maximum recommended clinical dose of 25 mg, based on AUC exposure.
Alogliptin was not mutagenic or clastogenic, with and without metabolic activation, in the Ames test with S. typhimurium and E. coli or the cytogenetic assay in mouse lymphoma cells. Alogliptin was negative in the in vivo mouse micronucleus study.
In a fertility study in rats, alogliptin had no adverse effects on early embryonic development, mating or fertility at doses up to 500 mg/kg, or approximately 172 times the clinical dose based on plasma drug exposure (AUC).
-
14 CLINICAL STUDIES
NESINA has been studied as monotherapy and in combination with metformin, a sulfonylurea, a thiazolidinedione (either alone or in combination with metformin or a sulfonylurea) and insulin (either alone or in combination with metformin).
A total of 14,053 patients with type 2 diabetes were randomized in 11 double-blind, placebo- or active-controlled clinical safety and efficacy studies conducted to evaluate the effects of NESINA on glycemic control. The racial distribution of patients exposed to study medication was 70% Caucasian, 17% Asian, 6% Black and 7% other racial groups. The ethnic distribution was 30% Hispanic. Patients had an overall mean age of 57 years (range 21 to 91 years).
In patients with type 2 diabetes, treatment with NESINA produced clinically meaningful and statistically significant improvements in hemoglobin A1c (A1C) compared to placebo. As is typical for trials of agents to treat type 2 diabetes, the mean reduction in A1C with NESINA appears to be related to the degree of A1C elevation at baseline.
NESINA had similar changes from baseline in serum lipids compared to placebo.
14.1 Patients with Inadequate Glycemic Control on Diet and Exercise
A total of 1768 patients with type 2 diabetes participated in three double-blind studies to evaluate the efficacy and safety of NESINA in patients with inadequate glycemic control on diet and exercise. All three studies had a four week, single-blind, placebo run-in period followed by a 26 week randomized treatment period. Patients who failed to meet prespecified hyperglycemic goals during the 26 week treatment periods received glycemic rescue therapy.
In a 26 week, double-blind, placebo-controlled study, a total of 329 patients (mean baseline A1C = 8%) were randomized to receive NESINA 12.5 mg, NESINA 25 mg or placebo once daily. Treatment with NESINA 25 mg resulted in statistically significant improvements from baseline in A1C and fasting plasma glucose (FPG) compared to placebo at Week 26 (Table 3). A total of 8% of patients receiving NESINA 25 mg and 30% of those receiving placebo required glycemic rescue therapy.
Improvements in A1C were not affected by gender, age or baseline body mass index (BMI).
The mean change in body weight with NESINA was similar to placebo.
Table 3. Glycemic Parameters at Week 26 in a Placebo-Controlled Monotherapy Study of NESINA* NESINA 25 mg Placebo - * Intent-to-treat population using last observation on study
- † Least squares means adjusted for treatment, baseline value, geographic region and duration of diabetes
- ‡ p<0.01 compared to placebo
A1C (%) N=128 N=63 Baseline (mean) 7.9 8.0 Change from baseline (adjusted mean†) -0.6 0 Difference from placebo (adjusted mean† with 95% confidence interval) -0.6‡ (-0.8, -0.3) ˗ % of patients (n/N) achieving A1C ≤7% 44% (58/131)‡ 23% (15/64) FPG (mg/dL) N=129 N=64 Baseline (mean) 172 173 Change from baseline (adjusted mean†) -16 11 Difference from placebo (adjusted mean† with 95% confidence interval) -28‡ (-40, -15) ˗ In a 26-week, double-blind, active-controlled study, a total of 655 patients (mean baseline A1C = 8.8%) were randomized to receive NESINA 25 mg alone, pioglitazone 30 mg alone, NESINA 12.5 mg with pioglitazone 30 mg or NESINA 25 mg with pioglitazone 30 mg once daily. Coadministration of NESINA 25 mg with pioglitazone 30 mg resulted in statistically significant improvements from baseline in A1C and FPG compared to NESINA 25 mg alone and to pioglitazone 30 mg alone (Table 4). A total of 3% of patients receiving NESINA 25 mg coadministered with pioglitazone 30 mg, 11% of those receiving NESINA 25 mg alone and 6% of those receiving pioglitazone 30 mg alone required glycemic rescue.
Improvements in A1C were not affected by gender, age or baseline BMI.
The mean increase in body weight was similar between pioglitazone alone and NESINA when coadministered with pioglitazone.
Table 4. Glycemic Parameters at Week 26 in an Active-Controlled Study of NESINA, Pioglitazone, and NESINA in Combination with Pioglitazone* NESINA 25 mg Pioglitazone 30 mg NESINA 25 mg + Pioglitazone 30 mg - * Intent-to-treat population using last observation carried forward
- † Least squares means adjusted for treatment, geographic region and baseline value
- ‡ p<0.01 compared to NESINA 25 mg or pioglitazone 30 mg
A1C (%) N=160 N=153 N=158 Baseline (mean) 8.8 8.8 8.8 Change from baseline (adjusted mean†) -1.0 -1.2 -1.7 Difference from NESINA 25 mg (adjusted mean† with 95% confidence interval) ˗ ˗ -0.8‡ (-1.0, -0.5) Difference from pioglitazone 30 mg (adjusted mean† with 95% confidence interval) ˗ ˗ -0.6‡ (-0.8, -0.3) % of patients (n/N) achieving A1C ≤7% 24% (40/164) 34% (55/163) 63% (103/164)‡ FPG (mg/dL) N=162 N=157 N=162 Baseline (mean) 189 189 185 Change from baseline (adjusted mean†) -26 -37 -50 Difference from NESINA 25 mg (adjusted mean† with 95% confidence interval) ˗ ˗ -24‡ (-34, -15) Difference from pioglitazone 30 mg (adjusted mean† with 95% confidence interval) ˗ ˗ -13‡ (-22, -4) In a 26 week, double-blind, placebo-controlled study, a total of 784 patients inadequately controlled on diet and exercise alone (mean baseline A1C = 8.4%) were randomized to one of seven treatment groups: placebo; metformin HCl 500 mg or metformin HCl 1000 mg twice daily; NESINA 12.5 mg twice daily; NESINA 25 mg daily; or NESINA 12.5 mg in combination with metformin HCl 500 mg or metformin HCl 1000 mg twice daily. Both coadministration treatment arms (NESINA 12.5 mg + metformin HCl 500 mg and NESINA 12.5 mg + metformin HCl 1000 mg) resulted in statistically significant improvements in A1C and FPG when compared with their respective individual alogliptin and metformin component regimens (Table 5). Coadministration treatment arms demonstrated improvements in two hour postprandial glucose (PPG) compared to NESINA alone or metformin alone (Table 5). A total of 12.3% of patients receiving NESINA 12.5 mg + metformin HCl 500 mg, 2.6% of patients receiving NESINA 12.5 mg + metformin HCl 1000 mg, 17.3% of patients receiving NESINA 12.5 mg, 22.9% of patients receiving metformin HCl 500 mg, 10.8% of patients receiving metformin HCl 1000 mg and 38.7% of patients receiving placebo required glycemic rescue.
Improvements in A1C were not affected by gender, age, race or baseline BMI. The mean decrease in body weight was similar between metformin alone and NESINA when coadministered with metformin.
Table 5. Glycemic Parameters at Week 26 for NESINA and Metformin Alone and in Combination in Patients with Type 2 Diabetes Placebo NESINA 12.5 mg Twice Daily Metformin HCl 500 mg Twice Daily Metformin HCl 1000 mg Twice Daily NESINA 12.5 mg + Metformin HCl 500 mg
Twice DailyNESINA 12.5 mg + Metformin HCl 1000 mg
Twice Daily- * Intent-to-treat population using last observation on study prior to discontinuation of double-blind study medication or sulfonylurea rescue therapy for patients needing rescue
- † Least squares means adjusted for treatment, geographic region and baseline value
- ‡ p<0.05 when compared to metformin and NESINA alone
- § Compared using logistic regression
- ¶ Intent-to-treat population using data available at Week 26
A1C (%)* N=102 N=104 N=103 N=108 N=102 N=111 Baseline (mean) 8.5 8.4 8.5 8.4 8.5 8.4 Change from baseline
(adjusted mean†)0.1 -0.6 -0.7 -1.1 -1.2 -1.6 Difference from metformin (adjusted mean† with 95% confidence interval) - - - - -0.6‡
(-0.9, -0.3)-0.4‡
(-0.7, -0.2)Difference from NESINA (adjusted mean† with 95% confidence interval) - - - - -0.7‡
(-1.0, -0.4)-1.0‡
(-1.3, -0.7)% of patients (n/N) achieving
A1C <7%§4%
(4/102)20%
(21/104)27%
(28/103)34%
(37/108)47%‡
(48/102)59%‡
(66/111)FPG (mg/dL)* N=105 N=106 N=106 N=110 N=106 N=112 Baseline (mean) 187 177 180 181 176 185 Change from baseline
(adjusted mean†)12 -10 -12 -32 -32 -46 Difference from metformin (adjusted mean† with 95% confidence interval) - - - - -20‡
(-33, -8)-14‡
(-26, -2)Difference from NESINA (adjusted mean† with 95% confidence interval) - - - - -22‡
(-35, -10)-36‡
(-49, -24)2-Hour PPG (mg/dL)¶ N=26 N=34 N=28 N=37 N=31 N=37 Baseline (mean) 263 272 247 266 261 268 Change from baseline
(adjusted mean†)-21 -43 -49 -54 -68 -86‡ Difference from metformin (adjusted mean† with 95% confidence interval) - - - - -19
(-49, 11)-32‡
(-58, -5)Difference from NESINA (adjusted mean† with 95% confidence interval) - - - - -25
(-53, -3)-43‡
(-70, -16)14.2 Combination Therapy
Add-On Therapy to Metformin
A total of 2081 patients with type 2 diabetes participated in two 26 week, double-blind, placebo-controlled studies to evaluate the efficacy and safety of NESINA as add-on therapy to metformin. In both studies, patients were inadequately controlled on metformin at a dose of at least 1500 mg per day or at the maximum tolerated dose. All patients entered a four week, single-blind placebo run-in period prior to randomization. Patients who failed to meet prespecified hyperglycemic goals during the 26 week treatment periods received glycemic rescue therapy.
In the first 26 week, placebo-controlled study, a total of 527 patients already on metformin (mean baseline A1C = 8%) were randomized to receive NESINA 12.5 mg, NESINA 25 mg or placebo. Patients were maintained on a stable dose of metformin (median dose = 1700 mg) during the treatment period. NESINA 25 mg in combination with metformin resulted in statistically significant improvements from baseline in A1C and FPG at Week 26, when compared to placebo (Table 6). A total of 8% of patients receiving NESINA 25 mg and 24% of patients receiving placebo required glycemic rescue.
Improvements in A1C were not affected by gender, age, baseline BMI or baseline metformin dose.
The mean decrease in body weight was similar between NESINA and placebo when given in combination with metformin.
Table 6. Glycemic Parameters at Week 26 in a Placebo-Controlled Study of NESINA as Add-On Therapy to Metformin* NESINA 25 mg + Metformin Placebo + Metformin - * Intent-to-treat population using last observation on study
- † Least squares means adjusted for treatment, baseline value, geographic region and baseline metformin dose
- ‡ p<0.001 compared to placebo
A1C (%) N=203 N=103 Baseline (mean) 7.9 8.0 Change from baseline (adjusted mean†) -0.6 -0.1 Difference from placebo (adjusted mean† with 95% confidence interval) -0.5‡ (-0.7, -0.3) ˗ % of patients (n/N) achieving A1C ≤7% 44% (92/207)‡ 18% (19/104) FPG (mg/dL) N=204 N=104 Baseline (mean) 172 180 Change from baseline (adjusted mean†) -17 0 Difference from placebo (adjusted mean† with 95% confidence interval) -17‡ (-26, -9) ˗ In the second 26 week, double-blind, placebo-controlled study, a total of 1554 patients already on metformin (mean baseline A1C = 8.5%) were randomized to one of 12 double-blind treatment groups: placebo; 12.5 mg or 25 mg of NESINA alone; 15 mg, 30 mg or 45 mg of pioglitazone alone; or 12.5 mg or 25 mg of NESINA in combination with 15 mg, 30 mg or 45 mg of pioglitazone. Patients were maintained on a stable dose of metformin (median dose = 1700 mg) during the treatment period. Coadministration of NESINA and pioglitazone provided statistically significant improvements in A1C and FPG compared to placebo, to NESINA alone or to pioglitazone alone when added to background metformin therapy (Table 7, Figure 3). In addition, improvements from baseline A1C were comparable between NESINA alone and pioglitazone alone (15 mg, 30 mg and 45 mg) at Week 26. A total of 4%, 5% or 2% of patients receiving NESINA 25 mg with 15 mg, 30 mg or 45 mg pioglitazone, 33% of patients receiving placebo, 13% of patients receiving NESINA 25 mg and 10%, 15% or 9% of patients receiving pioglitazone 15 mg, 30 mg or 45 mg alone required glycemic rescue.
Improvements in A1C were not affected by gender, age or baseline BMI.
The mean increase in body weight was similar between pioglitazone alone and NESINA when coadministered with pioglitazone.
Table 7. Glycemic Parameters in a 26-Week Study of NESINA, Pioglitazone and NESINA in Combination with Pioglitazone when Added to Metformin* Placebo NESINA 25 mg Pioglitazone 15 mg Pioglitazone 30 mg Pioglitazone 45 mg NESINA 25 mg + Pioglitazone 15 mg NESINA 25 mg + Pioglitazone 30 mg NESINA 25 mg + Pioglitazone 45 mg - * Intent-to-treat population using last observation on study
- † Least squares means adjusted for treatment, geographic region, metformin dose and baseline value
- ‡ p≤0.01 when compared to corresponding doses of pioglitazone and NESINA alone
A1C (%) N=126 N=123 N=127 N=123 N=126 N=127 N=124 N=126 Baseline (mean) 8.5 8.6 8.5 8.5 8.5 8.5 8.5 8.6 Change from baseline (adjusted mean†) -0.1 -0.9 -0.8 -0.9 -1.0 -1.3‡ -1.4‡ -1.6‡ Difference from pioglitazone
(adjusted mean† with 95% confidence interval)- - - - -0.5‡ (-0.7, -0.3) -0.5‡ (-0.7, -0.3) -0.6‡ (-0.8, -0.4) Difference from NESINA (adjusted mean† with 95% confidence interval) - - - - - -0.4‡ (-0.6, -0.1) -0.5‡ (-0.7, -0.3) -0.7‡ (-0.9, -0.5) Patients (%) achieving A1C ≤7% 6%
(8/129)27%
(35/129)26%
(33/129)30%
(38/129)36%
(47/129)55%
(71/130)‡53%
(69/130)‡60%
(78/130)‡FPG (mg/dL) N=129 N=126 N=127 N=125 N=129 N=130 N=126 N=127 Baseline (mean) 177 184 177 175 181 179 179 178 Change from baseline (adjusted mean†) 7 -19 -24 -29 -32 -38‡ -42‡ -53‡ Difference from pioglitazone
(adjusted mean† with 95% confidence interval)- - - - - -14‡ (-24, -5) -13‡ (-23, -3) -20‡ (-30, -11) Difference from NESINA (adjusted mean† with 95% confidence interval) - - - - - -19‡ (-29, -10) -23‡ (-33, -13) -34‡ (-44, -24) Figure 3. Change from Baseline in A1C at Week 26 with NESINA and Pioglitazone Alone and NESINA in Combination with Pioglitazone When Added to Metformin 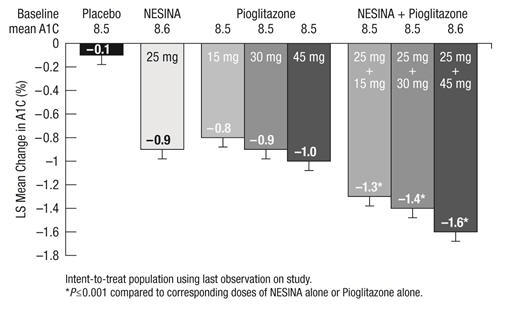
Add-On Therapy to a Thiazolidinedione
In a 26 week, placebo-controlled study, a total of 493 patients inadequately controlled on a thiazolidinedione alone or in combination with metformin or a sulfonylurea (10 mg) (mean baseline A1C = 8%) were randomized to receive NESINA 12.5 mg, NESINA 25 mg or placebo. Patients were maintained on a stable dose of pioglitazone (median dose = 30 mg) during the treatment period; those who were also previously treated on metformin (median dose = 2000 mg) or sulfonylurea (median dose = 10 mg) prior to randomization were maintained on the combination therapy during the treatment period. All patients entered into a four-week, single-blind placebo run-in period prior to randomization. Patients who failed to meet prespecified hyperglycemic goals during the 26 week treatment period received glycemic rescue therapy.
The addition of NESINA 25 mg once daily to pioglitazone therapy resulted in statistically significant improvements from baseline in A1C and FPG at Week 26, compared to placebo (Table 8). A total of 9% of patients who were receiving NESINA 25 mg and 12% of patients receiving placebo required glycemic rescue.
Improvements in A1C were not affected by gender, age, baseline BMI or baseline pioglitazone dose.
Clinically meaningful reductions in A1C were observed with NESINA compared to placebo regardless of whether subjects were receiving concomitant metformin or sulfonylurea (-0.2% placebo versus -0.9% NESINA) therapy or pioglitazone alone (0% placebo versus -0.52% NESINA).
The mean increase in body weight was similar between NESINA and placebo when given in combination with pioglitazone.
Table 8. Glycemic Parameters in a 26 Week, Placebo-Controlled Study of NESINA as Add-On Therapy to Pioglitazone* NESINA 25 mg + Pioglitazone ± Metformin ± Sulfonylurea Placebo + Pioglitazone ± Metformin ± Sulfonylurea - * Intent-to-treat population using last observation on study
- † Least squares means adjusted for treatment, baseline value, geographic region, baseline treatment regimen (pioglitazone, pioglitazone + metformin or pioglitazone + sulfonylurea) and baseline pioglitazone dose
- ‡ p<0.01 compared to placebo
A1C (%) N=195 N=95 Baseline (mean) 8 8 Change from baseline (adjusted mean†) -0.8 -0.2 Difference from placebo (adjusted mean† with 95% confidence interval) -0.6‡ (-0.8, -0.4) ˗ % of patients (n/N) achieving A1C ≤7% 49% (98/199)‡ 34% (33/97) FPG (mg/dL) N=197 N=97 Baseline (mean) 170 172 Change from baseline (adjusted mean†) -20 -6 Difference from placebo (adjusted mean† with 95% confidence interval) -14‡ (-23, -5) ˗ Add-on Combination Therapy with Pioglitazone and Metformin
In a 52 week, active-comparator study, a total of 803 patients inadequately controlled (mean baseline A1C = 8.2%) on a current regimen of pioglitazone 30 mg and metformin at least 1500 mg per day or at the maximum tolerated dose were randomized to either receive the addition of NESINA 25 mg or the titration of pioglitazone 30 mg to 45 mg following a four-week, single-blind placebo run-in period. Patients were maintained on a stable dose of metformin (median dose = 1700 mg). Patients who failed to meet prespecified hyperglycemic goals during the 52 week treatment period received glycemic rescue therapy.
In combination with pioglitazone and metformin, NESINA 25 mg was shown to be statistically superior in lowering A1C and FPG compared with the titration of pioglitazone from 30 mg to 45 mg at Week 26 and at Week 52 (Table 9; results shown only for Week 52). A total of 11% of patients in the NESINA 25 mg treatment group and 22% of patients in the pioglitazone up-titration group required glycemic rescue.
Improvements in A1C were not affected by gender, age, race or baseline BMI.
The mean increase in body weight was similar in both treatment arms.
Table 9. Glycemic Parameters in a 52 Week, Active-Controlled Study of NESINA as Add-On Combination Therapy to Metformin and Pioglitazone* NESINA 25 mg + Pioglitazone 30 mg + Metformin Pioglitazone 45 mg + Metformin - * Intent-to-treat population using last observation on study
- † Least squares means adjusted for treatment, baseline value, geographic region and baseline metformin dose.
- ‡ Noninferior and statistically superior to metformin + pioglitazone at the 0.025 one-sided significance level
- § p<0.001 compared to pioglitazone 45 mg + metformin
A1C (%) N=397 N=394 Baseline (mean) 8.2 8.1 Change from baseline (adjusted mean†) -0.7 -0.3 Difference from pioglitazone 45 mg + metformin (adjusted mean† with 95% confidence interval) -0.4‡ (-0.5, -0.3) ˗ % of patients (n/N) achieving A1C≤7% 33% (134/404)§ 21% (85/399) Fasting Plasma Glucose (mg/dL)‡ N=399 N=396 Baseline (mean) 162 162 Change from baseline (adjusted mean†) -15 -4 Difference from pioglitazone 45 mg + metformin (adjusted mean† with 95% confidence interval) -11§ (-16, -6) ˗ Add-On Therapy to a Sulfonylurea
In a 26 week, placebo-controlled study, a total of 500 patients inadequately controlled on a sulfonylurea (mean baseline A1C = 8.1%) were randomized to receive NESINA 12.5 mg, NESINA 25 mg or placebo. Patients were maintained on a stable dose of glyburide (median dose = 10 mg) during the treatment period. All patients entered into a four week, single-blind, placebo run-in period prior to randomization. Patients who failed to meet prespecified hyperglycemic goals during the 26 week treatment period received glycemic rescue therapy.
The addition of NESINA 25 mg to glyburide therapy resulted in statistically significant improvements from baseline in A1C at Week 26 when compared to placebo (Table 10). Improvements in FPG observed with NESINA 25 mg were not statistically significant compared with placebo. A total of 16% of patients receiving NESINA 25 mg and 28% of those receiving placebo required glycemic rescue.
Improvements in A1C were not affected by gender, age, baseline BMI or baseline glyburide dose.
The mean change in body weight was similar between NESINA and placebo when given in combination with glyburide.
Table 10. Glycemic Parameters in a 26 Week, Placebo-Controlled Study of NESINA as Add-On Therapy to Glyburide* NESINA 25 mg + Glyburide Placebo + Glyburide - * Intent-to-treat population using last observation on study
- † Least squares means adjusted for treatment, baseline value, geographic region and baseline glyburide dose
- ‡ p<0.01 compared to placebo
A1C (%) N=197 N=97 Baseline (mean) 8.1 8.2 Change from baseline (adjusted mean†) -0.5 0 Difference from placebo (adjusted mean† with 95% confidence interval) -0.5‡ (-0.7, -0.3) ˗ % of patients (n/N) achieving A1C ≤7% 35% (69/198)‡ 18% (18/99) FPG (mg/dL) N=198 N=99 Baseline (mean) 174 177 Change from baseline (adjusted mean†) -8 2 Difference from placebo (adjusted mean† with 95% confidence interval) -11 (-22, 1) ˗ Add-On Therapy to Insulin
In a 26 week, placebo-controlled study, a total of 390 patients inadequately controlled on insulin alone (42%) or in combination with metformin (58%) (mean baseline A1C = 9.3%) were randomized to receive NESINA 12.5 mg, NESINA 25 mg or placebo. Patients were maintained on their insulin regimen (median dose = 55 IU) upon randomization and those previously treated with insulin in combination with metformin (median dose = 1700 mg) prior to randomization continued on the combination regimen during the treatment period. Patients entered the trial on short-, intermediate- or long-acting (basal) insulin or premixed insulin. Patients who failed to meet prespecified hyperglycemic goals during the 26 week treatment period received glycemic rescue therapy.
The addition of NESINA 25 mg once daily to insulin therapy resulted in statistically significant improvements from baseline in A1C and FPG at Week 26, when compared to placebo (Table 11). A total of 20% of patients receiving NESINA 25 mg and 40% of those receiving placebo required glycemic rescue.
Improvements in A1C were not affected by gender, age, baseline BMI or baseline insulin dose. Clinically meaningful reductions in A1C were observed with NESINA compared to placebo regardless of whether subjects were receiving concomitant metformin and insulin (-0.2% placebo versus -0.8% NESINA) therapy or insulin alone (0.1% placebo versus -0.7% NESINA).
The mean increase in body weight was similar between NESINA and placebo when given in combination with insulin.
Table 11. Glycemic Parameters in a 26-Week, Placebo-Controlled Study of NESINA as Add-On Therapy to Insulin* NESINA 25 mg + Insulin ± Metformin Placebo + Insulin ± Metformin - * Intent-to-treat population using last observation on study
- † Least squares means adjusted for treatment, baseline value, geographic region, baseline treatment regimen (insulin or insulin + metformin) and baseline daily insulin dose
- ‡ p<0.05 compared to placebo
A1C (%) N=126 N=126 Baseline (mean) 9.3 9.3 Change from baseline (adjusted mean†) -0.7 -0.1 Difference from placebo (adjusted mean† with 95% confidence interval) -0.6‡ (-0.8, -0.4) ˗ % of patients (n/N) achieving A1C ≤7% 8% (10/129) 1% (1/129) FPG (mg/dL) N=128 N=127 Baseline (mean) 186 196 Change from baseline (adjusted mean†) -12 6 Difference from placebo (adjusted mean† with 95% confidence interval) -18‡ (-33, -2) ˗ 14.3 Cardiovascular Safety Trial
A randomized, double-blind, placebo-controlled cardiovascular outcomes trial (EXAMINE) was conducted to evaluate the cardiovascular risk of NESINA. The trial compared the risk of major adverse cardiovascular events (MACE) between NESINA (N=2701) and placebo (N=2679) when added to standard of care therapies for diabetes and atherosclerotic vascular disease (ASCVD). The trial was event driven and patients were followed until a sufficient number of primary outcome events accrued.
Eligible patients were adults with type 2 diabetes who had inadequate glycemic control at baseline (e.g., HbA1c >6.5%) and had been hospitalized for an acute coronary syndrome event (e.g., acute myocardial infarction or unstable angina requiring hospitalization) 15 to 90 days prior to randomization. The dose of NESINA was based on estimated renal function at baseline per dosage and administration recommendations [see Dosage and Administration (2.2)]. The average time between an acute coronary syndrome event and randomization was approximately 48 days.
The mean age of the population was 61 years. Most patients were male (68%), Caucasian (73%), and were recruited from outside of the United States (86%). Asian and Black patients contributed 20% and 4% of the total population, respectively. At the time of randomization patients had a diagnosis of type 2 diabetes mellitus for approximately 9 years, 87% had a prior myocardial infarction and 14% were current smokers. Hypertension (83%) and renal impairment (27% with an eGFR ≤60 ml/min/1.73 m2) were prevalent co-morbid conditions. Use of medications to treat diabetes (e.g., metformin 73%, sulfonylurea 54%, insulin 41%), and ASCVD (e.g., statin 94%, aspirin 93%, renin-angiotensin system blocker 88%, beta-blocker 87%) was similar between patients randomized to NESINA and placebo at baseline. During the trial, medications to treat diabetes and ASCVD could be adjusted to ensure care for these conditions adhered to standard of care recommendations set by local practice guidelines.
The primary endpoint in EXAMINE was the time to first occurrence of a MACE defined as the composite of cardiovascular death, nonfatal myocardial infarction (MI), or nonfatal stroke. The study was designed to exclude a pre-specified risk margin of 1.3 for the hazard ratio of MACE. The median exposure to study drug was 526 days and 95% of the patients were followed to study completion or death.
Table 12 shows the study results for the primary MACE composite endpoint and the contribution of each component to the primary MACE endpoint. The upper bound of the confidence interval was 1.16 and excluded a risk margin larger than 1.3.
Table 12. Patients with MACE in EXAMINE - * Patient Years (PY)
Composite of first event of CV death, nonfatal MI or nonfatal stroke (MACE) NESINA Placebo Hazard Ratio Number of Patients (%) Rate per 100 PY* Number of Patients (%) Rate per 100 PY* (98% CI) N=2701 N=2679 305 (11.3) 7.6 316 (11.8) 7.9 0.96 (0.80, 1.16) CV Death 89 (3.3) 2.2 111 (4.1) 2.8 Non-fatal MI 187 (6.9) 4.6 173 (6.5) 4.3 Non-fatal stroke 29 (1.1) 0.7 32 (1.2) 0.8 The Kaplan-Meier based cumulative event probability is presented in Figure 4 for the time to first occurrence of the primary MACE composite endpoint by treatment arm. The curves for placebo and NESINA overlap throughout the duration of the study. The observed incidence of MACE was highest within the first 60 days after randomization in both treatment arms (14.8 MACE per 100 PY), decreased from day 60 to the end of the first year (8.4 per 100 PY) and was lowest after one year of follow-up (5.2 per 100 PY).
Figure 4. Observed Cumulative Rate of MACE in EXAMINE 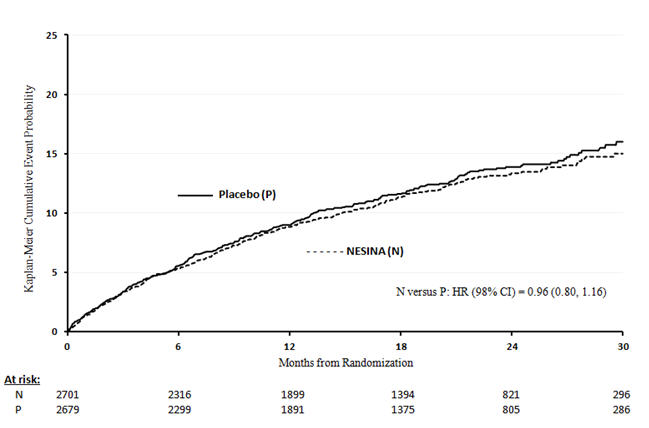
The rate of all cause death was similar between treatment arms with 153 (3.6 per 100 PY) recorded among patients randomized to NESINA and 173 (4.1 per 100 PY) among patients randomized to placebo. A total of 112 deaths (2.9 per 100 PY) among patients on NESINA and 130 among patients on placebo (3.5 per 100 PY) were adjudicated as cardiovascular deaths.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
NESINA tablets are available as film-coated tablets containing 25 mg, 12.5 mg or 6.25 mg of alogliptin as follows:
25 mg tablet: light red, oval, biconvex, film-coated, with "TAK ALG-25" printed on one side, available in:
NDC: 64764-250-30 Bottles of 30 tablets NDC: 64764-250-90 Bottles of 90 tablets NDC: 64764-250-50 Bottles of 500 tablets 12.5 mg tablet: yellow, oval, biconvex, film-coated, with "TAK ALG-12.5" printed on one side, available in:
NDC: 64764-125-30 Bottles of 30 tablets NDC: 64764-125-90 Bottles of 90 tablets NDC: 64764-125-50 Bottles of 500 tablets 6.25 mg tablet: light pink, oval, biconvex, film-coated, with "TAK ALG-6.25" printed on one side, available in:
NDC: 64764-625-30 Bottles of 30 tablets NDC: 64764-625-90 Bottles of 90 tablets -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Inform patients of the potential risks and benefits of NESINA.
Patients should be informed that acute pancreatitis has been reported during use of NESINA. Patients should be informed that persistent, severe abdominal pain, sometimes radiating to the back, which may or may not be accompanied by vomiting, is the hallmark symptom of acute pancreatitis. Patients should be instructed to promptly discontinue NESINA and contact their physician if persistent severe abdominal pain occurs.
Patients should be informed of the signs and symptoms of heart failure. Before initiating NESINA, patients should be asked about a history of heart failure or other risk factors for heart failure including moderate to severe renal impairment. Patients should be instructed to contact their healthcare providers as soon as possible if they experience symptoms of heart failure, including increasing shortness of breath, rapid increase in weight, or swelling of the feet.
Patients should be informed that allergic reactions have been reported during use of NESINA. If symptoms of allergic reactions (including skin rash, hives and swelling of the face, lips, tongue and throat that may cause difficulty in breathing or swallowing) occur, patients should be instructed to discontinue NESINA and seek medical advice promptly.
Patients should be informed that postmarketing reports of liver injury, sometimes fatal, have been reported during use of NESINA. If signs or symptoms of liver injury occur, patients should be instructed to discontinue NESINA and seek medical advice promptly.
Inform patients that hypoglycemia can occur, particularly when an insulin secretagogue or insulin is used in combination with NESINA. Explain the risks, symptoms and appropriate management of hypoglycemia.
Inform patients that severe and disabling joint pain may occur with this class of drugs. The time to onset of symptoms can range from one day to years. Instruct patients to seek medical advice if severe joint pain occurs.
Inform patients that bullous pemphigoid may occur with this class of drugs. Instruct patients to seek medical advice if blisters or erosions occur [see Warnings and Precautions (5.7)].
Instruct patients to take NESINA only as prescribed. If a dose is missed, advise patients not to double their next dose.
Instruct patients to read the Medication Guide before starting NESINA therapy and to reread each time the prescription is refilled. Instruct patients to inform their healthcare provider if an unusual symptom develops or if a symptom persists or worsens.
NES011 R8
-
MEDICATION GUIDENESINA (nes-see'-na)(alogliptin)tablets
Read this Medication Guide carefully before you start taking NESINA and each time you get a refill. There may be new information. This information does not take the place of talking with your doctor about your medical condition or treatment. If you have any questions about NESINA, ask your doctor or pharmacist.
What is the most important information I should know about NESINA?
Serious side effects can happen to people taking NESINA, including:
-
Inflammation of the pancreas (pancreatitis): NESINA may cause pancreatitis which may be severe.
Certain medical conditions make you more likely to get pancreatitis.
Before you start taking NESINA:
Tell your doctor if you have ever had:Stop taking NESINA and call your doctor right away if you have pain in your stomach area (abdomen) that is severe and will not go away. The pain may be felt going from your abdomen through to your back. The pain may happen with or without vomiting. These may be symptoms of pancreatitis.- pancreatitis
- kidney problems
- liver problems
-
Heart failure:
Before you start taking NESINA:
Tell your healthcare provider if you have ever had heart failure or have problems with your kidneys.
Contact your healthcare provider right away if you have any of the following symptoms:These may be symptoms of heart failure.- increasing shortness of breath or trouble breathing especially when lying down
- an unusually fast increase in weight
- swelling of feet, ankles, or legs
What is NESINA?
- NESINA is a prescription medicine used along with diet and exercise to improve blood sugar (glucose) control in adults with type 2 diabetes.
- NESINA is unlikely by itself to cause your blood sugar to be lowered to a dangerous level (hypoglycemia). However, hypoglycemia may still occur with NESINA.
- NESINA is not for people with type 1 diabetes.
- NESINA is not for people with diabetic ketoacidosis (increased ketones in blood or urine).
It is not known if NESINA is safe and effective in children under the age of 18.
Do not take NESINA if you:
- Are allergic to any ingredients in NESINA or have had a serious allergic (hypersensitivity) reaction to NESINA. See the end of this Medication Guide for a complete list of the ingredients in NESINA.
Symptoms of a serious allergic reaction to NESINA may include:If you have any of these symptoms, stop taking NESINA and contact your doctor or go to the nearest hospital emergency room right away.- swelling of your face, lips, throat and other areas on your skin
- raised, red areas on your skin (hives)
- difficulty with swallowing or breathing
- skin rash, itching, flaking or peeling
What should I tell my doctor before and during treatment with NESINA?
Before you take NESINA, tell your doctor if you:
- have or have had inflammation of your pancreas (pancreatitis)
- have kidney or liver problems
- have other medical conditions
- are pregnant or plan to become pregnant. It is not known if NESINA can harm your unborn baby. Talk with your doctor about the best way to control your blood sugar while you are pregnant or if you plan to become pregnant
- are breastfeeding or plan to breastfeed. It is not known whether NESINA passes into your breast milk. Talk with your doctor about the best way to feed your baby if you are taking NESINA
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements.
Know the medicines you take. Keep a list of them and show it to your doctor and pharmacist before you start any new medicine.
NESINA may affect the way other medicines work, and other medicines may affect how NESINA works. Contact your doctor before you start or stop other types of medicines.
How should I take NESINA?
- Take NESINA exactly as your doctor tells you to take it.
- Take NESINA 1 time each day with or without food.
- If you miss a dose, take it as soon as you remember. If you do not remember until it is time for your next dose, skip the missed dose, and take the next dose at your regular time. Do not take 2 doses of NESINA at the same time.
- If you take too much NESINA, call your doctor or go to the nearest hospital emergency room right away.
- If your body is under stress, such as from fever, infection, accident or surgery, the dose of your diabetes medicines may need to be changed. Call your doctor right away.
- Stay on your diet and exercise programs and check your blood sugar as your doctor tells you to.
- Your doctor may do certain blood tests before you start NESINA and during treatment as needed. Your doctor may change your dose of NESINA based on the results of your blood tests due to how well your kidneys are working.
- Your doctor will check your diabetes with regular blood tests, including your blood sugar levels and your hemoglobin A1C.
What are the possible side effects of NESINA?
NESINA can cause serious side effects, including:
See "What is the most important information I should know about NESINA?"
-
Allergic (hypersensitivity) reactions such as:
- swelling of your face, lips, throat and other areas on your skin
- raised, red areas on your skin (hives)
- difficulty swallowing or breathing
- skin rash, itching, flaking or peeling
If you have these symptoms, stop taking NESINA and contact your doctor right away.
-
Liver problems. Call your doctor right away if you have unexplained symptoms, such as:
- nausea or vomiting
- loss of appetite
- stomach pain
- dark urine
- unusual or unexplained tiredness
- yellowing of your skin or the whites of your eyes
-
Low blood sugar (hypoglycemia). If you take NESINA with another medicine that can cause low blood sugar, such as a sulfonylurea or insulin, your risk of getting low blood sugar is higher. The dose of your sulfonylurea medicine or insulin may need to be lowered while you take NESINA. If you have symptoms of low blood sugar, you should check your blood sugar and treat if low, then call your doctor. Signs and symptoms of low blood sugar include:
- shaking or feeling jittery
- fast heartbeat
- sweating
- change in vision
- hunger
- confusion
- headache
- dizziness
- change in mood
- Joint pain. Some people who take medicines called DPP-4 inhibitors like NESINA, may develop joint pain that can be severe. Call your doctor if you have severe joint pain.
- Skin reaction. Some people who take medicines called DPP-4 inhibitors, like NESINA, may develop a skin reaction called bullous pemphigoid that can require treatment in a hospital. Tell your doctor right away if you develop blisters or the breakdown of the outer layer of your skin (erosion). Your doctor may tell you to stop taking NESINA.
The most common side effects of NESINA include stuffy or runny nose and sore throat, headache, or cold-like symptoms (upper respiratory tract infection).
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of NESINA. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store NESINA?
Store NESINA at room temperature between 68°F to 77°F (20°C to 25°C).
Keep NESINA and all medicines out of the reach of children.
General information about the safe and effective use of NESINA.
Medicines are sometimes prescribed for purposes other than those listed in the Medication Guide. Do not take NESINA for a condition for which it was not prescribed. Do not give NESINA to other people, even if they have the same symptoms you have. It may harm them.
This Medication Guide summarizes the most important information about NESINA. If you would like to know more information, talk with your doctor. You can ask your doctor or pharmacist for information about NESINA that is written for health professionals.
For more information go to www.NESINA.com or call 1-877-TAKEDA-7 (1-877-825-3327).
What are the ingredients in NESINA?
Active ingredient: alogliptin
Inactive ingredients: mannitol, microcrystalline cellulose, hydroxypropyl cellulose, croscarmellose sodium and magnesium stearate. In addition, the film-coating contains the following inactive ingredients: hypromellose, titanium dioxide, ferric oxide (red or yellow) and polyethylene glycol and is marked with gray F1 printing ink
Distributed by: Takeda Pharmaceuticals America, Inc. Deerfield, IL 60015. NESINA is a trademark of Takeda Pharmaceutical Company Limited registered with the U.S. Patent and Trademark Office and is used under license by Takeda Pharmaceuticals America, Inc.
© 2013 - 2016 Takeda Pharmaceuticals America, Inc.
NES011 R8This Medication Guide has been approved by the U.S. Food and Drug Administration.
12/2016 -
Inflammation of the pancreas (pancreatitis): NESINA may cause pancreatitis which may be severe.
-
PRINCIPAL DISPLAY PANEL - 6.25 mg Tablet Bottle Label
NDC: 64764-625-30
Nesina
(alogliptin) tablets6.25 mg
Dispense with
Medication Guide30 Tablets
Takeda
Rx Only

-
PRINCIPAL DISPLAY PANEL - 12.5 mg Tablet Bottle Label
NDC: 64764-125-30
Nesina
(alogliptin) tablets12.5 mg
Dispense with
Medication Guide30 Tablets
Takeda
Rx Only

-
PRINCIPAL DISPLAY PANEL - 25 mg Tablet Bottle Label
NDC: 64764-250-30
Nesina
(alogliptin) tablets25 mg
Dispense with
Medication Guide30 Tablets
Takeda
Rx Only

-
INGREDIENTS AND APPEARANCE
NESINA
alogliptin tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 64764-625 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ALOGLIPTIN BENZOATE (UNII: EEN99869SC) (ALOGLIPTIN - UNII:JHC049LO86) ALOGLIPTIN 6.25 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYDROXYPROPYL CELLULOSE (90000 WAMW) (UNII: UKE75GEA7F) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) POLYETHYLENE GLYCOL 8000 (UNII: Q662QK8M3B) SHELLAC (UNII: 46N107B71O) FERROSOFERRIC OXIDE (UNII: XM0M87F357) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) ALCOHOL (UNII: 3K9958V90M) Product Characteristics Color PINK (light pink) Score no score Shape OVAL (biconvex) Size 9mm Flavor Imprint Code TAK;ALG;6;25 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 64764-625-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/25/2013 2 NDC: 64764-625-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 01/25/2013 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022271 01/25/2013 NESINA
alogliptin tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 64764-125 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ALOGLIPTIN BENZOATE (UNII: EEN99869SC) (ALOGLIPTIN - UNII:JHC049LO86) ALOGLIPTIN 12.5 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYDROXYPROPYL CELLULOSE (90000 WAMW) (UNII: UKE75GEA7F) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) POLYETHYLENE GLYCOL 8000 (UNII: Q662QK8M3B) SHELLAC (UNII: 46N107B71O) FERROSOFERRIC OXIDE (UNII: XM0M87F357) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) ALCOHOL (UNII: 3K9958V90M) Product Characteristics Color YELLOW Score no score Shape OVAL (biconvex) Size 9mm Flavor Imprint Code TAK;ALG;12;5 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 64764-125-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/25/2013 2 NDC: 64764-125-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 01/25/2013 3 NDC: 64764-125-50 500 in 1 BOTTLE; Type 0: Not a Combination Product 01/25/2013 4 NDC: 64764-125-02 4 in 1 TRAY 01/25/2013 4 1 in 1 CARTON 4 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022271 01/25/2013 NESINA
alogliptin tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 64764-250 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ALOGLIPTIN BENZOATE (UNII: EEN99869SC) (ALOGLIPTIN - UNII:JHC049LO86) ALOGLIPTIN 25 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYDROXYPROPYL CELLULOSE (90000 WAMW) (UNII: UKE75GEA7F) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) POLYETHYLENE GLYCOL 8000 (UNII: Q662QK8M3B) SHELLAC (UNII: 46N107B71O) FERROSOFERRIC OXIDE (UNII: XM0M87F357) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) ALCOHOL (UNII: 3K9958V90M) Product Characteristics Color RED (light red) Score no score Shape OVAL (biconvex) Size 9mm Flavor Imprint Code TAK;ALG;25 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 64764-250-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/25/2013 2 NDC: 64764-250-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 01/25/2013 3 NDC: 64764-250-50 500 in 1 BOTTLE; Type 0: Not a Combination Product 01/25/2013 4 NDC: 64764-250-02 4 in 1 TRAY 01/25/2013 4 1 in 1 CARTON 4 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022271 01/25/2013 Labeler - Takeda Pharmaceuticals America, Inc. (830134016)
Trademark Results [Nesina]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 NESINA 77446275 not registered Dead/Abandoned |
Takeda Pharmaceutical Company Limited 2008-04-11 |
 NESINA 77140293 3582114 Live/Registered |
Takeda Pharmaceutical Company Limited 2007-03-26 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.