NOURIANZ- istradefylline tablet, film coated
NOURIANZ by
Drug Labeling and Warnings
NOURIANZ by is a Prescription medication manufactured, distributed, or labeled by Kyowa Kirin, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NOURIANZ safely and effectively. See full prescribing information for NOURIANZ.
NOURIANZ™ (istradefylline) tablets, for oral use
Initial U.S. Approval: 2019INDICATIONS AND USAGE
NOURIANZ is an adenosine receptor antagonist indicated as adjunctive treatment to levodopa/carbidopa in adult patients with Parkinson's disease (PD) experiencing "off" episodes (1).
DOSAGE AND ADMINISTRATION
- The recommended dosage is 20 mg orally once daily. The dosage may be increased to a maximum of 40 mg once daily (2.1).
- May be taken with or without food (2.1).
- Patients with hepatic impairment: Maximum recommended dosage with moderate hepatic impairment is 20 mg once daily; use of NOURIANZ in patients with severe hepatic impairment should be avoided (2.4, 8.7).
- Patients who smoke 20 or more cigarettes per day (or the equivalent of another tobacco product): Recommended dosage is 40 mg once daily (2.5, 8.8).
DOSAGE FORMS AND STRENGTHS
Tablets: 20 mg and 40 mg (3).
CONTRAINDICATIONS
None (4).
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
The most common adverse reactions (at least 5% and more frequent than placebo) were dyskinesia, dizziness, constipation, nausea, hallucination, and insomnia (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Kyowa Kirin Inc. at 1-844-768-3544 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
2.2 Dosage Adjustment with Strong CYP 3A4 Inhibitors
2.3 Dosing with Strong CYP 3A4 Inducers
2.4 Dosage Adjustment in Patients with Hepatic Impairment
2.5 Dosage Adjustment for Tobacco Smokers
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Dyskinesia
5.2 Hallucinations / Psychotic Behavior
5.3 Impulse Control / Compulsive Behaviors
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on NOURIANZ
7.2 Effect of NOURIANZ on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
8.8 Tobacco Smokers
10 OVERDOSAGE
10.1 Human Experience
10.2 Management of Overdose
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
The recommended dosage of NOURIANZ is 20 mg administered orally once daily. The dosage may be increased to a maximum of 40 mg once daily, based on individual need and tolerability. Initial dose titration is not required.
NOURIANZ can be taken with or without food [see Clinical Pharmacology (12.3)].
2.2 Dosage Adjustment with Strong CYP 3A4 Inhibitors
The maximum recommended dosage of NOURIANZ with concomitant use of strong CYP3A4 inhibitors is 20 mg once daily [see Drug Interactions (7.1)].
2.3 Dosing with Strong CYP 3A4 Inducers
Avoid use of NOURIANZ with strong CYP3A4 inducers [see Drug Interactions (7.1)].
2.4 Dosage Adjustment in Patients with Hepatic Impairment
The maximum recommended dosage of NOURIANZ in patients with moderate hepatic impairment (Child-Pugh B) is 20 mg once daily. Closely monitor patients with moderate hepatic impairment for adverse reactions when on NOURIANZ treatment [see Adverse Reactions (6.1)]. Avoid use of NOURIANZ in patients with severe hepatic impairment (Child-Pugh C) [see Use in Specific Populations (8.7)].
2.5 Dosage Adjustment for Tobacco Smokers
The recommended dosage of NOURIANZ in patients who use tobacco in amounts of 20 or more cigarettes per day (or the equivalent of another tobacco product) is 40 mg once daily [see Use in Specific Populations (8.8) and Clinical Pharmacology (12.3)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Dyskinesia
NOURIANZ in combination with levodopa may cause dyskinesia or exacerbate pre-existing dyskinesia.
In controlled clinical trials (Studies 1, 2, 3, and 4) [see Clinical Studies (14)], the incidence of dyskinesia was 15% for NOURIANZ 20 mg, 17% for NOURIANZ 40 mg, and 8% for placebo, in combination with levodopa. One percent of patients treated with either NOURIANZ 20 mg or 40 mg discontinued treatment because of dyskinesia, compared to 0% for placebo.
5.2 Hallucinations / Psychotic Behavior
Because of the potential risk of exacerbating psychosis, patients with a major psychotic disorder should not be treated with NOURIANZ. Consider dosage reduction or discontinuation if a patient develops hallucinations or psychotic behaviors while taking NOURIANZ.
In controlled clinical trials (Studies 1, 2, 3, and 4) [see Clinical Studies (14)], the incidence of hallucinations was 2% for NOURIANZ 20 mg, 6% for NOURIANZ 40 mg, and 3% for placebo. In patients treated with NOURIANZ 40 mg, 1% discontinued because of hallucinations, compared to 0% for placebo and 0% for patients treated with NOURIANZ 20 mg. The incidence of "abnormal thinking and behavior" (paranoid ideation, delusions, confusion, mania, disorientation, aggressive behavior, agitation, or delirium) reported as an adverse reaction was 1% for NOURIANZ 20 mg, 2% for NOURIANZ 40 mg, and 1% for placebo.
5.3 Impulse Control / Compulsive Behaviors
Patients treated with NOURIANZ and one or more medication(s) for the treatment of Parkinson's disease (including levodopa) may experience intense urges to gamble, increased sexual urges, intense urges to spend money, binge or compulsive eating, and/or other intense urges, and the inability to control these urges. In controlled clinical trials (Studies 1, 2, 3 and 4) [see Clinical Studies (14)],one patient treated with NOURIANZ 40 mg was reported to have impulse control disorder, compared to no patient on placebo or NOURIANZ 20 mg.
In some postmarketing cases, these urges were reported to have stopped when the dose was reduced, or the medication was discontinued. Because patients may not recognize these behaviors as abnormal, it is important for prescribers to specifically ask patients or their caregivers about the development of new or increased gambling urges, sexual urges, uncontrolled spending, binge or compulsive eating, or other urges while being treated with NOURIANZ. Consider dose reduction or discontinuation if a patient develops such urges while taking NOURIANZ [see Adverse Reactions (6.2)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Dyskinesia [see Warnings and Precautions (5.1)]
- Hallucinations / Psychotic Behavior [see Warnings and Precautions (5.2)]
- Impulse Control / Compulsive Behaviors [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of NOURIANZ was evaluated in 734 patients with Parkinson's disease (PD) taking a stable dose of levodopa and a DOPA decarboxylase inhibitor, with or without other PD medications, in four randomized, multicenter, double-blind, placebo-controlled trials 12 weeks in duration (Studies 1, 2, 3 and 4) [see Clinical Studies (14)]. Of the patient population exposed to NOURIANZ, 50% were male, 32% White, 67% Asian, and the mean age was 65 years (range: 33 to 84 years). Of these patients, 356 received NOURIANZ 20 mg and 378 received NOURIANZ 40 mg.
Adverse Reactions Leading to Discontinuation of Treatment
The incidence of patients discontinuing for any adverse reaction was 5% for NOURIANZ 20 mg, 6% for NOURIANZ 40 mg, and 5% for placebo. The most frequently reported adverse reaction causing study discontinuation was dyskinesia [see Warnings and Precautions (5.1)].
Common Adverse Reactions in Pooled Placebo-Controlled Trials
Table 1 shows adverse reactions with a frequency of at least 2% in patients treated with NOURIANZ 20 mg or 40 mg once daily. The most common adverse reactions in which the frequency for NOURIANZ was at least 5%, and greater than the incidence on placebo, were dyskinesia, dizziness, constipation, nausea, hallucination, and insomnia.
Table 1: Adverse Reactions with an Incidence of at Least 2% in Patients Treated with NOURIANZ, and Greater than on Placebo, in Pooled Studies 1, 2, 3, and 4 Adverse Reactions NOURIANZ
20 mg/day
(N=356)
%NOURIANZ
40 mg/day
(N=378)
%Placebo
N=426
(%)- * Includes hallucinations, hallucinations visual, hallucinations olfactory, hallucinations somatic, hallucinations auditory.
Nervous system disorders Dyskinesia 15 17 8 Dizziness 3 6 4 Gastrointestinal disorders Constipation 5 6 3 Nausea 4 6 5 Diarrhea 1 2 1 Psychiatric disorders Hallucination* 2 6 3 Insomnia 1 6 4 Metabolism and nutrition disorders Decreased appetite 1 3 1 Investigations Blood alkaline phosphatase increased 1 2 1 Blood glucose increased 1 2 0 Blood urea increased 1 2 0 Respiratory, thoracic and mediastinal disorders Upper Respiratory Tract Inflammation 1 2 0 Skin and subcutaneous tissue disorders Rash 1 2 1 6.2 Postmarketing Experience
The following adverse reaction has been identified during post approval use of istradefylline outside of the United States. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure: increased libido.
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on NOURIANZ
Strong CYP3A4 Inhibitors
Coadministration of NOURIANZ with a strong CYP3A4 inhibitor (ketoconazole) increased istradefylline AUCinf by 2.5-fold [see Clinical Pharmacology (12.3)]. Therefore, the recommended maximum dosage of NOURIANZ in patients concomitantly using strong CYP3A4 inhibitors (e.g., itraconazole, ketoconazole, clarithromycin) is 20 mg once daily [see Dosage and Administration (2.2)].
Strong CYP3A4 Inducers
Coadministration of NOURIANZ with a strong CYP3A4 inducer (rifampin) decreased istradefylline Cmax and AUCinf by 45% and 81%, respectively [see Clinical Pharmacology (12.3)]. Therefore, it is recommended to avoid use of NOURIANZ with strong CYP3A4 inducers (e.g., carbamazepine, rifampin, phenytoin, St. John's wort) [see Dosage and Administration (2.3)].
7.2 Effect of NOURIANZ on Other Drugs
CYP3A4 Substrates
Coadministration of NOURIANZ 20 mg with a CYP3A4 substrate (midazolam) did not affect the CYP3A4 substrate exposure, while concomitant administration of NOURIANZ 40 mg increased the CYP3A4 substrate (atorvastatin) Cmax and AUCinf by 1.5-fold [see Clinical Pharmacology (12.3)]. Monitor for an increase in adverse reactions of concomitant drugs that are CYP3A4 substrates when coadministering with NOURIANZ 40 mg.
P-glycoprotein (P-gp) Substrates
Coadministration of NOURIANZ with a P-gp substrate (digoxin) increased the P-gp substrate Cmax and AUCinf by 33% and 21%, respectively [see Clinical Pharmacology (12.3)]. Monitor for an increase in adverse reactions of concomitant drugs that are P-gp substrates when coadministering with NOURIANZ.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with the use of NOURIANZ in pregnant women. In animal studies (see Data), oral administration of istradefylline during pregnancy resulted in teratogenicity (increased incidences of fetal structural abnormalities, embryofetal and offspring mortality and growth deficits) at clinically relevant exposures and in the absence of maternal toxicity. The teratogenic effects of istradefylline in pregnant rabbits were substantially greater when administered in combination with levodopa/carbidopa than when administered alone.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies are 2-4% and 15-20%, respectively.
Data
Animal Data
Oral administration of istradefylline (0, 40, 200, or 1000 mg/kg/day) to pregnant rats throughout organogenesis resulted in decreased fetal body weight and increased fetal skeletal and visceral variations at the highest dose tested. Plasma exposure (AUC) at the no-effect dose for adverse effects on embryofetal development in rats (200 mg/kg/day) is approximately 4 times that in humans at the maximum recommended human dose (MRHD) of 40 mg.
Oral administration of istradefylline (0, 50, 200, or 800 mg/kg/day) to pregnant rabbits throughout organogenesis resulted in increased embryofetal mortality at the mid and high doses, increased fetal malformations (external, visceral, skeletal) at all doses, and reduced fetal body weight at the highest dose tested. A no-effect dose for adverse effects on embryofetal development in rabbits was not identified. Plasma exposure (AUC) at the lowest dose tested (50 mg/kg/day) is less than that in humans at the MRHD.
In pregnant rabbits, oral administration of istradefylline (0, 50, 200, or 400 mg/kg/day) alone or in combination with oral levodopa/carbidopa (80/20 mg/kg/day) throughout the period of organogenesis resulted in an increase in embryofetal mortality and an increase (marked at the high dose) in malformations (including limb reduction, craniofacial, and cardiovascular) in fetuses from rats administered istradefylline at all doses in combination with levodopa/carbidopa. Istradefylline alone resulted in an increase in embryofetal mortality and visceral malformations; no increase in fetal malformations was observed with levodopa/carbidopa alone. Fetal body weight was reduced by istradefylline alone (400 mg/kg/day) and in combination (200 and 400 mg/kg/day) with levodopa/carbidopa. A no-effect dose for adverse effects on embryofetal development in rabbits when istradefylline was administered in combination with levodopa/carbidopa was not identified. Plasma exposure (AUC) at the lowest dose of istradefylline tested (50 mg/kg/day) in combination with levodopa/carbidopa is less than that in humans at the MRHD.
Oral administration of istradefylline (0, 6, 25, 100, or 400 mg/kg/day) to female rats throughout gestation and lactation resulted in decreased pup survival and reduced pup body weight (which persisted into adulthood) at all but the lowest dose tested. Exposure to drug in the milk may have contributed to these effects, as demonstrated in pups of untreated (control) dams reared by dams receiving istradefylline (400 mg/kg/day). No adverse effects were observed on physical or neurobehavioral development, or reproductive function. Plasma exposure at the no-effect dose for adverse effects on pre- and postnatal development in rats (6 mg/kg/day) is less than that in humans at the MRHD.
8.2 Lactation
Risk Summary
There are no data on the presence of istradefylline in human milk, the effects of istradefylline on the breastfed infant, or the effects of istradefylline on milk production. Istradefylline was present in the milk of lactating rats at concentrations up to 10 times that in maternal plasma.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for NOURIANZ, and any potential adverse effects on the breastfed infant from NOURIANZ or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Contraception
Use of NOURIANZ during pregnancy is not recommended. Women of childbearing potential should be advised to use contraception during treatment with NOURIANZ [see Use in Specific Populations (8.1)].
8.5 Geriatric Use
No adjustment of NOURIANZ dosage is recommended on the basis of age. Of the total number of PD patients who received NOURIANZ in clinical trials, 53% were ≥65 years and 13% were ≥75 years of age. No overall differences in effectiveness were observed between these patients and younger patients.
8.6 Renal Impairment
No adjustment of NOURIANZ dosage is needed in patients with mild renal impairment (estimated creatinine clearance (CrCL) by Cockcroft-Gault equation: 60-89 mL/min), moderate renal impairment (CrCL 30-59 mL/min), or severe renal impairment (CrCL 15-29 mL/min). NOURIANZ has not been evaluated in patients with end-stage renal disease (ESRD) (CrCL <15 mL/min) or ESRD requiring hemodialysis [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No adjustment of NOURIANZ dosage is needed in patients with mild hepatic impairment (Child-Pugh Class A).
In patients with moderate hepatic impairment (Child-Pugh B), the steady-state exposures (AUC0-24h) were predicted to be 3.3-fold higher than in healthy subjects, based on the estimated mean terminal half-life. Therefore, the maximum recommended dosage of NOURIANZ in patients with moderate hepatic impairment (Child-Pugh B) is 20 mg once daily [see Clinical Pharmacology (12.3)]. Closely monitor patients with moderate hepatic impairment for adverse events when on NOURIANZ treatment [see Adverse Reactions (6.1)].
NOURIANZ has not been studied in patients with severe hepatic impairment (Child-Pugh Class C). Avoid use of NOURIANZ in patients with severe hepatic impairment [see Clinical Pharmacology (12.3)].
8.8 Tobacco Smokers
Tobacco smoking decreased NOURIANZ steady-state systemic exposures by 38% to 54% [see Clinical Pharmacology (12.3)], which may decrease efficacy. Therefore, the recommended NOURIANZ dosage in patients who smoke 20 or more cigarettes per day (or the equivalent amount of another tobacco product) is 40 mg once daily.
-
10 OVERDOSAGE
10.1 Human Experience
There is limited clinical experience regarding human overdosage with NOURIANZ. In clinical trials, one patient took 6 tablets (120 mg, 3 times the maximum recommended dosage) of istradefylline with alcoholic beverages and developed hallucinations, agitation, and worsening dyskinesia.
10.2 Management of Overdose
There are no known specific antidotes for NOURIANZ nor any specific treatment for istradefylline overdose. If an overdose occurs, NOURIANZ treatment should be discontinued and supportive treatment should be administered as clinically indicated. Consider the long terminal half-life of istradefylline (about 83 hours) and the possibility of multiple drug involvement.
Consult a Certified Poison Control Center for up-to-date guidance and advice.
-
11 DESCRIPTION
NOURIANZ contains istradefylline, an adenosine receptor antagonist, which has a xanthine derivative structure. The chemical name is (E)-8-(3,4-dimethoxystyryl)-1,3-diethyl-7-methyl-3,7-dihydro-1H-purine-2,6-dione. Its molecular formula is C20H24N4O4. The molecular weight is 384.43. Istradefylline has the following structural formula:
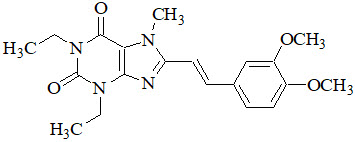
Istradefylline is a light yellow-green crystalline powder. Istradefylline has a dissociation constant (pKa) of 0.78. The aqueous solubility of istradefylline is ~0.5 µg/mL across the physiological pH range and 0.6 µg/mL in water.
NOURIANZ tablets are intended for oral administration only. Each tablet contains 20 mg or 40 mg of istradefylline and the following inactive ingredients: crospovidone, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and polyvinyl alcohol. The film coating contains hypromellose, lactose monohydrate, polyethylene glycol 3350, titanium dioxide, triacetin, and the following dyes: iron oxide red and iron oxide yellow. Carnauba wax is used for polishing.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The precise mechanism by which istradefylline exerts its therapeutic effect in Parkinson disease is unknown. In in vitro studies and in in vivo animal studies, istradefylline was demonstrated to be an adenosine A2A receptor antagonist.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of NOURIANZ (40 mg or 160 mg [4 times the maximum recommended dosage] once daily for 14 days) on the QTc interval was evaluated in a randomized, placebo and moxifloxacin-controlled, multiple-dose, blinded, parallel group study. There was no clinically significant prolongation of QTc interval or relationship between changes in QTc and concentrations of istradefylline.
12.3 Pharmacokinetics
Istradefylline exhibits dose-proportional pharmacokinetics after multiple oral doses from 20 mg to 80 mg (2 times the maximum recommended dosage). Steady-state was reached within 2 weeks of once-daily dosing. The pharmacokinetics of istradefylline were similar in PD patients and healthy subjects.
Absorption
The median time to reach the maximum concentration (Tmax) for istradefylline was about 4 hours under fasted dosing conditions.
Effect of Food
Istradefylline exposure, represented by the area under the curve over time to infinity (AUCinf), increased 1.25-fold when NOURIANZ was coadministered with a standard high-fat meal, compared with administration in a fasted state. Istradefylline maximum plasma concentrations (Cmax) increased by 1.64-fold and Tmax was shortened by 1 hour when NOURIANZ was administered with a high-fat meal. These differences in pharmacokinetic parameters are not expected to be clinically significant [see Dosage and Administration (2.1)].
Distribution
The plasma protein binding of istradefylline was approximately 98%. The apparent volume of distribution (Vd/F) of istradefylline is approximately 557 liters.
Elimination
The total clearance of istradefylline is approximately 4.6 L/hour. The mean terminal half-life (t1/2) for istradefylline at steady-state is approximately 83 hours.
Metabolism
In humans, istradefylline is exclusively eliminated via metabolism. In vitro studies indicate that istradefylline is primarily metabolized via CYP1A1 and CYP3A4, with minor contribution from CYP1A2, 2B6, 2C8, CYP2C9, CYP2C18, and 2D6. Six metabolites have been identified in human plasma. These metabolites each account for less than 10% of the exposure of the parent drug.
Specific Populations
In patients with moderate hepatic impairment (Child-Pugh B), the steady-state exposure (AUC0-24) of istradefylline is predicted to be 3.3-fold higher relative to healthy subjects, based on the estimated mean terminal half-life [see Use in Specific Populations (8.7)]. Based on population pharmacokinetic analyses, no clinically relevant changes in the pharmacokinetics of istradefylline were observed based on age, sex, weight, or race. No clinically relevant changes in istradefylline exposure were observed in patients with severe renal impairment (CrCL 15-29 mL/min) or mild hepatic impairment. NOURIANZ has not been studied in patients with ESRD (CrCL < 15 mL/min), ESRD patients requiring hemodialysis, or severe hepatic impairment (Child-Pugh C) [see Use in Specific Populations (8.6, 8.7)].
Steady-state systemic exposure to istradefylline (40 mg) is 38% to 54% lower in tobacco smokers (who smoke 20 or more cigarettes per day) when compared with non-smokers matched for age, gender, and body weight [see Specific Populations (8.8)].
Drug Interaction Studies
In Vitro Assessment of Drug Interactions
Drug-Metabolizing Enzyme Inhibition
Istradefylline is a weak inhibitor of CYP3A4, but not an inhibitor of CYP1A2, 2B6, 2C9, 2C19, or 2D6 in vitro.
In Vivo Assessment of Drug Interactions
Effect of Other Drugs on Istradefylline
Strong CYP3A4 Inhibitors
Coadministration of ketoconazole (200 mg twice daily for 4 days) with a single dose of istradefylline (40 mg) increased the AUCinf of istradefylline by 2.5-fold, but had no effect on Cmax [see Drug Interactions (7.1)].
Strong CYP3A4 Inducers
Coadministration of rifampin (600 mg daily for 20 days) with a single dose of istradefylline (40 mg) reduced the Cmax and AUCinf of istradefylline by 45% and 81% respectively, when compared with istradefylline administered alone [see Drug Interactions (7.1)].
Effect of Istradefylline on Other Drugs
CYP3A4 Substrates
Coadministration of istradefylline at higher than the recommended doses (80 mg for 14 days) with a single dose of midazolam (10 mg) increased midazolam AUCinf 2.4-fold, and Cmax by 1.6-fold, when compared with midazolam administered alone. Coadministration of lower doses of istradefylline (5 mg and 20 mg) with midazolam (7.5 mg) did not have these effects [see Drug Interactions (7.2)].
Coadministration of istradefylline (40 mg daily for 17 days) with a single dose of atorvastatin (40 mg) increased the Cmax and AUCinf of atorvastatin by 1.5-fold, compared with atorvastatin alone [see Drug Interactions (7.2)].
P-glycoprotein Substrates
Coadministration of istradefylline (40 mg daily for 21 days) with a single dose of digoxin (0.4 mg) increased the Cmax and AUCinf of digoxin by 33% and 21%, respectively, when compared with digoxin alone [see Drug Interactions (7.2)].
Carbidopa/Levodopa
Coadministration of istradefylline (80 mg [two times the recommended maximum dosage] daily for 14 days) with a single dose of carbidopa/levodopa (50/200 mg) did not affect the pharmacokinetics of carbidopa/levodopa. Also, coadministration of istradefylline (20 mg or 40 mg daily for 14 days) with carbidopa/levodopa (25/100 mg three times a day for 14 days) did not affect the systemic exposure of carbidopa/levodopa.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenesis
In lifetime oral carcinogenicity studies, there was no evidence of carcinogenicity in mouse (0, 25, 125, or 250 mg/kg) or rat (0, 30, 100, or 320 mg/kg). Plasma exposures (AUC) at the highest doses tested were approximately 20 (mouse) and 10 (rat) times that in humans at the maximum recommended human dose (MRHD) of 40 mg/day.
Mutagenesis
Istradefylline was negative in in vitro (bacterial reverse mutation assay, chromosomal aberration in mammalian cells) and in vivo (mouse bone marrow micronucleus) assays.
Impairment of Fertility
Oral administration of istradefylline (0, 160, 360, or 800 mg/kg/day) to male and female rats prior to and during mating and continuing in females to gestation day 7 resulted in a decrease in fertility at the highest dose tested and an increase in preimplantation loss at the mid and high doses. Sperm motility was reduced at the highest dose tested. Plasma exposure (AUC) at the no-effect dose (160 mg/kg for adverse effects on reproductive function is approximately 3 times that in humans at the MRHD.
13.2 Animal Toxicology and/or Pharmacology
Oral administration of istradefylline (0, 30, 100, or 320 mg/kg/day) to rats for two years resulted in an increase in the incidence and severity of vascular mineralization in the brain (including in the caudate/putamen, globus pallidus, thalamus, and nucleus accumbens) at all doses tested. The vascular mineralization was composed of calcium and phosphorus and, at higher doses, were reported to partially or completely occlude the blood vessels. There was no evidence of neuronal degeneration, inflammation, or glial response associated with the foci of mineralization.
Brain mineralization was not detected in mice administered istradefylline (0, 25, 125, or 250 mg/kg/day) orally for two years or in dogs administered istradefylline (0, 10, 30, or 100 mg/kg/day) orally for 52 weeks.
-
14 CLINICAL STUDIES
The efficacy of NOURIANZ for the adjunctive treatment to levodopa/carbidopa in patients with Parkinson's disease experiencing "off" episodes was shown in four randomized, multicenter, double-blind, 12-week, placebo-controlled studies (Study 1, NCT00456586; Study 2, NCT00199407; Study 3, NCT00455507; and Study 4, NCT00955526). The studies enrolled patients with a mean duration of Parkinson's disease of 9 years (range: 1 month to 37 years) that were Hoehn and Yahr Stage II to IV, experiencing at least 2 hours (mean approximately 6 hours) of "off" time per day, and were treated with levodopa for at least one year, with stable dosage for at least 4 weeks before screening (mean total daily dosage range: 416 to 785 mg). Patients continued levodopa treatment with or without concomitant PD medications, including dopamine agonists (85%), COMT inhibitors (38%), MAO-B inhibitors (40%), anticholinergics (13%), and/or amantadine (33%), provided the medications were stable for at least 4 weeks before screening and throughout the study period. The studies excluded patients who had received a neurosurgical treatment for PD (e.g., pallidotomy, thalamotomy, deep brain stimulation).
The primary efficacy endpoint was the change from baseline in the daily awake percentage of "off" time, or the change from baseline in total daily "off" time, based on 24-hour diaries completed by patients. A change from baseline in "on" time without troublesome dyskinesia (i.e., "on" time without dyskinesia plus "on" time with non-troublesome dyskinesia) was a secondary efficacy endpoint.
Study 1 was conducted in the U.S. and Canada, and Study 2 was conducted in the U.S. In these studies, patients were randomized to once-daily treatment with NOURIANZ 20 mg, 40 mg, or placebo. Patients treated with NOURIANZ 20 mg or NOURIANZ 40 mg once daily experienced a statistically significant decrease from baseline in percentage of daily awake "off" time, compared with patients on placebo, as summarized in Table 2.
Table 2: Studies 1 and 2: Change From Baseline in Daily Awake OFF Time Baseline Change from Baseline to Endpoint N (mean ± SD)
% of awake "off" hoursN (LSMD* vs. placebo),
% awake "off" hours,
(p-value)SD: Standard Deviation - * LSMD: Least squares mean difference; a negative value indicates a greater reduction from baseline in Percentage Daily Awake "off" time for NOURIANZ, relative to placebo.
Study 1 Placebo 66 37.2 ± 13.8 65 -- NOURIANZ 40 mg 129 38.4 ± 16.2 126 - 6.78 (p=0.007) Study 2 Placebo 113 38.7 ± 11.6 113 -- NOURIANZ 20 mg 112 39.8 ± 14.0 112 - 4.57 (p=0.025) Compared with patients on placebo, patients treated with NOURIANZ experienced an additional increase from baseline in "on" time without troublesome dyskinesia of 0.96 hours (nominal p=0.026) in Study 1, and of 0.55 hours (nominal p=0.135) in Study 2.
Study 3 and Study 4 were conducted in Japan. In these studies, patients were randomized equally to treatment with NOURIANZ 20 mg, 40 mg, or placebo. Patients treated with NOURIANZ 20 mg or NOURIANZ 40 mg once daily experienced a statistically significant decrease from baseline in "off" time compared with patients on placebo, as summarized in Table 3.
Table 3: Studies 3 and 4: Change From Baseline in Daily OFF Time Baseline Change from Baseline to Endpoint N (mean ± SD)
hoursN (LSMD* vs. placebo)
hours
( p-value)SD: Standard Deviation - * LSMD: Least squares mean difference; a negative value indicates a greater reduction from baseline in "off" time for NOURIANZ, relative to placebo.
Study 3 Placebo 118 6.4 ± 2.7 118 -- NOURIANZ 20 mg 115 6.8 ± 2.9 115 -0.65 (p=0.028) NOURIANZ 40 mg 124 6.6 ± 2.5 124 -0.92 (p=0.002) Study 4 Placebo 123 6.3 ± 2.5 123 -- NOURIANZ 20 mg 120 6.6 ± 2.7 120 -0.76 (p=0.006) NOURIANZ 40 mg 123 6.0 ± 2.5 123 -0.74 (p=0.008) In Study 3, compared with placebo, an additional increase from baseline in "on" time without troublesome dyskinesia of 0.57 hours (nominal p=0.085) and of 0.65 hours (nominal p=0.048), respectively, were observed in patients treated with NOURIANZ 20 mg or NOURIANZ 40 mg. In Study 4, the corresponding increases in "on" time without troublesome dyskinesia were 0.83 hours (nominal p=0.008) for NOURIANZ 20 mg and 0.81 hours (nominal p=0.008) for NOURIANZ 40 mg.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
NOURIANZ (istradefylline) tablets are available as:
20 mg Tablets:
Peach-colored, pillow-shaped, film-coated tablets with "20" debossed on one side.
Bottle of 90: NDC: 42747-602-90
40 mg Tablets:
Peach-colored, almond-shaped, film-coated tablets with "40" debossed on one side.
Bottle of 90: NDC: 42747-604-90
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Dyskinesia
Advise patients that NOURIANZ may cause dyskinesia or exacerbate pre-existing dyskinesia [see Warnings and Precautions (5.1)].
Hallucinations / Psychotic Behavior
Advise patients that NOURIANZ may cause hallucinations or psychotic behavior and they should report any of these adverse reactions to their healthcare provider [see Warnings and Precautions (5.2)].
Impulse Control / Compulsive Behaviors
Inform patients that they may experience intense urges to gamble, increased sexual urges, and other intense urges and the inability to control these urges while taking NOURIANZ and one or more medication(s) for the treatment of Parkinson's disease (including levodopa). Advise patients that they should report any of these adverse reactions to their healthcare provider [see Warnings and Precautions (5.3)].
Concomitant Medications
Certain medications can cause an interaction with NOURIANZ. Advise patients to inform their healthcare provider about their smoking status and about all of the medicines they are taking or plan to take, including over-the-counter medicines, dietary supplements, and herbal products [see Drug Interactions (7.1, 7.2) and Use in Specific Populations (8.8)].
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 20 mg Tablet Bottle Label
- PRINCIPAL DISPLAY PANEL - 40 mg Tablet Bottle Label
-
INGREDIENTS AND APPEARANCE
NOURIANZ
istradefylline tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 42747-602 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength istradefylline (UNII: 2GZ0LIK7T4) (istradefylline - UNII:2GZ0LIK7T4) istradefylline 20 mg Inactive Ingredients Ingredient Name Strength Lactose Monohydrate (UNII: EWQ57Q8I5X) Microcrystalline Cellulose (UNII: OP1R32D61U) CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) Magnesium Stearate (UNII: 70097M6I30) Carnauba Wax (UNII: R12CBM0EIZ) Product Characteristics Color PINK (Peach) Score no score Shape RECTANGLE (pillow) Size 8mm Flavor Imprint Code 20 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 42747-602-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 08/27/2019 2 NDC: 42747-602-07 1 in 1 BOX 08/27/2019 2 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022075 08/27/2019 NOURIANZ
istradefylline tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 42747-604 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength istradefylline (UNII: 2GZ0LIK7T4) (istradefylline - UNII:2GZ0LIK7T4) istradefylline 40 mg Inactive Ingredients Ingredient Name Strength Lactose Monohydrate (UNII: EWQ57Q8I5X) Microcrystalline Cellulose (UNII: OP1R32D61U) CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) Magnesium Stearate (UNII: 70097M6I30) Carnauba Wax (UNII: R12CBM0EIZ) Product Characteristics Color PINK (Peach) Score no score Shape TEAR (almond) Size 10mm Flavor Imprint Code 40 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 42747-604-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 08/27/2019 2 NDC: 42747-604-07 1 in 1 BOX 08/27/2019 2 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022075 08/27/2019 Labeler - Kyowa Kirin, Inc. (014778321)


Trademark Results [NOURIANZ]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 NOURIANZ 87086056 not registered Live/Pending |
KYOWA KIRIN CO., LTD. 2016-06-28 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.