IDHIFA- enasidenib mesylate tablet, film coated
Idhifa by
Drug Labeling and Warnings
Idhifa by is a Prescription medication manufactured, distributed, or labeled by Celgene Corporation. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use IDHIFA safely and effectively. See full prescribing information for IDHIFA.
IDHIFA® (enasidenib) tablets, for oral use
Initial U.S. Approval: 2017WARNING: DIFFERENTIATION SYNDROME
See full prescribing information for complete boxed warning.
Patients treated with IDHIFA have experienced symptoms of differentiation syndrome, which can be fatal if not treated. If differentiation syndrome is suspected, initiate corticosteroid therapy and hemodynamic monitoring until symptom resolution (5.1, 6.1).
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
IDHIFA is an isocitrate dehydrogenase-2 inhibitor indicated for the treatment of adult patients with relapsed or refractory acute myeloid leukemia (AML) with an isocitrate dehydrogenase-2 (IDH2) mutation as detected by an FDA-approved test (1.1).
DOSAGE AND ADMINISTRATION
100 mg orally once daily until disease progression or unacceptable toxicity (2.2).
DOSAGE FORMS AND STRENGTHS
Tablets: 50 mg or 100 mg (3).
CONTRAINDICATIONS
None (4).
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
The most common adverse reactions (≥20%) included nausea, vomiting, diarrhea, elevated bilirubin, and decreased appetite (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Celgene Corporation at 1-888-423-5436 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
Lactation: Advise women not to breastfeed (8.2).
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 9/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: DIFFERENTIATION SYNDROME
1 INDICATIONS AND USAGE
1.1 Acute Myeloid Leukemia
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Dosage
2.3 Monitoring and Dosage Modifications for Toxicities
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Differentiation Syndrome
5.2 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Acute Myeloid Leukemia
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: DIFFERENTIATION SYNDROME
Patients treated with IDHIFA have experienced symptoms of differentiation syndrome, which can be fatal if not treated. Symptoms may include fever, dyspnea, acute respiratory distress, pulmonary infiltrates, pleural or pericardial effusions, rapid weight gain or peripheral edema, lymphadenopathy, bone pain, and hepatic, renal, or multi-organ dysfunction. If differentiation syndrome is suspected, initiate corticosteroid therapy and hemodynamic monitoring until symptom resolution [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for the treatment of AML with IDHIFA based on the presence of IDH2 mutations in the blood or bone marrow [see Indications and Usage (1.1) and Clinical Studies (14.1)]. Information on FDA-approved tests for the detection of IDH2 mutations in AML is available at http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage
The recommended starting dose of IDHIFA is 100 mg taken orally once daily with or without food until disease progression or unacceptable toxicity. For patients without disease progression or unacceptable toxicity, treat for a minimum of 6 months to allow time for clinical response.
Do not split or crush IDHIFA tablets. Administer IDHIFA tablets orally about the same time each day. If a dose of IDHIFA is vomited, missed, or not taken at the usual time, administer the dose as soon as possible on the same day, and return to the normal schedule the following day.
2.3 Monitoring and Dosage Modifications for Toxicities
Assess blood counts and blood chemistries for leukocytosis and tumor lysis syndrome prior to the initiation of IDHIFA and monitor at a minimum of every 2 weeks for at least the first 3 months during treatment. Manage any abnormalities promptly [see Adverse Reactions (6.1)].
Interrupt dosing or reduce dose for toxicities. See Table 1 for dosage modification guidelines.
Table 1: Dosage Modifications for IDHIFA-Related Toxicities *Grade 1 is mild, Grade 2 is moderate, Grade 3 is serious, Grade 4 is life-threatening. Adverse Reaction Recommended Action - Differentiation syndrome
- If differentiation syndrome is suspected, administer systemic corticosteroids and initiate hemodynamic monitoring [see Warnings and Precautions (5.1)].
- Interrupt IDHIFA if severe pulmonary symptoms requiring intubation or ventilator support, and/or renal dysfunction persist for more than 48 hours after initiation of corticosteroids [see Warnings and Precautions (5.1)].
- Resume IDHIFA when signs and symptoms improve to Grade 2* or lower.
- Noninfectious leukocytosis (white blood cell [WBC] count greater than 30 x 109/L)
- Initiate treatment with hydroxyurea, as per standard institutional practices.
- Interrupt IDHIFA if leukocytosis is not improved with hydroxyurea, and then resume IDHIFA at 100 mg daily when WBC is less than 30 x 109/L.
- Elevation of bilirubin greater than 3 times the upper limit of normal (ULN) sustained for ≥2 weeks without elevated transaminases or other hepatic disorders
- Reduce IDHIFA dose to 50 mg daily.
- Resume IDHIFA at 100 mg daily if bilirubin elevation resolves to less than 2 x ULN.
- Other Grade 3* or higher toxicity considered related to treatment including tumor lysis syndrome
- Interrupt IDHIFA until toxicity resolves to Grade 2* or lower.
- Resume IDHIFA at 50 mg daily; may increase to 100 mg daily if toxicities resolve to Grade 1* or lower.
- If Grade 3* or higher toxicity recurs, discontinue IDHIFA.
-
3 DOSAGE FORMS AND STRENGTHS
IDHIFA is available in the following tablet strengths:
- 50-mg tablet: Pale yellow to yellow oval-shaped film-coated tablet debossed "ENA" on one side and "50" on the other side.
- 100-mg tablet: Pale yellow to yellow capsule-shaped film-coated tablet debossed "ENA" on one side and "100" on the other side.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Differentiation Syndrome
In the clinical trial, 14% of patients treated with IDHIFA experienced differentiation syndrome, which may be life-threatening or fatal if not treated. Differentiation syndrome is associated with rapid proliferation and differentiation of myeloid cells. While there is no diagnostic test for differentiation syndrome, symptoms in patients treated with IDHIFA included acute respiratory distress represented by dyspnea and/or hypoxia (68%) and need for supplemental oxygen (76%); pulmonary infiltrates (73%) and pleural effusion (45%); renal impairment (70%); fever (36%); lymphadenopathy (33%); bone pain (27%); peripheral edema with rapid weight gain (21%); and pericardial effusion (18%). Hepatic, renal, and multi-organ dysfunction have also been observed.
Differentiation syndrome has been observed with and without concomitant hyperleukocytosis, in as early as 1 day and up to 5 months after IDHIFA initiation.
If differentiation syndrome is suspected, initiate oral or intravenous corticosteroids (e.g., dexamethasone 10 mg every 12 hours) and hemodynamic monitoring until improvement. Taper corticosteroids only after resolution of symptoms. Symptoms of differentiation syndrome may recur with premature discontinuation of corticosteroid treatment. If severe pulmonary symptoms requiring intubation or ventilator support, and/or renal dysfunction persist for more than 48 hours after initiation of corticosteroids, interrupt IDHIFA until signs and symptoms are no longer severe [see Dosage and Administration (2.3)]. Hospitalization for close observation and monitoring of patients with pulmonary and/or renal manifestation is recommended.
5.2 Embryo-Fetal Toxicity
Based on animal embryo-fetal toxicity studies, IDHIFA can cause embryo-fetal harm when administered to a pregnant woman. In animal embryo-fetal toxicity studies, enasidenib caused embryo-fetal toxicities starting at 0.1 times the steady state clinical exposure based on the area under the concentration-time curve (AUC) at the recommended human dose. Advise females of reproductive potential to use effective contraception during treatment with IDHIFA and for at least 2 months after the last dose of IDHIFA. Advise males with female partners of reproductive potential to use effective contraception during treatment with IDHIFA and for at least 2 months after the last dose of IDHIFA. Pregnant women, patients becoming pregnant while receiving IDHIFA, or male patients with pregnant female partners should be apprised of the potential risk to the fetus [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Differentiation Syndrome [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety evaluation of single-agent IDHIFA is based on 214 patients with relapsed or refractory AML who were assigned to receive 100 mg daily [see Clinical Studies (14.1)]. The median duration of exposure to IDHIFA was 4.3 months (range 0.3 to 23.6). The 30-day and 60-day mortality rates observed with IDHIFA were 4.2% (9/214) and 11.7% (25/214), respectively.
The most common adverse reactions (≥20%) of any grade were nausea, vomiting, diarrhea, elevated bilirubin and decreased appetite.
Serious adverse reactions were reported in 77.1% of patients. The most frequent serious adverse reactions (≥2%) were leukocytosis (10%), diarrhea (6%), nausea (5%), vomiting (3%), decreased appetite (3%), tumor lysis syndrome (5%), and differentiation syndrome (8%). Differentiation syndrome events characterized as serious included pyrexia, renal failure acute, hypoxia, respiratory failure, and multi-organ failure.
Overall, 92 of 214 patients (43%) required a dose interruption due to an adverse reaction; the most common adverse reactions leading to dose interruption were differentiation syndrome (4%) and leukocytosis (3%). Ten of 214 patients (5%) required a dose reduction due to an adverse reaction; no adverse reaction required dose reduction in more than 2 patients. Thirty-six of 214 patients (17%) permanently discontinued IDHIFA due to an adverse reaction; the most common adverse reaction leading to permanent discontinuation was leukocytosis (1%).
Adverse reactions reported in the trial are shown in Table 2.
Table 2: Adverse Reactions Reported in ≥10% (Any Grade) or ≥3% (Grade 3-5) of Patients with Relapsed or Refractory AML a Gastrointestinal disorders observed with IDHIFA treatment can be associated with other commonly reported events such as abdominal pain, and weight decreased.
b Tumor lysis syndrome observed with IDHIFA treatment can be associated with commonly reported uric acid increased.
c Differentiation syndrome can be associated with other commonly reported events such as respiratory failure, dyspnea, hypoxia, pyrexia, peripheral edema, rash, or renal insufficiency.IDHIFA (100 mg daily)
N=214Body System
Adverse ReactionAll Grades
N=214
n (%)≥Grade 3
N=214
n (%)Gastrointestinal Disordersa Nausea 107 (50) 11 (5) Diarrhea 91 (43) 17 (8) Vomiting 73 (34) 4 (2) Metabolism and Nutrition Disorders Decreased appetite 73 (34) 9 (4) Tumor lysis syndrome b 13 (6) 12 (6) Blood and Lymphatic System Disorders Differentiation syndrome c 29 (14) 15 (7) Noninfectious leukocytosis 26 (12) 12 (6) Nervous System Disorders Dysgeusia 25 (12) 0 (0) Other clinically significant adverse reactions occurring in ≤10% of patients included:
Respiratory, Thoracic, and Mediastinal Disorders: Pulmonary edema, acute respiratory distress syndromeChanges in selected post-baseline laboratory values that were observed in patients with relapsed or refractory AML are shown in Table 3.
Table 3: Most Common (≥20%) New or Worsening Laboratory Abnormalities Reported in Patients with Relapsed or Refractory AML a Includes abnormalities occurring up to 28 days after last IDHIFA dose, if new or worsened by at least one grade from baseline, or if baseline was unknown. The denominator varies based on data collected for each parameter (N=213 except phosphorous N=209). IDHIFA (100 mg daily)
N=214Parameter a All Grades
(%)Grade ≥3
(%)Total bilirubin increased 81 15 Calcium decreased 74 8 Potassium decreased 41 15 Phosphorus decreased 27 8 Elevated Bilirubin
IDHIFA may interfere with bilirubin metabolism through inhibition of UGT1A1 [see Clinical Pharmacology (12.3)]. Thirty-seven percent of patients (80/214) experienced total bilirubin elevations ≥2 x ULN at least one time. Of those patients who experienced total bilirubin elevations ≥2 x ULN, 35% had elevations within the first month of treatment, and 89% had no concomitant elevation of transaminases or other severe adverse events related to liver disorders. No patients required a dose reduction for hyperbilirubinemia; treatment was interrupted in 3.7% of patients, for a median of 6 days. Three patients (1.4%) discontinued IDHIFA permanently due to hyperbilirubinemia.
Noninfectious Leukocytosis
IDHIFA can induce myeloid proliferation resulting in a rapid increase in white blood cell count.
Tumor Lysis Syndrome
IDHIFA can induce myeloid proliferation resulting in a rapid reduction in tumor cells which may pose a risk for tumor lysis syndrome.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on animal embryo-fetal toxicity studies, IDHIFA can cause fetal harm when administered to a pregnant woman. There are no available data on IDHIFA use in pregnant women to inform a drug-associated risk of major birth defects and miscarriage. In animal embryo-fetal toxicity studies, oral administration of enasidenib to pregnant rats and rabbits during organogenesis was associated with embryo-fetal mortality and alterations to growth starting at 0.1 times the steady state clinical exposure based on the AUC at the recommended human dose (see Data). If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, advise the patient of the potential risk to a fetus.
Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively.
Animal Data
Enasidenib administered to pregnant rats at a dose of 30 mg/kg twice daily during organogenesis (gestation days 6-17) was associated with maternal toxicity and adverse embryo-fetal effects including post-implantation loss, resorptions, decreased viable fetuses, lower fetal birth weights, and skeletal variations. These effects occurred in rats at approximately 1.6 times the clinical exposure at the recommended human daily dose of 100 mg/day.
In pregnant rabbits treated during organogenesis (gestation days 7-19), enasidenib was maternally toxic at doses equal to 5 mg/kg/day or higher (exposure approximately 0.1 to 0.6 times the steady state clinical exposure at the recommended daily dose) and caused spontaneous abortions at 5 mg/kg/day (exposure approximately 0.1 times the steady state clinical exposure at the recommended daily dose).
8.2 Lactation
Risk Summary
There are no data on the presence of enasidenib or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Because many drugs are excreted in human milk and because of the potential for adverse reactions in breastfed children, advise women not to breastfeed during treatment with IDHIFA and for at least 2 months after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Based on animal embryo-fetal toxicity studies, IDHIFA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Obtain a pregnancy test on females of reproductive potential prior to starting treatment with IDHIFA.
Contraception
Females
Advise females of reproductive potential to avoid becoming pregnant while receiving IDHIFA. Advise females of reproductive potential to use effective contraception during treatment with IDHIFA and for at least 2 months after the last dose. Coadministration of IDHIFA may increase or decrease the concentrations of combined hormonal contraceptives. The clinical significance of this potential drug interaction is unknown at this time.
Males
Advise males with female partners of reproductive potential to use effective contraception during treatment with IDHIFA and for at least 2 months after the last dose of IDHIFA.
Infertility
Based on findings in animals, IDHIFA may impair fertility in females and males of reproductive potential. It is not known whether these effects on fertility are reversible [see Nonclinical Toxicology (13.1)].
8.5 Geriatric Use
No dosage adjustment is required for IDHIFA based on age. In the clinical study, 61% of 214 patients were aged 65 years or older, while 24% were older than 75 years. No overall differences in effectiveness or safety were observed between patients aged 65 years or older and younger patients.
-
11 DESCRIPTION
IDHIFA (enasidenib) is an inhibitor of isocitrate dehydrogenase-2 (IDH2) enzyme. Enasidenib is available as the mesylate salt with the chemical name:
2-methyl-1-[(4-[6-(trifluoromethyl)pyridin-2-yl]-6-{[2-(trifluoromethyl)pyridin-4-yl]amino}-1,3,5-triazin-2-yl)amino]propan-2-ol methanesulfonate.
Or
2-Propanol, 2-methyl-1-[[4-[6-(trifluoromethyl)-2-pyridinyl]-6-[[2-(trifluoromethyl)-4-pyridinyl]amino-1,3,5-triazin-2-yl]amino]-, methanesulfonate (1:1).
The chemical structure is:
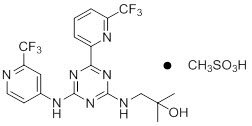
The empirical formula is C19H17F6N7O CH3SO3H (C20H21F6N7O4S), and the molecular weight is 569.48 g/mol. Enasidenib is practically insoluble (solubility ≤74 mcg/mL) in aqueous solutions across physiological pH range (pH 1.2 and 7.4).
IDHIFA (enasidenib) is available as a 50-mg tablet (equivalent to 60 mg enasidenib mesylate) and a 100-mg tablet (equivalent to 120 mg enasidenib mesylate) for oral administration. Each tablet contains inactive ingredients of colloidal silicon dioxide, hydroxypropyl cellulose, hypromellose acetate succinate, iron oxide yellow, magnesium stearate, microcrystalline cellulose, polyethylene glycol, polyvinyl alcohol, sodium lauryl sulfate, sodium starch glycolate, talc, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Enasidenib is a small molecule inhibitor of the isocitrate dehydrogenase 2 (IDH2) enzyme. Enasidenib targets the mutant IDH2 variants R140Q, R172S, and R172K at approximately 40-fold lower concentrations than the wild-type enzyme in vitro. Inhibition of the mutant IDH2 enzyme by enasidenib led to decreased 2-hydroxyglutarate (2-HG) levels and induced myeloid differentiation in vitro and in vivo in mouse xenograft models of IDH2 mutated AML. In blood samples from patients with AML with mutated IDH2, enasidenib decreased 2-HG levels, reduced blast counts and increased percentages of mature myeloid cells.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The potential for QTc prolongation with enasidenib was evaluated in an open-label study in patients with advanced hematologic malignancies with an IDH2 mutation. Based on the QTc data for a single dose of 30 mg to 650 mg and multiple doses of 100 mg daily in the fasted state, no large mean changes in the QTc interval (>20 ms) were observed following treatment with enasidenib.
12.3 Pharmacokinetics
The peak plasma concentration (Cmax) is 1.4 mcg/mL [% coefficient of variation (CV%) 50.2] after a single dose of 100 mg, and 13.1 mcg/mL (CV% 44.8) at steady state for 100 mg daily. The area under concentration time curve (AUC) of enasidenib increases in an approximately dose proportional manner from 50 mg (0.5 times approved recommended dosage) to 450 mg (4.5 times approved recommended dosage) daily dose. Steady-state plasma levels are reached within 29 days of once-daily dosing. Accumulation is approximately 10-fold when administered once daily.
Absorption
The absolute bioavailability after 100 mg oral dose of enasidenib is approximately 57%. After a single oral dose, the median time to Cmax (Tmax) is 4 hours.
Distribution
The mean volume of distribution (Vd) of enasidenib is 55.8 L (CV% 29). Human plasma protein binding of enasidenib is 98.5% and of its metabolite AGI-16903 is 96.6% in vitro.
Enasidenib is not a substrate for P-glycoprotein or BCRP, while AGI-16903 is a substrate of both P-glycoprotein and BCRP. Enasidenib and AGI-16903 are not substrates of MRP2, OAT1, OAT3, OATP1B1, OATP1B3, and OCT2.
Elimination
Enasidenib has a terminal half-life of 7.9 days and a mean total body clearance (CL/F) of 0.70 L/hour (CV% 62.5).
Metabolism
Enasidenib accounted for 89% of the radioactivity in circulation and AGI-16903, the N-dealkylated metabolite, represented 10% of the circulating radioactivity.
In vitro studies suggest that metabolism of enasidenib is mediated by multiple CYP enzymes (e.g., CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4), and by multiple UGTs (e.g., UGT1A1, UGT1A3, UGT1A4, UGT1A9, UGT2B7, and UGT2B15). Further metabolism of the metabolite AGI-16903 is also mediated by multiple enzymes (e.g., CYP1A2, CYP2C19, CYP3A4, UGT1A1, UGT1A3, and UGT1A9).
Excretion
Eighty-nine percent (89%) of enasidenib is eliminated in feces and 11% in the urine. Excretion of unchanged enasidenib accounts for 34% of the radiolabeled drug in the feces and 0.4% in the urine.
Specific Populations
No clinically meaningful effect on the pharmacokinetics of enasidenib was observed for the following covariates: age (19 years to 100 years), race (White, Black, or Asian), mild hepatic impairment [defined as total bilirubin ≤ upper limit of normal (ULN) and aspartate transaminase (AST) >ULN or total bilirubin 1 to 1.5 times ULN and any AST], renal impairment (defined as creatinine clearance ≥30 mL/min by Cockcroft-Gault formula), sex, body weight (39 kg to 136 kg), and body surface area.
Drug Interaction Studies
In vitro studies suggest that enasidenib inhibits the activity of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, and UGT1A1. Enasidenib inhibits P-gp, BCRP, OAT1, OATP1B1, OATP1B3, and OCT2, but not MRP2 or OAT3. Enasidenib induces CYP2B6 and CYP3A4.
In vitro studies suggest that the metabolite AGI-16903 inhibits the activity of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP2D6. AGI-16903 inhibits BCRP, OAT1, OAT3, OATP1B1, and OCT2, but not P-gp, MRP2, or OATP1B3.
Coadministration of IDHIFA may increase or decrease the concentrations of combined hormonal contraceptives. The clinical significance of this potential drug interaction is unknown at this time.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with enasidenib.
Enasidenib was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay. Enasidenib was not clastogenic in an in vitro human lymphocyte chromosomal aberration assay, or in an in vivo rat bone marrow micronucleus assay.
Fertility studies in animals have not been conducted with enasidenib. In repeat-dose toxicity studies with twice daily oral administration of enasidenib in rats up to 90-days in duration, changes were reported in male and female reproductive organs including seminiferous tubular degeneration, hypospermia, atrophy of the seminal vesicle and prostate, decreased corpora lutea and increased atretic follicles in the ovaries, and atrophy in the uterus.
-
14 CLINICAL STUDIES
14.1 Acute Myeloid Leukemia
The efficacy of IDHIFA was evaluated in an open-label, single-arm, multicenter, two-cohort clinical trial (Study AG221-C-001, NCT01915498) of 199 adult patients with relapsed or refractory AML and an IDH2 mutation, who were assigned to receive 100 mg daily dose. Cohort 1 included 101 patients and Cohort 2 included 98 patients. IDH2 mutations were identified by a local diagnostic test and retrospectively confirmed by the Abbott RealTime IDH2 assay, or prospectively identified by the Abbott RealTime IDH2 assay, which is the FDA-approved test for selection of patients with AML for treatment with IDHIFA. IDHIFA was given orally at starting dose of 100 mg daily until disease progression or unacceptable toxicity. Dose reductions were allowed to manage adverse events.
The baseline demographic and disease characteristics are shown in Table 4. The baseline demographics and disease characteristics were similar in both study cohorts.
Table 4: Baseline Demographic and Disease Characteristics in Patients with Relapsed or Refractory AML ECOG PS: Eastern Cooperative Oncology Group Performance Status.
a 1 patient had missing baseline ECOG PS.
b For 3 patients with different mutations detected in bone marrow compared to blood, the result of blood is reported.
c Patients were defined as transfusion dependent at baseline if they received any red blood cell or platelet transfusions within the 8-week baseline period.
d Includes intensive and/or nonintensive therapies.Demographic and Disease Characteristics IDHIFA (100 mg daily)
N=199Demographics Age (Years) Median (Min, Max) 68 (19, 100) Age Categories, n (%) <65 years 76 (38) ≥65 years to <75 years 74 (37) ≥75 years 49 (25) Sex, n (%) Male 103 (52) Female 96 (48) Race, n (%) White 153 (77) Black 10 (5) Asian 1 (1) Native Hawaiian/Other Pacific Islander 1 (1) Other / Not Provided 34 (17) Disease Characteristics, n (%) ECOG PS a, n (%) 0 46 (23) 1 124 (62) 2 28 (14) Relapsed AML, n (%) 95 (48) Refractory AML, n (%) 104 (52) IDH2 Mutation b, n (%) R140 155 (78) R172 44 (22) Time from Initial AML Diagnosis (months) Median (min, max) (172 patients) 11.3 (1.2, 129.1) Cytogenetic Risk Status, n (%) Intermediate 98 (49) Poor 54 (27) Missing /Failure 47 (24) Prior Stem Cell Transplantation for AML, n (%) 25 (13) Transfusion Dependent at Baseline c, n (%) 157 (79) Number of Prior Anticancer Regimens, n (%) d 1 89 (45) 2 64 (32) ≥3 46 (23) Median number of prior therapies (min, max) 2 (1, 6) Efficacy was established on the basis of the rate of complete response (CR)/complete response with partial hematologic recovery (CRh), the duration of CR/CRh, and the rate of conversion from transfusion dependence to transfusion independence. The efficacy results are shown in Table 5 and were similar in both cohorts. The median follow-up was 6.6 months (range, 0.4 to 27.7 months). Similar CR/CRh rates were observed in patients with either R140 or R172 mutation.
Table 5: Efficacy Results in Patients with Relapsed or Refractory AML CI: confidence interval, NA: not available.
a CR (complete remission) was defined as <5% of blasts in the bone marrow, no evidence of disease, and full recovery of peripheral blood counts (platelets >100,000/microliter and absolute neutrophil counts [ANC] >1,000/microliter).
b DOR (duration of response) was defined as time since first response of CR or CRh to relapse or death, whichever is earlier.
c CRh (complete remission with partial hematological recovery) was defined as <5% of blasts in the bone marrow, no evidence of disease, and partial recovery of peripheral blood counts (platelets >50,000/microliter and ANC >500/microliter).Endpoint IDHIFA (100 mg daily)
N=199CRa n (%) 37 (19) 95% CI (13, 25) Median DORb (months) 8.2 95% CI (4.7, 19.4) CRhc n (%) 9 (4) 95% CI (2, 8) Median DOR (months) 9.6 95% CI (0.7, NA) CR/CRh n (%) 46 (23) 95% CI (18, 30) Median DOR (months) 8.2 95% CI (4.3, 19.4) For patients who achieved a CR/CRh, the median time to first response was 1.9 months (range, 0.5 to 7.5 months) and the median time to best response of CR/CRh was 3.7 months (range, 0.6 to 11.2 months). Of the 46 patients who achieved a best response of CR/CRh, 39 (85%) did so within 6 months of initiating IDHIFA.
Among the 157 patients who were dependent on red blood cell (RBC) and/or platelet transfusions at baseline, 53 (34%) became independent of RBC and platelet transfusions during any 56-day post baseline period. Of the 42 patients who were independent of both RBC and platelet transfusions at baseline, 32 (76%) remained transfusion independent during any 56-day post baseline period.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
50-mg tablet: Pale yellow to yellow oval-shaped film-coated tablet debossed "ENA" on one side and "50" on the other side.
- 30-count bottles of 50-mg tablets with a desiccant canister (NDC: 59572-705-30)
100-mg tablet: Pale yellow to yellow capsule-shaped film-coated tablet debossed "ENA" on one side and "100" on the other side.
- 30-count bottles of 100-mg tablets with a desiccant canister (NDC: 59572-710-30)
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Differentiation Syndrome
Advise patients on the risks of developing differentiation syndrome as early as 1 day and during the first 5 months on treatment. Ask patients to immediately report any symptoms suggestive of differentiation syndrome, such as fever, cough or difficulty breathing, bone pain, rapid weight gain or swelling of their arms or legs, to their healthcare provider for further evaluation [see Boxed Warning and Warnings and Precautions (5.1)].
Tumor Lysis Syndrome
Advise patients on the risks of developing tumor lysis syndrome. Advise patients on the importance of maintaining high fluid intake, and the need for frequent monitoring of blood chemistry values [see Dosage and Administration (2.3) and Adverse Reactions (6.1)].
Gastrointestinal Adverse Reactions
Advise patients on risk of experiencing gastrointestinal reactions such as diarrhea, nausea, vomiting, decreased appetite, and changes in their sense of taste. Ask patients to report these events to their healthcare provider, and advise patients how to manage them [see Adverse Reactions (6.1)].
Elevated Blood Bilirubin
Inform patients that taking IDHIFA may cause elevated blood bilirubin, which is due to its mechanism of action, and not due to liver damage. Advise patients to report any changes to the color of their skin or the whites of their eyes to their healthcare provider for further evaluation [see Adverse Reactions (6.1)].
Embryo-Fetal Toxicity and Use of Contraceptives
Advise female patients with reproductive potential to use effective contraceptive methods while receiving IDHIFA and to avoid pregnancy while on treatment and for 2 months after completion of treatment. Advise patients to notify their healthcare provider immediately in the event of a pregnancy or if pregnancy is suspected during IDHIFA treatment. Advise males with female partners of reproductive potential to use effective contraception during treatment with IDHIFA and for at least 2 months after the last dose of IDHIFA. Coadministration of IDHIFA may increase or decrease the concentrations of combined hormonal contraceptives. The clinical significance of this potential drug interaction is unknown at this time [see Warnings and Precautions (5.2) and Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with IDHIFA and for at least 2 months after the final dose [see Use in Specific Populations (8.2)].
Dosing and Storage Instructions
- Advise patients not to chew or split the tablets but swallow whole with a cup of water.
- Instruct patients that if they miss a dose or vomit after a dose of IDHIFA, to take it as soon as possible on the same day and return to normal schedule the following day. Warn patients not to take 2 doses to make up for the missed dose [see Dosage and Administration (2.2)].
- Keep IDHIFA in the original container. Keep the container tightly closed with desiccant canister inside to protect the tablets from moisture [see How Supplied/Storage and Handling (16.2)].
Manufactured for and marketed by:
Celgene Corporation
Summit, NJ 07901Licensed from:
Agios Pharmaceuticals
Cambridge, MA 02139Trademarks are the property of their respective owners.
IDHIFA® is a registered trademark of Celgene Corporation.
Pat. www.celgene.com/therapies
© 2016-2019 Celgene Corporation
All Rights Reserved.IDHPI.003/MG.003
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration Revised: September 2019 MEDICATION GUIDE
IDHIFA® (eyed-HEE-fuh)
(enasidenib) tabletsWhat is the most important information I should know about IDHIFA?
IDHIFA may cause serious side effects, including:
-
Differentiation Syndrome. Differentiation syndrome is a condition that affects your blood cells which may be life-threatening or lead to death if not treated. Differentiation syndrome has happened within 1 day and up to 5 months after starting IDHIFA. Call your healthcare provider or go to the nearest hospital emergency room right away if you develop any of the following symptoms of differentiation syndrome while taking IDHIFA:
- fever
- cough
- shortness of breath
- swelling of arms and legs
- swelling around neck, groin, or underarm area
- fast weight gain (greater than 10 pounds within a week)
- bone pain
If you develop any of these symptoms of differentiation syndrome, your healthcare provider may start you on a medicine taken by mouth or given through a vein (intravenous) called corticosteroids and may monitor you in the hospital.
What is IDHIFA?
IDHIFA is a prescription medicine used to treat people with acute myeloid leukemia (AML) with an isocitrate dehydrogenase-2 (IDH2) mutation whose disease has come back or has not improved after previous treatment(s). It is not known if IDHIFA is safe and effective in children.
Before taking IDHIFA, tell your healthcare provider about all of your medical conditions, including if you:
- Are pregnant or plan to become pregnant. IDHIFA can cause harm to your unborn baby if taken during pregnancy.
- If you are able to become pregnant, your healthcare provider will do a pregnancy test before you start taking IDHIFA.
- Females who are able to become pregnant and who take IDHIFA should use effective birth control (contraception) during treatment with IDHIFA and for at least 2 months after your last dose of IDHIFA.
- Males who have female partners that are able to become pregnant should use effective birth control during treatment with IDHIFA and for at least 2 months after your last dose of IDHIFA.
- IDHIFA may affect how hormonal contraceptives work and may cause them to not work as well.
- Talk to your healthcare provider about birth control methods that may be right for you while taking IDHIFA.
- IDHIFA may cause fertility problems in females and males, which may affect your ability to have children. Talk to your healthcare provider if you have concerns about fertility.
- Are breastfeeding or plan to breastfeed. It is not known if IDHIFA passes into your breast milk. You should not breastfeed during your treatment with IDHIFA and for at least 2 months after your last dose of IDHIFA.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How should I take IDHIFA?
- Take IDHIFA exactly as your healthcare provider tells you to.
- Take IDHIFA 1 time a day at the same time each day.
- Swallow IDHIFA tablets whole. Do not chew or split the tablet.
- Swallow IDHIFA with 8 ounces (one cup) of water.
- IDHIFA can be taken with or without food.
- If you miss a dose of IDHIFA or vomit after taking a dose of IDHIFA, take the dose of IDHIFA as soon as possible on the same day. Then take your next dose the next day at your regularly scheduled time. Do not take 2 doses at the same time to make up for the missed dose.
- Your healthcare provider should do blood tests to check your blood counts before you start IDHIFA treatment and at a minimum of every 2 weeks for at least the first 3 months during treatment to check for side effects.
What are the possible side effects of IDHIFA?
IDHIFA may cause serious side effects, including:
See "What is the most important information I should know about IDHIFA?"
The most common side effects of IDHIFA include:
- nausea
- vomiting
- diarrhea
- jaundice
- decreased appetite
Tell your healthcare provider if you have any changes to the color of your skin or the whites of your eyes.
Your healthcare provider will monitor you for side effects during treatment and may tell you to stop taking IDHIFA if you develop certain side effects.These are not all the possible side effects of IDHIFA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store IDHIFA?
- Store IDHIFA at room temperature from 68°F to 77°F (20°C to 25°C).
- Keep IDHIFA in the original container.
- Keep the container tightly closed with desiccant canister inside to protect the tablets from moisture.
Keep IDHIFA and all medicines out of the reach of children.
General information about the safe and effective use of IDHIFA
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not take IDHIFA for conditions for which it was not prescribed. Do not give IDHIFA to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about IDHIFA that is written for health professionals.
What are the ingredients in IDHIFA?
Active ingredient: enasidenib
Inactive ingredients: colloidal silicon dioxide, hydroxypropyl cellulose, hypromellose acetate succinate, iron oxide yellow, magnesium stearate, microcrystalline cellulose, polyethylene glycol, polyvinyl alcohol, sodium lauryl sulfate, sodium starch glycolate, talc, and titanium dioxide
Manufactured for and marketed by: Celgene Corporation, Summit, NJ 07901
Licensed from: Agios Pharmaceuticals, Cambridge, MA 02139
IDHIFA® is a registered trademark of Celgene Corporation.
Pat. http://www.celgene.com/therapies
IDHMG.003 © 2016-2019 Celgene Corporation
All rights reserved.
For more information go to www.IDHIFA.com or call 1-888-423-5436.
-
Differentiation Syndrome. Differentiation syndrome is a condition that affects your blood cells which may be life-threatening or lead to death if not treated. Differentiation syndrome has happened within 1 day and up to 5 months after starting IDHIFA. Call your healthcare provider or go to the nearest hospital emergency room right away if you develop any of the following symptoms of differentiation syndrome while taking IDHIFA:
- PRINCIPAL DISPLAY PANEL - NDC: NDC: 59572-705-30 - 50 mg Label
- PRINCIPAL DISPLAY PANEL - NDC: NDC: 59572-710-30 - 100 mg Label
-
INGREDIENTS AND APPEARANCE
IDHIFA
enasidenib mesylate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59572-705 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ENASIDENIB MESYLATE (UNII: UF6PC17XAV) (ENASIDENIB - UNII:3T1SS4E7AG) ENASIDENIB 50 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) SODIUM LAURYL SULFATE (UNII: 368GB5141J) HYPROMELLOSE ACETATE SUCCINATE 06081224 (3 MM2/S) (UNII: 6N003M473W) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color YELLOW Score no score Shape OVAL Size 12mm Flavor Imprint Code ENA;50 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59572-705-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 08/01/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209606 08/01/2017 IDHIFA
enasidenib mesylate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59572-710 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ENASIDENIB MESYLATE (UNII: UF6PC17XAV) (ENASIDENIB - UNII:3T1SS4E7AG) ENASIDENIB 100 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) SODIUM LAURYL SULFATE (UNII: 368GB5141J) HYPROMELLOSE ACETATE SUCCINATE 06081224 (3 MM2/S) (UNII: 6N003M473W) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color YELLOW Score no score Shape OVAL Size 15mm Flavor Imprint Code ENA;100 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59572-710-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 08/01/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209606 08/01/2017 Labeler - Celgene Corporation (174201137)


Trademark Results [Idhifa]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 IDHIFA 87024412 5244018 Live/Registered |
Celgene Corporation 2016-05-04 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.