Irbesartan by Lupin Pharmaceuticals, Inc. / LUPIN LIMITED IRBESARTAN tablet
Irbesartan by
Drug Labeling and Warnings
Irbesartan by is a Prescription medication manufactured, distributed, or labeled by Lupin Pharmaceuticals, Inc., LUPIN LIMITED. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use IRBESARTAN TABLETS safely and effectively. See full prescribing information for IRBESARTAN TABLETS.
IRBESARTAN tablets, USP for oral use
Initial U.S. Approval: 1997
INDICATIONS AND USAGE
Irbesartan tablet USP is an angiotensin II receptor blocker (ARB) indicated for:
- Treatment of hypertension, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. (1.1)
- Treatment of diabetic nephropathy in hypertensive patients with type 2 diabetes, an elevated serum creatinine, and proteinuria. (1.2)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
- Tablets: 75 mg, 150 mg, 300 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
- Nephropathy in type 2 diabetic patients: The most common adverse reactions which were more frequent than placebo were hyperkalemia dizziness, orthostatic dizziness, and orthostatic hypotension. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Lupin Pharmaceuticals, Inc. at 1-800-399-2561 or FDA at 1-800-FDA-1088 or www.fda.gov/ medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
- Nursing Mothers: Potential for adverse effects in infant. (8.3)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Hypertension
1.2 Nephropathy in Type 2 Diabetic Patients
2 DOSAGE AND ADMINISTRATION
2.1 General Considerations
2.2 Hypertension
2.3 Nephropathy in Type 2 Diabetic Patients
2.4 Dose Adjustment in Volume and Salt-Depleted Patients
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Fetal Toxicity
5.2 Hypotension in Volume or Salt-Depleted Patients
5.3 Impaired Renal Function
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-Marketing Experience
7 DRUG INTERACTIONS
7.1 Agents Increasing Serum Potassium
7.2 Lithium
7.3 Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) Including Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors)
7.4 Dual Blockade of the Renin-Angiotensin System (RAS)
8 USE IN SPECIFIC POPULATIONS
8.1 PREGNANCY
8.3 NURSING MOTHERS
8.4 PEDIATRIC USE
8.5 GERIATRIC USE
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 MECHANISM OF ACTION
12.2 PHARMACODYNAMICS
12.3 PHARMACOKINETICS
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Hypertension
14.2 Nephropathy in Type 2 Diabetic Patients
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
- When pregnancy is detected, discontinue irbesartan tablets as soon as possible.
- Drugs that act directly on the renin-angiotensin system can cause injury and death to the developing fetus [see WARNINGS AND PRECAUTIONS (5.1)].
-
1 INDICATIONS AND USAGE
1.1 Hypertension
Irbesartan tablets USP are indicated for the treatment of hypertension, to lower blood pressure. Lowering blood pressure lowers the risk of fatal and non-fatal cardiovascular (CV) events, primarily strokes and myocardial infarction. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes including this drug.
Control of high blood pressure should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than 1 drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program's Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mmHg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (for example, patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in black patients, and many antihypertensive drugs have additional approved indications and effects (e.g., on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
Irbesartan tablets USP may be used alone or in combination with other antihypertensive agents.
1.2 Nephropathy in Type 2 Diabetic Patients
Irbesartan tablets USP are indicated for the treatment of diabetic nephropathy in patients with type 2 diabetes and hypertension, an elevated serum creatinine, and proteinuria (>300 mg/day). In this population, irbesartan tablet USP reduces the rate of progression of nephropathy as measured by the occurrence of doubling of serum creatinine or end-stage renal disease (need for dialysis or renal transplantation) [see CLINICAL STUDIES (14.2)].
-
2 DOSAGE AND ADMINISTRATION
2.1 General Considerations
Irbesartan tablet USP may be administered with other antihypertensive agents and with or without food.
2.2 Hypertension
The recommended initial dose of irbesartan tablet USP is 150 mg once daily. The dosage can be increased to a maximum dose of 300 mg once daily as needed to control blood pressure [see CLINICAL STUDIES (14.1)].
2.3 Nephropathy in Type 2 Diabetic Patients
The recommended dose is 300 mg once daily [see CLINICAL STUDIES (14.2)].
2.4 Dose Adjustment in Volume and Salt-Depleted Patients
The recommended initial dose is 75 mg once daily in patients with depletion of intravascular volume or salt (e.g., patients treated vigorously with diuretics or on hemodialysis) [see WARNINGS AND PRECAUTIONS (5.2)].
-
3 DOSAGE FORMS AND STRENGTHS
Irbesartan tablets USP, 75 mg are white to off-white, oval shaped, uncoated tablets, debossed with 'LU' on one side and 'M11' on the other side.
Irbesartan tablets USP, 150 mg are white to off-white, oval shaped, uncoated tablets, debossed with 'LU' on one side and 'M12' on the other side.
Irbesartan tablets USP, 300 mg are white to off-white, oval shaped, uncoated tablets, debossed with 'LU' on one side and 'M13' on the other side.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Fetal Toxicity
Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected, discontinue irbesartan as soon as possible [see USE IN SPECIFIC POPULATIONS (8.1)].
5.2 Hypotension in Volume or Salt-Depleted Patients
In patients with an activated renin-angiotensin system, such as volume- or salt-depleted patients (e.g. those being treated with high doses of diuretics), symptomatic hypotension may occur after initialization of treatment with irbesartan. Correct volume or salt depletion prior to administration of irbesartan or use a lower starting dose [see DOSAGE AND ADMINISTRATION (2.4)].
5.3 Impaired Renal Function
Changes in renal function including acute renal failure can be caused by drugs that inhibit the renin-angiotensin system. Patients whose renal function may depend in part on the activity of the renin-angiotensin system (e.g., patients with renal artery stenosis, chronic kidney disease, severe heart failure, or volume depletion) may be at particular risk of developing acute renal failure or death on irbesartan. Monitor renal function periodically in these patients. Consider withholding or discontinuing therapy in patients who develop a clinically significant decrease in renal function on irbesartan [see DRUG INTERACTIONS (7.3)].
-
6 ADVERSE REACTIONS
The following important adverse reactions are described elsewhere in the labeling:
- Hypotension in Volume- or Salt-depleted Patients [see WARNINGS AND PRECAUTIONS (5.2)]
- Impaired Renal Function [see WARNINGS AND PRECAUTIONS (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. The adverse reaction information from clinical trials does, however, provide a basis for identifying the adverse events that appear to be related to drug use and for approximating rates.
Hypertension
Irbesartan has been evaluated for safety in more than 4300 patients with hypertension and about 5000 subjects overall. This experience includes 1303 patients treated for over 6 months and 407 patients for 1 year or more.
In placebo-controlled clinical trials, the following adverse reactions were reported in at least 1% of patients treated with irbesartan (n=1965) and at a higher incidence versus placebo (n=641), excluding those too general to be informative and those not reasonably associated with the use of drug because they were associated with the condition being treated or are very common in the treated population, include: diarrhea (3% vs 2%), dyspepsia/heartburn (2% vs 1%), and fatigue (4% vs 3%).
Irbesartan use was not associated with an increased incidence of dry cough, as is typically associated with ACE inhibitor use. In placebo-controlled studies, the incidence of cough in irbesartan-treated patients was 2.8% versus 2.7% in patients receiving placebo.
Nephropathy in Type 2 Diabetic Patients
Hyperkalemia:
In the Irbesartan Diabetic Nephropathy Trial (IDNT) (proteinuria ≥900 mg/day, and serum creatinine ranging from 1.0 to 3.0 mg/dL), the percent of patients with potassium>6 mEq/L was 18.6% in the irbesartan group versus 6.0% in the placebo group. Discontinuations due to hyperkalemia in the irbesartan group were 2.1% versus 0.4% in the placebo group.
In IDNT, the adverse reactions were similar to those seen in patients with hypertension with the exception of an increased incidence of orthostatic symptoms which occurred more frequently in the irbesartan versus placebo group: dizziness (10.2% vs 6.0%), orthostatic dizziness (5.4% vs 2.7%) and orthostatic hypotension (5.4% vs 3.2%).
6.2 Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of irbesartan. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate reliably their frequency or to establish a causal relationship to drug exposure.
Urticaria; angioedema (involving swelling of the face, lips, pharynx, and/or tongue); anaphylactic reaction including anaphylactic shock;increased liver function tests; jaundice; hepatitis; hyperkalemia; thrombocytopenia; increased CPK; tinnitus.
-
7 DRUG INTERACTIONS
7.1 Agents Increasing Serum Potassium
Coadministration of irbesartan with other drugs that raise serum potassium levels may result in hyperkalemia, sometimes severe. Monitor serum potassium in such patients.
7.2 Lithium
Increases in serum lithium concentrations and lithium toxicity have been reported with concomitant use of irbesartan and lithium. Monitor lithium levels in patients receiving irbesartan and lithium.
7.3 Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) Including Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors)
In patients who are elderly, volume-depleted (including those on diuretic therapy), or with compromised renal function, co-administration of NSAIDs, including selective COX-2 inhibitors, with angiotensin II receptor antagonists (including irbesartan) may result in deterioration of renal function, including possible acute renal failure. These effects are usually reversible. Monitor renal function periodically in patients receiving irbesartan and NSAID therapy.
The antihypertensive effect of angiotensin II receptor antagonists, including irbesartan, may be attenuated by NSAIDs including selective COX-2 inhibitors.
7.4 Dual Blockade of the Renin-Angiotensin System (RAS)
Dual blockade of the RAS with angiotensin receptor blockers, ACE inhibitors, or aliskiren is associated with increased risks of hypotension, hyperkalemia, and changes in renal function (including acute renal failure) compared to monotherapy. Most patients receiving the combination of two RAS inhibitors do not obtain any additional benefit compared to monotherapy. In general, avoid combined use of RAS inhibitors. Closely monitor blood pressure, renal function and electrolytes in patients on irbesartan and other agents that affect the RAS.
Do not co-administer aliskiren with irbesartan in patients with diabetes. Avoid use of aliskiren with irbesartan in patients with renal impairment (GFR <60 mL/min).
-
8 USE IN SPECIFIC POPULATIONS
8.1 PREGNANCY
Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected, discontinue irbesartan as soon as possible. These adverse outcomes are usually associated with use of these drugs in the second and third trimesters of pregnancy. Most epidemiologic studies examining fetal abnormalities after exposure to antihypertensive use in the first trimester have not distinguished drugs affecting the renin-angiotensin system from other antihypertensive agents. Appropriate management of maternal hypertension during pregnancy is important to optimize outcomes for both mother and fetus.
In the unusual case that there is no appropriate alternative to therapy with drugs affecting the renin-angiotensin system for a particular patient, apprise the mother of the potential risk to the fetus. Perform serial ultrasound examinations to assess the intra-amniotic environment. If oligohydramnios is observed, discontinue irbesartan, unless it is considered lifesaving for the mother. Fetal testing may be appropriate, based on the week of pregnancy. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury. Closely observe infants with histories of in utero exposure to irbesartan for hypotension, oliguria, and hyperkalemia [see USE IN SPECIFIC POPULATIONS (8.4)].
Irbesartan crosses the placenta in rats and rabbits. In pregnant rats given irbesartan at doses greater than the maximum recommended human dose (MRHD), fetuses showed increased incidences of renal pelvic cavitation, hydroureter and/or absence of renal papilla. Subcutaneous edema also occurred in fetuses at doses about 4 times the MRHD (based on body surface area). These anomalies occurred when pregnant rats received irbesartan through Day 20 of gestation but not when drug was stopped on gestation Day 15. The observed effects are believed to be late gestational effects of the drug. Pregnant rabbits given oral doses of irbesartan equivalent to 1.5 times the MRHD experienced a high rate of maternal mortality and abortion. Surviving females had a slight increase in early resorptions and a corresponding decrease in live fetuses [see NONCLINICAL TOXICOLOGY (13.2)].
Radioactivity was present in the rat and rabbit fetus during late gestation and in rat milk following oral doses of radiolabeled irbesartan.
8.3 NURSING MOTHERS
It is not known whether irbesartan is excreted in human milk, but irbesartan or some metabolite of irbesartan is secreted at low concentration in the milk of lactating rats. Because of the potential for adverse effects on the nursing infant, discontinue nursing or discontinue irbesartan.
8.4 PEDIATRIC USE
In infants with histories of in utero exposure to an angiotensin II receptor antagonist observe for hypotension, oliguria, and hyperkalemia. If oliguria occurs, support blood pressure and renal perfusion. Exchange transfusion or dialysis may be required as means of reversing hypotension and/or substituting for disordered renal function.
Irbesartan, in a study at a dose of up to 4.5 mg/kg/day, once daily, did not appear to lower blood pressure effectively in pediatric patients ages 6 to 16 years.
Irbesartan has not been studied in pediatric patients less than 6 years old.
8.5 GERIATRIC USE
Of 4925 subjects receiving irbesartan in controlled clinical studies of hypertension, 911 (18.5%) were 65 years and over, while 150 (3.0%) were 75 years and over. No overall differences in effectiveness or safety were observed between these subjects and younger subjects, but greater sensitivity of some older individuals cannot be ruled out. [see CLINICAL PHARMACOLOGY (12.3) and CLINICAL STUDIES (14.1)].
-
10 OVERDOSAGE
No data are available in regard to overdosage in humans. However, daily doses of 900 mg for 8 weeks were well-tolerated. The most likely manifestations of overdosage are expected to be hypotension and tachycardia; bradycardia might also occur from overdose. Irbesartan is not removed by hemodialysis.
Acute oral toxicity studies with irbesartan in mice and rats indicated acute lethal doses were in excess of 2000 mg/kg, about 25-fold and 50-fold the MRHD (300 mg) on a mg/m2 basis, respectively.
-
11 DESCRIPTION
Irbesartan is an angiotensin II receptor (AT1 subtype) antagonist.
Irbesartan is a non-peptide compound, chemically described as a 2-butyl-3-[p-(o-1H-tetrazol-5 ylphenyl)benzyl]-1,3-diazaspiro[4.4]non-1-en-4-one.
Its empirical formula is C25 H28 N6 O, and the structural formula:
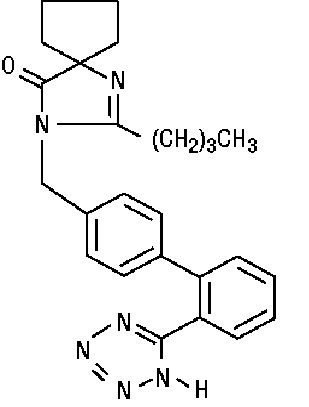
Irbesartan is a white to off-white crystalline powder with a molecular weight of 428.5. It is a nonpolar compound with a partition coefficient (octanol/water) of 10.1 at pH of 7.4. Irbesartan is slightly soluble in alcohol and methylene chloride and practically insoluble in water.
Irbesartan tablets USP are available for oral administration in unscored tablets containing 75 mg, 150 mg, or 300 mg of irbesartan. Inactive ingredients include: colloidal silicon dioxide, crospovidone, magnesium stearate, mannitol, povidone and sodium lauryl sulphate.
-
12 CLINICAL PHARMACOLOGY
12.1 MECHANISM OF ACTION
Angiotensin II is a potent vasoconstrictor formed from angiotensin I in a reaction catalyzed by angiotensin-converting enzyme (ACE, kininase II). Angiotensin II is the primary vasoactive hormone of the renin-angiotensin system, and an important component in the pathophysiology of hypertension. It also stimulates aldosterone secretion by the adrenal cortex. Irbesartan blocks the vasoconstrictor and aldosterone-secreting effects of angiotensin II by selectively binding to the AT1 angiotensin II receptor found in many tissues (e.g., vascular smooth muscle, adrenal gland). There is also an AT2 receptor in many tissues, but it is not involved in cardiovascular homeostasis.
Irbesartan is a specific competitive antagonist of AT1 receptors with a much greater affinity (more than 8500-fold) for the AT1 receptor than for the AT2 receptor and no agonist activity.
Blockade of the AT1 receptor removes the negative feedback of angiotensin II on renin secretion, but the resulting increased plasma renin activity and circulating angiotensin II do not overcome the effects of irbesartan on blood pressure.
Irbesartan does not inhibit ACE or renin or affect other hormone receptors or ion channels known to be involved in the cardiovascular regulation of blood pressure and sodium homeostasis.
12.2 PHARMACODYNAMICS
In healthy subjects, single oral irbesartan doses of up to 300 mg produced dose-dependent inhibition of the pressor effect of angiotensin II infusions. Inhibition was complete (100%) 4 hours following oral doses of 150 mg or 300 mg and partial inhibition was sustained for 24 hours (60% and 40% at 300 mg and 150 mg, respectively).
In hypertensive patients, angiotensin II receptor inhibition following chronic administration of irbesartan causes a 1.5-fold to 2-fold rise in angiotensin II plasma concentration and a 2-fold to 3-fold increase in plasma renin levels. Aldosterone plasma concentrations generally decline following irbesartan administration, but serum potassium levels are not significantly affected at recommended doses.
In hypertensive patients, chronic oral doses of irbesartan (up to 300 mg) had no effect on glomerular filtration rate, renal plasma flow, or filtration fraction. In multiple dose studies in hypertensive patients, there were no clinically important effects on fasting triglycerides, total cholesterol, HDL-cholesterol, or fasting glucose concentrations. There was no effect on serum uric acid during chronic oral administration, and no uricosuric effect.
12.3 PHARMACOKINETICS
The oral absorption of irbesartan is rapid and complete with an average absolute bioavailability of 60% to 80%. Following oral administration of irbesartan, peak plasma concentrations of irbesartan are attained at 1.5 to 2 hours after dosing. Food does not affect the bioavailability of irbesartan.
Irbesartan exhibits linear pharmacokinetics over the therapeutic dose range.
Distribution
Irbesartan is 90% bound to serum proteins (primarily albumin and α1-acid glycoprotein) with negligible binding to cellular components of blood. The average volume of distribution is 53 to 93 liters.
Studies in animals indicate that radiolabeled irbesartan weakly crosses the blood-brain barrier and placenta. Irbesartan is excreted in the milk of lactating rats.
Elimination
Total plasma and renal clearances are in the range of 157 to 176 mL/min and 3.0 to 3.5 mL/min, respectively. The terminal elimination half-life of irbesartan averages 11 to 15 hours. Steady-state concentrations are achieved within 3 days. Limited accumulation of irbesartan (<20%) is observed in plasma upon repeated once-daily dosing and is not clinically relevant.
Metabolism
Irbesartan is an orally active agent that does not require biotransformation into an active form. Irbesartan is metabolized via glucuronide conjugation and oxidation. Following oral or intravenous administration of 14C-labeled irbesartan, more than 80% of the circulating plasma radioactivity is attributable to unchanged irbesartan. The primary circulating metabolite is the inactive irbesartan glucuronide conjugate (approximately 6%). The remaining oxidative metabolites do not add appreciably to irbesartan's pharmacologic activity.
In vitro studies indicate irbesartan is oxidized primarily by CYP2C9; metabolism by CYP3A4 is negligible.
Excretion
Irbesartan and its metabolites are excreted by both biliary and renal routes. Following either oral or intravenous administration of 14C-labeled irbesartan, about 20% of radioactivity is recovered in the urine and the remainder in the feces, as irbesartan or irbesartan glucuronide.
Specific Populations
Sex:
No sex-related differences in pharmacokinetics are observed in healthy elderly (age 65 to 80 years) or in healthy young (age 18 to 40 years) subjects. In studies of hypertensive patients, there is no sex difference in half-life or accumulation, but somewhat higher plasma concentrations of irbesartan are observed in females (11% to 44%). No sex-related dosage adjustment is necessary.
Geriatrics:
In elderly subjects (age 65 to 80 years), irbesartan elimination half-life is not significantly altered, but AUC and Cmax values are about 20% to 50% greater than those of young subjects (age 18 to 40 years). No dosage adjustment is necessary in the elderly.
Race/Ethnicity:
In healthy black subjects, irbesartan AUC values are approximately 25% greater than whites; there is no difference in Cmax values.
Renal Impairment:
The pharmacokinetics of irbesartan is not altered in patients with renal impairment or in patients on hemodialysis. Irbesartan is not removed by hemodialysis. No dosage adjustment is necessary in patients with mild to severe renal impairment unless a patient with renal impairment is also volume depleted [see WARNINGS AND PRECAUTIONS (5.2) and DOSAGE AND ADMINISTRATION (2.4)].
Hepatic Insufficiency:
The pharmacokinetics of irbesartan following repeated oral administration are not significantly affected in patients with mild to moderate cirrhosis of the liver. No dosage adjustment is necessary in patients with hepatic insufficiency.
Drug-Drug Interactions
In vitro studies show significant inhibition of the formation of oxidized irbesartan metabolites with the known cytochrome CYP 2C9 substrates/inhibitors sulphenazole, tolbutamide and nifedipine. However, in clinical studies the consequences of concomitant irbesartan on the pharmacodynamics of warfarin were negligible. Based on in vitro data, no interaction would be expected with drugs whose metabolism is dependent upon cytochrome P450 isoenzymes 1A1, 1A2, 2A6, 2B6, 2D6, 2E1, or 3A4.
In separate studies of patients receiving maintenance doses of warfarin, hydrochlorothiazide, or digoxin, irbesartan administration for 7 days has no effect on the pharmacodynamics of warfarin (prothrombin time) or pharmacokinetics of digoxin. The pharmacokinetics of irbesartan are not affected by coadministration of nifedipine or hydrochlorothiazide.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No evidence of carcinogenicity was observed when irbesartan was administered at dosages of up to 500/1000 mg/kg/day (males/females, respectively) in rats and 1000 mg/kg/day in mice for up to 2 years. For male and female rats, 500 mg/kg/day provided an average systemic exposure to irbesartan (AUC0 to 24 hour, bound plus unbound) about 3 and 11 times, respectively, the average systemic exposure in humans receiving the maximum recommended dose (MRD) of 300 mg irbesartan/day, whereas 1000 mg/kg/day (administered to females only) provided an average systemic exposure about 21 times that reported for humans at the MRD. For male and female mice, 1000 mg/kg/day provided an exposure to irbesartan about 3 and 5 times, respectively, the human exposure at 300 mg/day.
Irbesartan was not mutagenic in a battery of in vitro tests (Ames microbial test, rat hepatocyte DNA repair test, V79 mammalian-cell forward gene-mutation assay). Irbesartan was negative in several tests for induction of chromosomal aberrations (in vitro—human lymphocyte assay; in vivo—mouse micronucleus study).
Irbesartan had no adverse effects on fertility or mating of male or female rats at oral dosages ≤650 mg/kg/day, the highest dose providing a systemic exposure to irbesartan (AUC0 to 24 hour, bound plus unbound) about 5 times that found in humans receiving the MRD of 300 mg/day.
13.2 Animal Toxicology and/or Pharmacology
When pregnant rats were treated with irbesartan from Day 0 to Day 20 of gestation (oral doses of 50 mg/kg/day, 180 mg/kg/day, and 650 mg/kg/day), increased incidences of renal pelvic cavitation, hydroureter and/or absence of renal papilla were observed in fetuses at dosages ≥50 mg/kg/day (approximately equivalent to the maximum recommended human dose [MRHD], 300 mg/day, on a body surface area basis). Subcutaneous edema was observed in fetuses at dosages ≥180 mg/kg/day (about 4 times the MRHD on a body surface area basis). As these anomalies were not observed in rats in which irbesartan exposure (oral doses of 50, 150, and 450 mg/kg/day) was limited to gestation days 6 to 15, they appear to reflect late gestational effects of the drug. In pregnant rabbits, oral doses of 30 mg irbesartan/kg/day were associated with maternal mortality and abortion. Surviving females receiving this dose (about 1.5 times the MRHD on a body surface area basis) had a slight increase in early resorptions and a corresponding decrease in live fetuses. Irbesartan was found to cross the placental barrier in rats and rabbits.
-
14 CLINICAL STUDIES
14.1 Hypertension
The antihypertensive effects of irbesartan were examined in 7 placebo-controlled 8- to 12-week trials in patients with baseline diastolic blood pressures of 95 to 110 mmHg. Doses of 1 to 900 mg were included in these trials in order to fully explore the dose-range of irbesartan. These studies allowed comparison of once- or twice-daily regimens at 150 mg/day, comparisons of peak and trough effects, and comparisons of response by sex, age, and race. Two of the seven placebo-controlled trials identified above examined the antihypertensive effects of irbesartan and hydrochlorothiazide in combination.
The 7 studies of irbesartan monotherapy included a total of 1915 patients randomized to irbesartan (1 to 900 mg) and 611 patients randomized to placebo. Once-daily doses of 150 mg and 300 mg provided statistically and clinically significant decreases in systolic and diastolic blood pressure with trough (24 hours post dose) effects after 6 to 12 weeks treatment compared to placebo, of about 8 to 10/5 to 6 mmHg and 8 to 12/5 to 8 mmHg, respectively. No further increase in effect was seen at dosages greater than 300 mg. The dose-response relationships for effects on systolic and diastolic pressure are shown in Figures 1 and 2.
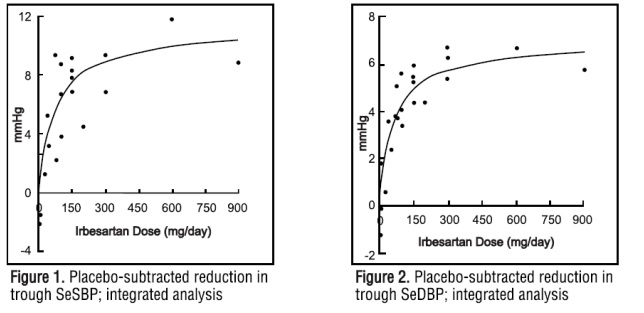
Once-daily administration of therapeutic doses of irbesartan gave peak effects at around 3 to 6 hours and, in one ambulatory blood pressure monitoring study, again around 14 hours. This was seen with both once-daily and twice-daily dosing. Trough-to-peak ratios for systolic and diastolic response were generally between 60% and 70%. In a continuous ambulatory blood pressure monitoring study, once-daily dosing with 150 mg gave trough and mean 24-hour responses similar to those observed in patients receiving twice-daily dosing at the same total daily dose.
In controlled trials, the addition of irbesartan to hydrochlorothiazide doses of 6.25 mg, 12.5 mg, or 25 mg produced further dose-related reductions in blood pressure similar to those achieved with the same monotherapy dose of irbesartan. HCTZ also had an approximately additive effect.
Analysis of age, sex, and race subgroups of patients showed that men and women, and patients over and under 65 years of age, had generally similar responses. Irbesartan was effective in reducing blood pressure regardless of race, although the effect was somewhat less in blacks (usually a low-renin population).
The effect of irbesartan is apparent after the first dose, and it is close to its full observed effect at 2 weeks. At the end of an 8-week exposure, about 2/3 of the antihypertensive effect was still present one week after the last dose. Rebound hypertension was not observed. There was essentially no change in average heart rate in irbesartan-treated patients in controlled trials.
14.2 Nephropathy in Type 2 Diabetic Patients
The Irbesartan Diabetic Nephropathy Trial (IDNT) was a randomized, placebo- and active- controlled, double-blind, multicenter study conducted worldwide in 1715 patients with type 2 diabetes, hypertension (SeSBP >135 mmHg or SeDBP >85 mmHg), and nephropathy (serum creatinine 1.0 to 3.0 mg/dL in females or 1.2 to 3.0 mg/dL in males and proteinuria ≥900 mg/day). Patients were randomized to receive irbesartan 75 mg, amlodipine 2.5 mg, or matching placebo once-daily. Patients were titrated to a maintenance dose of irbesartan 300 mg, or amlodipine 10 mg, as tolerated. Additional antihypertensive agents (excluding ACE inhibitors, angiotensin II receptor antagonists and calcium channel blockers) were added as needed to achieve blood pressure goal (≤135/85 or 10 mmHg reduction in systolic blood pressure if higher than 160 mmHg) for patients in all groups.
The study population was 66.5% male, 72.9% below 65 years of age, and 72% White (Asian/Pacific Islander 5.0%, Black 13.3%, Hispanic 4.8%). The mean baseline seated systolic and diastolic blood pressures were 159 mmHg and 87 mmHg, respectively. The patients entered the trial with a mean serum creatinine of 1.7 mg/dL and mean proteinuria of 4144 mg/day.
The mean blood pressure achieved was 142/77 mmHg for irbesartan, 142/76 mmHg for amlodipine, and 145/79 mmHg for placebo. Overall, 83.0% of patients received the target dose of irbesartan more than 50% of the time. Patients were followed for a mean duration of 2.6 years.
The primary composite endpoint was the time to occurrence of any one of the following events: doubling of baseline serum creatinine, end-stage renal disease (ESRD; defined by serum creatinine ≥6 mg/dL, dialysis, or renal transplantation), or death. Treatment with irbesartan resulted in a 20% risk reduction versus placebo (p=0.0234) (see Figure 3 and Table 1). Treatment with irbesartan also reduced the occurrence of sustained doubling of serum creatinine as a separate endpoint (33%), but had no significant effect on ESRD alone and no effect on overall mortality (see Table 1).
Figure 3: IDNT: Kaplan-Meier Estimates of Primary Endpoint (Doubling of Serum Creatinine, End-Stage Renal Disease or All-Cause Mortality
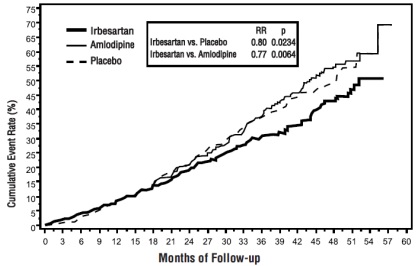
The percentages of patients experiencing an event during the course of the study can be seen in Table 1 below:
Table 1: IDNT: Components of Primary Composite Endpoint
Comparison With Placebo
Comparison With Amlodipine
Irbesartan N=579
(%)
Placebo
N=569
(%)
Hazard Ratio
95% CI
Amlodipine
N=567 (%)
Hazard Ratio
95% CI
Primary
Composite
Endpoint
32.6
39.0
0.80
0.66 to 0.97 (p=0.0234)
41.1
0.77
0.63 to 0.93
Breakdown of first occurring event contributing to primary endpoint
2x creatinine
14.2
19.5
---
---
22.8
---
---
ESRD
7.4
8.3
---
---
8.8
---
---
Death
11.1
11.2
---
---
9.5
---
---
Incidence of total events over entire period of follow-up
2x creatinine
16.9
23.7
0.67
0.52 to 0.87
25.4
0.63
0.49 to 0.81
ESRD
14.2
17.8
0.77
0.57 to 1.03
18.3
0.77
0.57 to 1.03
Death
15.0
16.3
0.92
0.69 to 1.23
14.6
1.04
0.77 to 1.40
The secondary endpoint of the study was a composite of cardiovascular mortality and morbidity (myocardial infarction, hospitalization for heart failure, stroke with permanent neurological deficit, amputation). There were no statistically significant differences among treatment groups in these endpoints. Compared with placebo, irbesartan significantly reduced proteinuria by about 27%, an effect that was evident within 3 months of starting therapy. Irbesartan significantly reduced the rate of loss of renal function (glomerular filtration rate), as measured by the reciprocal of the serum creatinine concentration, by 18.2%.
Table 2 presents results for demographic subgroups. Subgroup analyses are difficult to interpret, and it is not known whether these observations represent true differences or chance effects. For the primary endpoint, irbesartan's favorable effects were seen in patients also taking other antihypertensive medications (angiotensin II receptor antagonists, angiotensin-converting enzyme inhibitors, and calcium channel blockers were not allowed), oral hypoglycemic agents, and lipid-lowering agents.
Table 2: IDNT: Primary Efficacy Outcome Within Subgroups
Comparison With Placebo
Baseline Factors
Irbesartan
N=579
(%)
Placebo
N=569
(%)
Hazard Ratio
95% CI
Sex
Male
27.5
36.7
0.68
0.53 to 0.88
Female
42.3
44.6
0.98
0.72 to 1.34
Race
White
29.5
37.3
0.75
0.60 to 0.95
Non-White
42.6
43.5
0.95
0.67 to 1.34
Age (years)
<65
31.8
39.9
0.77
0.62 to 0.97
≥65
35.1
36.8
0.88
0.61 to 1.29
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Irbesartan Tablets USP are available as white to off-white, oval shaped, uncoated tablets, debossed with a LU on one side and a code on the other (see Table below). Unit-of-use bottles contain 30 or 90 or 500 tablets as follows:
75 mg
150 mg
300 mg
Debossing
M11
M12
M13
Bottle of 30
68180-410-06
68180-411-06
68180-412-06
Bottle of 90
68180-410-09
68180-411-09
68180-412-09
Bottle of 500
-
68180-411-02
68180-412-02
Store at 25°C (77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise female patients of childbearing age about the consequences of exposure to irbesartan during pregnancy. Discuss treatment options with women planning to become pregnant. Patients should be asked to report pregnancies to their physicians as soon as possible.
Potassium Supplements
Advise patients receiving irbesartan not to use potassium supplements or salt substitutes containing potassium without consulting their healthcare provider [see DRUG INTERACTIONS (7.1)].
Manufactured for:
Lupin Pharmaceuticals, Inc.
Baltimore, Maryland 21202
United States.
Manufactured by:
Lupin Limited
Pithampur (M.P.) - 454 775
INDIA.
Revised: November 2018 ID#: 257343
-
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
Rx Only
75 mg
NDC: 68180-410-06
30's Tablets

Rx Only
150 mg
NDC: 68180-411-06
30's Tablets

Rx Only
300 mg
NDC: 68180-412-06
30's Tablets

-
INGREDIENTS AND APPEARANCE
IRBESARTAN
irbesartan tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68180-410 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IRBESARTAN (UNII: J0E2756Z7N) (IRBESARTAN - UNII:J0E2756Z7N) IRBESARTAN 75 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) SODIUM LAURYL SULFATE (UNII: 368GB5141J) POVIDONE (UNII: FZ989GH94E) MAGNESIUM STEARATE (UNII: 70097M6I30) CROSPOVIDONE (UNII: 2S7830E561) Product Characteristics Color WHITE (White to off-white) Score no score Shape OVAL (Oval) Size 11mm Flavor Imprint Code LU;M11 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68180-410-06 30 in 1 BOTTLE; Type 0: Not a Combination Product 10/23/2012 2 NDC: 68180-410-09 90 in 1 BOTTLE; Type 0: Not a Combination Product 10/23/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA201531 10/23/2012 IRBESARTAN
irbesartan tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68180-411 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IRBESARTAN (UNII: J0E2756Z7N) (IRBESARTAN - UNII:J0E2756Z7N) IRBESARTAN 150 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) SODIUM LAURYL SULFATE (UNII: 368GB5141J) POVIDONE (UNII: FZ989GH94E) MAGNESIUM STEARATE (UNII: 70097M6I30) CROSPOVIDONE (UNII: 2S7830E561) Product Characteristics Color WHITE (White to off-white) Score no score Shape OVAL (Oval) Size 14mm Flavor Imprint Code LU;M12 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68180-411-06 30 in 1 BOTTLE; Type 0: Not a Combination Product 10/23/2012 2 NDC: 68180-411-02 500 in 1 BOTTLE; Type 0: Not a Combination Product 10/23/2012 3 NDC: 68180-411-09 90 in 1 BOTTLE; Type 0: Not a Combination Product 10/23/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA201531 10/23/2012 IRBESARTAN
irbesartan tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68180-412 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IRBESARTAN (UNII: J0E2756Z7N) (IRBESARTAN - UNII:J0E2756Z7N) IRBESARTAN 300 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) SODIUM LAURYL SULFATE (UNII: 368GB5141J) POVIDONE (UNII: FZ989GH94E) MAGNESIUM STEARATE (UNII: 70097M6I30) CROSPOVIDONE (UNII: 2S7830E561) Product Characteristics Color WHITE (White to off-white) Score no score Shape OVAL (Oval) Size 19mm Flavor Imprint Code LU;M13 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68180-412-06 30 in 1 BOTTLE; Type 0: Not a Combination Product 10/23/2012 2 NDC: 68180-412-02 500 in 1 BOTTLE; Type 0: Not a Combination Product 10/23/2012 3 NDC: 68180-412-09 90 in 1 BOTTLE; Type 0: Not a Combination Product 10/23/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA201531 10/23/2012 Labeler - Lupin Pharmaceuticals, Inc. (089153071) Registrant - LUPIN LIMITED (675923163) Establishment Name Address ID/FEI Business Operations LUPIN LIMITED 863645527 MANUFACTURE(68180-410, 68180-411, 68180-412)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.