ORILISSA- elagolix tablet, film coated
Orilissa by
Drug Labeling and Warnings
Orilissa by is a Prescription medication manufactured, distributed, or labeled by AbbVie Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ORILISSA safely and effectively. See full prescribing information for ORILISSA.
ORILISSA® (elagolix) tablets, for oral use
Initial U.S. Approval: 2018INDICATIONS AND USAGE
ORILISSA is a gonadotropin-releasing hormone (GnRH) receptor antagonist indicated for the management of moderate to severe pain associated with endometriosis. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Oral tablets: 150 mg and 200 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Bone Loss: Dose- and duration-dependent decreases in bone mineral density (BMD) that may not be completely reversible. Assess BMD in women with additional risk factors for bone loss (5.1)
- Reduced Ability to Recognize Pregnancy: ORILISSA may alter menstrual bleeding, which may reduce the ability to recognize pregnancy. Perform testing if pregnancy is suspected. Discontinue if pregnancy is confirmed (5.2)
- Suicidal Ideation and Mood Disorders: Advise patients to seek medical attention for suicidal ideation, suicidal behavior, new onset or worsening depression, anxiety, or other mood changes (5.3)
- Hepatic Transaminase Elevations: Dose-dependent elevations in serum alanine aminotransferase (ALT). Counsel patients on signs and symptoms of liver injury (5.4)
- Potential for Reduced Efficacy with Estrogen-Containing Contraceptives: Use non-hormonal contraception during treatment and for one week after discontinuing ORILISSA (5.5)
ADVERSE REACTIONS
Most common adverse reactions (>5%) in clinical trials included hot flushes and night sweats, headache, nausea, insomnia, amenorrhea, anxiety, arthralgia, depression-related adverse reactions and mood changes (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact AbbVie Inc. at 1-800-633-9110 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
See full prescribing information for a list of clinically important drug interactions (7).
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 8/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing Information
2.2 Hepatic Impairment
2.3 Missed Dose
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Bone Loss
5.2 Change in Menstrual Bleeding Pattern and Reduced Ability to Recognize Pregnancy
5.3 Suicidal Ideation, Suicidal Behavior, and Exacerbation of Mood Disorders
5.4 Hepatic Transaminase Elevations
5.5 Reduced Efficacy with Estrogen-Containing Contraceptives
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Potential for ORILISSA to Affect Other Drugs
7.2 Potential for Other Drugs to Affect ORILISSA
7.3 Drug Interactions- Examples and Clinical Management
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing Information
- Exclude pregnancy before starting ORILISSA or start ORILISSA within 7 days from the onset of menses.
- Take ORILISSA at approximately the same time each day, with or without food.
- Use the lowest effective dose, taking into account the severity of symptoms and treatment objectives [see Warnings and Precautions (5.1, 5.3, 5.4) and Clinical Studies (14)].
- Limit the duration of use because of bone loss (Table 1) [see Warnings and Precautions (5.1)].
Table 1. Recommended Dosage and Duration of Use Dosing Regimen Maximum
Treatment
DurationCoexisting Condition Initiate treatment with
ORILISSA 150 mg once daily24 months None Consider initiating treatment with
ORILISSA 200 mg twice daily6 months Dyspareunia Initiate treatment with ORILISSA
150 mg once daily. Use of 200 mg
twice daily is not recommended.6 months Moderate hepatic impairment
(Child-Pugh Class B)2.2 Hepatic Impairment
No dosage adjustment of ORILISSA is required in women with mild hepatic impairment (Child-Pugh A).
Compared to women with normal liver function, those with moderate hepatic impairment had approximately 3-fold higher elagolix exposures and those with severe hepatic impairment had approximately 7-fold higher elagolix exposures. Because of these increased exposures and risk for bone loss:
- ORILISSA 150 mg once daily is recommended for women with moderate hepatic impairment (Child-Pugh B) with the duration of treatment limited to 6 months. Use of ORILISSA 200 mg twice daily is not recommended for women with moderate hepatic impairment [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
- ORILISSA is contraindicated in women with severe hepatic impairment (Child-Pugh C) [see Contraindications (4), Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
-
3 DOSAGE FORMS AND STRENGTHS
The 150 mg tablets are light pink, oblong, film-coated tablets with “EL 150” debossed on one side. Each tablet contains 155.2 mg of elagolix sodium equivalent to 150 mg of elagolix.
The 200 mg tablets are light orange, oblong, film-coated tablets with “EL 200” debossed on one side. Each tablet contains 207.0 mg of elagolix sodium equivalent to 200 mg of elagolix.
-
4 CONTRAINDICATIONS
ORILISSA is contraindicated in women:
- Who are pregnant [see Use in Specific Populations (8.1)]. Exposure to ORILISSA early in pregnancy may increase the risk of early pregnancy loss.
- With known osteoporosis because of the risk of further bone loss [see Warnings and Precautions (5.1)]
- With severe hepatic impairment [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3)]
- With concomitant use of strong organic anion transporting polypeptide (OATP) 1B1 inhibitors (e.g., cyclosporine and gemfibrozil) [see Drug Interactions (7.2)]
-
5 WARNINGS AND PRECAUTIONS
5.1 Bone Loss
ORILISSA causes a dose-dependent decrease in bone mineral density (BMD). BMD loss is greater with increasing duration of use and may not be completely reversible after stopping treatment [see Adverse Reactions (6.1)]. The impact of these BMD decreases on long-term bone health and future fracture risk are unknown. Consider assessment of BMD in patients with a history of a low-trauma fracture or other risk factors for osteoporosis or bone loss, and do not use in women with known osteoporosis. Limit the duration of use to reduce the extent of bone loss [see Dosage and Administration (2.2)].
Although the effect of supplementation with calcium and vitamin D was not studied, such supplementation may be beneficial for all patients.
5.2 Change in Menstrual Bleeding Pattern and Reduced Ability to Recognize Pregnancy
Women who take ORILISSA may experience a reduction in the amount, intensity or duration of menstrual bleeding, which may reduce the ability to recognize the occurrence of a pregnancy in a timely manner [see Adverse Reactions (6.1)]. Perform pregnancy testing if pregnancy is suspected, and discontinue ORILISSA if pregnancy is confirmed.
5.3 Suicidal Ideation, Suicidal Behavior, and Exacerbation of Mood Disorders
Suicidal ideation and behavior, including one completed suicide, occurred in subjects treated with ORILISSA in the endometriosis clinical trials. ORILISSA subjects had a higher incidence of depression and mood changes compared to placebo, and ORILISSA subjects with a history of suicidality or depression had a higher incidence of depression compared to subjects without such a history [see Adverse Reactions (6.1)]. Promptly evaluate patients with depressive symptoms to determine whether the risks of continued therapy outweigh the benefits [see Adverse Reactions (6.1)]. Patients with new or worsening depression, anxiety or other mood changes should be referred to a mental health professional, as appropriate. Advise patients to seek immediate medical attention for suicidal ideation and behavior. Reevaluate the benefits and risks of continuing ORILISSA if such events occur.
5.4 Hepatic Transaminase Elevations
In clinical trials, dose-dependent elevations of serum alanine aminotransferase (ALT) at least 3-times the upper limit of the reference range occurred with ORILISSA. Use the lowest effective dose of ORILISSA and instruct patients to promptly seek medical attention in case of symptoms or signs that may reflect liver injury, such as jaundice. Promptly evaluate patients with elevations in liver tests to determine whether the benefits of continued therapy outweigh the risks [see Adverse Reactions (6.1)].
5.5 Reduced Efficacy with Estrogen-Containing Contraceptives
Based on the mechanism of action of ORILISSA, estrogen containing contraceptives are expected to reduce the efficacy of ORILISSA. The effect of progestin-only contraceptives on the efficacy of ORILISSA is unknown. Advise women to use non-hormonal contraceptives during treatment with ORILISSA and for one week after discontinuing ORILISSA [see Use in Specific Populations (8.1, 8.3), Drug Interactions (7.3), Clinical Pharmacology (12.3)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed elsewhere in labeling:
- Bone loss [see Warnings and Precautions (5.1)]
- Change in menstrual bleeding pattern and reduced ability to recognize pregnancy [see Warnings and Precautions (5.2)]
- Suicidal ideation, suicidal behavior, and exacerbation of mood disorders [see Warnings and Precautions (5.3)]
- Hepatic transaminase elevations [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of ORILISSA was evaluated in two six-month, randomized, double-blind, placebo-controlled clinical trials [EM-1 (NCT01620528) and EM-2 (NCT01931670)] in which a total of 952 adult women with moderate to severe pain associated with endometriosis were treated with ORILISSA (475 with 150 mg once daily and 477 with 200 mg twice daily) and 734 were treated with placebo. The population age range was 18-49 years old. Women who completed six months of treatment and met eligibility criteria continued treatment in two uncontrolled, blinded six-month extension trials [EM-3 (NCT01760954) and EM-4 (NCT02143713)], for a total treatment duration of up to 12 months.
Overall, the most common serious adverse events reported for subjects treated with ORILISSA in the two placebo-controlled clinical trials (Studies EM-1 and EM-2) included appendicitis (0.3%), abdominal pain (0.2%), and back pain (0.2%). In these trials, 0.2% of subjects treated with ORILISSA 150 mg once daily and 0.2% of subjects treated with ORILISSA 200 mg twice daily discontinued therapy due to serious adverse reactions compared to 0.5% of those given placebo.
Adverse Reactions Leading to Study Discontinuation
In the two placebo-controlled clinical trials (Studies EM-1 and EM-2), 5.5% of subjects treated with ORILISSA 150 mg once daily and 9.6% of subjects treated with ORILISSA 200 mg twice daily discontinued therapy due to adverse reactions compared to 6.0% of those given placebo. Discontinuations were most commonly due to hot flushes or night sweats (1.1% with 150 mg once daily and 2.5% with 200 mg twice daily) and nausea (0.8% with 150 mg once daily and 1.5% with 200 mg twice daily) and were dose-related. The majority of discontinuations due to hot flushes or night sweats (10 of 17, 59%) and nausea (7 of 11, 64%) occurred within the first 2 months of therapy.
In the two extension trials (Studies EM-3 and EM-4), discontinuations were most commonly due to decreased BMD and were dose-related. In these trials, 0.3% of subjects treated with ORILISSA 150 mg once daily and 3.6% of subjects treated with ORILISSA 200 mg twice daily discontinued therapy due to decreased BMD.
Adverse reactions reported in ≥ 5% of women in the two placebo-controlled trials in either ORILISSA dose group and at a greater frequency than placebo are noted in the following table.
Table 2. Percentage of Subjects in Studies EM-1 and EM-2 with Treatment-Emergent Adverse Reactions Occurring in at Least 5% of Subjects (either ORILISSA Dose Group) and at a Greater Incidence than with Placebo ORILISSA
150 mg Once Daily
N=475ORILISSA
200 mg Twice Daily
N=477Placebo
N=734% % % Hot Flush or Night Sweats 24 46 9 Headache 17 20 12 Nausea 11 16 13 Insomnia 6 9 3 Mood altered, mood swings 6 5 3 Amenorrhea 4 7 <1 Depressed mood, depression, depressive
symptoms and/or tearfulness3 6 2 Anxiety 3 5 3 Arthralgia 3 5 3 Less Common Adverse Reactions:
In Study EM-1 and Study EM-2, adverse reactions reported in ≥ 3% and < 5% in either ORILISSA dose group and greater than placebo included: decreased libido, diarrhea, abdominal pain, weight gain, dizziness, constipation and irritability.
The most commonly reported adverse reactions in the extension trials (EM-3 and EM-4) were similar to those in the placebo-controlled trials.
The effect of ORILISSA on BMD was assessed by dual-energy X-ray absorptiometry (DXA).
In Studies EM-1 and EM-2, there was a dose-dependent decrease in BMD in ORILISSA-treated subjects compared to an increase in placebo-treated subjects.
In Study EM-1, compared to placebo, the mean change from baseline in lumbar spine BMD at 6 months was -0.9% (95% CI: -1.3, -0.4) with ORILISSA 150 mg once daily and -3.1% (95% CI: -3.6, -2.6) with ORILISSA 200 mg twice daily (Table 3). The percentage of subjects with greater than 8% BMD decrease in lumbar spine, total hip or femoral neck at any time point during the placebo-controlled treatment period was 2% with ORILISSA 150 mg once daily, 7% with ORILISSA 200 mg twice daily and < 1% with placebo. In the blinded extension Study EM-3, continued bone loss was observed with 12 months of continuous treatment with ORILISSA. The percentage of subjects with greater than 8% BMD decrease in lumbar spine, total hip or femoral neck at any time point during the extension treatment period was 8% with continuous ORILISSA 150 mg once daily and 21% with continuous ORILISSA 200 mg twice daily.
In Study EM-2, compared to placebo, the mean change from baseline in lumbar spine BMD at 6 months was -1.3% (95% CI: -1.8, -0.8) with ORILISSA 150 mg once daily and -3.0% (95% CI: -3.5, -2.6) with ORILISSA 200 mg twice daily (Table 3). The percentage of subjects with greater than 8% BMD decrease in lumbar spine, total hip or femoral neck at any time point during the placebo-controlled treatment period was < 1% with ORILISSA 150 mg once daily, 6% with ORILISSA 200 mg twice daily and 0% with placebo. In the blinded extension Study EM-4, continued bone loss was observed with 12 months of continuous treatment with ORILISSA. The percentage of subjects with greater than 8% BMD decrease in lumbar spine, total hip or femoral neck at any time point during the extension treatment period was 2% with continuous ORILISSA 150 mg once daily and 21% with continuous ORILISSA 200 mg twice daily.
Table 3. Percent Change from Baseline in Lumbar Spine BMD at Month 6 ORILISSA
150 mg
Once DailyORILISSA
200 mg
Twice DailyPlacebo EM-1 N 183 180 277 Percent Change from Baseline, % -0.3 -2.6 0.5 Treatment Difference, % (95% CI) -0.9
(-1.3, -0.4)-3.1
(-3.6, -2.6)EM-2 N 174 183 271 Percent Change from Baseline, % -0.7 -2.5 0.6 Treatment Difference, % (95% CI) -1.3
(-1.8, -0.8)-3.0
(-3.5, -2.6)To assess for recovery, the change in lumbar spine BMD over time was analyzed for subjects who received continuous treatment with ORILISSA 150 mg once daily or ORILISSA 200 mg twice daily for up to 12 months and who were then followed after cessation of therapy for an additional 6 months. Partial recovery of BMD was seen in these subjects (Figure 1).
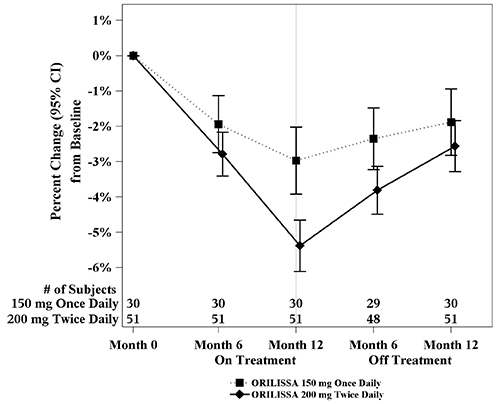
In Study EM-3, if a subject had BMD loss of more than 1.5% at the lumbar spine or more than 2.5% at the total hip at the end of treatment, follow-up DXA was required after 6 months off-treatment. In Study EM-4, all subjects were required to have a follow-up DXA 6 months off treatment regardless of change in BMD and if a subject had BMD loss of more than 1.5% at the lumbar spine or more than 2.5% at the total hip after 6 months off treatment, follow-up DXA was required after 12 months off-treatment. Figure 2 shows the change in lumbar spine BMD for the subjects in Study EM-2/EM-4 who completed 12 months of treatment with ORILISSA and who had a follow-up DXA 12-months off treatment.
Figure 1. Percent Change from Baseline in Lumbar Spine BMD in Subjects Who Received 12 Months of ORILISSA and Had Follow-up BMD 6 Months off Therapy in Studies EM-2/EM-4
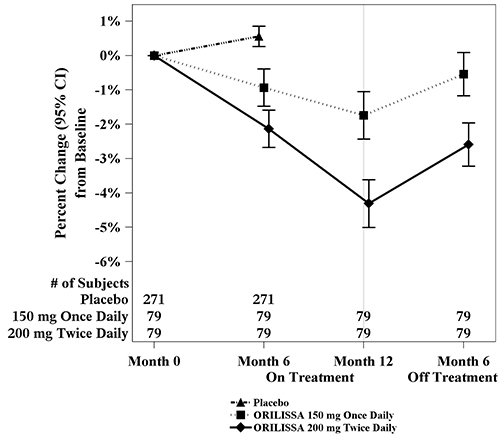
Figure 2. Percent Change from Baseline in Lumbar Spine BMD in Subjects Who Received 12 Months of ORILISSA and Had Follow-up BMD 12 Months off Therapy in Studies EM-2/EM-4
Suicidal Ideation, Suicidal Behavior and Exacerbation of Mood Disorders
In the placebo-controlled trials (Studies EM-1 and EM-2), ORILISSA was associated with adverse mood changes (see Table 2 and Table 4), particularly in those with a history of depression.
Table 4. Suicidal Ideation and Suicidal Behavior in Studies EM-1 and EM-2 Adverse Reactions ORILISSA Placebo
(N=734)
n (%)150 mg
Once Daily
(N=475)
n (%)200 mg
Twice Daily
(N=477)
n (%)Completed suicide 1 (0.2) 0 0 Suicidal ideation 1 (0.2) 1 (0.2) 0 A 44-year-old woman received 31 days of ORILISSA 150 mg once daily then completed suicide 2 days after ORILISSA discontinuation. She had no relevant past medical history; life stressors were noted.
Among the 2090 subjects exposed to ORILISSA in the endometriosis Phase 2 and Phase 3 studies, there were four reports of suicidal ideation. In addition to the two subjects in Table 4, there were two additional reports of suicidal ideation: one subject in EM-3 (150 mg once daily) and one in a Phase 2 study (75 mg once daily, an unapproved dose). Three of these subjects had a history of depression. Two subjects discontinued ORILISSA and two completed the clinical trial treatment periods.
Hepatic Transaminase Elevations
In the placebo-controlled clinical trials (Studies EM-1 and EM-2), dose-dependent asymptomatic elevations of serum ALT to at least 3-times the upper limit of the reference range occurred during treatment with ORILISSA (150 mg once daily – 1/450, 0.2%; 200 mg twice daily – 5/443, 1.1%; placebo – 1/696, 0.1%). Similar increases were seen in the extension trials (Studies EM-3 and EM-4).
Dose-dependent increases in total cholesterol, low-density lipoprotein cholesterol (LDL-C), high density lipoprotein cholesterol (HDL-C), and serum triglycerides were noted during ORILISSA treatment in EM-1 and EM-2. In EM-1 and EM-2, 12% and 1% of subjects with mildly elevated LDL-C (130-159 mg/dL) at baseline had an increase in LDL-C concentrations to 190 mg/dL or higher during treatment with ORILISSA and placebo, respectively. In EM-1 and EM-2, 4% and 1% of subjects with mildly elevated serum triglycerides (150-300 mg/dL) at baseline had an increase in serum triglycerides to at least 500 mg/dL during treatment with ORILISSA and placebo, respectively. The highest measured serum triglyceride concentration during treatment with ORILISSA was 982 mg/dL.
Table 5. Mean Change and Maximum Increase from Baseline in Serum Lipids in Studies EM-1 and EM-2 ORILISSA
150 mg
Once Daily
N=475ORILISSA
200 mg
Twice Daily
N=477Placebo
N=734LDL-C (mg/dL) Mean change at Month 6 5 13 -3 Maximum increase during
Treatment Period137 107 122 HDL-C (mg/dL) Mean change at Month 6 2 4 1 Maximum increase during
Treatment Period43 52 45 Triglycerides (mg/dL) Mean change at Month 6 <1 11 -3 Maximum increase during
Treatment Period624 484 440 Lipid increases occurred within 1 to 2 months after the start of ORILISSA and remained stable thereafter over 12 months.
In Studies EM-1 and EM-2, non-serious hypersensitivity reactions including rash occurred in 5.8% of ORILISSA treated-subjects and 6.1% of placebo-treated subjects. These events led to study drug discontinuation in 0.4% of ORILISSA-treated subjects and 0.5% of placebo-treated subjects.
Endometrial biopsies were performed in subjects in Study EM-1 and its extension at Month 6 and Month 12. These biopsies showed a dose-dependent decrease in proliferative and secretory biopsy patterns and an increase in quiescent/minimally stimulated biopsy patterns. There were no abnormal biopsy findings on treatment, such as endometrial hyperplasia or cancer.
Based on transvaginal ultrasound, during the course of a 3-menstrual cycle study in healthy women, ORILISSA 150 mg once daily and 200 mg twice daily resulted in a dose-dependent decrease from baseline in mean endometrial thickness.
Effects on menstrual bleeding patterns
The effects of ORILISSA on menstrual bleeding were evaluated for up to 12 months using an electronic daily diary where subjects classified their flow of menstrual bleeding (if present in the last 24 hours) as spotting, light, medium, or heavy. ORILISSA led to a dose-dependent reduction in mean number of bleeding and spotting days and bleeding intensity in those subjects who reported menstrual bleeding.
Table 6. Mean Bleeding/Spotting Days and Mean Intensity Scores at Month 3 ORILISSA
150mg Once DailyORILISSA
200mg Twice DailyPlacebo Baseline Month 3 Baseline Month 3 Baseline Month 3 Mean bleeding/
spotting days
in prior 28 days5.3 2.8 5.7 0.8 5.4 4.6 Mean Intensity
scorea2.6 2.2 2.5 2.0 2.6 2.4 aIntensity for subjects who reported at least 1 day of bleeding or spotting during 28 day interval. Scale ranges from 1 to 4, 1 = spotting, 2 = light, 3 = medium, 4 = heavy ORILISSA also demonstrated a dose-dependent increase in the percentage of women with amenorrhea (defined as no bleeding or spotting in a 56-day interval) over the treatment period. The incidence of amenorrhea during the first six months of treatment ranged from 6-17% for ORILISSA 150 mg once daily, 13-52% for ORILISSA 200 mg twice daily and less than 1% for placebo. During the second 6 months of treatment, the incidence of amenorrhea ranged from 11-15% for ORILISSA 150 mg once daily and 46-57% for ORILISSA 200 mg twice daily.
After 6 months of therapy with ORILISSA 150 mg once daily, resumption of menses after stopping treatment was reported by 59%, 87% and 95% of women within 1, 2, and 6 months, respectively. After 6 months of therapy with ORILISSA 200 mg twice daily, resumption of menses after stopping treatment was reported by 60%, 88%, and 97% of women within 1, 2, and 6 months, respectively.
After 12 months of therapy with ORILISSA 150 mg once daily resumption of menses after stopping treatment was reported by 77%, 95% and 98% of women within 1, 2, and 6 months respectively. After 12 months of therapy with ORILISSA 200 mg twice daily resumption of menses after stopping treatment was reported by 55%, 91% and 96% of women within 1, 2, and 6 months respectively.
-
7 DRUG INTERACTIONS
7.1 Potential for ORILISSA to Affect Other Drugs
Elagolix is a weak to moderate inducer of cytochrome P450 (CYP) 3A. Co-administration with ORILISSA may decrease plasma concentrations of drugs that are substrates of CYP3A.
Elagolix is a weak inhibitor of CYP 2C19. Co-administration with ORILISSA may increase plasma concentrations of drugs that are substrates of CYP2C19 (e.g., omeprazole).
Elagolix is an inhibitor of efflux transporter P-glycoprotein (P-gp). Co-administration with ORILISSA may increase plasma concentrations of drugs that are substrates of P-gp (e.g., digoxin).
7.2 Potential for Other Drugs to Affect ORILISSA
Elagolix is a substrate of CYP3A, P-gp, and OATP1B1.
Concomitant use of ORILISSA 200 mg twice daily and strong CYP3A inhibitors for more than 1 month is not recommended. Limit concomitant use of ORILISSA 150 mg once daily and strong CYP3A inhibitors to 6 months.
Co-administration of ORILISSA with drugs that induce CYP3A may decrease elagolix plasma concentrations.
The effect of concomitant use of P-gp inhibitors or inducers on the pharmacokinetics of ORILISSA is unknown. Co-administration of ORILISSA with drugs that inhibit OATP1B1 may increase elagolix plasma concentrations. Concomitant use of ORILISSA and strong OATP1B1 inhibitors (e.g., cyclosporine and gemfibrozil) is contraindicated.
7.3 Drug Interactions- Examples and Clinical Management
Table 7 summarizes the effect of co-administration of ORILISSA on concentrations of concomitant drugs and the effect of concomitant drugs on ORILISSA.
Table 7. Established Drug Interactions Based on Drug Interaction Trials Concomitant Drug
Class:
Drug NameEffect on
Plasma
Exposure of
Elagolix or
Concomitant
DrugClinical Recommendations Antiarrhythmics
digoxin↑ digoxin Clinical monitoring is recommended for digoxin when co-administered with ORILISSA. Antimycobacterial
rifampin↑ elagolix Concomitant use of ORILISSA 200 mg twice daily and rifampin is not recommended. Limit concomitant use of ORILISSA 150 mg once daily and rifampin to 6 months. Benzodiazepines
oral midazolam↓ midazolam Consider increasing the dose of midazolam and individualize therapy based on the patient’s response. Statins
rosuvastatin↓ rosuvastatin Consider increasing the dose of rosuvastatin. Proton pump
inhibitors
omeprazole↑ omeprazole No dose adjustments are needed for omeprazole at doses of 40 mg once daily or lower. When ORILISSA is used concomitantly with higher doses of omeprazole, e.g. in patients with Zollinger-Ellison syndrome, consider dosage reduction of omeprazole. See Clinical Pharmacology, Tables 10 and 11.
The direction of the arrow indicates the direction of the change in the area under the curve (AUC) (↑= increase, ↓ = decrease). -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy registry that monitors outcomes in women who become pregnant while treated with ORILISSA. Patients should be encouraged to enroll by calling 1-833-782-7241.Exposure to ORILISSA early in pregnancy may increase the risk of early pregnancy loss. Use of ORILISSA is contraindicated in pregnant women. Discontinue ORILISSA if pregnancy occurs during treatment.
The limited human data with the use of ORILISSA in pregnant women are insufficient to determine whether there is a risk for major birth defects or miscarriage. Although two cases of congenital malformations were reported in clinical trials with ORILISSA, no pattern was identified and miscarriages were reported at a similar incidence across treatment groups (see Data).
When pregnant rats and rabbits were orally dosed with elagolix during the period of organogenesis, postimplantation loss was observed in pregnant rats at doses 20 times the maximum recommended human dose (MRHD). Spontaneous abortion and total litter loss was observed in rabbits at doses 7 and 12 times the MRHD. There were no structural abnormalities in the fetuses at exposures up to 40 and 12 times the MRHD for the rat and rabbit, respectively (see Data).
The background risk for major birth defects and miscarriage in the indicated population are unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
There were 49 pregnancies reported in clinical trials of more than 3,500 women (of whom more than 2,000 had endometriosis) treated with ORILISSA for up to 12 months. These pregnancies occurred while the women were receiving ORILISSA or within 30 days after stopping ORILISSA. Among these 49 pregnancies, two major congenital malformations were reported. In one case of infant cleft palate, the mother was treated with ORILISSA 150 mg daily and the estimated fetal exposure to ORILISSA occurred during the first 30 days of pregnancy. In one case of infant tracheoesophageal fistula, the mother was treated with ORILISSA 150 mg daily and the estimated fetal exposure to ORILISSA occurred during the first 15 days of pregnancy.
Among these 49 pregnancies, there were five cases of spontaneous abortion (miscarriage) compared to five cases among the 20 pregnancies that occurred in more than 1100 women treated with placebo. Although the duration of fetal exposure was limited in ORILISSA clinical trials, there were no apparent decreases in birth weights associated with ORILISSA in comparison to placebo.
Embryofetal development studies were conducted in the rat and rabbit. Elagolix was administered by oral gavage to pregnant rats (25 animals/dose) at doses of 0, 300, 600 and 1200 mg/kg/day and to rabbits (20 animals/dose) at doses of 0, 100, 150, and 200 mg/kg/day, during the period of organogenesis (gestation day 6-17 in the rat and gestation day 7-20 in the rabbit).
In rats, maternal toxicity was present at all doses and included six deaths and decreases in body weight gain and food consumption. Increased postimplantation losses were present in the mid dose group, which was 20 times the MRHD based on AUC. In rabbits, three spontaneous abortions and a single total litter loss were observed at the highest, maternally toxic dose, which was 12 times the MRHD based on AUC. A single total litter loss occurred at a lower non-maternally toxic dose of 150 mg/kg/day, which was 7 times the MRHD.
No fetal malformations were present at any dose level tested in either species even in the presence of maternal toxicity. At the highest doses tested, the exposure margins were 40 and 12 times the MRHD for the rat and rabbit, respectively. However, because elagolix binds poorly to the rat gonadotropin-releasing hormone (GnRH) receptor (~1000 fold less than to the human GnRH receptor), the rat study is unlikely to identify pharmacologically mediated effects of elagolix on embryofetal development. The rat study is still expected to provide information on potential non-target-related effects of elagolix.
In a pre- and postnatal development study in rats, elagolix was given in the diet to achieve doses of 0, 100 and 300 mg/kg/day (25 per dose group) from gestation day 6 to lactation day 20. There was no evidence of maternal toxicity. At the highest dose, two dams had total litter loss, and one failed to deliver. Pup survival was decreased from birth to postnatal day 4. Pups had lower birth weights and lower body weight gains were observed throughout the pre-weaning period at 300 mg/kg/day. Smaller body size and effect on startle response were associated with lower pup weights at 300 mg/kg/day. Post-weaning growth, development and behavioral endpoints were unaffected.
Maternal plasma concentrations in rats on lactation day 21 at 100 and 300 mg/kg/day (47 and 125 ng/mL) were 0.06-fold and 0.16-fold the maximal elagolix concentration (Cmax) in humans at the MRHD. Because the exposures achieved in rats were much lower than the human MRHD, this study is not predictive of potentially higher lactational exposure in humans.
8.2 Lactation
There is no information on the presence of elagolix or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. There are no adequate animal data on the excretion of ORILISSA in milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ORILISSA and any potential adverse effects on the breastfed child from ORILISSA.
There are no adequate animal data on excretion of ORILISSA in milk.
8.3 Females and Males of Reproductive Potential
Based on the mechanism of action, there is a risk of early pregnancy loss if ORILISSA is administered to a pregnant woman [see Use in Specific Populations (8.1), Clinical Pharmacology (12.1)].
Exclude pregnancy before initiating treatment with ORILISSA. Perform pregnancy testing if pregnancy is suspected during treatment with ORILISSA [see Warnings and Precautions (5.2)].
Advise women to use effective non-hormonal contraception during treatment with ORILISSA and for one week after discontinuing ORILISSA [see Warnings and Precautions (5.2) and Drug Interactions (7.3)].
8.4 Pediatric Use
Safety and effectiveness of ORILISSA in patients less than 18 years of age have not been established.
8.6 Renal Impairment
No dose adjustment of ORILISSA is required in women with any degree of renal impairment or end-stage renal disease (including women on dialysis) [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dosage adjustment of ORILISSA is required for women with mild hepatic impairment (Child-Pugh A). Only the 150 mg once daily regimen is recommended for women with moderate hepatic impairment (Child-Pugh B) and the duration of treatment should be limited to 6 months.
ORILISSA is contraindicated in women with severe hepatic impairment (Child-Pugh C) [see Contraindications (4), and Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
ORILISSA (elagolix) tablets for oral administration contain elagolix sodium, the sodium salt of the active moiety elagolix. Elagolix sodium is a nonpeptide small molecule, GnRH receptor antagonist. Elagolix sodium is chemically described as sodium 4-({(1R)-2-[5-(2-fluoro-3-methoxyphenyl)-3-{[2-fluoro-6-(trifluoromethyl)phenyl]methyl}-4-methyl-2,6-dioxo-3,6-dihydropyrimidin-1(2H)-yl]-1-phenylethyl}amino)butanoate. Elagolix sodium has a molecular formula of C32H29F5N3O5Na and a molecular weight of 653.58. Elagolix free acid has a molecular weight of 631.60.
Elagolix sodium has the following structural formula:
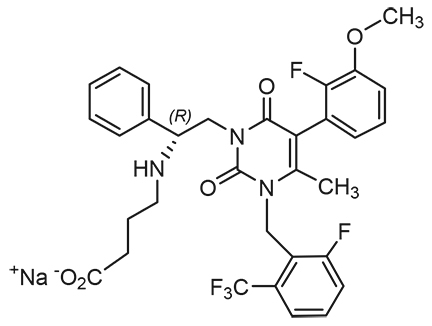
Elagolix sodium is a white to off white to light yellow powder and is freely soluble in water.
ORILISSA 150 mg tablets are light pink, oblong, film-coated tablets with “EL 150” debossed on one side. Each tablet contains 155.2 mg of elagolix sodium (equivalent to 150 mg of elagolix) as the active ingredient and the following inactive ingredients: mannitol, sodium carbonate monohydrate, pregelatinized starch, povidone, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and carmine high tint.
ORILISSA 200 mg tablets are light orange, oblong, film-coated tablets with “EL 200” debossed on one side. Each tablet contains 207.0 mg of elagolix sodium (equivalent to 200 mg of elagolix) as the active ingredient and the following inactive ingredients: mannitol, sodium carbonate monohydrate, pregelatinized starch, povidone, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide red.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
ORILISSA is a GnRH receptor antagonist that inhibits endogenous GnRH signaling by binding competitively to GnRH receptors in the pituitary gland. Administration of ORILISSA results in dose-dependent suppression of luteinizing hormone (LH) and follicle-stimulating hormone (FSH), leading to decreased blood concentrations of the ovarian sex hormones, estradiol and progesterone.
12.2 Pharmacodynamics
Effect on Ovulation and Estradiol
In a 3-menstrual cycle study in healthy women, ORILISSA 150 mg once daily and 200 mg twice daily resulted in an ovulation rate of approximately 50% and 32%, respectively. In the Phase 3 trials in women with endometriosis, ORILISSA caused a dose-dependent reduction in median estradiol concentrations to approximately 42 pg/mL for 150 mg once daily regimen and 12 pg/mL for the 200 mg twice daily regimen.
The effect of elagolix on the QTc interval was evaluated in a randomized, placebo- and positive-controlled, open-label, single-dose, crossover thorough QTc study in 48 healthy adult premenopausal women. Elagolix concentrations in subjects given a single dose of 1200 mg was 17-times higher than the concentration in subjects given elagolix 200 mg twice daily. There was no clinically relevant prolongation of the QTc interval.
12.3 Pharmacokinetics
The pharmacokinetic properties of ORILISSA in healthy subjects are summarized in Table 8. The steady state pharmacokinetic parameters under fasting conditions are summarized in Table 9.
Table 8. Pharmacokinetic Properties of ORILISSA in Healthy Subjects Absorption Tmax (h) 1.0 Effect of high-fat meal (relative to fasting) AUC: ↓24%, Cmax: ↓36% Distribution % Bound to human plasma proteins 80 Blood-to-plasma ratio 0.6 Metabolism Metabolism CYP3A (major)
Minor pathways include: CYP2D6, CYP2C8, and uridine glucuronosyl transferases (UGTs)Elimination Major route of elimination Hepatic metabolism Terminal phase elimination
half-life (t1/2) (h)4-6 % of dose excreted in urine <3 % of dose excreted in feces 90 Table 9. Mean (%CV) Steady State Pharmacokinetic Parameters of ORILISSA Pharmacokinetic Parameter
(Units)150 mg Once Daily
N = 6200 mg Twice Daily
N = 7Cmax (ng/mL) 574 (29) 774 (68) AUCτ (ng●hr/mL) 1292 (31) 1725 (57) CL/F (L/hr) 123 (21) 144 (43) Vdss/F 1674 (94) 881 (38) Rac 0.98 (7) 0.89 (19) CV: Coefficient of variation
Cmax: peak concentration
AUCτ: area under the plasma concentration-time curve during the dosing interval (τ) i.e., 12 hours for twice daily regimen, 24 hours for once daily regimen.
CL/F: oral clearance
Vdss/F: apparent volume of distribution at steady state
Rac: drug accumulation ratioElagolix exposures (Cmax and AUC) are not altered by renal impairment. The mean exposures are similar for women with moderate to severe or end stage renal disease (including women on dialysis) compared to women with normal renal function.
Elagolix exposures (Cmax and AUC) are similar between women with normal hepatic function and women with mild hepatic impairment. Elagolix exposures in women with moderate and severe hepatic impairment are approximately 3-fold and 7-fold, respectively, higher than exposures from women with normal hepatic function [see Use in Specific Populations (8.7)].
No clinically meaningful difference in the pharmacokinetics of ORILISSA between White and Black subjects or between Hispanics and others was observed. There is no clinically meaningful difference in the pharmacokinetics of ORILISSA between Japanese and Han Chinese subjects.
Body weight or body mass index does not affect the pharmacokinetics of ORILISSA.
Drug interaction studies were performed with ORILISSA and other drugs that are likely to be co-administered and with drugs commonly used as probes for pharmacokinetic interactions. Tables 10 and 11 summarize the pharmacokinetic effects when elagolix was co-administered with these drugs.
Table 10. Drug Interactions: Change in Pharmacokinetics of Elagolix in the Presence of Co-administered Drugs Co-
administered
DrugRegimen
of Co-
administered
DrugRegimen
of
ElagolixN Ratio (90% CI)* Ketoconazole 400 mg once
daily150 mg single
dose11 Cmax AUC 1.77
(1.48 – 2.12)2.20
(1.98 – 2.44)Rifampin 600 mg single
dose150 mg single
dose12 4.37
(3.62 – 5.28)5.58
(4.88 – 6.37)600 mg once
daily2.00
(1.66 – 2.41)1.65
(1.45 – 1.89)CI: Confidence interval
*ratios for Cmax and AUC compare co-administration of the medication with elagolix vs. administration of elagolix alone.No clinically significant changes in elagolix exposures were observed when co-administered with rosuvastatin (20 mg once daily), sertraline (25 mg once daily) or fluconazole (200 mg single dose).
Table 11. Drug Interactions: Change in Pharmacokinetics of Co-administered Drug in the Presence of Elagolix Co-
administered
DrugRegimen
of Co-
administered
DrugRegimen
of
ElagolixN Ratio (90% CI)* Digoxin 0.5 mg single
dose200 mg
twice daily
x 10 days11 Cmax AUC 1.71
(1.53 – 1.91)1.26
(1.17 – 1.35)Rosuvastatin 20 mg once
daily300 mg
twice daily
x 7 days10 0.99
(0.73 – 1.35)0.60
(0.50 – 0.71)Midazolam 2 mg single
dose300 mg
twice daily
x 11 days20 0.56
(0.51 – 0.62)0.46
(0.41 – 0.50)150 mg
once daily
x 13 days11 0.81
(0.74 – 0.89)0.65
(0.58 – 0.72)Norethindrone 0.35 mg once
daily x 112 days150 mg
once daily
x 56 days32 0.95
(0.86 – 1.05)0.88
(0.79 – 0.99)Ethinyl Estradiol Ethinyl estradiol
35 mcg and
triphasic
norgestimate
0.18/0.215/0.25
mg once daily150 mg
once daily21 1.15
(1.07 – 1.25)1.30
(1.19 – 1.42)Norelgestromina 0.87
(0.78 – 0.97)0.85
(0.78 – 0.92)Norgestrela 0.89
(0.78 – 1.00)0.92
(0.84 – 1.01)Omeprazole 40 mg single
dose300 mg
twice daily
x 9 days20 1.95
(1.50 – 2.53)1.78
(1.39 – 2.27)CI: Confidence interval
*ratios for Cmax and AUC compare co-administration of the medication with elagolix vs. administration of the medication alone.
a metabolite of norgestimateNo clinically significant changes in sertraline or fluconazole exposures were observed when co-administered with elagolix.
12.5 Pharmacogenomics
Disposition of elagolix involves the OATP 1B1 transporter protein. Higher plasma concentrations of elagolix have been observed in groups of patients who have two reduced function alleles of the gene that encodes OATP 1B1 (SLCO1B1 521T>C). The frequency of this SLCO1B1 521 C/C genotype is generally less than 5% in most racial/ethnic groups. Subjects with this genotype are expected to have a 78% mean increase in elagolix concentrations compared to subjects with normal transporter function (i.e., SLCO1B1 521T/T genotype).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year carcinogenicity studies conducted in mice (50, 150, or 500 mg/kg/day) and rats (150, 300, or 800 mg/kg/day) that administered elagolix by the dietary route revealed no increase in tumors in mice at up to 19-fold the MRHD based on AUC. In the rat, there was an increase in thyroid (male and female) and liver (males only) tumors at the high dose (12 to 13-fold the MRHD). The rat tumors were likely species-specific and of negligible relevance to humans.
Elagolix was not genotoxic or mutagenic in a battery of tests, including the in vitro bacterial reverse mutation assay, the in vitro mammalian cell forward mutation assay at the thymidine kinase (TK+/-) locus in L5178Y mouse lymphoma cells, and the in vivo mouse micronucleus assay.
In a fertility study conducted in the rat, there was no effect of elagolix on fertility at any dose (50, 150, or 300 mg/kg/day). Based on AUC, the exposure multiple for the MRHD in women compared to the highest dose of 300 mg/kg/day in female rats is approximately 5-fold. However, because elagolix has low affinity for the GnRH receptor in the rat [see Use in Specific Populations (8.1)], and because effects on fertility are most likely to be mediated via the GnRH receptor, these data have low relevance to humans.
-
14 CLINICAL STUDIES
The efficacy of ORILISSA 150 mg once daily and 200 mg twice daily for the management of moderate to severe pain associated with endometriosis was demonstrated in two multinational double-blind, placebo-controlled trials in 1686 premenopausal women [Study EM-1 (NCT01620528) and Study EM-2 (NCT01931670)]. The median age of women in the trials was 32 years; 88% were White, 9% were Black or African American and 3% were other races. Each placebo-controlled trial assessed the reduction in endometriosis-associated pain over 6 months of treatment.
Moderate to severe pain associated with endometriosis was required for entry into the trials and was assessed during screening using the composite pelvic signs and symptoms score (CPSSS) and other baseline criteria.
The CPSSS is based on a modified Biberoglu and Behrman scale with five elements: three responses reported by study subjects (dysmenorrhea, dyspareunia, and non-menstrual pelvic pain) and two findings based on investigator assessment during physical examination (rating of pelvic tenderness and induration). Each element is scored from 0 (absent) to 3 (severe) for a maximum total score of 15. A total score of at least 6, with a score of at least 2 for dysmenorrhea and at least 2 for non-menstrual pelvic pain was required to qualify for randomization. Subjects were also required to have non-menstrual pelvic pain for at least four days in the preceding calendar month, defined as 35 days. Other criteria to determine eligibility for randomization included subject responses in a daily electronic diary (Endometriosis Daily Pain Impact Scale, described below) for both dysmenorrhea and non-menstrual pelvic pain in the 35 days prior to randomization.
Dysmenorrhea and Non-Menstrual Pelvic Pain
The co-primary efficacy endpoints were (1) the proportion of subjects whose dysmenorrhea responded to treatment at Month 3 and (2) the proportion of subjects whose pelvic pain not related to menses (also known as non-menstrual pelvic pain) responded to treatment at Month 3. Dysmenorrhea and non-menstrual pelvic pain were evaluated daily using the Endometriosis Daily Pain Impact Scale that asked subjects to rate their pain severity and its impact on daily activities during the prior 24 hours as none, mild, moderate or severe (correlating with a score of 0 to 3, respectively, where higher scores indicated greater severity). Scores at baseline and at each month were averaged over a 35-day interval.
Women were defined as responders if they experienced a reduction in dysmenorrhea and non-menstrual pelvic pain as defined in Table 12 with no increase in analgesic use (nonsteroidal anti-inflammatory drug or opioid) for endometriosis-associated pain. The threshold for defining responders was based on a receiver operating characteristic (ROC) analysis using the patient global impression of change as an anchor. A higher proportion of women treated with ORILISSA 150 mg once daily or 200 mg twice daily were responders for dysmenorrhea and non-menstrual pelvic pain compared to placebo in a dose-dependent manner at Month 3 [see Table 12].
Table 12. Proportion of Responders† for Dysmenorrhea and Non-Menstrual Pelvic Pain at Month 3 in Studies EM-1 and EM-2, Using the Endometriosis Daily Pain Impact Scale Study EM-1 Study EM-2 ORILISSA Placebo ORILISSA Placebo 150 mg
Once Daily
N=248200 mg
Twice Daily
N=244N=373 150 mg
Once Daily
N=221200 mg
Twice Daily
N=225N=353 Dysmenorrhea
Difference from placebo46%
27%**76%
56%**20% 43%
21%**72%
50%**23% Non-Menstrual
Pelvic Pain
Difference from placebo50%
14%**55%
18%**36% 50%
13%*58%
21%**37% † Study EM-1-Dysmenorrhea responder threshold: at least 0.81 point decrease from baseline in dysmenorrhea score; Non-Menstrual Pelvic Pain responder threshold: at least 0.36 point decrease from baseline in Non-Menstrual Pelvic Pain score
Study EM-2 - Dysmenorrhea responder threshold: at least 0.85 point decrease from baseline in dysmenorrhea score; Non-Menstrual Pelvic Pain responder threshold: at least 0.43 point decrease from baseline in Non-Menstrual Pelvic Pain score
*p ≤0.01 for test of difference from placebo
**p≤0.001 for test of difference from placeboWomen in these studies also provided a daily self-assessment of their endometriosis pain using a numeric rating scale (NRS) that asked subjects to rate their endometriosis pain at its worst over the last 24 hours on a scale from 0 (no pain) to 10 (worst pain ever). In Study EM-1, baseline NRS scores were 5.7 for ORILISSA 150 mg once daily, 5.5 for ORILISSA 200 mg twice daily and 5.6 for placebo. In Study EM-2, baseline NRS scores were 5.7 for ORILISSA 150 mg once daily, 5.3 for ORILISSA 200 mg twice daily and 5.6 for placebo. Women taking ORILISSA 150 mg once daily and 200 mg twice daily reported a statistically (p <0.001) significant reduction from baseline in NRS scores compared to placebo at Month 3 in both Studies EM-1 and EM-2 (Study EM-1: 0.7 points for ORILISSA 150 mg once daily and 1.3 points for ORILISSA 200 mg twice daily; Study EM-2: 0.6 points for ORILISSA 150 mg once daily and 1.2 points for ORILISSA 200 mg twice daily).
In addition, both ORILISSA treatment groups showed statistically significantly greater mean decreases from baseline compared to placebo in dysmenorrhea and non-menstrual pelvic pain scores at Month 6. Figures 3 through 6 show the mean scores for dysmenorrhea and non-menstrual pelvic pain over time for Study EM-1 and EM-2.
Figure 3. Mean Dysmenorrhea Pain Scoresa in Study EM-1 Over 6 Months
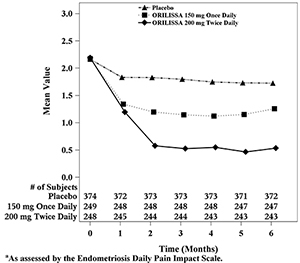
Figure 4. Mean Dysmenorrhea Pain Scoresa in Study EM-2 Over 6 Months
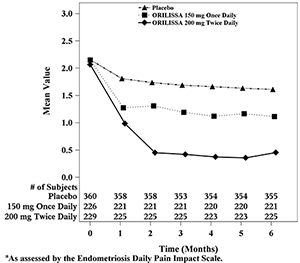
Figure 5. Mean Non-Menstrual Pelvic Paina Scores in Study EM-1 Over 6 Months
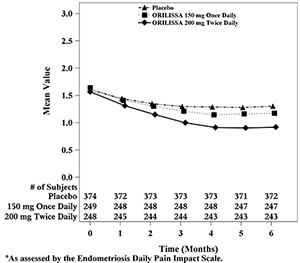
Figure 6. Mean Non-Menstrual Pelvic Paina Scores in Study EM-2 Over 6 Months
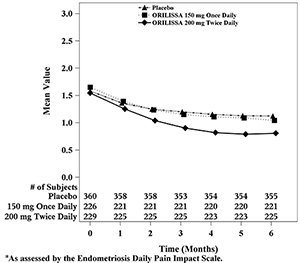
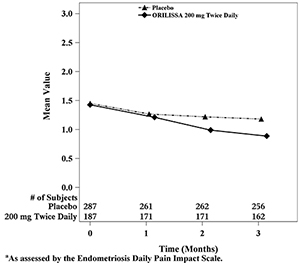
Dyspareunia associated with endometriosis was evaluated as a secondary endpoint using the Endometriosis Daily Pain Impact Scale that asked subjects to rate their pain during sexual intercourse in the prior 24 hours as none, mild, moderate, severe (correlating with a score of 0 to 3, respectively, where higher scores indicated greater severity), or not applicable. In both Studies EM-1 and EM-2, women treated with ORILISSA 200 mg twice daily showed statistically significantly greater reduction in dyspareunia from baseline to Month 3 than women given placebo (Study EM-1: 0.2; Study EM-2: 0.3). Figures 7 and 8 show the mean scores over time for Study EM-1 and EM-2.
Figure 7. Mean Dyspareunia Scoresa in Study EM-1 Over 3 Months
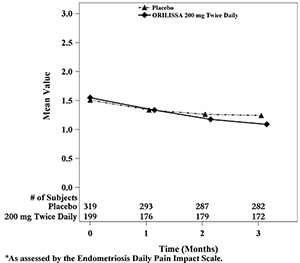
Figure 8. Mean Dyspareunia Scoresa in Study EM-2 Over 3 Months
In EM-1 and EM-2, 59% and 60% of patients used an opioid rescue analgesic for pain at baseline. The opioid rescue analgesics used at baseline were predominantly hydrocodone/acetaminophen (HC/APAP) and codeine/APAP at strengths of 5/300-325 mg and 30/300-500 mg. In EM-1, of all patients on an opioid at baseline, 98% and 2% were on HC/APAP and codeine/APAP, respectively. In EM-2, of all patients on an opioid at baseline, 50% were on HC/APAP and 16% were on codeine/APAP.
Other data related to opioid rescue analgesic use are summarized in Table 13.
Table 13. Opioid Rescue Analgesic Use in EM-1 and EM-2 Study EM-1 Study EM-2 ORILISSA
150 mg
Once DailyORILISSA
200 mg
Twice DailyPlacebo ORILISSA
150 mg
Once DailyORILISSA
200 mg
Twice DailyPlacebo Tablets per month at baseline
(mean±SD)15
±2415
±2513
±2113
±2912
±2612
±21Tablets per month at baseline
[Median (Min, Max)]4
(0, 184)4
(0, 195)4
(0, 146)4
(0, 236)3
(0, 214)4
(0, 152)Tablets per month at Month 3
(mean±SD)12
±297
±1810
±178
±225
±148
±15Tablets per month at Month 3
[Median (Min, Max)]0
(0, 251)0
(0, 162)2
(0, 144)0
(0, 168)0
(0, 136)2
(0, 142)Tablets per month at Month 6
(mean±SD)11
±267
±1711
±197
±195
±148
±15Tablets per month at Month 6
[Median (Min, Max)]0
(0, 224)0
(0, 157)3
(0, 185)0
(0, 185)0
(0, 157)2
(0, 142)Number and % of patients on any dose of opioid rescue at baseline who were off opioid at Month 3* 46/150
(31%)59/151
(39%)36/211
(17%)44/124
(35%)68/134
(51%)54/220
(25%)Number and % of patients on any dose of opioid rescue at baseline who were off opioid at Month 6* 43/149
(29%)66/150
(44%)36/211
(17%)50/124
(40%)78/134
(58%)70/222
(32%)Number and % of patients not on opioid rescue at baseline who were on any opioid at Month 3^ 9/98
(9%)6/93
(6%)17/162
(10%)10/97
(10%)10/91
(11%)29/133
(22%)Number and % of patients not on opioid rescue at baseline who were on any opioid at Month 6^ 16/98
(16%)6/93
(6%)32/161
(20%)13/97
(13%)6/91
(7%)32/133
(24%)Min = minimum; Max = maximum; SD = standard deviation
Monthly calculations are based on a 35-day interval.
*Denominator is the number of subjects on opioid rescue at baseline.
^Denominator is the number of subjects not on opioid rescue at baseline.The clinical relevance of these data has not been demonstrated.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
ORILISSA tablets are available in two strengths: 150 mg and 200 mg, which are equivalent to 155.2 mg and 207.0 mg of elagolix sodium, respectively.
ORILISSA 150 mg tablets are light pink, oblong, film-coated tablets with “EL 150” debossed on one side. ORILISSA 150 mg tablets are packaged in weekly blister packs. Each blister pack contains 7 tablets supplying the drug product for one week. Four blister packs (a total of 28 tablets) are packaged into a carton that provides the drug product for 4 weeks (NDC: 0074-0038-28).
ORILISSA 200 mg tablets are light orange, oblong, film-coated tablets with “EL 200” debossed on one side. The 200 mg tablets are packaged in weekly blister packs. Each blister pack contains 14 tablets supplying the drug product for one week. Four blister packs (a total of 56 tablets) are packaged in a carton that provides the drug product for 4 weeks (NDC: 0074-0039-56).
Store at 2°C to 30°C (36°F to 86°F).
Dispose unused medication via a take-back option if available. Otherwise, follow FDA instructions for disposing medication in the household trash, www.fda.gov/drugdisposal. Do NOT flush down the toilet.
-
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Medication Guide).
- Advise patients on contraceptive options, not to get pregnant while using ORILISSA, to be mindful that menstrual changes could reflect pregnancy and to discontinue ORILISSA if pregnancy occurs [see Contraindications (4), and Warnings and Precautions (5.2)].
- There is a pregnancy registry that monitors outcomes in women who become pregnant while treated with ORILISSA. Inform patients they can enroll by calling 1-833-782-7241 [see Use in Specific Populations (8.1)].
- Inform patients that estrogen containing contraceptives are expected to reduce the efficacy of ORILISSA.
- Inform patients about the risk of bone loss. Advise adequate intake of calcium and vitamin D [see Warnings and Precautions (5.1)].
- Advise patients to seek immediate medical attention for suicidal ideation and behavior. Instruct patients with new onset or worsening depression, anxiety, or other mood changes to promptly seek medical attention [see Warnings and Precautions (5.3)].
- Counsel patients on signs and symptoms of liver injury [see Warnings and Precautions (5.4)].
- Instruct patients who miss a dose of ORILISSA to take the missed dose on the same day as soon as she remembers and then resume the regular dosing schedule:
- 150 mg once daily: no more than 1 tablet each day should be taken.
- 200 mg twice daily: no more than 2 tablets each day should be taken.
- Instruct patients to dispose of unused medication via a take-back option if available or to otherwise follow FDA instructions for disposing of medication in the household trash, www.fda.gov/drugdisposal, and not to flush down the toilet.
Manufactured by AbbVie Inc. North Chicago, IL 60064
- Advise patients on contraceptive options, not to get pregnant while using ORILISSA, to be mindful that menstrual changes could reflect pregnancy and to discontinue ORILISSA if pregnancy occurs [see Contraindications (4), and Warnings and Precautions (5.2)].
-
MEDICATION GUIDE
MEDICATION GUIDE
ORILISSA® (awr-ah-lih-sah)
(elagolix)
tablets, for oral useWhat is the most important information I should know about ORILISSA?
ORILISSA may cause serious side effects, including:
-
bone loss (decreased bone mineral density).
- While you are taking ORILISSA, your estrogen levels will be low. Low estrogen levels can lead to bone mineral density loss.
- Your bone density may improve after you stop taking ORILISSA but complete recovery may not occur. It is unknown if these bone changes could increase your risk for broken bones as you age.
- Your healthcare provider may advise you to take vitamin D and calcium supplements as part of a healthy lifestyle that promotes bone health.
- If you have conditions or take other medicines that can cause bone loss, or if you have broken a bone with minimal or no injury, your healthcare provider may order an X-ray test called a DXA scan to check your bone mineral density.
-
effects on pregnancy
- Do not take ORILISSA if you are trying to become or are pregnant. It may increase the risk of early pregnancy loss.
-
If you think you are pregnant, stop taking ORILISSA right away and call your healthcare provider.
- If you become pregnant while taking ORILISSA, you are encouraged to enroll in the Pregnancy Registry. The purpose of the pregnancy registry is to collect information about the health of you and your baby. Talk to your healthcare provider or call 1-833-782-7241 to enroll in this registry.
- ORILISSA may change your menstrual periods (irregular bleeding or spotting, a decrease in menstrual bleeding, or no bleeding at all), making it hard to know if you are pregnant. Watch for other signs of pregnancy such as breast tenderness, weight gain and nausea.
- ORILISSA does not prevent pregnancy. You will need to use effective methods of birth control that do not contain hormones such as condoms or spermicide while taking ORILISSA and for 1 week after you stop taking ORILISSA. Birth control pills that contain estrogen may make ORILISSA less effective. It is not known how well ORILISSA will work while you are taking progestin-only birth control such as injections or implants.
- Talk to your healthcare provider about which birth control to use during treatment with ORILISSA. Your healthcare provider may change the birth control you were on before you start taking ORILISSA.
ORILISSA is a prescription medicine used to treat moderate to severe pain associated with endometriosis. It is not known if ORILISSA is safe and effective in children under 18 years of age.
- are or may be pregnant
- have osteoporosis
- have severe liver disease
- are taking medicines known as strong OATP1B1 inhibitors such as cyclosporine or gemfibrozil. Ask your healthcare provider if you are not sure if you are taking one of these medicines.
Before you take ORILISSA, tell your healthcare provider about all of your medical conditions, including if you:
- have or have had broken bones
- have other conditions or take medicines that may cause bone problems
- have or have had depression, mood problems or suicidal thoughts or behavior
- have liver problems
- think you may be pregnant. You should avoid becoming pregnant while taking ORILISSA
- are breastfeeding or plan to breastfeed. It is not known if ORILISSA passes into your breastmilk. Talk to your healthcare provider about the best way to feed your baby if you take ORILISSA.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Especially tell your healthcare provider if you take:
- birth control pills. Your healthcare provider may advise you to change the pills you take, or your method of birth control.
Know the medicines you take. Keep a list of your medicines with you to show to your healthcare provider and pharmacist when you get a new medicine.
- Take ORILISSA exactly as your healthcare provider tells you to take it.
- Your healthcare provider will give you a pregnancy test before you start taking ORILISSA or will have you start taking ORILISSA within 7 days after you start your period.
- If your healthcare provider prescribes: ORILISSA 150 mg (a pink tablet), take it 1 time each day
- ORILISSA 200 mg (an orange tablet), take it 2 times each day
- Take ORILISSA at about the same time each day with or without food.
- If you miss a dose of ORILISSA:
- 150 mg (1 time each day), take it as soon as you remember as long as it is on the same day. Do not take more than 1 tablet each day.
- 200 mg (2 times each day), take it as soon as you remember as long as it is on the same day. Do not take more than 2 tablets each day.
- If you take too much ORILISSA, call your healthcare provider or go to the nearest hospital.
What are the possible side effects of ORILISSA?
ORILISSA can cause serious side effects including:
- See “What is the most important information I should know about ORILISSA?”
-
suicidal thoughts, suicidal behavior, and worsening of mood. ORILISSA may cause suicidal thoughts or actions. Call your healthcare provider right away if you have any of these symptoms or call 911 if an emergency, especially if they are new, worse, or bother you:
- thoughts about suicide or dying
- try to commit suicide
- new or worse depression
- new or worse anxiety
- other unusual changes in behavior or mood
-
abnormal liver tests. Call your healthcare provider right away if you have any of these signs and symptoms of liver problems:
- yellowing of the skin or the whites of the eyes (jaundice)
- dark amber-colored urine
- feeling tired (fatigue or exhaustion)
- nausea and vomiting
- generalized swelling
- right upper stomach area (abdomen) pain
- bruising easily
The most common side effects of ORILISSA include: hot flashes or night sweats, headache, nausea, difficulty sleeping, absence of periods, anxiety, joint pain, depression and mood changes.
These are not all the possible side effects of ORILISSA. Call your healthcare provider for medical advice about side effects.
- Store ORILISSA between 36°F to 86°F (2°C to 30°C).
- Do not keep medicine that is out of date or that you no longer need. Dispose of unused medicines through community take-back disposal programs when available or place ORILISSA in an unrecognizable closed container in the household trash. Do NOT flush ORILISSA down the toilet. See www.fda.gov/drugdisposal for more information.
- Keep ORILISSA and all medicines out of the reach of children.
General information about the safe and effective use of ORILISSA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use ORILISSA for a condition for which it was not prescribed. Do not give ORILISSA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about ORILISSA that is written for health professionals.
What are the ingredients in ORILISSA?
Inactive ingredients 150 mg tablets: mannitol, sodium carbonate monohydrate, pregelatinized starch, povidone, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and carmine high tint.
Inactive ingredients 200 mg tablets: mannitol, sodium carbonate monohydrate, pregelatinized starch, povidone, magnesium stearate, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide red.
For more information, go to www.orilissa.com or call 1-844-674-3676.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: August, 2019 -
bone loss (decreased bone mineral density).
-
PRINCIPAL DISPLAY PANEL
NDC: 0074-0038-01
150 mg equivalent to 155.2 mg elagolix sodium

NDC: 0074-0038-07
150 mg equivalent to 155.2 mg elagolix sodium
PROFESSIONAL SAMPLE. NOT FOR SALE.

NDC: 0074-0038-28
150 mg equivalent to 155.2 mg elagolix sodium
Each carton contains 28 tablets in 4 weekly blister packs
Each weekly blister pack contains 7 tablets of elagolix 150 mg
NDC: 0074-0039–01
200 mg equivalent to 207.0 mg elagolix sodium

NDC: 0074-0039-14
200 mg equivalent to 207.0 mg elagolix sodium
PROFESSIONAL SAMPLE. NOT FOR SALE.

NDC: 0074-0039-56
200 mg equivalent to 207.0 mg elagolix sodium
Each carton contains 56 tablets in 4 weekly blister packs
Each weekly blister pack contains 14 tablets of elagolix 200 mg
-
INGREDIENTS AND APPEARANCE
ORILISSA
elagolix tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0074-0038 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ELAGOLIX SODIUM (UNII: 5948VUI423) (ELAGOLIX - UNII:5B2546MB5Z) ELAGOLIX SODIUM 150 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) SODIUM CARBONATE MONOHYDRATE (UNII: 2A1Q1Q3557) STARCH, CORN (UNII: O8232NY3SJ) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) CARMINIC ACID (UNII: CID8Z8N95N) Product Characteristics Color PINK (LIGHT PINK) Score no score Shape OVAL (oblong) Size 14mm Flavor Imprint Code EL;150 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0074-0038-28 4 in 1 CARTON 07/23/2018 1 7 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC: 0074-0038-01 1 in 1 CARTON 07/23/2018 2 7 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC: 0074-0038-07 1 in 1 CARTON 07/23/2018 3 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA210450 07/23/2018 ORILISSA
elagolix tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0074-0039 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ELAGOLIX SODIUM (UNII: 5948VUI423) (ELAGOLIX - UNII:5B2546MB5Z) ELAGOLIX SODIUM 200 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) SODIUM CARBONATE MONOHYDRATE (UNII: 2A1Q1Q3557) STARCH, CORN (UNII: O8232NY3SJ) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color ORANGE (LIGHT ORANGE) Score no score Shape OVAL (oblong) Size 15mm Flavor Imprint Code EL;200 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0074-0039-56 4 in 1 CARTON 07/23/2018 1 14 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC: 0074-0039-01 1 in 1 CARTON 07/23/2018 2 14 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC: 0074-0039-14 1 in 1 CARTON 07/23/2018 3 14 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA210450 07/23/2018 Labeler - AbbVie Inc. (078458370)
Trademark Results [Orilissa]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ORILISSA 87104202 5617902 Live/Registered |
AbbVie Inc. 2016-07-14 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.