CAPRELSA® (vandetanib) Tablets
CAPRELSA by
Drug Labeling and Warnings
CAPRELSA by is a Prescription medication manufactured, distributed, or labeled by Genzyme Corporation, Lonza AG, IPR Pharmaceuticals, Inc, Penn Pharmaceutical Services Limited, AstraZeneca UK Ltd, AstraZeneca Pharmaceuticals LP, EUROAPI UK LIMITED. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
CAPRELSA- vandetanib tablet, film coated
Genzyme Corporation
Disclaimer: This drug has not been found by FDA to be safe and effective, and this labeling has not been approved by FDA. For further information about unapproved drugs, click here.
----------
CAPRELSA® (vandetanib) Tablets
IMPORTANT PRESCRIBING INFORMATION
| Subject: Temporary importation of CAPRELSA® (vandetanib) Tablets to address drug shortage |
Dear Healthcare Provider,
As customers were informed on June 17, 2022, Sanofi is currently experiencing global supply disruptions for CAPRELSA® (vandetanib) tablets, 100 mg and 300 mg. We anticipate that supply disruptions will be resolved by September 2022 for the 100mg and October 2022 for the 300 mg. To that end, Sanofi has implemented a mitigation plan to address supply chain issues and as part of that plan, Sanofi is coordinating with the U.S. Food and Drug Administration (FDA) to allow the temporary importation of CAPRELSA 300 mg tablets from the United Kingdom (U.K.) into the U.S. market. The imported tablets are identical in composition and dosing regimen to that of the FDA-approved CAPRELSA 300 mg tablets and will still be available to you through the CAPRELSA REMS Program. Due to current market supply, Sanofi is only able to import the 300 mg.
| NDC Number | PROPRIETARY NAME | ESTABLISHED NAME | STRENGTH | DOSAGE FORM | PACK SIZE | |
|---|---|---|---|---|---|---|
| U.K. | NDC: 58468-7860-3 | CAPRELSA | Vandetanib | 300mg | Tablets | 3 Blisters of 10 (30 total) |
What actions are required of the HCP?
- Advise patients to continue to take CAPRELSA as prescribed
- Educate patients that CAPRELSA from the U.K. product is identical to the U.S. product even though it comes in different packaging
- Refer patients to the Medication Guide. This can be downloaded from: https://www.caprelsa.com/files/caprelsa-medication-guide.pdf
- Ensure you are certified to prescribe CAPRELSA and follow the REMS guidelines
What should you expect?
It is important to note the following differences between the two products:
| CAPRELSA 300 mg Tablets | ||
|---|---|---|
| U.S. | U.K. | |
| Dosing (Pediatrics) | Not approved | Approved ≥5 years of age |
| Primary container and carton labeling | Bottle inside a carton | Blister inside a carton |
 |  |
|
 |  |
|
Please note that the U.K. barcode may not register accurately on the U.S. scanning systems. Institutions should manually input the product into their systems to confirm that barcode systems do not provide incorrect information when the product is scanned. Alternative procedures should be followed to assure that the correct drug product is being used and administered to individual patients.
| WARNING: QT PROLONGATION, TORSADES DE POINTES, AND SUDDEN DEATH |
| See full prescribing information for complete boxed warning. |
| CAPRELSA can prolong the QT interval. Torsades de pointes and sudden death have occurred in patients receiving CAPRELSA. Do not use CAPRELSA in patients with hypocalcemia, hypokalemia, hypomagnesemia, or long QT syndrome. Correct hypocalcemia, hypokalemia and/or hypomagnesemia prior to CAPRELSA administration. Monitor electrolytes periodically. Avoid drugs known to prolong the QT interval. Only prescribers and pharmacies certified with the restricted distribution program are able to prescribe and dispense CAPRELSA (5.1, 5.15). |
As a reminder, the following information is taken from the United States Prescribing Information and should continue to be your source of information for CAPRELSA tablets:
Highlights from Section 5: WARNINGS AND PRECAUTIONS
- Prolonged QT interval, torsades de pointes, and sudden death: Monitor electrocardiograms and levels of serum potassium, calcium, magnesium and TSH. Reduce CAPRELSA dose as appropriate.
- Severe skin reactions, including toxic epidermal necrolysis and Stevens-Johnson syndrome, some fatal. Discontinue CAPRELSA for severe skin reactions.
- Interstitial lung disease (ILD), including fatalities: investigate unexplained nonspecific respiratory signs and symptoms. Discontinue CAPRELSA for confirmed ILD.
- Ischemic cerebrovascular events, hemorrhage, heart failure, diarrhea, hypertension, and reversible posterior leukoencephalopathy syndrome: Discontinue or interrupt CAPRELSA.
- Risk of impaired wound healing: Withhold for at least 1 month prior to elective surgery. Do not administer CAPRELSA for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of treatment with CAPRELSA after resolution of wound healing complications has not been established.
- Embryo-fetal toxicity: Can cause fetal harm. Advise women of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment with CAPRELSA and for 4 months following the last dose.
- REMS: CAPRELSA is available only through a restricted distribution program called the CAPRELSA REMS Program.
Enclosed please find the full U.S. prescribing information for CAPRELSA tablets as well as a more comprehensive table comparing the U.S. and U.K. labels.
Reporting Adverse Events
Healthcare providers and patients are encouraged to report adverse events or quality problems experienced with the use of this product by calling Sanofi Genzyme Customer Service by phone at: 1-800-633-1610.
Adverse events, medication errors, or quality problems experienced with the use of this product may also be reported to FDA’s MedWatch Adverse Reporting Program either online, by regular mail or by fax:
- Complete and submit the report Online: www.fda.gov/medwatch/report.htm
- Regular Mail or Fax: Download form www.fda.gov/MedWatch/getforms.htm or call 1-800-3321088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178 (1-800-332-0178).
Yours sincerely,
George Dickens, Pharm.D.
Acting Medical Head U.S. General Medicines
Head, of U.S. Field Base Medical
General Medicine U.S. Business Unit, Medical
Genzyme Corporation, A Sanofi Company
©2022 Genzyme Corporation. All rights reserved.
Highlights of differences and similarities between U.S. and U.K. CAPRELSA tablets
| CAPRELSA 300 mg Tablets | ||
|---|---|---|
| U.S. | U.K. | |
| Dosing (Adults) | 300 mg once daily (starting dose is 200 mg once daily in renal impairment) | Same as the U.S. approved dose |
| Tablet Description | White, Oval, biconvex, film-coated, and intagliated with 'Z 300' on one side and plain on the reverse side | Same as the U.S. tablet |
| Quantity: 30 Tablets | Same as U.S. quantity | |
| Storage Requirements | Store at 25°C (77°F); excursions permitted to (15°C-30°C) | Do not store above 30°C |
| Information for Patients | Yes, Medication Guide included in all packaging | Yes, Patient Information Leaflet is included in all packaging |
| Risk Strategy | Yes, Risk Evaluation and Mitigation Strategy (REMS) that requires certification of all prescribing HCPs | Yes, risk minimization materials that include dosing and monitoring guide for HCPs |
| Sections | U.S. Prescribing Information | U.K. SmPC |
|---|---|---|
| INDICATIONS | CAPRELSA is indicated for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease. Use CAPRELSA in patients with indolent, asymptomatic or slowly progressing disease only after careful consideration of the treatment related risks of CAPRELSA. | Caprelsa is indicated for the treatment of aggressive and symptomatic medullary thyroid cancer (MTC) in patients with unresectable locally advanced or metastatic disease. Caprelsa is indicated in adults, children and adolescents aged 5 years and older. For patients in whom Rearranged during Transfection (RET) mutation is not known or is negative, a possible lower benefit should be taken into account before individual treatment decision (see important information in sections 4.4 and 5.1). |
| DOSAGE | The recommended dose of CAPRELSA is 300 mg taken orally once daily until disease progression or unacceptable toxicity occurs. CAPRELSA may be taken with or without food. Do not take a missed dose within 12 hours of the next dose. Do not crush CAPRELSA tablets. The tablets can be dispersed in 2 ounces of water by stirring for approximately 10 minutes (will not completely dissolve). Do not use other liquids for dispersion. Swallow immediately after dispersion. Mix any remaining residue with 4 additional ounces of water and swallow. The dispersion can also be administered through nasogastric or gastrostomy tubes. | Treatment should be initiated and supervised by a physician experienced in treatment of MTC and in the use of anticancer medicinal products and experienced in the assessment of electrocardiogram (ECG). Patients treated with Caprelsa must be given the patient alert card and be informed about the risks of Caprelsa (see also package leaflet). Posology in paediatric patients with MTC Dosing for paediatric patients should be on the basis of BSA in mg/m2. Paediatric patients treated with Caprelsa and patients' caregivers must be given the dosing guide and be informed on the correct dose to be taken with the initial prescription and each subsequent dose adjustment. (make a ref to the U.K. SPC) Special patient populations Paediatric population Caprelsa should not be given to children below 5 years of age. The safety and efficacy of Caprelsa in children below 5 years of age have not been established. No data are available. There is no experience in paediatric patients with hereditary MTC below 9 years of age (see section 5.1). Patients aged 5-18 years should be dosed according to the nomogram in Table 1. Vandetanib doses higher than 150 mg/m2 have not been used in clinical studies in paediatric patients. Elderly No adjustment in starting dose is required for elderly patients. There is limited clinical data with vandetanib in patients with MTC aged over 75. Renal impairement in adult patients with MTC: The starting dose could be reduced to 200 mg in patients with moderate renal impairment; safety and efficacy have however not been established with 200 mg (see section 4.4). Vandetanib is not recommended for use in patients with severe renal impairment since there is limited data in patients with severe renal impairment, and safety and efficacy have not been established. |
| CONTRAINDICATIONS | Do not use in patients with congenital long QT syndrome [see Boxed Warning]. |
|
| WARNINGS & PRECAUTIONS | QT Prolongation and Torsades de Pointes CAPRELSA can prolong the QT interval in a concentration-dependent manner [see Clinical Pharmacology (12.2)]. Torsades de pointes, ventricular tachycardia and sudden deaths have occurred in patients treated with CAPRELSA. Do not start CAPRELSA treatment in patients whose QTcF interval is greater than 450 ms. Do not administer CAPRELSA to patients who have a history of Torsades de pointes, congenital long QT syndrome, bradyarrhythmias or uncompensated heart failure. CAPRELSA has not been studied in patients with ventricular arrhythmias or recent myocardial infarction. Vandetanib exposure is increased in patients with impaired renal function. Reduce the starting dose to 200 mg in patients with moderate to severe renal impairment and monitor QT interval frequently. Obtain an ECG and serum potassium, calcium, magnesium and TSH at baseline, 2 to 4 weeks and 8 to 12 weeks after starting treatment with CAPRELSA, and every 3 months thereafter. Monitor electrolytes and ECGs more frequently in patients who experience diarrhea. Following any dose reduction for QT prolongation or any dose interruption greater than 2 weeks, conduct QT assessments as described above. Maintain serum potassium levels of 4 mEq/L or higher (within normal range) and maintain serum magnesium and calcium levels within normal ranges to reduce the risk of QT prolongation. Avoid using CAPRELSA with drugs known to prolong the QT interval [see Warnings and Precautions (5.11) and Drug Interactions (7.4)]. If such drugs are given to patients already receiving CAPRELSA and no alternative therapy exists, perform ECG monitoring of the QT interval more frequently. Stop CAPRELSA in patients who develop a QTcF greater than 500 ms until the QTcF returns to less than 450 ms. Dosing of CAPRELSA can then be resumed at a reduced dose [see Dosage and Administration (2.1)]. | In view of the associated risks, it is important to limit treatment with vandetanib to patients who are in real need for treatment, i.e. with a symptomatic aggressive course of the disease. Either symptomatic disease or progressive disease alone is not enough to prompt the need of treatment with vandetanib. Rate of change in biomarker levels such as of calcitonin (CTN) and/or carcinoembryonic antigen (CEA) as well as the rate of change of tumour volume during watchful waiting might help to identify not only patients in need for treatment but also the optimal moment to commence treatment with vandetanib. QTc prolongation and Torsades de Pointes Vandetanib at a dose of 300 mg is associated with a substantial and concentration dependent prolongation in QTc (mean 28 msec, median 35 msec). First QTc prolongations occurred most often in the first 3 months of treatment, but continued to first occur after this time. The half life of vandetanib (19 days) renders this prolongation in QTc interval particularly problematic (see section 4.8). At a dose of 300 mg per day in MTC, ECG QTc prolongation to above 500 msec was observed in a phase III study in 11% of patients. ECG QTc prolongation appears to be dose-dependent. Torsades de pointes and ventricular tachycardia have been uncommonly reported in patients administered vandetanib 300 mg daily. The risk of Torsades may be increased in patients with electrolyte imbalance (see section 4.8). Vandetanib treatment must not be started in patients whose ECG QTc interval is greater than 480 msec. Vandetanib should not be given to patients who have a history of Torsades de pointes. Vandetanib has not been studied in patients with ventricular arrhythmias or recent myocardial infarction. An ECG, and levels of serum potassium, calcium and magnesium and thyroid stimulating hormone (TSH) should be obtained at baseline, at 1, 3, 6 and 12 weeks after starting treatment and every 3 months for at least a year thereafter. This schedule should apply to the period after dose reduction due to QTc prolongation and after dose interruption for more than two weeks. ECGs and blood tests should also be obtained as clinically indicated during this period and afterwards. Frequent ECG monitoring of the QTc interval should be continued. Serum potassium, serum magnesium and serum calcium should be kept within normal range to reduce the risk of ECG QTc prolongation. Additional monitoring of QTc, electrolytes and renal function are required especially in case of diarrhoea, increase in diarrhoea/dehydration, electrolyte imbalance and/or impaired renal function. If QTc increases markedly but stays below 500 msec, cardiologist advice should be sought. The administration of vandetanib with substances known to prolong the ECG QTc interval is contraindicated or not recommended (see section 4.3 and 4.5). The concomitant use of vandetanib with ondansetron is not recommended (see section 4.5) Patients who develop a single value of a QTc interval of ≥500 msec should stop taking vandetanib. Dosing can be resumed at a reduced dose after return of the QTc interval to pretreatment status has been confirmed and correction of possible electrolyte imbalance has been made. |
| Severe Skin Reactions Severe and sometimes fatal skin reactions, including toxic epidermal necrolysis (TEN) and Stevens-Johnson syndrome, have occurred in patients treated with CAPRELSA. Permanently discontinue CAPRELSA for severe skin reactions and refer the patient for urgent medical evaluation. Systemic therapies such as corticosteroids may be required. Photosensitivity reactions can occur during CAPRELSA treatment and up to 4 months after treatment discontinuation | Skin reactions Rash and other skin reactions including photosensitivity reactions and palmar plantar erythrodysaesthesia syndrome have been observed in patients who have received vandetanib. Mild to moderate skin reactions can be managed by symptomatic treatment, or by dose reduction or interruption. For more severe skin reactions (such as Stevens Johnson syndrome), referral of the patient to seek urgent medical advice is recommended. Care should be taken with sun exposure by wearing protective clothing and/or sunscreen due to the potential risk of phototoxicity reactions associated with vandetanib treatment. |
|
| Interstitial Lung Disease Interstitial Lung Disease (ILD) or pneumonitis, including fatalities, has occurred in patients treated with CAPRELSA. Consider a diagnosis of ILD in patients presenting with non-specific respiratory signs and symptoms. Interrupt CAPRELSA for acute or worsening pulmonary symptoms. Discontinue CAPRELSA if ILD is confirmed. | Interstitial lung disease Interstitial Lung Disease (ILD) has been observed in patients receiving vandetanib and some cases have been fatal. If a patient presents with respiratory symptoms such as dyspnoea, cough and fever, vandetanib treatment should be interrupted and prompt investigation initiated. If ILD is confirmed, vandetanib should be permanently discontinued and the patient treated appropriately. |
|
| Ischemic Cerebrovascular Events Ischemic cerebrovascular events, including fatalities, occurred in patients treated with CAPRELSA. In the randomized medullary thyroid cancer (MTC) study, ischemic cerebrovascular events occurred more frequently with CAPRELSA compared to placebo (1.3% compared to 0%). The safety of resumption of CAPRELSA therapy after resolution of an ischemic cerebrovascular event has not been studied. Discontinue CAPRELSA in patients who experience a severe ischemic cerebrovascular event. | Section not mentioned in U.K. SmPC | |
| Hemorrhage Serious hemorrhagic events, including fatalities, occurred in patients treated with CAPRELSA. Do not administer CAPRELSA to patients with a recent history of hemoptysis of ≥1/2 teaspoon of red blood. Discontinue CAPRELSA in patients with severe hemorrhage. | Haemorrhage Caution should be used when administering vandetanib to patients with brain metastases, as intracranial haemorrhage has been reported. |
|
| Heart Failure Heart failure, including fatalities, occurred in patients treated with CAPRELSA. Monitor for signs and symptoms of heart failure. Consider discontinuation of CAPRELSA in patients with heart failure. Heart failure may not be reversible upon stopping CAPRELSA. | Heart failure Heart failure has been observed in patients who received vandetanib. Temporary or permanent discontinuation of therapy may be necessary in patients with heart failure. It may not be reversible on stopping vandetanib. Some cases have been fatal. |
|
| Diarrhea Diarrhea of Grade 3 or greater severity occurred in 11% of patients receiving CAPRELSA in the randomized MTC study. If diarrhea occurs, carefully monitor serum electrolytes and ECGs to reduce the risk and enable early detection of QT prolongation resulting from dehydration [see Warnings and Precautions (5.1)]. Interrupt CAPRELSA for severe diarrhea. Upon improvement, resume CAPRELSA at a reduced dose [see Dosage and Administration (2.1)]. | Diarrhoea Diarrhoea is a disease related symptom as well as a known undesirable effect of vandetanib. Roe anti diarrhoeal agents are recommended for the treatment of diarrhoea. QTc and serum electrolytes should be monitored more frequently. If severe diarrhoea (CTCAE grade 3 4) develops, vandetanib should be stopped until diarrhoea improves. Upon improvement, treatment should be resumed at a reduced dose (see sections 4.2 and 4.8). |
|
| Hypothyroidism In the randomized MTC study in which 90% of the patients enrolled had prior thyroidectomy, increased dosing of thyroid replacement therapy was required in 49% of CAPRELSA-treated patients compared to 17% of placebo-treated patients. Obtain Thyroid-stimulating hormone (TSH) at baseline, at 2 to 4 weeks and 8 to 12 weeks after starting treatment with CAPRELSA, and every 3 months thereafter. If signs or symptoms of hypothyroidism occur, examine thyroid hormone levels and adjust thyroid replacement therapy accordingly. | Section not mentioned in U.K. SmPC | |
| Hypertension Hypertension, including hypertensive crisis, has occurred in patients treated with CAPRELSA. Monitor all patients for hypertension. Dose reduction or interruption for hypertension may be necessary. If hypertension cannot be controlled, do not resume CAPRELSA [see Dosage and Administration (2.1)]. | Hypertension Hypertension, including hypertensive crisis, has been observed in patients treated with vandetanib. Patients should be monitored for hypertension and controlled as appropriate. If high blood pressure cannot be controlled with medical management, vandetanib should not be restarted until the blood pressure is controlled medically. Reduction in dose may be necessary (see section 4.8). |
|
| Reversible Posterior Leukoencephalopathy Syndrome Reversible posterior leukoencephalopathy syndrome (RPLS), a syndrome of subcortical vasogenic edema diagnosed by an MRI of the brain, has occurred in patients treated with CAPRELSA. Consider this syndrome in any patient presenting with seizures, headache, visual disturbances, confusion or altered mental function. In clinical studies, three of four patients who developed RPLS while taking CAPRELSA also had hypertension. Discontinue CAPRELSA treatment in patients with RPLS. | Posterior reversible encephalopathy syndrome, PRES (Reversible posterior leukoencephalopathy syndrome RPLS) PRES is a syndrome of subcortical vasogenic oedema diagnosed by a MRI of the brain, has been observed infrequently with vandetanib treatment in combination with chemotherapy. PRES has also been observed in patients receiving vandetanib as monotherapy. This syndrome should be considered in any patient presenting with seizures, headache, visual disturbances, confusion or altered mental function. Brain MRI should be performed in any patient presenting with seizures, confusion or altered mental status. |
|
| Drug Interactions Avoid administration of CAPRELSA with anti-arrhythmic drugs (including but not limited to amiodarone, disopyramide, procainamide, sotalol, dofetilide) and other drugs that may prolong the QT interval (including but not limited to chloroquine, clarithromycin, dolasetron, granisetron, haloperidol, methadone, moxifloxacin, and pimozide) [see Drug Interactions (7.4) and Clinical Pharmacology (12.2)]. | Medicinal products known to prolong QTc interval Vandetanib has been shown to prolong the ECG QTc interval; Torsades de pointes have been uncommonly reported. Therefore, the concomitant use of vandetanib with medicinal products known to also prolong the QTc interval and/or induce Torsades de pointes is either contraindicated or not recommended depending on existing alternative therapies.
Results of a pharmacodynamic and pharmacokinetic interaction study indicated that co-administration with ondansetron in healthy patients appeared to have little effect on the pharmacokinetics of vandetanib, but had a small additive effect on the prolongation of the QTc interval of approximately 10 ms. Therefore, the concomitant use of ondansetron with vandetanib is not recommended. If ondansetron is administered with vandetanib, closer monitoring of serum electrolytes and ECGs and aggressive management of any abnormalities is required. |
|
| Renal Failure Renal failure occurred in patients treated with CAPRELSA [see Adverse Reactions (6.1)]. Withhold, reduce the dose or permanently discontinue based on severity [see Dosage and Administration (2.1)]. Vandetanib exposure is increased in patients with impaired renal function. Reduce the starting dose to 200 mg in patients with moderate renal impairment and monitor the QT interval closely [see Dosage and Administration (2.1)]. Vandetanib is not recommended for use in patients with severe renal impairment (clearance below 30 mL/min). There is no information available for patients with end-stage renal disease requiring dialysis [see Boxed Warning, Dosage and Administration (2.1), Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)]. | Patients with renal impairment Vandetanib is not recommended for use in adult and paediatric patients with moderate or severe renal impairment since there is limited data, and safety and efficacy have not been established (see sections 4.2, 5.1, and 5.2). |
|
| Hepatic Impairment CAPRELSA is not recommended for use in patients with moderate and severe hepatic impairment, as safety and efficacy have not been established [see Dosage and Administration (2.1)]. | Patients with hepatic impairment Vandetanib is not recommended for use in patients with hepatic impairment (serum bilirubin greater than 1.5 times upper limit of normal), since there is limited data in patients with hepatic impairment, and safety and efficacy have not been established. Pharmacokinetic data from volunteers, suggests that no change in starting dose is required in patients with mild, moderate or severe hepatic impairment (see sections 4.2 and 5.2). |
|
| Impaired Wound Healing Impaired wound healing can occur in patients who receive drugs that inhibit the VEGF signaling pathway. Impaired wound healing has occurred in patients treated with CAPRELSA. Withhold CAPRELSA for at least 1 month prior to elective surgery. Do not administer CAPRELSA for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of treatment with CAPRELSA after resolution of wound healing complications has not been established. | Section not mentioned in U.K. SmPC | |
| Embryo-Fetal Toxicity Based on its mechanism of action, CAPRELSA can cause fetal harm when administered to a pregnant woman. In rats, vandetanib was embryotoxic, fetotoxic, and induced fetal malformations at exposures equivalent to or lower than those expected at the 300 mg clinical dose and had adverse effects on female fertility, embryofetal development, and postnatal development of pups. There are no data on the presence of vandetanib or its metabolites in human milk or the effects of vandetanib on the breastfed child or on milk production. Vandetanib was present in the milk of lactating rats. Because of the potential for serious adverse reactions from CAPRELSA in breastfed children, advise women not to breastfeed during treatment with CAPRELSA and for 4 months after the final dose. Verify the pregnancy status of females of reproductive potential prior to initiating treatment with CAPRELSA Advise women of the potential hazard to a fetus. Advise women of reproductive potential to use effective contraception during treatment with CAPRELSA and for at least 4 months following the last dose [see Use in Specific Populations (8.1), (8.3)]. | 4.6 Fertility, pregnancy and lactation Women of childbearing potential Women of childbearing potential must use effective contraception during therapy and for at least four months following the last dose. Pregnancy There is a limited amount of data on the use of vandetanib during pregnancy. As expected from its pharmacological actions, vandetanib has shown significant effects on all stages of female reproduction in rats (see section 5.3). If vandetanib is used during pregnancy or if the patient becomes pregnant while receiving vandetanib, she should be apprised of the potential for foetal abnormalities or loss of the pregnancy. Treatment should only be continued in pregnant women if the potential benefit to the mother outweighs the risk to the foetus. Breast feeding There are no data on the use of vandetanib in breast feeding women. Vandetanib and/or its metabolites is excreted into milk in rats and found in plasma of pups following dosing to lactating rats (see section 5.3). Breast feeding is contraindicated while receiving vandetanib therapy. Fertility In rats, vandetanib had no effect on male fertility but impaired female fertility (see section 5.3). Effects on reproduction in paediatric patients treated with vandetanib are not known. |
|
| CAPRELSA REMS (Risk Evaluation and Mitigation Strategy) Program Because of the risk of QT prolongation, Torsades de pointes, and sudden death, CAPRELSA is available only through a restricted distribution program called the CAPRELSA REMS Program. Only prescribers and pharmacies certified with the program are able to prescribe and dispense CAPRELSA. | Section not mentioned in U.K. SmPC (U.S. specific only). Risk Management Plan is approved in EU annexes | |
| Section not mentioned in USPI | Rearranged during transfection (RET) status Patients without RET mutation may have a decreased benefit from vandetanib treatment and the benefit/risk balance for this group of patients may therefore differ from that of the group with RET mutations. For patients whose RET mutation status could be negative, a possible lower benefit should be taken into account before individual treatment decisions and the use of vandetanib should be carefully considered because of the treatment related risks. Therefore, RET mutation testing is recommended. When establishing RET mutation status, tissue samples should be obtained if possible, at the time of initiation of treatment rather than at the time of diagnosis (see sections 4.1 and 5.1). |
|
| Section not mentioned in USPI | Aneurysms and artery dissections The use of VEGF pathway inhibitors in patients with or without hypertension may promote the formation of aneurysms and/or artery dissections. Before initiating vandetanib, this risk should be carefully considered in patients with risk factors such as hypertension or history of aneurysm. |
|
| Section not mentioned in USPI | Alanine aminotransferase elevations Alanine aminotransferase elevations occur commonly in patients treated with vandetanib. The majority of elevations resolve while continuing treatment, others usually resolve after a 1-2 week interruption in therapy. Periodic monitoring of alanine aminotransferase is recommended. |
|
| Strong CYP3A4 inducers: In a cross-over study in 12 healthy volunteers, a single oral 300 mg dose of CAPRELSA was administered alone on day 1 and on day 10 in combination with daily doses of 600 mg of rifampicin (a strong CYP3A4 inducer) given on days 1 to 31. The coadministration of rifampicin with CAPRELSA decreased the geometric mean AUC0–504h of vandetanib by 40% (90% confidence interval (CI): 56%, 63%) compared to vandetanib alone. No clinically meaningful change in the mean Cmax of vandetanib was observed. The geometric mean AUC0–504h and Cmax of N-desmethylvandetanib increased by 266% and 414%, respectively, in the presence of rifampicin compared with vandetanib alone | CYP3A4 inducers The concomitant use of vandetanib with strong CYP3A4 inducers (such as rifampicin, St John's Wort, carbamazepine, phenobarbital) should be avoided (see section 4.5). |
|
| Section not mentioned in USPI | CTN less than 500 pg/ml The benefit of vandetanib in patients with CTN less than 500 pg/ml has not been determined, therefore use in patients with CTN < 500 pg/ml should be carefully considered because of the treatment related risks of vandetanib. |
|
| Section not mentioned in USPI | Pediatric population Based on height measurements at all visits, all children and adolescents in a paediatric study demonstrated linear growth while receiving vandetanib. However, long term safety data in paediatric patients are not available. |
THIS MEDICINAL PRODUCT IS SUBJECT TO ADDITIONAL MONITORING. THIS WILL ALLOW QUICK IDENTIFICATION OF NEW SAFETY INFORMATION. HEALTHCARE PROFESSIONALS ARE ASKED TO REPORT ANY SUSPECTED ADVERSE REACTIONS. SEE SECTION 4.8 FOR HOW TO REPORT ADVERSE REACTIONS.
2. QUALITATIVE AND QUANTITATIVE COMPOSITION
Each film-coated tablet contains 300 mg of vandetanib.
For a full list of excipients, see section 6.1.
3. PHARMACEUTICAL FORM
The Caprelsa 300 mg tablet is an oval-shaped, biconvex, white film-coated tablet with 'Z300' impressed on one side.
4. CLINICAL PARTICULARS
4.1 Therapeutic indications
Caprelsa is indicated for the treatment of aggressive and symptomatic medullary thyroid cancer (MTC) in patients with unresectable locally advanced or metastatic disease.
Caprelsa is indicated in adults, children and adolescents aged 5 years and older.
For patients in whom Rearranged during Transfection (RET) mutation is not known or is negative, a possible lower benefit should be taken into account before individual treatment decision (see important information in sections 4.4 and 5.1).
4.2 Posology and method of administration
Treatment should be initiated and supervised by a physician experienced in treatment of MTC and in the use of anticancer medicinal products and experienced in the assessment of electrocardiogram (ECG).
Only one supply per prescription is allowed. For a further supply, a new prescription is required.
If a dose is missed, it should be taken as soon as the patient remembers. If it is less than 12 hours to the next dose, the patient should not take the missed dose. Patients should not take a double dose (two doses at the same time) to make up for a forgotten dose.
Patients treated with Caprelsa must be given the patient alert card and be informed about the risks of Caprelsa (see also package leaflet).
Posology for MTC in adult patients
The recommended dose is 300 mg once a day, taken with or without food at about the same time each day.
Dose adjustments in adult patients with MTC
QTc interval should be carefully assessed prior to initiation of treatment. In the event of common terminology criteria for adverse events (CTCAE) grade 3 or higher toxicity or prolongation of the ECG QTc interval, dosing with vandetanib should be at least temporarily stopped and resumed at a reduced dose when toxicity has resolved or improved to CTCAE grade 1 (see section 4.4). The 300 mg daily dose can be reduced to 200 mg (two 100 mg tablets), and then to 100 mg if necessary. The patient must be monitored appropriately. Due to the 19-day half-life, adverse reactions including a prolonged QTc interval may not resolve quickly (see section 4.4).
Posology in paediatric patients with MTC
Dosing for paediatric patients should be on the basis of BSA in mg/m2. Paediatric patients treated with Caprelsa and patients' caregivers must be given the dosing guide and be informed on the correct dose to be taken with the initial prescription and each subsequent dose adjustment. Recommended dosing regimens and dose modifications are presented in Table 1.
| BSA (m2) | Start dose (mg)* | Dose increase (mg)† when tolerated well after 8 weeks at starting dose | Dose reduction (mg) ‡ |
|---|---|---|---|
|
|
|||
| 0.7 – <0.9 | 100 every other day | 100 daily | - |
| 0.9 – <1.2 | 100 daily | 7 day schedule: 100-200-100-200-100-200-100 | 100 every other day |
| 1.2 – <1.6 | 7 day schedule: 100-200-100-200-100-200-100 | 200 daily | 100 daily |
| ≥ 1.6 | 200 daily | 300 daily | 7 day schedule: 100-200-100-200-100-200-100 |
Dose adjustments in paediatric patients with MTC
- In the event of CTCAE grade 3 or higher toxicity or prolongation of the ECG QTc interval, dosing with vandetanib should be at least temporarily stopped and resumed at a reduced dose when toxicity has resolved or improved to CTCAE grade 1.
- Patients who are on the starting dose (* in Table 1), should be recommenced at the reduced dose (‡ in Table 1).
- Patients who are on the increased dose († in Table 1), should be recommenced at the starting dose (* in Table 1). If another event of common terminology criteria for adverse events (CTCAE) grade 3 or higher toxicity or prolongation of the ECG QTc interval occurs, dosing with Caprelsa should be at least temporarily stopped and resumed at a reduced dose (‡ in Table 1) when toxicity has resolved or improved to CTCAE grade 1.
- If a further event of CTCAE grade 3 or higher toxicity or prolongation of the ECG QTc interval occurs, dosing with vandetanib should be permanently stopped.
The patient must be monitored appropriately. Due to the 19-day half-life, adverse reactions including a prolonged QTc interval may not resolve quickly (see section 4.4).
Duration
Vandetanib may be administered until disease progression or until the benefits of treatment continuation do no longer outweigh its risk, thereby considering the severity of adverse events (see sections 4.8) in relation to the degree of clinical stabilization of the tumour status.
Special patient populations
Paediatric population
Caprelsa should not be given to children below 5 years of age. The safety and efficacy of Caprelsa in children below 5 years of age have not been established. No data are available.
There is no experience in paediatric patients with hereditary MTC below 9 years of age (see section 5.1). Patients aged 5–18 years should be dosed according to the nomogram in Table 1. Vandetanib doses higher than 150 mg/m2 have not been used in clinical studies in paediatric patients.
Elderly
No adjustment in starting dose is required for elderly patients. There is limited clinical data with vandetanib in patients with MTC aged over 75.
Renal impairment in adult patients with MTC
A pharmacokinetic study in volunteers with mild, moderate and severe renal impairment shows that exposure to vandetanib after single dose is increased up to 1.5, 1.6 and 2-fold respectively in patients with mild, moderate (creatinine clearance ≥ 30 to < 50 ml/min) and severe (clearance below 30 ml/min) renal impairment at baseline (see section 5.2). Clinical data suggest that no change in starting dose is required in patients with mild renal impairment. There is limited data with 300 mg in patients with moderate renal impairment: the dose needed to be lowered to 200 mg in 5 out of 6 patients. The starting dose could be reduced to 200 mg in patients with moderate renal impairment; safety and efficacy have however not been established with 200 mg (see section 4.4). Vandetanib is not recommended for use in patients with severe renal impairment since there is limited data in patients with severe renal impairment, and safety and efficacy have not been established.
Renal impairment in paediatric patients with MTC
There is no experience with the use of vandetanib in paediatric patients with renal impairment.
Considering the data available in adult patients with renal impairment:
- No change in starting dose is recommended in paediatric patients with mild renal impairment
- The reduced dose as specified in Table 1 could be used in paediatric patients with moderate renal impairment. Individual patient management will be required by the physician, especially in paediatric patients with low BSA.
- Vandetanib is not recommended in paediatric patients with severe renal impairment
Hepatic impairment
Vandetanib is not recommended for use in adult and paediatric patients with hepatic impairment (serum bilirubin greater than 1.5 times upper limit of reference range (ULRR), this criterion does not apply to patients with Gilbert's Disease and alanine aminotransferase (ALT), aspartate aminotransferase (AST), or alkaline phosphatase (ALP) greater than 2.5 times ULRR, or greater than 5.0 times ULRR if judged by the physician to be related to liver metastases), since there is limited data in patients with hepatic impairment, and safety and efficacy have not been established (see section 4.4).
Pharmacokinetic data from volunteers, suggests that no change in starting dose is required in patients with mild, moderate or severe hepatic impairment (see section 5.2).
Method of administration
For patients who have difficulty swallowing, vandetanib tablets may be dispersed in half a glass of non-carbonated drinking water. No other liquids should be used. The tablet is to be dropped in water, without crushing, stirred until dispersed (approximately 10 minutes) and the resultant dispersion swallowed immediately. Any residues in the glass are to be mixed with half a glass of water and swallowed. The liquid can also be administered through nasogastric or gastrostomy tubes.
4.3 Contraindications
- Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
- Congenital long QTc syndrome.
- Patients with a QTc interval over 480 msec.
- Concomitant use of vandetanib with the following medicinal products known to also prolong the QTc interval and/or induce Torsades de pointes: Arsenic, cisapride, erythromycin intravenous (IV), toremifene, mizolastine, moxifloxacin, Class IA and III antiarrhythmics (see section 4.5).
- Breast-feeding (see section 4.6).
4.4 Special warnings and precautions for use
In view of the associated risks, it is important to limit treatment with vandetanib to patients who are in real need for treatment, i.e. with a symptomatic-aggressive course of the disease. Either symptomatic disease or progressive disease alone is not enough to prompt the need of treatment with vandetanib. Rate of change in biomarker levels such as of calcitonin (CTN) and/or carcinoembryonic antigen (CEA) as well as the rate of change of tumour volume during watchful waiting might help to identify not only patients in need for treatment but also the optimal moment to commence treatment with vandetanib.
| QTc prolongation and Torsades de Pointes |
| Vandetanib at a dose of 300 mg is associated with a substantial and concentration dependent prolongation in QTc (mean 28 msec, median 35 msec). First QTc prolongations occurred most often in the first 3 months of treatment, but continued to first occur after this time. The half-life of vandetanib (19 days) renders this prolongation in QTc interval particularly problematic (see section 4.8). At a dose of 300 mg per day in MTC, ECG QTc prolongation to above 500 msec was observed in a phase III study in 11% of patients. ECG QTc prolongation appears to be dose-dependent. Torsades de pointes and ventricular tachycardia have been uncommonly reported in patients administered vandetanib 300 mg daily. The risk of Torsades may be increased in patients with electrolyte imbalance (see section 4.8). |
| Vandetanib treatment must not be started in patients whose ECG QTc interval is greater than 480 msec. Vandetanib should not be given to patients who have a history of Torsades de pointes. Vandetanib has not been studied in patients with ventricular arrhythmias or recent myocardial infarction. |
| An ECG, and levels of serum potassium, calcium and magnesium and thyroid stimulating hormone (TSH) should be obtained at baseline, at 1, 3, 6 and 12 weeks after starting treatment and every 3 months for at least a year thereafter. This schedule should apply to the period after dose reduction due to QTc prolongation and after dose interruption for more than two weeks. ECGs and blood tests should also be obtained as clinically indicated during this period and afterwards. Frequent ECG monitoring of the QTc interval should be continued. |
| Serum potassium, serum magnesium and serum calcium should be kept within normal range to reduce the risk of ECG QTc prolongation. Additional monitoring of QTc, electrolytes and renal function are required especially in case of diarrhoea, increase in diarrhoea/dehydration, electrolyte imbalance and/or impaired renal function. If QTc increases markedly but stays below 500 msec, cardiologist advice should be sought. |
| The administration of vandetanib with substances known to prolong the ECG QTc interval is contraindicated or not recommended (see section 4.3 and 4.5). |
| The concomitant use of vandetanib with ondansetron is not recommended (see section 4.5) |
| Patients who develop a single value of a QTc interval of ≥500 msec should stop taking vandetanib. Dosing can be resumed at a reduced dose after return of the QTc interval to pretreatment status has been confirmed and correction of possible electrolyte imbalance has been made. |
Posterior reversible encephalopathy syndrome, PRES (Reversible posterior leukoencephalopathy syndrome-RPLS)
PRES is a syndrome of subcortical vasogenic oedema diagnosed by a MRI of the brain, has been observed infrequently with vandetanib treatment in combination with chemotherapy. PRES has also been observed in patients receiving vandetanib as monotherapy. This syndrome should be considered in any patient presenting with seizures, headache, visual disturbances, confusion or altered mental function. Brain MRI should be performed in any patient presenting with seizures, confusion or altered mental status.
Rearranged during transfection (RET) status
Patients without RET mutation may have a decreased benefit from vandetanib treatment and the benefit/risk balance for this group of patients may therefore differ from that of the group with RET mutations. For patients whose RET mutation status could be negative, a possible lower benefit should be taken into account before individual treatment decisions and the use of vandetanib should be carefully considered because of the treatment related risks. Therefore, RET mutation testing is recommended. When establishing RET mutation status, tissue samples should be obtained if possible at the time of initiation of treatment rather than at the time of diagnosis (see sections 4.1 and 5.1).
Skin reactions
Rash and other skin reactions including photosensitivity reactions and palmar-plantar erythrodysaesthesia syndrome have been observed in patients who have received vandetanib.
Mild to moderate skin reactions can be managed by symptomatic treatment, or by dose reduction or interruption. For more severe skin reactions (such as Stevens-Johnson syndrome), referral of the patient to seek urgent medical advice is recommended.
Care should be taken with sun exposure by wearing protective clothing and/or sunscreen due to the potential risk of phototoxicity reactions associated with vandetanib treatment.
Diarrhoea
Diarrhoea is a disease related symptom as well as a known undesirable effect of vandetanib. Routine anti-diarrhoeal agents are recommended for the treatment of diarrhoea. QTc and serum electrolytes should be monitored more frequently. If severe diarrhoea (CTCAE grade 3–4) develops, vandetanib should be stopped until diarrhoea improves. Upon improvement, treatment should be resumed at a reduced dose (see sections 4.2 and 4.8).
Haemorrhage
Caution should be used when administering vandetanib to patients with brain metastases, as intracranial haemorrhage has been reported.
Heart failure
Heart failure has been observed in patients who received vandetanib. Temporary or permanent discontinuation of therapy may be necessary in patients with heart failure. It may not be reversible on stopping vandetanib. Some cases have been fatal.
Hypertension
Hypertension, including hypertensive crisis, has been observed in patients treated with vandetanib. Patients should be monitored for hypertension and controlled as appropriate. If high blood pressure cannot be controlled with medical management, vandetanib should not be restarted until the blood pressure is controlled medically. Reduction in dose may be necessary (see section 4.8).
Aneurysms and artery dissections
The use of VEGF pathway inhibitors in patients with or without hypertension may promote the formation of aneurysms and/or artery dissections. Before initiating vandetanib, this risk should be carefully considered in patients with risk factors such as hypertension or history of aneurysm.
Patients with renal impairment
Vandetanib is not recommended for use in adult and paediatric patients with moderate or severe renal impairment since there is limited data, and safety and efficacy have not been established (see sections 4.2, 5.1, and 5.2).
Patients with hepatic impairment
Vandetanib is not recommended for use in patients with hepatic impairment (serum bilirubin greater than 1.5 times upper limit of normal), since there is limited data in patients with hepatic impairment, and safety and efficacy have not been established. Pharmacokinetic data from volunteers, suggests that no change in starting dose is required in patients with mild, moderate or severe hepatic impairment (see sections 4.2 and 5.2).
Alanine aminotransferase elevations
Alanine aminotransferase elevations occur commonly in patients treated with vandetanib. The majority of elevations resolve while continuing treatment, others usually resolve after a 1–2 week interruption in therapy. Periodic monitoring of alanine aminotransferase is recommended.
Interstitial lung disease
Interstitial Lung Disease (ILD) has been observed in patients receiving vandetanib and some cases have been fatal. If a patient presents with respiratory symptoms such as dyspnoea, cough and fever, vandetanib treatment should be interrupted and prompt investigation initiated. If ILD is confirmed, vandetanib should be permanently discontinued and the patient treated appropriately.
CYP3A4 inducers
The concomitant use of vandetanib with strong CYP3A4 inducers (such as rifampicin, St John's Wort, carbamazepine, phenobarbital) should be avoided (see section 4.5).
CTN less than 500 pg/ml
The benefit of vandetanib in patients with CTN less than 500 pg/ml has not been determined, therefore use in patients with CTN < 500 pg/ml should be carefully considered because of the treatment related risks of vandetanib.
4.5 Interaction with other medicinal products and other forms of interaction
Pharmacokinetic interactions
Effect of vandetanib on other medicinal products
In healthy subjects, the exposure for midazolam (CYP3A4 substrate) was not affected when given together with a single dose of vandetanib at 800 mg.
Vandetanib is an inhibitor of the organic cation 2 (OCT2) transporter. In healthy subjects with wild type for OCT2, the AUC(0-t) and Cmax for metformin (OCT2 substrate) were increased by 74% and 50%, respectively and CLR of metformin was decreased by 52% when given together with vandetanib. Appropriate clinical and/or laboratory monitoring is recommended for patients receiving concomitant metformin and vandetanib, and such patients may require a lower dose of metformin.
In healthy subjects, the AUC(0-t) and Cmax for digoxin (P-gp substrate) were increased by 23% and 29% respectively, when given together due to P-gp inhibition by vandetanib. Furthermore, the bradycardiac effect of digoxin may increase the risk of vandetanib QTc interval prolongation and Torsade de Pointes. Therefore, an appropriate clinical (e.g. ECG) and/or laboratory monitoring is recommended for patients receiving concomitant digoxin and vandetanib, and such patients may require a lower dose of digoxin. (For vandetanib monitoring, see section 4.2 Posology and Method of administration and section 4.4 Special warnings and precautions for use).
As regards other P-gp substrates such as dabigatran, a clinical monitoring is recommended in case of combination with vandetanib.
Effect of other medicinal products on vandetanib
In healthy subjects, no clinically significant interaction was shown between vandetanib (a single dose of 300mg) and the potent CYP3A4 inhibitor, itraconazole (repeated doses of 200mg once daily). In healthy male subjects, the exposure to vandetanib was reduced by 40% when given together with the potent CYP3A4 inducer, rifampicin. Administration of vandetanib with potent CYP3A4 inducers should be avoided.
In healthy subjects, the Cmax for vandetanib was decreased by 15% while the AUC(0-t) for vandetanib was not affected when given together with omeprazole. Neither the Cmax nor the AUC(0-t) for vandetanib was affected when given together with ranitidine. Therefore, no change in dose of vandetanib is required when vandetanib is given with either omeprazole or ranitidine.
Pharmacodynamic interactions
Biliary excretion of unchanged vandetanib is one of the excretion pathways for vandetanib. Vandetanib is not a substrate of multidrug resistance protein 2 (MRP2), p-glycoprotein (P-gp) or breast cancer resistance protein (BCRP).
Medicinal products known to prolong QTc interval
Vandetanib has been shown to prolong the ECG QTc interval; Torsades de pointes have been uncommonly reported. Therefore, the concomitant use of vandetanib with medicinal products known to also prolong the QTc interval and/or induce Torsades de pointes is either contraindicated or not recommended depending on existing alternative therapies.
- Combinations contraindicated (see section 4.3): Cisapride, erythromycin intravenous (IV), toremifene, mizolastine, moxifloxacin, arsenic, Class IA and III antiarrhythmics
- Combinations not recommended: Methadone, haloperidol, amisulpride, chlorpromazine, sulpiride, zuclopenthixol, halofantrine, pentamidine and lumefantrine.
If there is no appropriate alternative therapy, not recommended combinations with vandetanib may be made with additional ECG monitoring of the QTc interval, evaluation of electrolytes and further control at onset or worsening of diarrhoea.
Results of a pharmacodynamic and pharmacokinetic interaction study indicated that co-administration with ondansetron in healthy patients appeared to have little effect on the pharmacokinetics of vandetanib, but had a small additive effect on the prolongation of the QTc interval of approximately 10 ms. Therefore, the concomitant use of ondansetron with vandetanib is not recommended. If ondansetron is administered with vandetanib, closer monitoring of serum electrolytes and ECGs and aggressive management of any abnormalities is required.
Vitamin K antagonists
Due to the increased thrombotic risk in patients with cancer, the use of anticoagulation is frequent. In consideration of the high intra-individual variability of the response to anticoagulation, and the possibility of interaction between vitamin K antagonists and chemotherapy, an increased frequency of the INR (International Normalised Ratio) monitoring is recommended, if it is decided to treat the patient with vitamin K antagonists.
4.6 Fertility, pregnancy and lactation
Women of childbearing potential
Women of childbearing potential must use effective contraception during therapy and for at least four months following the last dose.
Pregnancy
There is a limited amount of data on the use of vandetanib during pregnancy. As expected from its pharmacological actions, vandetanib has shown significant effects on all stages of female reproduction in rats (see section 5.3).
If vandetanib is used during pregnancy or if the patient becomes pregnant while receiving vandetanib, she should be apprised of the potential for foetal abnormalities or loss of the pregnancy. Treatment should only be continued in pregnant women if the potential benefit to the mother outweighs the risk to the foetus.
Breast-feeding
There are no data on the use of vandetanib in breast-feeding women. Vandetanib and/or its metabolites is excreted into milk in rats and found in plasma of pups following dosing to lactating rats (see section 5.3).
Breast-feeding is contraindicated while receiving vandetanib therapy.
Fertility
In rats, vandetanib had no effect on male fertility but impaired female fertility (see section 5.3).
Effects on reproduction in paediatric patients treated with vandetanib are not known.
4.7 Effects on ability to drive and use machines
No studies to establish the effects of vandetanib on ability to drive and use machines have been conducted. However, fatigue and blurred vision have been reported and those patients who experience these symptoms should observe caution when driving or using machines.
4.8 Undesirable effects
Summary of the safety profileThe most commonly reported adverse drug reactions have been diarrhoea, rash, nausea, hypertension, and headache.
Tabulated list of adverse reactions
The following adverse reactions have been identified in clinical studies with patients receiving vandetanib as treatment for MTC. Their frequency is presented in Table 2, adverse reactions using Council for International Organizations of Medical Sciences (CIOMS III), listed by MedDRA System Organ Class (SOC) and at the preferred term level and then by frequency classification. Frequencies of occurrence of undesirable effects are defined as: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1000); very rare (< 1/10,000) and not known (cannot be estimated from the available data). This section includes only data derived from completed studies where patient exposure is known.
| System Organ Class | Very common | Common | Uncommon | Not known |
|---|---|---|---|---|
|
|
||||
| Infection and infestation disorders | Nasopharyngitis bronchitis, upper respiratory tract infections, urinary tract infections | Pneumonia, sepsis, influenza, cystitis, sinusitis, laryngitis, folliculitis, furuncle, fungal infection, pyelonephritis | Appendicitis, staphylococcal infection, diverticulitis, cellulitis, abdominal wall abscess | |
| Endocrine disorders | Hypothyroidism | |||
| Metabolism and nutrition disorders | Appetite decreased, Hypocalcaemia | Hypokalaemia, hypercalcaemia, hyperglycemia, dehydration, hyponatremia | Malnutrition | |
| Psychiatric disorders | Insomnia, Depression | Anxiety | ||
| Nervous system disorders | Headache, paraesthesia, dysaesthesia, dizziness | Tremor, lethargy, loss of consciousness, balance disorders, dysgeusia | Convulsion, clonus, brain oedema | |
| Eye disorders | Vision blurred, corneal structural change (including corneal deposits and corneal opacity) | Visual impairment, halo vision, photopsia, glaucoma, conjunctivitis, dry eye, keratopathy | Cataract, accommodation disorders | |
| Cardiac disorders | Prolongation of ECG QTc interval(*) (†) | Heart failure, acute heart failure, rate and rhythm disorders, cardiac conduction disorders, ventricular arrhythmia and cardiac arrest | ||
| Vascular disorders | Hypertension | Hypertensive crisis, ischaemic cerebrovascular conditions | Aneurysms and artery dissections | |
| Respiratory, thoracic and mediastinal disorders | Epistaxis, haemoptysis, pneumonitis | Respiratory failure, pneumonia aspiration | ||
| Gastrointestinal disorders | Abdominal pain, diarrhoea, nausea, vomiting, dyspepsia | Colitis, dry mouth, stomatitis, dysphagia, constipation, gastritis, gastrointestinal haemorrhage | Pancreatitis, peritonitis, ileus, intestinal perforation, faecal incontinence | |
| Hepatobiliary disorders | Cholelithiasis | |||
| Skin and subcutaneous tissue disorders | Photosensitivity reaction, rash and other skin rections (including acne, dry skin, dermatitis, pruritus), nail disorders | Palmar-plantar erythrodysaesthiesia syndrome, alopecia | Bullous dermatitis | |
| Renal and urinary disorders | Proteinuria, nephrolithiasis | Dysuria, hematuria, renal failure, pollakiuria, micturition urgency | Chromaturia, anuria | |
| General disorders and administration site conditions | Asthenia, fatigue, pain, oedema | Pyrexia | Impaired healing | |
| Investigations | ECG QTc interval prolonged | Increase of serum ALT and AST, weight decreased blood creatinine increased | Increased haemoglobin,serum amylase increased | |
Description of selected adverse reactions
Events such as Torsades de pointes, Stevens-Johnson syndrome, erythema multiforme, interstitial lung disease (sometimes fatal) and PRES (RPLS) have occurred in patients treated with vandetanib monotherapy. It is expected that these would be uncommon adverse reactions in patients receiving vandetanib for MTC.
Ocular events such as blurred vision are common in patients who received vandetanib for MTC. Scheduled slit lamp examinations have revealed corneal opacities (vortex keratopathies) in treated patients; however, routine slit lamp examinations are not required for patients receiving vandetanib.
At various exposure durations, median haemoglobin levels in patients treated with vandetanib were increased by 0.5–1.5 g/dl compared to baseline.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed below:
United Kingdom
Yellow Card Scheme
Website: www.mhra.gov.uk/yellowcard or search for MHRA Yellow Card in the Google Play or
Apple App Store
Paediatric population
Paediatric clinical trial data with vandetanib in MTC (see section 5.1) obtained during drug development is limited to 16 patients aged 9 years to 17 years with hereditary medullary thyroid carcinoma (Study IRUSZACT0098). Whilst the study size is small owing to the rarity of MTC in children, it is considered representative of the target population. The safety findings in this study are consistent with the safety profile of vandetanib in adult patients with MTC. Long term safety data in paediatric patients are not available.
4.9 Overdose
There is no specific treatment in the event of overdose with vandetanib and possible symptoms of overdose have not been established. An increase in the frequency and severity of some adverse reactions, like rash, diarrhoea and hypertension was observed at multiple doses at and above 300 mg in healthy volunteer studies and in patients. In addition, the possibility of QTc prolongation and Torsades de pointes should be considered. Vandetanib doses higher than 150 mg/m2 have not been used in clinical studies in paediatric patients.
Adverse reactions associated with overdose are to be treated symptomatically; in particular, severe diarrhoea must be managed appropriately. In the event of an overdose, further doses must be interrupted, and appropriate measures taken to assure that an adverse event has not occurred, i.e. ECG within 24 hours to determine QTc prolongation. Adverse reactions associated with overdose may be prolonged due to the long half-life of vandetanib (see section 5.2).
5. PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic Group: antineoplastic agent, protein kinase inhibitor, ATC Code: L01XE12
Mechanism of action and pharmacodynamic effects
Vandetanib is a potent inhibitor of vascular endothelial growth factor receptor-2 (VEGFR-2 also known as kinase insert domain containing receptor [KDR]), epidermal growth factor receptor (EGFR) and RET tyrosine kinases. Vandetanib is also a sub-micromolar inhibitor of vascular endothelial receptor-3 tyrosine kinase.
Vandetanib inhibits VEGF-stimulated endothelial cell migration, proliferation, survival and new blood vessel formation in in vitro models of angiogenesis. In addition, vandetanib inhibits epidermal growth factor (EGF)-stimulated EGF receptor tyrosine kinase in tumour cells and endothelial cells. Vandetanib inhibits EGFR-dependent cell proliferation and cell survival in vitro. Vandetanib also inhibits both wild type and the majority of mutated, activated forms of RET, and significantly inhibits the proliferation of MTC cell lines in vitro.
In vivo vandetanib administration reduced tumour cell-induced angiogenesis, tumour vessel permeability, tumour microvessel density, and inhibited tumour growth of a range of human xenograft tumour models in athymic mice. Vandetanib also inhibited the growth of MTC xenograft tumours in vivo.
The precise mechanism of action of vandetanib in locally advanced or metastatic MTC is unknown.
Clinical efficacy in adults
Clinical data from MTC
A randomised, double-blind, placebo-controlled study (Study 58) was conducted to demonstrate safety and efficacy of vandetanib 300 mg versus placebo. This study included 331 patients with unresectable locally advanced or metastatic MTC. Only patients with CTN ≥ 500 pg/mL (conventional units) or ≥ 146.3 pmol/L (international standard units) were enrolled. Of the patients enrolled in the study 10 patients on vandetanib and 4 on placebo (4% of all patients) had a World Health Organization performance status (WHO PS) score of ≥ 2 and 28 (12.1%) patients on vandetanib and 10 (10.1%) on placebo had cardiac impairment. Cardiac impairment was defined as patients with previous cardiovascular abnormality.
The primary objective of this study was to demonstrate an improvement in progression-free survival (PFS) with vandetanib compared to placebo. The secondary endpoints were evaluation of overall objective response rate (ORR), disease control rate (DCR) defined as, partial response (PR) or complete response (CR) or stable disease (SD) lasting at least 24 weeks, duration of response (DOR), time to worsening of pain based on Brief Pain Inventory (BPI) worst pain scale, and overall survival (OS). The PFS primary endpoint, ORR and DCR were based on centralized, independent blinded review of the imaging data. Biochemical response with vandetanib as compared to placebo as measured by CTN and CEA was also assessed as secondary endpoints.
Patients were treated with vandetanib or placebo until they reached objective disease progression. Upon objective disease progression based on the investigator's assessment, patients were discontinued from blinded study treatment and given the option to receive open-label vandetanib. Twenty-eight of the 231 patients (12.1%) on vandetanib and 3 of the 99 (3.0%) on placebo discontinued treatment because of an adverse event. Fourteen of the 28 patients (50%) who stopped vandetanib for an adverse event discontinued without a dose reduction. Five out of 6 patients (83%) with moderate renal failure who were treated with vandetanib had a dose reduction to 200 mg for adverse reaction; 1 patient required a further reduction to 100 mg.
The result of the primary analysis of PFS showed a statistically significant improvement in PFS for patients randomised to vandetanib compared to placebo (Hazard Ratio (HR) = 0.46; 95% Confidence Interval (CI) = 0.31–0.69; p=0.0001).
The median PFS for patients randomised to vandetanib has not been reached; however, based on statistical modelling of data observed up to the 43rd percentile, the median PFS is predicted to be 30.5 months with 95% confidence interval 25.5 to 36.5 months. The median PFS for patients randomised to placebo was 19.3 months. At 12 months, the proportion of patients alive and progression-free was 192 (83%) for patients randomised to vandetanib and 63 (63%) for patients randomised to placebo. In the vandetanib arm, a total of 73 (32%) patients progressed: 64 (28%) by response evaluation criteria in solid tumours (RECIST) progression and 9 (4%) by death in the absence of progression. The remaining 158 patients (68%) were censored in the analysis of PFS. In the placebo arm, a total of 51 (51%) of patients had progressed: 46 (46%) by RECIST progression and 5 (5%) by death in the absence of progression. The remaining 49 patients (49%) were censored in the analysis of PFS.
Figure 1: Kaplan Meier plot of PFS
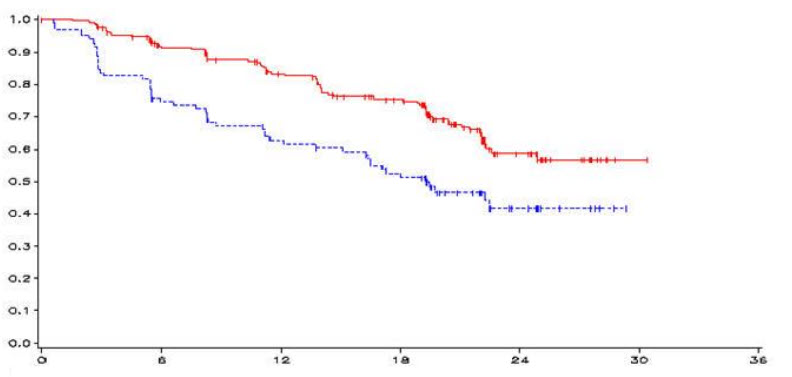
| ___vandetanib 300 mg, ------ placebo, y-axis=PFS, x-axis=time in months, n-vandetanib=number of patients at risk-vandetanib, n-placebo=number of patients at risk-placebo |
|||||||
| months | 0 | 6 | 12 | 18 | 24 | 30 | 36 |
| n-vandetanib | 231 | 196 | 169 | 140 | 40 | 1 | 0 |
| n-placebo | 100 | 71 | 57 | 45 | 13 | 0 | 0 |
| PFS | N | Median PFS | HR | 95% CI | p-value |
|---|---|---|---|---|---|
| HR = 0.46, 95%CI (0.31–0.69), p = 0.0001 | |||||
| Vandetanib 300 mg | 73/231 (32%) | Not reached (predicted 30.5 months) | 0.46 | 0.31, 0.69 | 0.0001 |
| Placebo | 51/100 (51%) | 19.3 months | |||
Survival status and the median final overall survival (81.6 months in the vandetanib arm and 80.4 months in the placebo arm) were similar across both treatment arms. There was no statistically significant difference in final OS (HR 0.99, 95.002% CI 0.72, 1.38, p=0.9750). Results should be interpreted with caution due to the high percentage of patients in the placebo arm switching to open-label vandetanib (79.0% [79/100] of patients).
Most (95% of the patients) had metastatic disease. Fourteen patients treated with vandetanib, and 3 with placebo had unresectable locally advanced disease only. There is limited clinical experience with vandetanib in patients with unresectable locally advanced disease and without metastasis.
Statistically significant advantages were seen for vandetanib for the secondary endpoints of response rate, disease control rate, and biochemical response.
| N=Number of events/number of randomised patients | |||||
|
|
|||||
| ORR* | N | Response rate | OR† | 95% CI | p-value |
| Vandetanib 300 mg | 104/231 | 45% | |||
| 5.48 | 2.99, 10.79 | < 0.0001 | |||
| Placebo | 13/100 | 13% | |||
| DCR* | N | Response rate | OR† | 95% CI | p-value |
| Vandetanib 300 mg | 200/231 | 87% | |||
| 2.64 | 1.48, 4.69 | 0.001 | |||
| Placebo | 71/100 | 71% | |||
| CTN Response | N | Response rate | OR† | 95% CI | p-value |
| Vandetanib 300 mg | 160/231 | 69% | |||
| 72.9 | 26.2, 303.2 | < 0.0001 | |||
| Placebo | 3/100 | 3% | |||
| CEA Response | N | Response rate | OR† | 95% CI | p-value |
| Vandetanib 300 mg | 119/231 | 52% | |||
| 52.0 | 16.0, 320.3 | < 0.0001 | |||
| Placebo | 2/100 | 2% | |||
| OVERALL SURVIVAL | N | Median OS | HR‡ | 95% CI | p-value |
| Vandetanib 300 mg | 116/231 | 81.6 months | 0.99 | ||
| 0.72, 1.38 | 0.9750 | ||||
| Placebo | 52/100 | 80.4 months | |||
A statistically significant advantage was seen for vandetanib for the secondary endpoint of time to worsening of pain (derived as a composite endpoint using the worst pain score from BPI and patient reported opioid analgesic use) (vandetanib 49%, placebo 57%, HR 0.61, 97.5%CI 0.43–0.87, p< 0.006: 8 vs. 3 months). There were no statistically significant differences observed for the exploratory endpoint of diarrhoea (reported as stool frequency).
RET mutation status in Study 58
In Study 58, RET mutation testing was performed by using the polymerase chain reaction (PCR) based Amplification Refractory Mutation System (ARMS) assay for the M918T mutation, and direct sequencing of DNA for mutations in exons 10, 11, 13, 14, 15 and 16 (site of M918T mutation) on all sporadic patients where DNA was available (297/298).
However, RET status could not be tested in a large proportion of patients (mainly because of unavailable results for direct sequencing of DNA) and response rate was somewhat lower in the patients with unknown RET status compared with RET mutation positive status: 51.8% vs. 35.9 % respectively. In the blinded comparison of vandetanib vs. placebo, only 2 patients known to be RET negative at all 6 exons received vandetanib and none demonstrated responses.
A post-hoc subgroup analysis of RET negative status based on absence of M918T mutation of the pivotal study 58 was performed. A patient was considered to have a RET mutation if either an M918T mutation by the ARMS assay, or a RET mutation in any exons sequenced was present in the tumour. Actually 79 patients were identified by absence of an M918T mutation and no RET mutation identified at any of the other 6 exons tested but in 71 of such patients sequencing of the 6 exons was incomplete. M918T mutation is the most frequent mutation observed in patients with sporadic MTC; however it cannot be ruled out that some patients tested RET negative for M918T mutation might be positive for mutation on other exons.
Results according to RET status (positive, unknown and RET M918T mutation negative definition) are presented in Table 4.
| Patients with documented RET mutation (n=187) | Patients with no M918T mutation and other mutations not tested or negative (n=79)* |
|
|---|---|---|
|
|
||
| Objective response rate (vandetanib arm) | 52% | 35% |
| Efficacy endpoint PFS HR (95%) confidence interval | 0.45 (0.26, 0.78) | 0.57 (0.29, 1.13) |
Clinical efficacy in paediatric patients:
A Phase I/II single-center open-label, single-arm study (Study IRUSZACT0098) assessed the activity of vandetanib in 16 patients with unresectable locally advanced or metastatic hereditary MTC. Characteristics of the patients at study entry were the following: mean age 14.2 years (range 9–17 years), 50% female, 50% male, 93.8% White, 26.7% Hispanic and 6.3% were Black. Most patients (81.3%) had undergone partial or total thyroidectomy prior to study entry. Starting vandetanib dose was 100mg/m2/day for all patients except for one who started at 150mg/m2/day. After having well tolerated the first 1 or 2 cycles of therapy (1 cycle = 28 days), the remaining patients continued on 100 mg/m2 of treatment. The primary efficacy outcome was ORR according to RECIST v 1.0. The objective response rate observed was 43.8%, all of which were partial responses. 31.3% of patients had stable disease for at least 8 weeks. Disease Control Rate including best response or Stable Disease ≥24 weeks was 75.0%. There is no experience with Caprelsa in patients 5–8 years of age in this study.
This medicinal product has been authorized under a so-called "conditional approval" scheme. This means that further evidence on this medicinal product is awaited. The European Medicines Agency (EMA) will review new information on the product every year and this SmPC will be updated as necessary.
5.2 Pharmacokinetic properties
Absorption
Following oral administration of vandetanib absorption is slow with peak plasma concentrations typically achieved at a median of 6 hours, range 4–10 hours, after dosing. Vandetanib accumulates approximately 8-fold on multiple dosing with steady state achieved from approximately 2 months.
Distribution
Vandetanib binds to human serum albumin and alpha-1-acid-glycoprotein with in vitro protein binding being approximately 90%. In ex vivo plasma samples from colorectal cancer patients at steady state exposure after 300 mg once daily, the mean percentage protein binding was 93.7% (range 92.2 to 95.7%). The pharmacokinetics of vandetanib at the 300 mg dose in MTC patients are characterised by a volume of distribution of approximately 7450 l.
Biotransformation
Following oral dosing of 14C- vandetanib, unchanged vandetanib and metabolites vandetanib N-oxide and N-desmethyl vandetanib were detected in plasma, urine and feces. A glucuronide conjugate was seen as a minor metabolite in excreta only. N-desmethyl-vandetanib is primarily produced by CYP3A4, and vandetanib-N-oxide by flavin-containing monooxygenase enzymes (FM01 and FMO3). N-desmethyl-vandetanib and vandetanib-N-oxide circulate at concentrations of approximately 11% and 1.4% of those of vandetanib.
Elimination
The pharmacokinetics of vandetanib at the 300 mg dose in MTC patients are characterised by a clearance of approximately 13.2 l/h. and plasma half-life of approximately 19 days. Within a 21 day collection period after a single dose of 14C-vandetanib, approximately 69% was recovered with 44% in faeces and 25% in urine. Excretion of the dose was slow and further excretion beyond 21 days would be expected based on the plasma half-life.
Special populations
Food effect
Exposure to vandetanib is not affected by food.
Pharmacokinetics in paediatric population
The pharmacokinetic parameters of vandetanib in paediatrics MTC patients aged 9–17 years were similar to those in adults. Vandetanib exposure in children between 5–8 years old with glioma-related indications was comparable to MTC patients aged 9–18 years. Dosing at 100mg/m2/day of the indicated posology (function of BSA) in paediatrics delivers similar exposure to that achieved in adults at 300 mg daily.
5.3 Preclinical safety data
Vandetanib has shown no mutagenic or clastogenic potential.
In repeat-dose toxicity studies of up to 9 months duration, effects included emesis, body weight loss and diarrhoea in dogs and physeal dysplasia in young dogs and rats with open growth plates. In rats, effects on teeth, kidney and skin were noted. These findings occurred at clinically-relevant plasma concentrations, were largely reversible within 4 weeks of cessation of dosing and were attributable to inhibition of vascular endothelial growth factor receptor (VEGFR) or EGFR.
Effects noted in other studies included inhibition of human ether-à-go-go related gene (hERG) current and prolongation of QTc interval in dogs. Elevation of systolic and diastolic blood pressure was observed in rats and dogs. In mice, vandetanib was shown to delay but not prevent wound healing. Vandetanib also showed evidence of phototoxic potential in an in vitro cytotoxicity assay. In an animal model of wound-healing, mice dosed with vandetanib had reduced skin-breaking strength compared with controls. This suggests that vandetanib slows but does not prevent wound healing. The appropriate interval between discontinuation of vandetanib and subsequent elective surgery required to avoid the risks of impaired wound healing has not been determined. In clinical studies, a small number of patients had surgery while receiving vandetanib and there were no reported wound healing complications.
Reproductive toxicology
Vandetanib had no effect on fertility in male rats. In a female fertility study, there was a trend towards increased oestrus cycle irregularity, a slight reduction in pregnancy incidence and increase in implantation loss. In a repeat-dose toxicity study in rats, there was a decrease in the number of corpora lutea in the ovaries of rats given vandetanib for 1 month.
In rats, embryofoetal toxicity was evident as foetal loss, delayed foetal development, heart vessel abnormalities and precocious ossification of some skull bones. In a rat pre- and post-natal development study, at doses producing maternal toxicity during gestation and/or lactation, vandetanib increased pre-birth loss and reduced post-natal pup growth. Vandetanib was excreted into milk in rat and found in plasma of pups following dosing to lactating rats.
Carcinogenicity
Vandetanib has shown no carcinogenic potential effect in a 6 month carcinogenicity study in rasH2 transgenic mice. A 2-year carcinogenicity study in rats was impaired by low survival in the high dose female group and limited exposure of the animals to vandetanib; however, no carcinogenic effects were observed in the remaining animals.
6. PHARMACEUTICAL PARTICULARS
6.1 List of excipients
7. MARKETING AUTHORISATION HOLDER
Aventis Pharma Limited
410 Thames Valley Park Drive
Reading
Berkshire
RG6 1PT
UK
Trading as:
Sanofi Genzyme
410 Thames Valley Park Drive
Reading
Berkshire
RG6 1PT
UK
9. DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION
Date of first authorisation: 17 December 2012
Date of CAP conversion: 01 January 2021
Date of last renewal: 09 December 2020
Package leaflet: Information for the patient
Caprelsa 100 mg film-coated tablets
Caprelsa 300 mg film-coated tablets
vandetanib
Is this leaflet hard to see or read?
Phone 0800 035 2525 for help
This medicine is subject to additional monitoring. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. See the end of section 4 for how to report side effects.
| In addition to this leaflet you will be given the Patient Alert Card, which contains important safety information that you need to know before you are given Caprelsa and during treatment with Caprelsa. |
Read all of this leaflet carefully before you start taking this medicine because it contains important information for you.
- - Keep this leaflet and the patient alert card. You may need to read it again.
- - It is important that you keep the Alert Card with you during treatment.
- - If you have any further questions, ask your doctor or pharmacist.
- - This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours.
- - If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4.
What is in this leaflet:
- What Caprelsa is and what it is used for
- What you need to know before you take Caprelsa
- How to take Caprelsa
- Possible side effects
- How to store Caprelsa
- Contents of the pack and other information
1. What Caprelsa is and what it is used for
Caprelsa is a treatment for adults and children aged 5 years and above with:
Medullary thyroid cancer that cannot be removed by surgery or has spread to other parts of the body.
Caprelsa works by slowing down the growth of new blood vessels in tumours (cancers). This cuts off the supply of food and oxygen to the tumour. Caprelsa may also act directly on cancer cells to kill them or slow down their growth.
2. What you need to know before you take Caprelsa
Do not take Caprelsa:
- - if you are allergic to vandetanib or any of the other ingredients of this medicine (listed in Section 6).
- - if you have a heart problem that you were born with called 'congenital long QTc syndrome'. This is seen on an electrocardiogram (ECG).
- - if you are breast-feeding.
- - if you are taking any of the following medicines: arsenic, cisapride (used to treat heartburn), erythromycin intravenous and moxifloxacin (used to treat infection), toremifene (used to treat breast cancer), mizolastine (used to treat allergies), Class IA and III antiarrhythmics (used to control heart rhythm).
Do not take Caprelsa if any of the above applies to you. If you are not sure, talk to your doctor.
Warnings and precautions
Talk to your doctor or pharmacist before taking Caprelsa:
- If you are sensitive to the sun. Some people who are taking Caprelsa become more sensitive to the sun. This can cause sunburn. While you are taking Caprelsa, protect yourself when you go outside by always using sunscreen and wearing clothes to avoid exposure to the sun.
- If you have high blood pressure.
- If you have or have had an aneurysm (enlargement and weakening of a blood vessel wall) or a tear in a blood vessel wall.
Monitoring of your blood and your heart:
Your doctor or nurse should perform tests to check the levels of your blood potassium, calcium, magnesium, and thyroid-stimulating hormone (TSH) as well as the electrical activity of your heart with a test called an electrocardiogram (ECG). You should have these tests:
- Before starting Caprelsa
- Regularly during Caprelsa treatment
- 1, 3 and 6 weeks after starting Caprelsa
- 12 weeks after starting Caprelsa
- Every 3 months thereafter
- If your doctor or pharmacist changes your dose of Caprelsa
- If you start taking medicines that affect your heart
- As instructed by your doctor or pharmacist
Children
Caprelsa should not be given to children below 5 years of age.
Other medicines and Caprelsa
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines, including medicines that you buy without a prescription and herbal medicines.
This is because Caprelsa can affect the way some medicines work and some medicines can have an effect on Caprelsa.
Tell your doctor or pharmacist if you are taking any of the following medicines:
- itraconazole, ketoconazole, ritonavir, clarithromycin, rifampicin and moxifloxacin (medicines used to treat infections)
- carbamazepine and phenobarbital (used to control seizures)
- ondansetron (used to treat nausea and vomiting)
- cisapride (used to treat heart burn), pimozide (used to treat uncontrolled repeated movements of the body and verbal outbursts) and halofantrine and lumefantrine (used to treat malaria)
- methadone (used to treat addiction), haloperidol, chlorpromazine, sulpiride, amisulpride, and zuclopenthixol, (used to treat mental illness)
- pentamidine (used to treat infection)
- vitamin K antagonists and dabigatran often referred to as 'blood thinners'
- cyclosporine and tacrolimus (used to treat transplant rejection), digoxin (used to treat irregular heart rate), and metformin (used to control your blood sugar)
- proton pump inhibitors (used to treat heartburn)
You will also find this information in the Patient Alert Card you have been given by your doctor. It is important that you keep this Alert Card and show it to your partner or caregivers.
Pregnancy and breast-feeding
If you are pregnant or breast-feeding, think you may be pregnant or are planning to have a baby, ask your doctor for advice before taking this medicine. This is because Caprelsa may harm an unborn child. Your doctor will discuss with you the benefits and risks of taking Caprelsa during this time.
- If you may become pregnant you must use effective contraception when you are taking Caprelsa and for at least four months after the last dose of Caprelsa.
You must not breast-feed during treatment with Caprelsa for the safety of your baby.
Driving and using machines
Use caution before driving or using machines. Keep in mind Caprelsa may make you feel tired, weak, or cause blurred vision.
3. How to take Caprelsa
Use in adults
Always take this medicine exactly as your doctor has told you. Check with your doctor or pharmacist if you are not sure.
- The recommended dose is 300 mg each day.
- Take Caprelsa about the same time each day.
- Caprelsa may be taken with or without food.
Use in children
The doctor will tell you how many tablets of Caprelsa to give to your child. The amount of Caprelsa given will depend on your child's body weight and height. The total daily dose in children must not exceed 300 mg. The treatment may either be given to your child as a once-daily dose, an every other day dosing or a repeating 7-day schedule as indicated in the dosing guide that has been given to you by your doctor. It is important that you keep this dosing guide and show it to your caregiver.
If you have trouble swallowing the tablet
If you have trouble swallowing the tablet, you can mix it with water as follows:
- Take half a glass of still (non-carbonated) water. Only use water, do not use any other liquids.
- Put the tablet into the water.
- Stir the tablet until it has dispersed into the water. This may take about 10 minutes.
- Then drink it straight away.
To make sure there is no medicine left, refill the glass halfway with water and drink it.
If you get side effects
If you get side effects always tell your doctor. Your doctor may tell you to take Caprelsa at a lower or increased dose (such as two 100 mg tablets or one 100 mg tablet). Your doctor may also prescribe other medicines to help control your side effects.
The side effects of Caprelsa are listed in Section 4.
If you take more Caprelsa than you should
If you take more Caprelsa than you have been prescribed, talk to a doctor or go to a hospital straight away.
If you forget to take Caprelsa
What to do if you forget to take a tablet depends on how long it is until your next dose.
- If it is 12 hours or more until your next dose: Take the missed tablet as soon as you remember. Then take the next dose at the normal time.
- If it is less than 12 hours until your next dose: Skip the missed dose. Then take the next dose at the normal time.
Do not take a double dose (two doses at the same time) to make up for a forgotten tablet.
If you have any further questions on the use of this medicine, ask your doctor or pharmacist.
4. Possible side effects
Like all medicines, this medicine can cause side effects, although not everybody gets them. If you get side effects, your doctor may tell you to take Caprelsa at a lower dose. Your doctor may also prescribe other medicines to help control your side effects.
Tell your doctor straight away if you notice any of the following side effects – you may need urgent medical treatment:
- Fainting, dizziness or heart rhythm changes. These may be signs of a change in the electrical activitiy of your heart. They are seen in 8% of people taking Caprelsa for medullary thyroid cancer. Your doctor may recommend you take Caprelsa at a lower dose or stop taking Caprelsa. Caprelsa has uncommonly been associated with life-threatening changes in heart rhythm.
- Severe skin reactions affecting large areas of your body. The signs may include redness, pain, ulcers, blisters and shedding of the skin. The lips, nose, eyes and genitals may also be affected. These may be common (affecting less than 1 in 10 people) or uncommon (affects less than 1 in 100 people) depending on the type of skin reaction.
- Severe diarrhoea.
- Serious breathlessness, or sudden worsening breathlessness, possibly with a cough or a high temperature (fever). This may mean that you have an inflammation of the lungs called 'interstitial lung disease'. This is uncommon (affects less than 1 in 100 people) but can be life-threatening.
- Seizures, headache, confusion or finding it difficult to concentrate. These may be signs of a condition called RPLS (Reversible Posterior Leukoencephalopathy Syndrome). These usually go away when Caprelsa is stopped. RPLS is uncommon (affects less than 1 in 100 people).
Tell your doctor straight away if you notice any of the side effects above.
Other side effects include:
Very common (affects more than 1 in 10 people):
- Diarrhoea. Your doctor may prescribe a medicine to treat this. If it gets severe, tell your doctor straight away.
- Abdominal pain.
- Skin rash or acne.
- Depression.
- Tiredness.
- Feeling sick (nausea).
- Upset stomach (dyspepsia).
- Nail disorders.
- Being sick (vomiting).
- Loss of appetite (anorexia).
- Weakness (asthenia).
- High blood pressure.Your doctor may prescribe a medicine to treat this.
- Headache.
- Fatigue.
- Trouble sleeping (insomnia).
- Inflammation of the nasal passages.
- Inflammation of the main air passages to the lungs.
- Upper respiratory tract infections.
- Urinary tract infections.
- Numbness or tingling of the skin.
- Abnormal sensation of the skin.
- Dizziness.
- Pain.
- Swelling caused by excess fluid (oedema).
- Stones or calcium deposits in the urinary tract (nephrolithiasis).
- Blurred vision, including mild changes in the eye which can lead to blurred vision (corneal opacity).
- Sensitivity of the skin to sunlight. While you are taking Caprelsa, protect yourself when you go outside by always using sun cream and wearing clothes to avoid exposure to the sun.
Common (affects less than 1 in 10 people)
- Dehydration.
- Severe high blood pressure.
- Weight loss.
- Stroke or other conditions where the brain may not get enough blood.
- A type of rash that affects the hands and feet (hand foot syndrome).
- Sore mouth (stomatitis).
- Dry mouth.
- Pneumonia.
- Toxins in the blood as a complication of infection.
- Flu.
- Inflammation of the urinary bladder.
- Inflammation of the sinuses.
- Inflammation of the voice box (larynx).
- Inflammation of a follicle, especially a hair follicle.
- Boil.
- Fungal infection.
- Kidney infection.
- Loss of body fluid (dehydration).
- Anxiety.
- Tremor.
- Drowsiness.
- Fainting.
- Feeling unsteady.
- Increased pressure in the eye (glaucoma).
- Coughing up of blood.
- Inflammation of the lung tissue.
- Difficulty swallowing.
- Constipation.
- Inflammation of the lining of the stomach (gastritis).
- Gastrointestinal bleeding.
- Gallstones (cholelithiasis).
- Painful urination.
- Kidney failure.
- Frequent urination.
- Urgent desire to urinate.
- Fever.
- Nose bleed (epistaxis).
- Dry eye.
- An irritation of the eyes (conjunctivitis).
- Visual impairment.
- Halo vision.
- Seeing flashes of light (photopsia).
- Disorder of the cornea of the eye (keratopathy).
- A type of diarrhoea (colitis).
- Loss of hair from the head or body (alopecia).
- Changes in taste of foods (dysgeusia).
Uncommon (affects less than 1 in 100 people)
- Heart failure.
- Inflammation of the appendix (appendicitis).
- Bacterial infection.
- Inflammation of the diverticula (small bulging pouches that can form in your digestive system).
- Bacterial skin infection.
- Abdominal wall abscess.
- Malnutrition.
- Involuntary muscle contraction (convulsions).
- Rapidly alternating muscular contraction and relaxation (clonus).
- Swelling of the brain.
- Clouding of the lens of the eye.
- Heart rate and rhythm disorders.
- Loss of heart function.
- Failure of the lungs to function properly.
- Pneumonia that happens when you breathe in foreign matter into your lungs.
- Bowel obstruction.
- Hole in your bowel.
- Inability to control your bowel movements.
- Abnormal color of urine.
- Lack of urine.
- Inability to heal properly.
- Inflammation of the pancreas (pancreatitis).
- Blistering of skin (bullous dermatitis).
Frequency: Not known
An enlargement and weakening of a blood vessel wall or a tear in a blood vessel wall (aneurysms and artery dissections).
The following side effects may be shown in tests that may be carried out by your doctor:
- Protein or blood in your urine (shown in a urine test).
- Heart rhythm changes (shown in an ECG). Your doctor may tell you to stop taking Caprelsa or take Caprelsa at a lower dose.
- Abnormalities in your liver or pancreas (shown in blood tests). These do not usually cause symptoms but your doctor may want to monitor them.
- Decreased levels of calcium in your blood. Your doctor may need to prescribe or change your thyroid hormone treatment.
- Decreased levels of potassium in your blood.
- Increased levels of calcium in your blood.
- Increased levels of glucose in your blood.
- Decreased levels of sodium in your blood.
- Decrease in thyroid function.
- Increased levels of red cells in your blood.
If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet, please tell your doctor or pharmacist straight away.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly via the contact details below. By reporting side effects you can help provide more information on the safety of this medicine.
United Kingdom
Yellow Card Scheme
Website: www.mhra.gov.uk/yellowcard or search for MHRA
Yellow Card in the Google Play or Apple App Store
5. How to store Caprelsa
Keep this medicine out of the sight and reach of children.
Do not use this medicine after the expiry date which is stated on the blister and the carton after EXP. The expiry date refers to the last day of that month.
Do not store above 30°C.
Do not throw away medicines via wastewater or household waste. Ask your pharmacist how to throw away medicines you no longer use. These measures will help protect the environment.
6. Contents of the pack and other information
What Caprelsa contains
- The active substance is vandetanib. Each tablet contains 100 or 300 mg of vandetanib.
- The other ingredients are calcium hydrogen phosphate dihydrate, microcrystalline cellulose, crospovidone (type A), povidone (K29-32), magnesium stearate, hypromellose, macrogol and titanium dioxide (E171).
What Caprelsa looks like and contents of the pack
Caprelsa 100 mg is a white round film-coated tablet with "Z100" imprinted on one side.
Caprelsa 300 mg is a white oval-shaped film-coated tablet with "Z300" imprinted on one side.
Caprelsa comes in blister packs of 30 tablets.
Marketing Authorisation Holder
Genzyme Europe B.V.,
Paasheuvelweg 25
1105 BP Amsterdam
The Netherlands
Manufacturer
EUROAPI UK Limited, 37 Hollands Road, Haverhill, Suffolk, CB9 8PU, United Kingdom
For any information about this medicine, please contact the local representative of the Marketing Authorisation Holder:
United Kingdom
Sanofi
Tel: +44 (0) 800 035 2525
Email: uk-medicalinformation@sanofi.com
This leaflet was last revised in September 2021.
This medicine has been given 'conditional approval'. This means that there is more evidence to come about this medicine.
The European Medicines Agency will review new information on the medicine at least every year and this leaflet will be updated as necessary.
Other sources of information
Detailed information on this medicine is available on the European Medicines Agency web site:
http://www.ema.europa.eu
830248

| CAPRELSA
vandetanib tablet, film coated |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Genzyme Corporation (025322157) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Lonza AG | 480007517 | API MANUFACTURE(58468-7860) , ANALYSIS(58468-7860) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| IPR Pharmaceuticals, Inc | 156931248 | MANUFACTURE(58468-7860) , ANALYSIS(58468-7860) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Penn Pharmaceutical Services Limited | 226277259 | ANALYSIS(58468-7860) , MANUFACTURE(58468-7860) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| AstraZeneca UK Ltd | 232235408 | ANALYSIS(58468-7860) , MANUFACTURE(58468-7860) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| AstraZeneca Pharmaceuticals LP | 054743190 | LABEL(58468-7860) , PACK(58468-7860) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| EUROAPI UK LIMITED | 229522842 | PACK(58468-7860) , LABEL(58468-7860) | |
Trademark Results [CAPRELSA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 CAPRELSA 85360880 4211262 Live/Registered |
GENZYME CORPORATION 2011-06-30 |
 CAPRELSA 78588611 3092679 Live/Registered |
GENZYME CORPORATION 2005-03-16 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.