KISQALI FEMARA CO-PACK- letrozole and ribociclib kit
KISQALI FEMARA CO-PACK by
Drug Labeling and Warnings
KISQALI FEMARA CO-PACK by is a Prescription medication manufactured, distributed, or labeled by Novartis Pharmaceuticals Corporation. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use KISQALI® FEMARA® CO-PACK safely and effectively. See full prescribing information for KISQALI® FEMARA® CO-PACK.
KISQALI® FEMARA® CO-PACK (ribociclib tablets; letrozole tablets), co-packaged for oral use
Initial U.S. Approval: 2017
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
KISQALI FEMARA CO-PACK, a co-packaged product containing ribociclib, a kinase inhibitor, and letrozole, an aromatase inhibitor, is indicated as initial endocrine-based therapy for the treatment of pre/perimenopausal or postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer. (1)
DOSAGE AND ADMINISTRATION
KISQALI FEMARA CO-PACK tablets are taken in combination orally with or without food. (2)
- KISQALI recommended starting dose: 600 mg orally (three 200 mg tablets) taken once daily for 21 consecutive days followed by 7 days off KISQALI treatment. (2.1)
- KISQALI dose interruption, reduction, and/or discontinuation may be required based on individual safety and tolerability. (2.2)
- FEMARA: 2.5 mg (one tablet) continuously for a 28-day cycle. (2.1)
CONTRAINDICATIONS
Known hypersensitivity to letrozole, or to any excipients of FEMARA. (4)
WARNINGS AND PRECAUTIONS
- Interstitial Lung Disease (ILD)/pneumonitis: Patients treated with CDK 4/6 inhibitors should be monitored for pulmonary symptoms indicative of ILD/pneumonitis. Interrupt and evaluate patients with new or worsening respiratory symptoms suspected to be due to ILD/pneumonitis. Permanently discontinue KISQALI in patients with recurrent symptomatic or severe ILD/pneumonitis (2.2, 5.1).
- QT Interval Prolongation: Monitor electrocardiograms (ECGs) and electrolytes prior to initiation of treatment with KISQALI. Repeat ECGs at approximately Day 14 of the first cycle and at the beginning of the second cycle, and as clinically indicated. Monitor electrolytes at the beginning of each cycle for 6 cycles, and as clinically indicated. Avoid using KISQALI with drugs known to prolong QT interval and/or strong CYP3A inhibitors. (2.2, 5.2, 7.1, 7.4)
- Hepatobiliary Toxicity: Increases in serum transaminase levels have been observed. Perform Liver Function Tests (LFTs) before initiating treatment with KISQALI FEMARA CO-PACK. Monitor LFTs every 2 weeks for the first 2 cycles, at the beginning of each subsequent 4 cycles, and as clinically indicated. (2.2, 5.3)
- Neutropenia: Perform Complete Blood Count (CBC) before initiating therapy with KISQALI FEMARA CO-PACK. Monitor CBC every 2 weeks for the first 2 cycles, at the beginning of each subsequent 4 cycles, and as clinically indicated. (2.2, 5.4)
- Embryo-Fetal Toxicity: Can cause fetal harm when administered to pregnant women. Advise women of child-bearing potential of the potential risk to a fetus and to use effective contraception during therapy. (5.5, 8.1, 8.3)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥ 20%) are neutropenia, nausea, infections, fatigue, leukopenia, diarrhea, alopecia, vomiting, headache, constipation, back pain, rash, and anemia. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
- CYP3A4 Inhibitors: Avoid concomitant use of KISQALI FEMARA CO-PACK with strong CYP3A inhibitors. If strong inhibitors cannot be avoided, reduce KISQALI dose. (2.2, 7.1)
- CYP3A4 Inducers: Avoid concomitant use of KISQALI FEMARA CO-PACK with strong CYP3A inducers. (7.2)
- CYP3A Substrates: The dose of sensitive CYP3A substrates with narrow therapeutic indices may need to be reduced when given concurrently with KISQALI FEMARA CO-PACK. (7.3)
- Drugs Known to Prolong QT interval: Avoid concomitant use of drugs known to prolong QT interval, such as anti-arrhythmic medicines. (7.4)
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 1/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosing and Administration
2.2 Dose Modifications
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease/Pneumonitis
5.2 QT Interval Prolongation
5.3 Hepatobiliary Toxicity
5.4 Neutropenia
5.5 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Drugs That May Increase Ribociclib Plasma Concentrations
7.2 Drugs That May Decrease Ribociclib Plasma Concentrations
7.3 Effect of KISQALI on Other Drugs
7.4 Drugs That Prolong the QT Interval
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing and Administration
The KISQALI FEMARA CO-PACK is comprised of ribociclib tablets copackaged with letrozole tablets, to provide a 28-day treatment regimen.
The KISQALI FEMARA CO-PACK should be coadministered, with or without food, as follows:
- KISQALI: The recommended starting dose for KISQALI is 600 mg (three 200 mg tablets) taken orally, once daily for 21 consecutive days followed by 7 days off KISQALI treatment resulting in a complete cycle of 28 days.
- FEMARA: 2.5 mg (one tablet) taken once daily throughout the 28-day cycle.
Patients should take their doses of KISQALI FEMARA CO-PACK at approximately the same time each day, preferably in the morning.
Pre/perimenopausal women treated with KISQALI FEMARA CO-PACK should be treated with a luteinizing hormone-releasing hormone (LHRH) agonist according to current clinical practice standards.
If the patient vomits after taking the dose or misses a dose, no additional dose should be taken that day. The next prescribed dose should be taken at the usual time. Tablets should be swallowed whole (tablets should not be chewed, crushed or split prior to swallowing). No tablet should be ingested if it is broken, cracked, or otherwise not intact.
For additional information on KISQALI® and FEMARA®, refer to the Full Prescribing Information for each product.
2.2 Dose Modifications
Dose Modifications for Adverse Reactions
The recommended dose modifications of KISQALI for adverse reactions are listed in Table 1.
Dose modifications are not recommended for FEMARA when administered with KISQALI for the adverse reactions of KISQALI, including: neutropenia, hepatobiliary toxicity, or QT prolongation [see Dosage and Administration (2)].
Table 1: Recommended Dose Modification of KISQALI FEMARA CO-PACK for Adverse Reactions Level KISQALI FEMARA Dose Number of Tablets Dose Number of Tablets Starting dose 600 mg/day three 200 mg tablets 2.5 mg/day one 2.5 mg tablet First dose reduction 400 mg/day two 200 mg tablets 2.5 mg/day one 2.5 mg tablet Second dose reduction 200 mg/day* one 200 mg tablet 2.5 mg/day one 2.5 mg tablet *If further dose reduction below 200 mg/day is required, discontinue KISQALI. Tables 2, 3, 4, 5, and 6 summarize recommendations for dose interruption, reduction, or discontinuation of KISQALI in the management of specific adverse reactions. Dose modifications of KISQALI FEMARA CO-PACK are recommended based on individual safety and tolerability.
Table 2: Dose Modification and Management of KISQALI for Neutropenia Neutropenia Grade 1 or 2
(ANC 1000/mm3 – < LLN)Grade 3
(ANC 500 - < 1000/mm3)Grade 3 febrile* neutropenia Grade 4
(ANC < 500/mm3)No dose adjustment is required. Dose interruption until recovery to Grade ≤ 2. Resume KISQALI at the same dose level. If toxicity recurs at Grade 3, dose interruption until recovery, then resume KISQALI at the next lower dose level. Dose interruption until recovery of neutropenia to Grade ≤ 2. Resume KISQALI at the next lower dose level. Dose interruption until recovery to Grade ≤ 2. Resume KISQALI at the next lower dose level. Perform Complete Blood Counts (CBCs) before initiating treatment with KISQALI. Monitor CBC every 2 weeks for the first 2 cycles, at the beginning of each subsequent 4 cycles, and as clinically indicated. Abbreviations: ANC, absolute neutrophil count; LLN, lower limit of normal.
*Grade 3 neutropenia with single episode of fever > 38.3°C (or) above 38°C for more than one hour and/or concurrent infection.
Grading according to Common Terminology Criteria for Adverse Events (CTCAE) Version 4.03.Table 3: Dose Modification and Management of KISQALI for Hepatobiliary Toxicity AST and/or ALT elevations from baseline*, WITHOUT increase in total bilirubin above 2 x ULN Grade 1
(> ULN – 3 x ULN)Grade 2
(> 3 to 5 x ULN)Grade 3
(> 5 to 20 x ULN)Grade 4
(> 20 x ULN)No dose adjustment is required. Baseline* at < Grade 2:
Dose interruption until recovery to ≤ baseline grade, and then resume KISQALI at same dose level. If Grade 2 recurs, resume KISQALI at next lower dose level.
-----------------------------
Baseline* at Grade 2:
No dose interruption.Dose interruption until recovery to ≤ baseline* grade, then resume at next lower dose level. If Grade 3 recurs, discontinue KISQALI. Discontinue KISQALI. Combined elevations in AST and/or ALT WITH total bilirubin increase, in the absence of cholestasis If patients develop ALT and/or AST > 3 x ULN along with total bilirubin > 2 x ULN irrespective of baseline grade, discontinue KISQALI. Perform Liver Function Tests (LFTs) before initiating treatment with KISQALI.
Monitor LFTs every 2 weeks for the first 2 cycles, at the beginning of each subsequent 4 cycles, and as clinically indicated.
If Grade ≥ 2 abnormalities are noted, more frequent monitoring is recommended.Abbreviations: AST, aspartate aminotransferase; ALT, alanine aminotransferase; ULN, upper limit of normal.
*Baseline = prior to treatment initiation.
Grading according to CTCAE Version 4.03.Table 4: Dose Modification and Management of KISQALI for QT Prolongation ECGs with QTcF* > 480 ms - Interrupt KISQALI Treatment.
- If QTcF prolongation resolves to < 481 ms, resume treatment at the next lower dose level;
- If QTcF ≥ 481 ms recurs, interrupt dose until QTcF resolves to < 481 ms; then resume KISQALI at next lower dose level.
ECGs with QTcF > 500 ms - Interrupt KISQALI treatment if QTcF greater than 500 ms.
- If QTcF prolongation resolves to < 481 ms, resume treatment at the next lower dose level
Electrocardiograms (ECGs) should be assessed prior to initiation of treatment.
Repeat ECGs at approximately Day 14 of the first cycle and at the beginning of the second cycle, and as clinically indicated.
In case of (QTcF) prolongation at any given time during treatment, more frequent ECG monitoring is recommended.
*QTcF = QT interval corrected by Fridericia’s formula.Table 5: Dose Modification and Management for Interstitial Lung Disease/Pneumonitis Grade 1
(asymptomatic)Grade 2
(symptomatic)Grade 3 (severe symptomatic)
or 4 (life-threatening)ILD/pneumonitis No dose interruption or adjustment is required. Initiate appropriate medical therapy and monitor as clinically indicated. Dose interruption until recovery
to Grade ≤ 1 then consider resuming KISQALI at the next lower dose level*.
If Grade 2 recurs, discontinue KISQALI.Discontinue KISQALI Abbreviation: ILD, interstitial lung disease.
Grading according to CTCAE Version 4.03.
*An individualized benefit-risk assessment should be performed when considering resuming KISQALI.Table 6: Dose Modification and Management of KISQALI for Other Toxicities* Other toxicities Grade 1 or 2 Grade 3 Grade 4 No dose adjustment is required. Initiate appropriate medical therapy and monitor as clinically indicated. Dose interruption until recovery to Grade ≤ 1 then resume KISQALI at same dose level. If Grade 3 recurs, resume KISQALI at the next lower dose level. Discontinue KISQALI. *Excluding neutropenia, hepatobiliary toxicity, and QT interval prolongation.
Grading according to CTCAE Version 4.03.Dose Modification for Use With Strong CYP3A Inhibitors
Avoid concomitant use of KISQALI FEMARA CO-PACK with strong CYP3A inhibitors and consider an alternative concomitant medication with less potential for CYP3A inhibition [see Drug Interactions (7.1)]. If a strong CYP3A inhibitor must be coadministered, reduce the KISQALI dose to 400 mg once daily. If the strong inhibitor is discontinued, change the KISQALI dose (after at least 5 half-lives of the strong CYP3A inhibitor) to the dose used prior to the initiation of the strong CYP3A inhibitor [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Dose Modification for Hepatic Impairment
No dose adjustment of KISQALI is necessary in patients with mild hepatic impairment (Child-Pugh class A). The recommended starting dose is 400 mg KISQALI once daily for patients with moderate (Child-Pugh class B) and severe hepatic impairment (Child-Pugh class C) [see Clinical Pharmacology (12.3)].
No dose adjustment of FEMARA is necessary in patients with mild (Child-Pugh class A) to moderate (Child-Pugh class B) hepatic impairment. The dose of FEMARA in patients with cirrhosis and severe hepatic dysfunction should be reduced by 50% [see Clinical Pharmacology (12.3)]. The recommended dose of FEMARA for such patients is 2.5 mg administered every other day. The effect of hepatic impairment on FEMARA exposure in noncirrhotic cancer patients with elevated bilirubin levels has not been determined [see Clinical Pharmacology (12.3)].
Dose Modification for Renal Impairment
No dose adjustment is necessary in patients with mild or moderate renal impairment. The recommended starting dose is 200 mg KISQALI once daily for patients with severe renal impairment [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
No dosage adjustment of FEMARA is required for patients with renal impairment if creatinine clearance is greater than or equal to 10 mL/min [see Clinical Pharmacology (12.3)].
-
3 DOSAGE FORMS AND STRENGTHS
KISQALI FEMARA CO-PACK is KISQALI (ribociclib) tablets copackaged with FEMARA (letrozole) tablets.
- Tablets 200 mg ribociclib (equivalent to 254.40 mg ribociclib succinate): Film coated, light greyish violet, round, curved with beveled edges, debossed with “RIC” on one side and “NVR” on the other side.
- Tablets 2.5 mg letrozole: Dark yellow, film-coated, round, slightly biconvex, with beveled edges (imprinted with the letters FV on one side and CG on the other side).
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease/Pneumonitis
Severe, life-threatening, or fatal interstitial lung disease (ILD) and/or pneumonitis can occur in patients treated with KISQALI and other CDK4/6 inhibitors.
Across clinical trials (MONALEESA-2, MONALEESA-3, MONALEESA-7), 1.1% of KISQALI-treated patients had ILD/pneumonitis of any grade, 0.3% had Grade 3 or 4, and 0.1% had a fatal outcome. Additional cases of ILD/pneumonitis have been observed in the postmarketing setting, with fatalities reported [see Adverse Reactions (6.2)].
Monitor patients for pulmonary symptoms indicative of ILD/pneumonitis which may include hypoxia, cough, and dyspnea. In patients who have new or worsening respiratory symptoms suspected to be due to ILD or pneumonitis, interrupt KISQALI immediately and evaluate the patient. Permanently discontinue KISQALI in patients with recurrent symptomatic or severe ILD/pneumonitis [see Dosage and Administration (2.2)]. Refer to the Full Prescribing Information for KISQALI®.
5.2 QT Interval Prolongation
KISQALI has been shown to prolong the QT interval in a concentration-dependent manner [see Clinical Pharmacology (12.2)]. Based on the observed QT prolongation during treatment, KISQALI may require dose interruption, reduction or discontinuation as described in Table 4 [see Dosage and Administration (2.2), Drug Interactions (7.4)]. In MONALEESA-2 and MONALEESA-7, in patients with advanced or metastatic breast cancer who received the combination of KISQALI plus an aromatase inhibitor, 6 out of 574 patients (1%) had > 500 ms post-baseline QTcF value, and 28 out of 574 patients (5%) had a > 60 ms increase from baseline in QTcF intervals. These ECG changes were reversible with dose interruption and the majority occurred within the first four weeks of treatment. There were no reported cases of Torsades de Pointes. In MONALEESA-2, there was one (0.3%) sudden death in a patient with Grade 3 hypokalemia and Grade 2 QT prolongation. No cases of sudden death were reported in MONALEESA-7 [see Adverse Reactions (6)].
Assess ECG prior to initiation of treatment. Initiate treatment with KISQALI FEMARA CO-PACK only in patients with QTcF values less than 450 ms. Repeat ECG at approximately Day 14 of the first cycle and the beginning of the second cycle, and as clinically indicated.
Monitor serum electrolytes (including potassium, calcium, phosphorous, and magnesium) prior to the initiation of treatment, at the beginning of the first 6 cycles, and as clinically indicated. Correct any abnormality before starting KISQALI FEMARA CO-PACK therapy [see Dosage and Administration (2.2)].
Avoid the use of KISQALI FEMARA CO-PACK in patients who already have or who are at significant risk of developing QT prolongation, including patients with:
- long QT syndrome
- uncontrolled or significant cardiac disease including recent myocardial infarction, congestive heart failure, unstable angina and bradyarrhythmias
- electrolyte abnormalities
Avoid using KISQALI FEMARA CO-PACK with drugs known to prolong QT interval and/or strong CYP3A inhibitors as this may lead to prolongation of the QTcF interval [see Drug Interactions (7.4), Clinical Pharmacology (12.3)].
Based on the observed QT prolongation during treatment, KISQALI may require dose interruption, reduction, or discontinuation as described in Table 4 [see Dosage and Administration (2.2), Drug Interactions (7.4)]. KISQALI is not indicated for concomitant use with tamoxifen. Refer to the Full Prescribing Information for KISQALI®.
5.3 Hepatobiliary Toxicity
In MONALEESA-2 and MONALEESA-7, increases in transaminases were observed, with Grade 3 or 4 increases in ALT (9% versus 1%) and AST (6% versus 2%) reported in the KISQALI plus aromatase inhibitor and placebo arms respectively.
Among the patients who had Grade ≥ 3 ALT/AST elevation, the median time-to-onset was 85 days for the KISQALI plus aromatase inhibitor treatment group. The median time to resolution to Grade ≤ 2 was 23 days in the KISQALI plus aromatase inhibitor treatment group.
Concurrent elevations in ALT or AST greater than three times the ULN and total bilirubin greater than two times the ULN, with normal alkaline phosphatase, in the absence of cholestasis occurred in 4 (1%) patients in MONALEESA-2 and all patients recovered after discontinuation of KISQALI. No cases occurred in MONALEESA-7.
Perform LFTs before initiating therapy with KISQALI FEMARA CO-PACK. Monitor LFTs every 2 weeks for first 2 cycles, at the beginning of each subsequent 4 cycles, and as clinically indicated [see Dosage and Administration (2.2)].
Based on the severity of the transaminase elevations, KISQALI may require dose interruption, reduction, or discontinuation as described in Table 3 (Dose Modification and Management for Hepatobiliary toxicity) [see Dosage and Administration (2.2)]. Recommendations for patients who have elevated AST/ALT Grade ≥ 3 at baseline have not been established.
5.4 Neutropenia
In MONALEESA-2 and MONALEESA-7, neutropenia was the most frequently reported adverse reaction (77%) and a Grade 3/4 decrease in neutrophil count (based on laboratory findings) was reported in 63% of patients receiving KISQALI plus an aromatase inhibitor. Among the patients who had Grade 2, 3, or 4 neutropenia, the median time to Grade > 2 neutropenia was 16 days. The median time to resolution of Grade ≥ 3 (to normalization or Grade < 3) was 15 days in the KISQALI plus aromatase inhibitor treatment group. Febrile neutropenia was reported in 2% of patients receiving KISQALI plus an aromatase inhibitor. Treatment discontinuation due to neutropenia was 0.7%.
Perform CBC before initiating therapy with KISQALI FEMARA CO-PACK. Monitor CBC every 2 weeks for the first 2 cycles, at the beginning of each subsequent 4 cycles, and as clinically indicated.
Based on the severity of the neutropenia, KISQALI may require dose interruption, reduction or discontinuation as described in Table 2 [see Dosage and Administration (2.2)].
5.5 Embryo-Fetal Toxicity
Based on findings from animal studies and the mechanisms of action, KISQALI FEMARA CO-PACK can cause fetal harm when administered to a pregnant woman.
In animal reproduction studies, administration of ribociclib to pregnant rats and rabbits during organogenesis caused embryo-fetal toxicities at maternal exposures that were 0.6 and 1.5 times the human clinical exposure, respectively, based on area under the curve (AUC).
Letrozole caused embryo-fetal toxicities in rats and rabbits at maternal exposures that were below the maximum recommended human dose (MRHD) on a mg/m2 basis.
Advise pregnant women of the potential risk to a fetus. Advise women of reproductive potential to use effective contraception during therapy with KISQALI FEMARA CO-PACK and for at least 3 weeks after the last dose [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- ILD/Pneumonitis [see Warnings and Precautions (5.1)]
- QT Interval Prolongation [see Warnings and Precautions (5.2)]
- Hepatobiliary Toxicity [see Warnings and Precautions (5.3)]
- Neutropenia [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
MONALEESA-2: KISQALI in Combination With Letrozole
Postmenopausal Women with HR-positive, HER2-negative Advanced or Metastatic Breast Cancer for Initial Endocrine-Based Therapy
The safety data reported below are based on MONALEESA-2, a clinical study of 668 postmenopausal women receiving KISQALI plus letrozole or placebo plus letrozole. The median duration of exposure to KISQALI plus letrozole was 13 months with 58% of patients exposed for ≥ 12 months.
Dose reductions due to adverse reactions (ARs) occurred in 45% of patients receiving KISQALI plus letrozole and in 3% of patients receiving placebo plus letrozole. Among patients receiving KISQALI plus letrozole, 7% were reported to have permanently discontinued both KISQALI and letrozole, and 7% were reported to have permanently discontinued KISQALI alone due to ARs. Among patients receiving placebo plus letrozole, 2% were reported to have permanently discontinued both, and 0.9% were reported to have permanently discontinued placebo alone due to ARs. Adverse reactions leading to treatment discontinuation of both KISQALI and letrozole in patients receiving KISQALI plus letrozole were ALT increased (4%), AST increased (3%), and vomiting (2%). Antiemetics and anti-diarrhea medications were used to manage symptoms as clinically indicated.
On-treatment deaths, regardless of causality, were reported in three cases (0.9%) of KISQALI plus letrozole treated patients vs. one case (0.3%) of placebo plus letrozole treated patients. Causes of death on KISQALI plus letrozole included one case each of the following: progressive disease, death (cause unknown), and sudden death (in the setting of Grade 3 hypokalemia and Grade 2 QT prolongation).
The most common ARs (reported at a frequency ≥ 20% on the KISQALI arm and ≥ 2% higher than placebo) were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain.
The most common Grade 3/4 ARs (reported at a frequency ≥ 5%) were neutropenia, leukopenia, abnormal liver function tests, and lymphopenia.
In MONALEESA-2, syncope occurred in 9 patients (3%) in the KISQALI plus letrozole arm versus 3 (1%) in placebo plus letrozole arm.
Adverse Reactions and laboratory abnormalities occurring in patients in MONALEESA-2 are listed in Table 7 and Table 8, respectively.
Table 7: Adverse Reactions Occurring in ≥ 10% and ≥ 2% Higher Than Placebo Arm in MONALEESA-2 (All Grades) KISQALI + letrozole Placebo + letrozole N = 334 N = 330 All Grades Grade 3 Grade 4 All Grades Grade 3 Grade 4 Adverse drug reactions % % % % % % Infections and infestations Urinary tract infection 11 1 0 8 0 0 Blood and lymphatic system disorders Neutropenia 75 50 10 5 1 0 Leukopenia 33 20 1 1 < 1 0 Anemia 18 1 < 1 5 1 0 Lymphopenia 11 6 1 2 1 0 Metabolism and nutrition disorders Decreased appetite 19 2 0 15 < 1 0 Nervous system disorders Headache 22 < 1 0 19 < 1 0 Insomnia 12 < 1 0 9 0 0 Respiratory, thoracic, and mediastinal disorders Dyspnea 12 1 0 9 1 0 Musculoskeletal and connective tissue disorders Back pain 20 2 0 18 < 1 0 Gastrointestinal disorders Nausea 52 2 0 29 1 0 Diarrhea 35 1 0 22 1 0 Vomiting 29 4 0 16 1 0 Constipation 25 1 0 19 0 0 Stomatitis 12 < 1 0 7 0 0 Abdominal pain 11 1 0 8 0 0 Skin and subcutaneous tissue disorders Alopecia 33 0 0 16 0 0 Rash 17 1 0 8 0 0 Pruritus 14 1 0 6 0 0 General disorders and administration-site conditions Fatigue 37 2 < 1 30 1 0 Pyrexia 13 < 1 0 6 0 0 Edema peripheral 12 0 0 10 0 0 Investigations Abnormal liver function tests1 18 8 2 6 2 0 Grading according to CTCAE 4.03 (Common Terminology Criteria for Adverse Events).
1abnormal liver function tests: ALT increased, AST increased, blood bilirubin increased.Additional adverse reactions in MONALEESA-2 for patients receiving KISQALI plus letrozole included interstitial lung disease (0.3%), lung infiltration (0.3%), pneumonitis (0.3%), and pulmonary fibrosis (0.6%).
Table 8: Laboratory Abnormalities Occurring in ≥ 10% of Patients in MONALEESA-2 KISQALI + letrozole Placebo + letrozole N = 334 N = 330 All Grades Grade 3 Grade 4 All Grades Grade 3 Grade 4 Laboratory parameters % % % % % % HEMATOLOGY Leukocyte count decreased 93 31 3 29 1 < 1 Neutrophil count decreased 93 49 11 24 1 < 1 Hemoglobin decreased 57 2 0 26 1 0 Lymphocyte count decreased 51 12 2 22 3 1 Platelet count decreased 29 1 < 1 6 0 < 1 CHEMISTRY Alanine aminotransferase increased 46 8 2 36 1 0 Aspartate aminotransferase increased 44 6 1 32 2 0 Creatinine increased 20 1 0 6 0 0 Phosphorous decreased 13 5 1 4 1 0 Potassium decreased 11 1 1 7 1 0 Adverse Reactions listed are based on the data of KISQALI in combination with letrozole (FEMARA). For the complete list of known ARs with KISQALI or FEMARA, see the Full Prescribing Information of KISQALI or FEMARA.
Bone Effects:
In MONALEESA-2, with a median duration of safety follow-up of 20.1 months, 12 patients (4%) in the ribociclib plus letrozole arm and 18 patients (6%) in the placebo plus letrozole arm experienced fractures. Osteoporosis (all Grades) was experienced in three patients (0.9%) in the ribociclib plus letrozole arm and 2 patients (0.6%) in the placebo plus letrozole arm.
MONALEESA-7: KISQALI in Combination With an Aromatase Inhibitor
Pre/perimenopausal Patients with HR-positive, HER2-negative Advanced or Metastatic Breast Cancer for Initial Endocrine-Based Therapy
MONALEESA-7 was conducted in 672 pre/perimenopausal patients with HR-positive, HER2-negative advanced or metastatic breast cancer receiving either KISQALI plus a non-steroidal aromatase inhibitors (NSAI) or tamoxifen plus goserelin or placebo plus NSAI or tamoxifen plus goserelin. The median duration of exposure on the KISQALI arm was 15.2 months with 66% of patients exposed for ≥ 12 months. The safety data reported below are based on 495 pre/perimenopausal patients receiving KISQALI plus NSAI plus goserelin or placebo plus NSAI plus goserelin.
Dose reductions due to ARs occurred in 33% of patients receiving KISQALI plus NSAI plus goserelin and in 4% of patients receiving placebo plus NSAI plus goserelin. Among patients receiving KISQALI plus NSAI, 3% were reported to have permanently discontinued both KISQALI and NSAI and 3% were reported to have permanently discontinued KISQALI alone due to ARs. Among patients receiving placebo plus NSAI, 2% were reported to have permanently discontinued both, and 0.8% were reported to have permanently discontinued placebo alone due to ARs. Adverse reactions leading to treatment discontinuation on KISQALI in patients receiving KISQALI plus NSAI (as compared to the placebo arm) were ALT increased (2% vs. 0.8%), AST increased (2% vs. 0.8%), drug-induced liver injury (1% vs. 0.4%). One patient (0.4%) died while on treatment with KISQALI plus NSAI plus goserelin, due to the underlying malignancy.
The most common ARs (reported at a frequency ≥ 20% on the KISQALI arm and ≥ 2% higher than placebo) were neutropenia, infections, leukopenia, arthralgia, nausea, and alopecia. The most common Grade 3/4 ARs (reported at a frequency ≥ 5%) were neutropenia, leukopenia, and abnormal liver function tests. See Table 9 below.
Adverse reactions and laboratory abnormalities occurring in patients in MONALEESA-7 are listed in Table 9 and Table 10, respectively.
Table 9: Adverse Reactions Occurring in ≥ 10% and ≥ 2% Higher Than Placebo Arm in MONALEESA-7 (NSAI) (All Grades) Grading according to CTCAE 4.03 (Common Terminology Criteria for Adverse Events).
1Infections: urinary tract infections; respiratory tract infections; gastroenteritis; sepsis (< 1%).KISQALI + NSAI + goserelin Placebo + NSAI + goserelin N = 248 N = 247 All Grades Grade 3 Grade 4 All Grades Grade 3 Grade 4 Adverse drug reactions % % % % % % Infections and Infestations Infections1 35 2 0 24 < 1 0 Blood and lymphatic system disorders Neutropenia 78 55 10 7 2 < 1 Leukopenia 29 13 < 1 3 < 1 0 Anemia 19 3 0 8 1 0 Respiratory, thoracic and mediastinal disorders Cough 15 0 0 10 0 0 Musculoskeletal and connective tissue disorders Arthralgia 33 < 1 0 29 1 0 Gastrointestinal disorders Nausea 31 0 0 20 0 0 Constipation 16 0 0 12 0 0 Stomatitis 10 0 0 8 < 1 0 Skin and subcutaneous tissue disorders Alopecia 21 0 0 13 0 0 Rash 17 < 1 0 9 0 0 Pruritus 10 0 0 4 0 0 General disorders and administration site conditions Pyrexia 17 < 1 0 6 0 0 Pain in extremity 10 0 0 8 1 0 Investigations Alanine aminotransferase increased 13 5 0 9 1 0 Aspartate aminotransferase increased 13 4 0 10 1 0 Additional adverse reactions in MONALEESA-7 for patients receiving KISQALI plus NSAI included asthenia (12%), thrombocytopenia (9%), dry skin (8%), oropharyngeal pain (7%), dyspepsia (5%), lacrimation increased (4%), dry eye (4%), vitiligo (3%), hypocalcemia, (2%), blood bilirubin increased (1%), syncope (0.4%), and pneumonitis (0.4%).
Table 10: Laboratory Abnormalities Occurring in ≥ 10% of Patients in MONALEESA-7 KISQALI + NSAI +
goserelinPlacebo + NSAI +
goserelinN = 248 N = 247 All Grades Grade 3 Grade 4 All Grades Grade 3 Grade 4 Laboratory parameters % % % % % % HEMATOLOGY Leukocyte count decreased 93 34 2 30 < 1 < 1 Neutrophil count decreased 92 54 9 27 2 0 Hemoglobin decreased 84 2 0 51 < 1 0 Lymphocyte count decreased 55 12 2 18 2 < 1 Platelet count decreased 26 < 1 0 9 0 < 1 CHEMISTRY Alanine aminotransferase increased 33 6 0 31 1 < 1 Aspartate aminotransferase increased 37 5 0 35 1 < 1 Creatinine increased 21 2 < 1 20 < 1 < 1 Phosphorous decreased 14 2 0 11 < 1 < 1 Potassium decreased 11 < 1 < 1 14 < 1 < 1 Gamma-glutamyl transferase increased 42 5 2 42 8 1 Glucose serum decreased 10 < 1 0 10 < 1 0 Adverse Reactions listed are based on the data of KISQALI in combination with NSAI [anastrozole or letrozole (FEMARA)]. For the complete list of known ARs with KISQALI or FEMARA, see the Full Prescribing Information of KISQALI or FEMARA.
Bone Effects:
In MONALEESA-7, with a median duration of safety follow-up of 26.5 months, 4 patients (2%) in the ribociclib plus NSAI subgroup and 7 patients (3%) in the placebo plus NSAI subgroup experienced fractures. No osteoporosis (all Grades) was reported in the ribociclib plus NSAI subgroup, and 1 patient (0.4%) experienced osteoporosis in the placebo plus NSAI subgroup.
6.2 Postmarketing Experience
The following adverse events have been reported during post-approval use of KISQALI. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Respiratory disorders: Interstitial lung disease (ILD)/pneumonitis
-
7 DRUG INTERACTIONS
Ribociclib
7.1 Drugs That May Increase Ribociclib Plasma Concentrations
CYP3A4 Inhibitors
Coadministration of a strong CYP3A4 inhibitor (ritonavir) increased ribociclib exposure in healthy subjects by 3.2-fold [see Clinical Pharmacology (12.3)]. Avoid concomitant use of strong CYP3A inhibitors (e.g., boceprevir, clarithromycin, conivaptan, grapefruit juice, indinavir, itraconazole, ketoconazole, lopinavir/ritonavir, nefazodone, nelfinavir, posaconazole, ritonavir, saquinavir, and voriconazole) and consider alternative concomitant medications with less potential for CYP3A inhibition.
If coadministration of KISQALI with a strong CYP3A inhibitor cannot be avoided, reduce the dose of KISQALI to 400 mg once daily [see Dosage and Administration (2.2)].
Instruct patients to avoid grapefruit or grapefruit juice, which are known to inhibit cytochrome CYP3A enzymes and may increase the exposure to ribociclib [see Patient Counseling Information (17)].
7.2 Drugs That May Decrease Ribociclib Plasma Concentrations
CYP3A4 Inducers
Coadministration of a strong CYP3A4 inducer (rifampin) decreased the plasma exposure of ribociclib in healthy subjects by 89% [see Clinical Pharmacology (12.3)]. Avoid concomitant use of strong CYP3A inducers and consider an alternate concomitant medication with no or minimal potential to induce CYP3A (e.g., phenytoin, rifampin, carbamazepine and St. John’s Wort (Hypericum perforatum)).
7.3 Effect of KISQALI on Other Drugs
CYP3A Substrates With Narrow Therapeutic Index:
Coadministration of midazolam (a sensitive CYP3A4 substrate) with multiple doses of KISQALI (400 mg) increased the midazolam exposure by 3.8-fold in healthy subjects, compared with administration of midazolam alone [see Clinical Pharmacology (12.3)]. KISQALI given at the clinically relevant dose of 600 mg is predicted to increase the midazolam AUC by 5.2-fold. Therefore, caution is recommended when KISQALI is administered with CYP3A substrates with a narrow therapeutic index. The dose of a sensitive CYP3A substrate with a narrow therapeutic index, including but not limited to alfentanil, cyclosporine, dihydroergotamine, ergotamine, everolimus, fentanyl, pimozide, quinidine, sirolimus, and tacrolimus may need to be reduced as ribociclib can increase their exposure.
7.4 Drugs That Prolong the QT Interval
Avoid coadministration of KISQALI with medicinal products with a known potential to prolong QT, such as antiarrhythmic medicines (including, but not limited to amiodarone, disopyramide, procainamide, quinidine, and sotalol), and other drugs that are known to prolong the QT interval (including, but not limited to, chloroquine, halofantrine, clarithromycin, haloperidol, methadone, moxifloxacin, bepridil, pimozide, and ondansetron) [see Warnings and Precautions (5.1), Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and the mechanisms of action, KISQALI FEMARA CO-PACK can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available human data informing the drug-associated risk. Advise pregnant women of the potential risk to a fetus.
In animal reproduction studies, administration of ribociclib to pregnant animals during organogenesis resulted in increased incidences of postimplantation loss and reduced fetal weights in rats and increased incidences of fetal abnormalities in rabbits at exposures 0.6 or 1.5 times the exposure in humans, respectively, at the highest recommended dose of 600 mg/day based on AUC (see Data).
In animal reproduction studies, administration of letrozole to pregnant animals during organogenesis resulted in increased post-implantation pregnancy loss and resorptions, fewer live fetuses, and fetal malformations affecting the renal and skeletal systems in rats and rabbits at doses approximately 0.1 times the daily MRHD on a mg/m2 basis (see Data).
The background risk of major birth defects for the indicated population is unknown. However, the background risk of major birth defects is 2%-4% and of miscarriage is 15%-20% of clinically recognized pregnancies in the U.S. general population.
Data
Animal Data - Ribociclib
In embryo-fetal development studies in rats and rabbits, pregnant animals received oral doses of ribociclib up to 1000 mg/kg/day and 60 mg/kg/day, respectively, during the period of organogenesis.
In rats, 300 mg/kg/day resulted in reduced maternal body weight gain and reduced fetal weights accompanied by skeletal changes related to the lower fetal weights. There were no significant effects on embryo-fetal viability or fetal morphology at 50 or 300 mg/kg/day.
In rabbits at doses ≥ 30 mg/kg/day, there were adverse effects on embryo-fetal development, including increased incidences of fetal abnormalities (malformations and external, visceral and skeletal variants) and fetal growth (lower fetal weights). These findings included reduced/small lung lobes, additional vessel on the descending aorta, additional vessel on the aortic arch, small eyes, diaphragmatic hernia, absent accessory lobe or (partly) fused lung lobes, reduced/small accessory lung lobe, extra/rudimentary 13th ribs, misshapen hyoid bone, bent hyoid bone alae and reduced number of phalanges in the pollex. There was no evidence of increased incidence of embryo-fetal mortality. There was no maternal toxicity observed at 30 mg/kg/day.
At 300 mg/kg/day in rats and 30 mg/kg/day in rabbits, the maternal systemic exposures (AUC) were approximately 0.6 and 1.5 times, respectively, the exposure in patients at the highest recommended dose of 600 mg/day.
Animal Data - Letrozole
In a fertility and early embryonic developmental toxicity study in female rats, oral administration of letrozole starting 2 weeks before mating until pregnancy day 6 resulted in an increase in pre-implantation loss at doses ≥ 0.003 mg/kg/day (approximately 0.01 times the MRHD on a mg/m2 basis).
In an embryo-fetal developmental toxicity study in rats, daily administration of oral letrozole during the period of organogenesis at doses ≥ 0.003 mg/kg (approximately 0.01 times the MRHD on a mg/m2 basis) resulted in embryo-fetal toxicity, including intrauterine mortality, increased resorptions and postimplantation loss, decreased numbers of live fetuses and fetal anomalies, including absence and shortening of renal papilla, dilation of ureter, edema and incomplete ossification of frontal skull and metatarsals. Letrozole was teratogenic to rats at a dose of 0.03 mg/kg (approximately 0.1 times the MRHD on a mg/m2 basis) and caused fetal domed head and cervical/centrum vertebral fusion.
In an embryo-fetal developmental toxicity study in rabbits, daily administration of oral letrozole during the period of organogenesis at doses ≥ 0.002 mg/kg (approximately 0.01 the MRHD on a mg/m2 basis) resulted in embryo-fetal toxicity, including increased resorptions, increased postimplantation loss and decreased numbers of live fetuses. Fetal anomalies included incomplete ossification of the skull, sternebrae, and fore- and hind legs.
8.2 Lactation
Risk Summary
It is not known if ribociclib and letrozole are present in human milk. There are no data on the effects of ribociclib and/or letrozole on the breastfed infant or milk production. Ribociclib and its metabolites readily passed into the milk of lactating rats (see Data). Exposure of lactating rats to letrozole was associated with an impaired reproductive performance of the male offspring (see Data). Because of the potential for serious adverse reactions in breastfed infants from KISQALI FEMARA CO-PACK, advise lactating women not to breastfeed while taking KISQALI FEMARA CO-PACK and for at least 3 weeks after the last dose.
Data
Animal Data - Ribociclib
In lactating rats administered a single dose of 50 mg/kg, exposure to ribociclib was 3.56-fold higher in milk compared to maternal plasma.
Animal Data - Letrozole
In a postnatal developmental toxicity study in lactating rats, letrozole was administered orally at doses of 1, 0.003, 0.03 or 0.3 mg/kg/day on day 0 through Day 20 of lactation. The reproductive performance of the male offspring was impaired at a letrozole dose as low as 0.003 mg/kg/day (approximately 0.01 the MRHD on a mg/m2 basis), as reflected by decreased mating and pregnancy ratios. There were no effects on the reproductive performance of female offspring.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Based on animal studies and mechanisms of action, KISQALI FEMARA CO-PACK can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Females of reproductive potential should have a pregnancy test prior to starting treatment with KISQALI FEMARA CO-PACK.
Contraception
Females
Based on animal studies and mechanisms of action, KISQALI FEMARA CO-PACK can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception (methods that result in less than 1% pregnancy rates) during treatment with KISQALI FEMARA CO-PACK and for at least 3 weeks after the last dose.
Infertility
Females
Based on studies with letrozole in female animals, KISQALI FEMARA CO-PACK may impair fertility in females of reproductive potential [see Nonclinical Toxicology (13.1)].
Males
Based on animal studies in males, KISQALI FEMARA CO-PACK may impair fertility in males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and efficacy of KISQALI FEMARA CO-PACK in pediatric patients have not been established.
Letrozole administration to young (postnatal Day 7) rats for 12 weeks duration at 0.003, 0.03, 0.3 mg/kg/day by oral gavage resulted in adverse skeletal/growth effects (bone maturation, bone mineral density) and neuroendocrine and reproductive developmental perturbations of the hypothalamic-pituitary axis. Administration of 0.3 mg/kg/day resulted in AUC values that were similar to the AUC in adult patients receiving the recommended dose of 2.5 mg/day. Decreased fertility was accompanied by hypertrophy of the hypophysis and testicular changes that included degeneration of the seminiferous tubular epithelium and atrophy of the female reproductive tract. Young rats in this study were allowed to recover following discontinuation of letrozole treatment for 42 days. Histopathological changes were not reversible at clinically relevant exposures.
8.5 Geriatric Use
Of 334 patients who received KISQALI plus letrozole in MONALEESA-2, 150 patients (45%) were ≥ 65 years of age and 35 patients (11%) were ≥ 75 years of age. No overall differences in safety or effectiveness of KISQALI plus letrozole were observed between these patients and younger patients.
8.6 Hepatic Impairment
No dose adjustment of KISQALI is necessary in patients with mild hepatic impairment (Child-Pugh class A). The recommended starting dose is 400 mg KISQALI once daily for patients with moderate (Child-Pugh class B) and severe hepatic impairment (Child-Pugh class C) [see Clinical Pharmacology (12.3)].
No dose adjustment of FEMARA is necessary in patients with mild (Child-Pugh class A) to moderate (Child-Pugh class B) hepatic impairment. The dose of FEMARA in patients with cirrhosis and severe hepatic dysfunction should be reduced by 50% [see Clinical Pharmacology (12.3)]. The recommended dose of FEMARA for such patients is 2.5 mg administered every other day. The effect of hepatic impairment on FEMARA exposure in noncirrhotic cancer patients with elevated bilirubin levels has not been determined [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
Based on a population pharmacokinetic analysis, no dose adjustment of KISQALI is necessary in patients with mild (60 mL/min/1.73 m2 ≤ estimated glomerular filtration rate (eGFR) < 90 mL/min/1.73 m2) or moderate (30 mL/min/1.73 m2 ≤ eGFR < 60 mL/min/1.73 m2) renal impairment. Based on a ribociclib renal impairment study in healthy subjects and non-cancer subjects with severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2), a starting dose of 200 mg of KISQALI is recommended. KISQALI has not been studied in breast cancer patients with severe renal impairment [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
No dosage adjustment of FEMARA is required for patients with renal impairment if creatinine clearance is greater than or equal to 10 mL/min [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There is limited experience with reported cases of overdose with ribociclib and letrozole in MONALEESA-2 and MONALEESA-7. General symptomatic and supportive measures should be initiated in all cases of overdose where necessary.
Refer to the Full Prescribing Information of FEMARA for additional overdosage information.
-
11 DESCRIPTION
KISQALI FEMARA CO-PACK consists of ribociclib 200 mg film coated tablets copackaged with letrozole 2.5 mg tablets.
Ribociclib:
KISQALI (ribociclib) is a kinase inhibitor.
Ribociclib succinate is a light yellow to yellowish brown crystalline powder. The chemical name of ribociclib succinate is: Butanedioic acid—7-cyclopentyl-N,N-dimethyl-2-{[5-(piperazin-1-yl) pyridin-2-yl]amino}-7H-pyrrolo[2,3-d]pyrimidine-6-carboxamide (1/1).
The molecular formula for ribociclib succinate is C23H30N8O.C4H6O4 and the molecular weight is 552.64 g/mol [Free base: 434.55 g/mol].
The chemical structure of ribociclib is shown below:
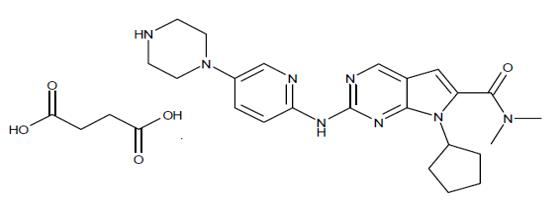
KISQALI film-coated tablets are supplied for oral use and contain 200 mg of ribociclib free base (equivalent to 254.40 mg ribociclib succinate). The tablets also contain colloidal silicon dioxide, crospovidone, hydroxypropylcellulose, magnesium stearate, and microcrystalline cellulose. The film-coating contains iron oxide black, iron oxide red, lecithin (soya), polyvinyl alcohol (partially hydrolyzed), talc, titanium dioxide, and xanthan gum as inactive ingredients.
Letrozole:
FEMARA (letrozole) is a nonsteroidal aromatase inhibitor (inhibitor of estrogen synthesis).
Letrozole is a white to yellowish crystalline powder, practically odorless, freely soluble in dichloromethane, slightly soluble in ethanol, and practically insoluble in water. It has a molecular weight of 285.31 g/mol, empirical formula C17H11N5, and a melting range of 184°C to 185°C.
The chemical name of letrozole is 4,4'-(1H-1,2,4-Triazol-1ylmethylene) dibenzonitrile, and its structural formula is
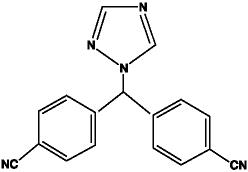
FEMARA is available as 2.5 mg tablets for oral administration.
Inactive Ingredients: Colloidal silicon dioxide, ferric oxide, hydroxypropyl methylcellulose, lactose monohydrate, magnesium stearate, maize starch, microcrystalline cellulose, polyethylene glycol, sodium starch glycolate, talc, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ribociclib is an inhibitor of cyclin-dependent kinase (CDK) 4 and 6. These kinases are activated upon binding to D-cyclins and play a crucial role in signaling pathways which lead to cell cycle progression and cellular proliferation. The cyclin D-CDK4/6 complex regulates cell cycle progression through phosphorylation of the retinoblastoma protein (pRb).
In vitro, ribociclib decreased pRb phosphorylation leading to arrest in the G1 phase of the cell cycle and reduced cell proliferation in breast cancer cell lines. In vivo, treatment with single agent ribociclib in a rat xenograft model with human tumor cells led to decreased tumor volumes which correlated with inhibition of pRb phosphorylation.
Letrozole is a nonsteroidal competitive inhibitor of the aromatase enzyme system by competitively binding to the heme of the cytochrome P450 subunit of the enzyme, resulting in a reduction of estrogen biosynthesis in all tissues. In postmenopausal women, estrogens are mainly derived from the action of the aromatase enzyme, which converts adrenal androgens (primarily androstenedione and testosterone) to estrone and estradiol. The suppression of estrogen biosynthesis in peripheral tissues and in the cancer tissue itself can therefore be achieved by specifically inhibiting the aromatase enzyme.
In vivo studies, using patient-derived estrogen receptor positive breast cancer xenograft models, combination of ribociclib and antiestrogen (e.g., letrozole) resulted in increased tumor growth inhibition compared to each drug alone.
12.2 Pharmacodynamics
Ribociclib
Cardiac Electrophysiology
Serial, triplicate ECGs were collected following a single dose and at steady-state to evaluate the effect of ribociclib on the QTcF interval in patients with advanced cancer. A pharmacokinetic-pharmacodynamic analysis included a total of 997 patients treated with ribociclib at doses ranging from 50 to 1200 mg. The analysis suggested that ribociclib causes concentration-dependent increases in the QTcF interval. The estimated mean change from baseline in QTcF for KISQALI 600 mg in combination with aromatase inhibitors was 22.0 ms (90% CI: 22.0, 23.4) at the geometric mean Cmax at steady-state [see Warnings and Precautions (5.2)].
Letrozole
In postmenopausal patients with advanced breast cancer, daily doses of 0.1 mg to 5 mg FEMARA (letrozole) suppress plasma concentrations of estradiol, estrone, and estrone sulfate by 75% to 95% from baseline with maximal suppression achieved within two-three days. Suppression is dose-related, with doses of 0.5 mg and higher giving many values of estrone and estrone sulfate that were below the limit of detection in the assays. Estrogen suppression was maintained throughout treatment in all patients treated at 0.5 mg or higher.
Letrozole is highly specific in inhibiting aromatase activity. There is no impairment of adrenal steroidogenesis. No clinically-relevant changes were found in the plasma concentrations of cortisol, aldosterone, 11-deoxycortisol, 17-hydroxy-progesterone, ACTH or in plasma renin activity among postmenopausal patients treated with a daily dose of FEMARA 0.1 mg to 5 mg. The ACTH stimulation test performed after 6 and 12 weeks of treatment with daily doses of 0.1, 0.25, 0.5, 1, 2.5, and 5 mg did not indicate any attenuation of aldosterone or cortisol production. Glucocorticoid or mineralocorticoid supplementation is, therefore, not necessary.
No changes were noted in plasma concentrations of androgens (androstenedione and testosterone) among healthy postmenopausal women after 0.1, 0.5, and 2.5 mg single doses of FEMARA or in plasma concentrations of androstenedione among postmenopausal patients treated with daily doses of 0.1 mg to 5 mg. This indicates that the blockade of estrogen biosynthesis does not lead to accumulation of androgenic precursors. Plasma levels of LH and FSH were not affected by letrozole in patients, nor was thyroid function as evaluated by TSH levels, T3 uptake, and T4 levels.
12.3 Pharmacokinetics
Ribociclib exhibited over-proportional increases in exposure (peak plasma concentrations (Cmax) and area under the time concentration curve (AUC)) across the dose range of 50 mg to 1200 mg following both single dose and repeated doses. Following repeated 600 mg once daily administration, steady-state was generally achieved after 8 days and ribociclib accumulated with a geometric mean accumulation ratio of 2.51 (range, 0.972 to 6.40).
Letrozole’s terminal elimination half-life is about 2 days and steady-state plasma concentration after daily 2.5 mg dosing is reached in 2-6 weeks. Plasma concentrations at steady state are 1.5 to 2 times higher than predicted from the concentrations measured after a single dose, indicating a slight non-linearity in the pharmacokinetics of letrozole upon daily administration of 2.5 mg. These steady-state levels are maintained over extended periods, however, and continuous accumulation of letrozole does not occur.
Absorption and Distribution
Ribociclib
The time to reach Cmax (Tmax) following ribociclib administration was between 1 and 4 hours.
Binding of ribociclib to human plasma proteins in vitro was approximately 70% and independent of concentration (10 to 10,000 ng/mL). Ribociclib was equally distributed between red blood cells and plasma with a mean in vivo blood-to-plasma ratio of 1.04. The apparent volume of distribution at steady-state (Vss/F) was 1090 L based on population PK analysis.
Food Effect: Compared to the fasted state, oral administration of a single 600 mg dose of KISQALI film-coated tablet with a high-fat, high-calorie meal (approximately 800 to 1000 calories with ~50% calories from fat, ~35% calories from carbohydrates, and ~15% calories from protein) had no effect on the rate and extent of absorption of ribociclib (Cmax GMR: 1.00; 90% CI: 0.898, 1.11; AUCinf GMR: 1.06; 90% CI: 1.01, 1.12) .
Letrozole
Letrozole is rapidly and completely absorbed from the gastrointestinal tract and absorption is not affected by food. It is metabolized slowly to an inactive metabolite whose glucuronide conjugate is excreted renally, representing the major clearance pathway. About 90% of radiolabeled letrozole is recovered in urine.
Letrozole is weakly protein bound and has a large volume of distribution (approximately 1.9 L/kg).
Metabolism and Elimination/Excretion
Ribociclib
In vitro and in vivo studies indicated ribociclib undergoes extensive hepatic metabolism mainly via CYP3A4 in humans. Following oral administration of a single 600 mg dose of radio-labeled ribociclib to humans, the primary metabolic pathways for ribociclib involved oxidation (dealkylation, C and/or N-oxygenation, oxidation (-2H)) and combinations thereof. Phase II conjugates of ribociclib Phase I metabolites involved N-acetylation, sulfation, cysteine conjugation, glycosylation and glucuronidation. Ribociclib was the major circulating drug-derived entity in plasma (44%). The major circulating metabolites included metabolite M13 (CCI284, N-hydroxylation), M4 (LEQ803, N-demethylation), and M1 (secondary glucuronide), each representing an estimated 9%, 9%, and 8% of total radioactivity, and 22%, 20%, and 18% of ribociclib exposure. Clinical activity (pharmacological and safety) of ribociclib was due primarily to parent drug, with negligible contribution from circulating metabolites.
Ribociclib was extensively metabolized with unchanged drug accounting for 17% and 12% in feces and urine, respectively. Metabolite LEQ803 was a significant metabolite in excreta and represented approximately 14% and 4% of the administered dose in feces and urine, respectively. Numerous other metabolites were detected in both feces and urine in minor amounts (≤ 3% of the administered dose).
The geometric mean plasma effective half-life (based on accumulation ratio) was 32.0 hours (63% CV) and the geometric mean apparent oral clearance (CL/F) was 25.5 L/hr (66% CV) at steady-state at 600 mg in patients with advanced cancer. The geometric mean apparent plasma terminal half-life (T½) of ribociclib ranged from 29.7 to 54.7 hours and geometric mean CL/F of ribociclib ranged from 39.9 to 77.5 L/hr at 600 mg across studies in healthy subjects.
Ribociclib is eliminated mainly via feces, with a small contribution of the renal route. In 6 healthy male subjects, following a single oral dose of radio-labeled ribociclib, 92% of the total administered radioactive dose was recovered within 22 days; feces was the major route of excretion (69%), with 23% of the dose recovered in urine.
Letrozole
Metabolism to a pharmacologically-inactive carbinol metabolite (4,4'methanol-bisbenzonitrile) and renal excretion of the glucuronide conjugate of this metabolite is the major pathway of letrozole clearance. Of the radiolabel recovered in urine, at least 75% was the glucuronide of the carbinol metabolite, about 9% was two unidentified metabolites, and 6% was unchanged letrozole.
In human microsomes with specific CYP isozyme activity, CYP3A4 metabolized letrozole to the carbinol metabolite while CYP2A6 formed both this metabolite and its ketone analog. In human liver microsomes, letrozole inhibited CYP2A6 and inhibited CYP2C19, however, the clinical significance of these findings is unknown.
Specific Populations
Patients With Hepatic Impairment
Ribociclib
Based on a pharmacokinetic trial in patients with hepatic impairment, mild (Child-Pugh class A) hepatic impairment had no effect on the exposure of ribociclib. The mean exposure for ribociclib was increased less than 2-fold in patients with moderate (Child-Pugh class B; geometric mean ratio [GMR]: 1.44 for Cmax; 1.28 for AUCinf) or severe (Child-Pugh class C; GMR: 1.32 for Cmax; 1.29 for AUCinf) hepatic impairment. Based on a population pharmacokinetic analysis that included 160 patients with normal hepatic function and 47 patients with mild hepatic impairment, mild hepatic impairment had no effect on the exposure of ribociclib, further supporting the findings from the dedicated hepatic impairment study.
Letrozole
The effect of hepatic impairment on FEMARA exposure in noncirrhotic cancer patients with elevated bilirubin levels has not been determined.
In a study of subjects with mild to moderate non-metastatic hepatic dysfunction (e.g., cirrhosis, Child-Pugh classification A and B), the mean AUC values of the volunteers with moderate hepatic impairment were 37% higher than in normal subjects, but still within the range seen in subjects without impaired function.
In a pharmacokinetic study, subjects with liver cirrhosis and severe hepatic impairment (Child-Pugh classification C, which included bilirubins about 2-11 times ULN with minimal to severe ascites) had 2-fold increase in exposure (AUC) and 47% reduction in systemic clearance. Breast cancer patients with severe hepatic impairment are thus expected to be exposed to higher levels of letrozole than patients with normal liver function receiving similar doses of this drug [see Clinical Pharmacology (12.3)].
Patients With Renal Impairment
Ribociclib
The effect of renal impairment on the pharmacokinetics of ribociclib was assessed in a renal impairment study in non-cancer subjects with normal renal function (eGFR ≥ 90 mL/min/1.73 m2, n = 9), severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2, n = 6), and End Stage Renal Disease (ESRD) (eGFR < 15 mL/min/1.73 m2, n = 4) at a single ribociclib dose of 400 mg/day. In subjects with severe renal impairment and ESRD, AUCinf increased 2.37-fold and 3.81-fold, and Cmax increased 2.10-fold and 2.68-fold relative to the exposure in non-cancer study participants with normal renal function.
Mild (60 mL/min/1.73 m2 ≤ eGFR < 90 mL/min/1.73 m2) or moderate (30 mL/min/1.73 m2 ≤ eGFR < 60 mL/min/1.73 m2) renal impairment had no effect on the exposure of ribociclib based on a population PK analysis that included 438 cancer patients with normal renal function, 488 patients with mild renal impairment and 113 patients with moderate renal impairment. In addition, in a sub-group analysis of data from studies following oral administration of ribociclib 600 mg as a single dose or repeat doses in cancer patients with mild or moderate renal impairment, AUC and Cmax were comparable to patients with normal renal function, suggesting no clinically meaningful effect of mild or moderate renal impairment on ribociclib exposure.
Letrozole
In a study of volunteers with varying renal function (24-hour creatinine clearance: 9 to 116 mL/min), no effect of renal function on the pharmacokinetics of single doses of 2.5 mg of FEMARA was found. In addition, in a study of 347 patients with advanced breast cancer, about half of whom received 2.5 mg FEMARA and half 0.5 mg FEMARA, renal impairment (calculated creatinine clearance: 20 to 50 mL/min) did not affect steady-state plasma letrozole concentrations.
Additional pharmacokinetic information on ribociclib:
The pharmacokinetics of ribociclib was investigated in patients with advanced cancer following oral daily doses ranging from 50 mg to 1200 mg. Healthy subjects received single oral doses of 400 or 600 mg or repeated daily oral doses (8 days) at 400 mg.
Effect of Age, Weight, Gender, and Race
Population PK analysis showed that there are no clinically relevant effects of age, body weight, gender, or race on the systemic exposure of ribociclib.
Drug Interaction Studies
Drugs That Affect Ribociclib Plasma Concentrations
CYP3A inhibitors: A drug interaction trial in healthy subjects was conducted with ritonavir (a strong CYP3A inhibitor). Compared to ribociclib alone, ritonavir (100 mg twice a day for 14 days) increased ribociclib Cmax and AUCinf by 1.7-fold and 3.2-fold, respectively, following a single 400 mg ribociclib dose. Cmax and AUC for LEQ803 (a prominent metabolite of LEE011, accounting for less than 10% of parent exposure) decreased by 96% and 98%, respectively. A moderate CYP3A4 inhibitor (erythromycin) is predicted to increase ribociclib Cmax and AUC by 1.3-fold and 1.9-fold, respectively.
CYP3A Inducers: A drug interaction trial in healthy subjects was conducted with rifampicin (a strong CYP3A4 inducer). Compared to ribociclib alone, rifampicin (600 mg daily for 14 days) decreased ribociclib Cmax and AUCinf by 81% and 89%, respectively, following a single 600 mg ribociclib dose. LEQ803 Cmax increased 1.7-fold and AUCinf decreased by 27%, respectively. A moderate CYP3A inducer (efavirenz) is predicted to decrease ribociclib Cmax and AUC by 37% and 60%, respectively.
Drugs That Are Affected by KISQALI
CYP3A4 and CYP1A2 Substrates: A drug interaction trial in healthy subjects was conducted as a cocktail study with midazolam (sensitive CYP3A4 substrate) and caffeine (sensitive CYP1A2 substrate). Compared to midazolam and caffeine alone, multiple doses of ribociclib (400 mg once daily for 8 days) increased midazolam Cmax and AUCinf by 2.1-fold and 3.8-fold, respectively. Administration of ribociclib at 600 mg once daily is predicted to increase midazolam Cmax and AUC by 2.4-fold and 5.2-fold, respectively. The effect of multiple doses of 400 mg ribociclib on caffeine was minimal, with Cmax decreased by 10% and AUCinf increased slightly by 20%. Only weak inhibitory effects on CYP1A2 substrates are predicted at 600 mg ribociclib once daily dose.
Gastric pH-elevating Agents: Coadministration of ribociclib with drugs that elevate the gastric pH was not evaluated in a clinical trial; however, altered ribociclib absorption was not identified in a population PK analysis and was not predicted using physiology based PK models.
Letrozole: Data from a clinical trial in patients with breast cancer and population PK analysis indicated no drug interaction between ribociclib and letrozole following coadministration of the drugs.
Anastrozole: Data from a clinical trial in patients with breast cancer indicated no clinically relevant drug interaction between ribociclib and anastrozole following coadministration of the drugs.
Exemestane: Data from a clinical trial in patients with breast cancer indicated no clinically relevant drug interaction between ribociclib and exemestane following coadministration of the drugs.
Fulvestrant: Data from a clinical trial in patients with breast cancer indicated no clinically relevant effect of fulvestrant on ribociclib exposure following coadministration of the drugs.
Tamoxifen: KISQALI is not indicated for concomitant use with tamoxifen. Data from a clinical trial in patients with breast cancer indicated that tamoxifen Cmax and AUC increased approximately 2-fold following coadministration of 600 mg ribociclib.
In vitro Studies
Effect of Ribociclib on CYP Enzymes: In vitro, ribociclib was a reversible inhibitor of CYP1A2, CYP2E1, and CYP3A4/5 and a time-dependent inhibitor of CYP3A4/5, at clinically relevant concentrations. In vitro evaluations indicated that KISQALI has no potential to inhibit the activities of CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP2D6 at clinically relevant concentrations. It has no potential for time-dependent inhibition of CYP1A2, CYP2C9, and CYP2D6, and no induction of CYP1A2, CYP2B6, CYP2C9, and CYP3A4 at clinically relevant concentrations.
Effect of Ribociclib on Transporters: In vitro evaluations indicated that KISQALI has a low potential to inhibit the activities of drug transporters P-gp, OATP1B1/B3, OCT1, MATEK2 at clinically relevant concentrations. KISQALI may inhibit BCRP, OCT2, MATE1, and human BSEP at clinically relevant concentrations.
Effect of Transporters on Ribociclib: Based on in vitro data, P-gp and BCRP mediated transport are unlikely to affect the extent of oral absorption of ribociclib at therapeutic doses. Ribociclib is not a substrate for hepatic uptake transporters OATP1B1/1B3 or OCT-1 in vitro.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Ribociclib
Carcinogenesis studies have not been conducted with ribociclib.
Ribociclib was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay or clastogenic in an in vitro human lymphocyte chromosomal aberration assay or an in vivo rat bone marrow micronucleus assay.
In a fertility and early embryonic development study, female rats received oral doses of ribociclib for 14 days prior to mating through the first week of pregnancy. Ribociclib did not affect reproductive function, fertility or early embryonic development at doses up to 300 mg/kg/day (approximately 0.6 times the clinical exposure in patients at the highest recommended dose of 600 mg/day based on AUC).
A fertility study in male rats has not been performed with ribociclib. In repeat-dose toxicity studies with oral administration of ribociclib daily for 3 weeks on/1 week off in rats up to 26 weeks duration and dogs up to 39 weeks duration, atrophic changes in testes were reported. Findings included degeneration of seminiferous tubular epithelia in the testes and hypospermia and luminal cellular debris in the epididymides of rats and dogs and vacuolation of epithelia in the epididymides of rats. These findings were observed at doses ≥ 75 mg/kg in rats and ≥ 1 mg/kg in dogs which resulted in systemic exposures that were 1.4 and 0.03 times the human exposure at the highest recommended daily dose of 600 mg/day based on AUC, respectively. These effects can be linked to a direct anti-proliferative effect on the testicular germ cells resulting in atrophy of the seminiferous tubules and showed a trend towards reversibility in rats and dogs after a four-week non-dosing period.
Letrozole
A conventional carcinogenesis study in mice at doses of 0.6 to 60 mg/kg/day (about 1 to 100 times the daily MRHD on a mg/m2 basis) administered by oral gavage for up to 2 years revealed a dose-related increase in the incidence of benign ovarian stromal tumors. The incidence of combined hepatocellular adenoma and carcinoma showed a significant trend in females when the high dose group was excluded due to low survival. In a separate study, plasma AUC0-12hr levels in mice at 60 mg/kg/day were 55 times higher than the AUC0-24hr level in breast cancer patients at the recommended dose. The carcinogenicity study in rats at oral doses of 0.1 to 10 mg/kg/day (about 0.4 to 40 times the daily MRHD on a mg/m2 basis) for up to 2 years also produced an increase in the incidence of benign ovarian stromal tumors at 10 mg/kg/day. Ovarian hyperplasia was observed in females at doses equal to or greater than 0.1 mg/kg/day. At 10 mg/kg/day, plasma AUC0-24hr levels in rats were 80 times higher than the level in breast cancer patients at the recommended dose. The benign ovarian stromal tumors observed in mice and rats were considered to be related to the pharmacological inhibition of estrogen synthesis and may be due to increased luteinizing hormone resulting from the decrease in circulating estrogen.
Letrozole was not mutagenic in in vitro tests (Ames and E.coli bacterial tests) but was observed to be a potential clastogen in in vitro assays (CHO K1 and CCL 61 Chinese hamster ovary cells). Letrozole was not clastogenic in vivo (micronucleus test in rats).
In a fertility and early embryonic developmental toxicity study in female rats, oral administration of letrozole starting 2 weeks before mating until pregnancy Day 6 resulted in decreases in the incidence of successful mating and pregnancy at doses ≥ 0.03 mg/kg/day (approximately 0.1 times the recommended human dose on a mg/m2 basis). In repeat-dose toxicity studies, administration of letrozole caused sexual inactivity in females and atrophy of the reproductive tract in males and females at doses of 0.6, 0.1 and 0.03 mg/kg in mice, rats and dogs, respectively (approximately 1, 0.4 and 0.4 times the daily MRHD on a mg/m2 basis, respectively).
13.2 Animal Toxicology and/or Pharmacology
Ribociclib
In vivo cardiac safety studies in dogs demonstrated dose and concentration related QTc interval prolongation at an exposure similar to patients receiving the recommended dose of 600 mg. There is a potential to induce incidences of premature ventricular contractions (PVCs) at elevated exposures (approximately 5-fold the anticipated clinical Cmax).
-
14 CLINICAL STUDIES
MONALEESA-2: KISQALI in Combination With Letrozole
Postmenopausal Women With HR-positive, HER2-negative Advanced or Metastatic Breast Cancer for Initial Endocrine-Based Therapy
MONALEESA-2 (NCT01958021) was a randomized, double-blind, placebo-controlled, multicenter clinical study of KISQALI plus letrozole versus placebo plus letrozole conducted in postmenopausal women with HR-positive, HER2-negative, advanced breast cancer who received no prior therapy for advanced disease.
A total of 668 patients were randomized to receive either KISQALI and letrozole (n = 334) or placebo and letrozole (n = 334), stratified according to the presence of liver and/or lung metastases. Letrozole 2.5 mg was given orally once daily for 28 days, with either KISQALI 600 mg or placebo orally once daily for 21 consecutive days followed by 7 days off until disease progression or unacceptable toxicity. The major efficacy outcome measure for the study was investigator-assessed progression-free survival (PFS) using Response Evaluation Criteria in Solid Tumors (RECIST v1.1).
Patients enrolled in MONALEESA-2 had a median age of 62 years (range, 23 to 91) and 45% of patients were older than 65. The majority of patients were White (82%), and all patients had an ECOG performance status of 0 or 1. A total of 47% of patients had received chemotherapy and 51% had received anti-hormonal therapy in the neoadjuvant or adjuvant setting. Thirty-four percent (34%) of patients had de novo metastatic disease, 21% had bone only disease, and 59% had visceral disease.
The efficacy results from MONALEESA-2 are summarized in Table 11 and Figure 1. The results shown are from a pre-planned interim efficacy analysis of PFS. Results were consistent across patient subgroups of prior adjuvant or neoadjuvant chemotherapy or hormonal therapies, liver and/or lung involvement, and bone-only metastatic disease. The PFS assessment based on a blinded independent central radiological review was consistent with investigator assessment. At the time of the PFS analysis, 6.5% of patients had died, and overall survival data were immature.
Table 11: Efficacy Results – MONALEESA-2 (Investigator Assessment, Intent-to-Treat Population) KISQALI + letrozole Placebo + letrozole Progression-free survival N = 334 N = 334 Events (%) 93 (27.8) 150 (44.9) Median (months, 95% CI) NR (19.3 – NR) 14.7 (13.0 – 16.5) Hazard Ratio (95% CI) 0.556 (0.429 to 0.720) p-value < 0.0001a Overall Response Rate N = 256 N = 245 Patients with measurable disease (95% CI) 52.7 (46.6, 58.9) 37.1 (31.1, 43.2) Abbreviations: CI, confidence interval; NR, not reached.
ap-value estimated from one-sided log-rank test.Figure 1: Kaplan-Meier Progression Free Survival Curves – MONALEESA-2 (Intent-to-Treat Population)
MONALEESA-7: KISQALI in Combination with an Aromatase Inhibitor
Pre/perimenopausal Patients With HR-positive, HER2-negative Advanced or Metastatic Breast Cancer for Initial Endocrine-Based Therapy
MONALEESA-7 (NCT02278120) was a randomized, double-blind, placebo-controlled study of KISQALI plus either a non-steroidal aromatase inhibitor (NSAI) or tamoxifen and goserelin vs. placebo plus either a NSAI or tamoxifen and goserelin conducted in pre/perimenopausal women with HR-positive, HER2-negative, advanced breast cancer who received no prior endocrine therapy for advanced disease.
A total of 672 patients were randomized to receive KISQALI plus NSAI or tamoxifen plus goserelin (n = 335) or placebo plus NSAI or tamoxifen plus goserelin (n = 337), stratified according to the presence of liver and/or lung metastases, prior chemotherapy for advanced disease, and endocrine combination partner (tamoxifen and goserelin vs. NSAI and goserelin). Among 248 patients who received KISQALI plus NSAI plus goserelin, 211 (85%) received letrozole and 37 (15%) received anastrozole.
NSAI (letrozole 2.5 mg or anastrozole 1 mg) or tamoxifen 20 mg or were given orally once daily on a continuous daily schedule, goserelin was administered as a sub-cutaneous injection on Day 1 of each 28-day cycle, with either KISQALI 600 mg or placebo orally once daily for 21 consecutive days followed by 7 days off until disease progression or unacceptable toxicity. The major efficacy outcome measure for the study was investigator-assessed progression-free survival (PFS) using Response Evaluation Criteria in Solid Tumors (RECIST) v1.1.
Patients enrolled in MONALEESA-7 had a median age of 44 years (range, 25 to 58) and were primarily Caucasian (58%), Asian (29%), or Black (3%). Nearly all patients (99%) had an ECOG performance status of 0 or 1. Of the 672 patients, 33% had received chemotherapy in the adjuvant vs. 18% in the neoadjuvant setting and 40% had received endocrine therapy in the adjuvant vs. 0.7% in the neoadjuvant setting prior to study entry. Forty percent (40%) of patients had de novo metastatic disease, 24% had bone only disease, and 57% had visceral disease. Demographics and baseline disease characteristics were balanced and comparable between study arms, and endocrine combination partner.
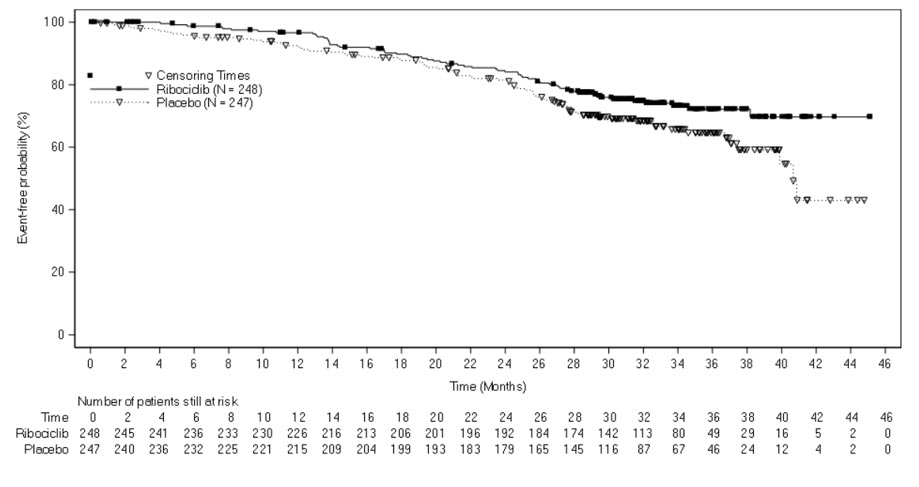
The efficacy results from a pre-specified subgroup analysis of 495 patients who had received KISQALI or placebo with NSAI plus goserelin are summarized in Table 12, Figure 2, and Figure 3. Consistent results were observed in the stratification factor subgroups of disease site and prior chemotherapy for advanced disease.
Table 12: Efficacy Results – MONALEESA-7 (NSAI) Abbreviations: CI, confidence interval; NR, not reached.
*Based on confirmed responses.
1Investigator Assessment.KISQALI + NSAI +
goserelinPlacebo + NSAI +
goserelinProgression-free survival1 N = 248 N = 247 Events (n, %) 92 (37.1%) 132 (53.4%) Median (months, 95% CI) 27.5 (19.1, NR) 13.8 (12.6, 17.4) Hazard Ratio (95% CI) 0.569 (0.436, 0.743) Overall survival N = 248 N = 247 Events (n, %) 61 (24.6%) 80 (32.4%) Median (months, 95% CI) NR (NR, NR) 40.7 (37.4, NR) Hazard Ratio (95% CI) 0.699 (0.501, 0.976) Overall Response Rate*1 N = 192 N = 199 Patients with measurable disease (95% CI) 50.5 (43.4, 57.6) 36.2 (29.5, 42.9) Figure 2: Kaplan-Meier Progression Free Survival Curves – MONALEESA-7 (NSAI, Investigator Assessment)
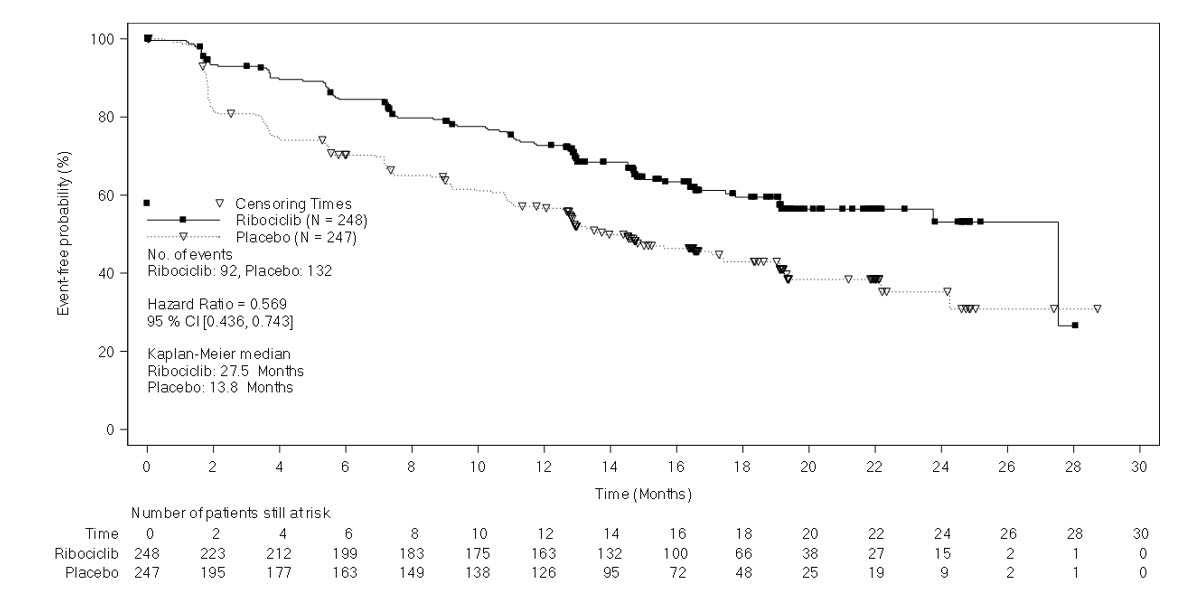
Figure 3 Kaplan-Meier Overall Survival Curves- MONALEESA-7 (NSAI)
-
16 HOW SUPPLIED/STORAGE AND HANDLING
KISQALI FEMARA CO-PACK is dispensed in a carton for a total of 28 days of therapy.
Each carton contains individual ribociclib and letrozole drug products as follows:
KISQALI (ribociclib) Tablets:
200 mg tablets.
Light greyish violet, round, curved with beveled edge, debossed with “RIC” on one side and “NVR” on the other side.
FEMARA (letrozole) Tablets:
2.5 mg tablets.
Dark yellow, round, slightly biconvex, with beveled edges, imprinted with the letters “FV” on one side and “CG” on the other side.
KISQALI FEMARA CO-PACK Cartons:
- NDC: 0078-0923-61 - 3 Blister packs, containing 21 tablets (200 mg per tablet) (600 mg daily dose) of KISQALI plus one 28-tablet count bottle of FEMARA
- NDC: 0078-0916-61 - 3 Blister packs, containing 14 tablets (200 mg per tablet) (400 mg daily dose) of KISQALI plus one 28-tablet count bottle of FEMARA
- NDC: 0078-0909-61 - 1 Blister pack, containing 21 tablets (200 mg per tablet) (200 mg daily dose) of KISQALI plus one 28-tablet count bottle of FEMARA
Store KISQALI FEMARA CO-PACK at 20°C to 25°C (68°F-77°F). Store in the original package.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
For additional information on KISQALI and FEMARA, refer to the full prescribing information for each product.
Interstitial Lung Disease/Pneumonitis
Advise patients to immediately report new or worsening respiratory symptoms [see Warnings and Precautions (5.1)].
QT Prolongation
Inform patients of the signs and symptoms of QT prolongation. Advise patients to contact their healthcare provider immediately for signs or symptoms of QT prolongation [see Warnings and Precautions (5.2)].
Hepatobiliary Toxicity
Inform patients of the signs and symptoms of hepatobiliary toxicity. Advise patients to contact their healthcare provider immediately for signs or symptoms of hepatobiliary toxicity [see Warnings and Precautions (5.3)].
Neutropenia
Advise patients of the possibility of developing neutropenia and to immediately contact their healthcare provider should they develop a fever, particularly in association with any suggestion of infection [see Warnings and Precautions (5.4)].
Embryo-Fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during KISQALI FEMARA CO-PACK therapy and for at least 3 weeks after the last dose. Advise females to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, during treatment with KISQALI FEMARA CO-PACK [see Warnings and Precautions (5.5), Use in Specific Populations (8.1, 8.3)].
Lactation
Advise women not to breastfeed during KISQALI FEMARA CO-PACK treatment and for at least 3 weeks after the last dose [see Use in Specific Populations (8.2)].
Drug Interactions
- Inform patients to avoid grapefruit or grapefruit juice while taking KISQALI FEMARA CO-PACK [see Drug Interactions (7.1)].
- Inform patients to avoid strong CYP3A inhibitors, strong CYP3A inducers, and drugs known to prolong the QT interval [see Drug Interactions (7.1, 7.2, 7.4)].
Dosing
- Instruct patients to take the doses of KISQALI FEMARA CO-PACK at approximately the same time every day and to swallow tablets whole (do not chew, crush, or split them prior to swallowing) [see Dosage and Administration (2.1)].
- If patient vomits or misses a dose, advise the patient to take the next prescribed dose at the usual time [see Dosage and Administration (2.1)].
- Advise the patient that KISQALI FEMARA CO-PACK may be taken with or without food [see Dosage and Administration (2.1)].
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936© Novartis
T2020-06
- Inform patients to avoid grapefruit or grapefruit juice while taking KISQALI FEMARA CO-PACK [see Drug Interactions (7.1)].
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: January 2020 PATIENT INFORMATION
KISQALI® FEMARA® CO-PACK (kis kah' lee fe ma' ra koe' pak)
(ribociclib tablets; letrozole tablets)
co-packaged for oral useWhat is the most important information I should know about KISQALI FEMARA CO-PACK?
KISQALI FEMARA CO-PACK may cause serious side effects, including:
-
Lung problems. KISQALI may cause severe or life-threatening inflammation of the lungs during treatment that may lead to death. Tell your healthcare provider right away if you have any new or worsening symptoms, including:
- trouble breathing or shortness of breath
- cough with or without mucus
- chest pain
- Heart rhythm problems (QT prolongation). KISQALI FEMARA CO-PACK can cause a heart problem known as QT prolongation. This condition can cause an abnormal heartbeat and may lead to death. Your healthcare provider should check your heart and do blood tests before and during treatment with KISQALI FEMARA CO-PACK. Tell your healthcare provider right away if you have a change in your heartbeat (a fast or irregular heartbeat), or if you feel dizzy or faint.
- Liver problems. KISQALI FEMARA CO-PACK can cause serious liver problems. Your healthcare provider will do blood tests to check your liver before you start and while you take KISQALI FEMARA CO-PACK. Tell your healthcare provider right away if you get any of the following signs and symptoms of liver problems:
o yellowing of your skin or the whites of your eyes (jaundice)
o dark or brown (tea-colored) urine
o feeling very tiredo loss of appetite
o pain on the upper right side of your stomach area (abdomen)
o bleeding or bruising more easily than normal- Low white blood cell counts (neutropenia). Low white blood cell counts are very common when taking KISQALI FEMARA CO-PACK and may result in infections that may be severe. Your healthcare provider should check your white blood cell counts before and during treatment with KISQALI FEMARA CO-PACK. Tell your healthcare provider right away if you have signs and symptoms of low white blood cell counts or infections, such as fever and chills.
Your healthcare provider may tell you to decrease your dose, temporarily stop or completely stop taking KISQALI if you develop certain serious side effects during treatment with KISQALI.
See “What are the possible side effects of KISQALI FEMARA CO-PACK?” for more information about side effects.What is KISQALI FEMARA CO-PACK?
- KISQALI FEMARA CO-PACK is a prescription medicine used as the first hormonal based therapy to treat pre/perimenopausal or postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative breast cancer that has spread to other parts of the body (metastatic).
- KISQALI FEMARA CO-PACK contains 2 different types of medicines:
- The violet tablet contains the medicine KISQALI (ribociclib).
- The yellow tablet contains the medicine FEMARA (letrozole).
It is not known if KISQALI FEMARA CO-PACK is safe and effective in children.
Do not take KISQALI FEMARA CO-PACK if you are allergic to letrozole or any of the ingredients of FEMARA.
See the end of this Patient Information for a list of the ingredients in KISQALI FEMARA CO-PACK.
Before you take KISQALI FEMARA CO-PACK, tell your healthcare provider about all your medical conditions, including if you:
- have any heart problems, including heart failure, irregular heartbeats, and QT prolongation
- have ever had a heart attack
- have a slow heartbeat (bradycardia)
- have problems with the amount of potassium, calcium, phosphorus, or magnesium in your blood
- have fever, chills, or any other signs or symptoms of infection
- have liver problems
- are pregnant, or plan to become pregnant. KISQALI FEMARA CO-PACK can harm your unborn baby.
- If you are able to become pregnant, your healthcare provider should do a pregnancy test before you start treatment with KISQALI FEMARA CO-PACK.
- Females who are able to become pregnant and who take KISQALI FEMARA CO-PACK should use effective birth control during treatment and for at least 3 weeks after the last dose of KISQALI FEMARA CO-PACK.
- Talk to your healthcare provider about birth control methods that may be right for you during this time.
- If you become pregnant or think you are pregnant, tell your healthcare provider right away.
- are breastfeeding or plan to breastfeed. It is not known if KISQALI FEMARA CO-PACK passes into your breast milk. Do not breastfeed during treatment with KISQALI FEMARA CO-PACK and for at least 3 weeks after the last dose of KISQALI FEMARA CO-PACK.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. KISQALI FEMARA CO-PACK and other medicines may affect each other causing side effects. Know the medicines you take. Keep a list of them to show your healthcare provider or pharmacist when you get a new medicine.
How should I take KISQALI FEMARA CO-PACK?
- Take KISQALI FEMARA CO-PACK exactly as your healthcare provider tells you.
- Do not change your dose or stop taking KISQALI FEMARA CO-PACK unless your healthcare provider tells you.
- KISQALI FEMARA CO-PACK comes in a carton that contains enough KISQALI tablets and FEMARA tablets for 28 days of treatment.
- Take KISQALI FEMARA CO-PACK each day at about the same time, preferably in the morning.
- You may take KISQALI FEMARA CO-PACK with food or without food.
- Swallow KISQALI tablets and FEMARA tablets whole. Do not chew, crush or split tablets before swallowing them.
- Do not take any KISQALI tablets and FEMARA tablets that are broken, cracked, or that look damaged.
- If you miss a dose or vomit after taking a dose of KISQALI FEMARA CO-PACK, do not take another dose on that day. Take your next dose at your regular time.
- If you take too much KISQALI FEMARA CO-PACK, call your healthcare provider right away or go to the nearest hospital emergency room.
- Inform your healthcare provider if you are pre- or peri-menopausal.
What should I avoid while taking KISQALI FEMARA CO-PACK?
- Avoid eating grapefruit and avoid drinking grapefruit juice during treatment with KISQALI FEMARA CO-PACK since these may increase the amount of KISQALI in your blood.
What are the possible side effects of KISQALI FEMARA CO-PACK?
KISQALI FEMARA CO-PACK may cause serious side effects.
- See "What is the most important information I should know about KISQALI FEMARA CO-PACK?"
The most common side effects of KISQALI FEMARA CO-PACK include:
neutropenia
leukopenia
headache
anemianausea
diarrhea
constipationinfections
hair loss (alopecia)
back painfatigue
vomiting
rashKISQALI FEMARA CO-PACK may cause fertility problems in females and males, which may affect your ability to have children. Talk to your healthcare provider if you have concerns about fertility.
These are not all the possible side effects of KISQALI FEMARA CO-PACK.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store KISQALI FEMARA CO-PACK? - Store KISQALI FEMARA CO-PACK at 68°F to 77°F (20°C to 25°C).
- Keep KISQALI FEMARA CO-PACK in its original container.
Keep KISQALI FEMARA CO-PACK and all medicines out of the reach of children.
General information about the safe and effective use of KISQALI FEMARA CO-PACK.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use KISQALI FEMARA CO-PACK for a condition for which it was not prescribed. Do not give KISQALI FEMARA CO-PACK to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for more information about KISQALI FEMARA CO-PACK that is written for health professionals.
What are the ingredients in KISQALI FEMARA CO-PACK?
KISQALI (ribociclib) tablets:
Active ingredient: ribociclib
Inactive ingredients: colloidal silicon dioxide, crospovidone, hydroxypropylcellulose, magnesium stearate and microcrystalline cellulose. The film-coating contains iron oxide black, iron oxide red, lecithin (soya), polyvinyl alcohol (partially hydrolyzed), talc, titanium dioxide, and xanthan gum
FEMARA (letrozole) tablets:Active ingredient: letrozole
Inactive ingredients: colloidal silicon dioxide, ferric oxide, hydroxypropyl methylcellulose, lactose monohydrate, magnesium stearate, maize starch, microcrystalline cellulose, polyethylene glycol, sodium starch glycolate, talc, and titanium dioxide
Distributed by: Novartis Pharmaceuticals Corporation, East Hanover, New Jersey 07936
© Novartis
T2020-07
For more information, go to www.KISQALI.COM or call 1-844-KISQALI (1-844-547-7254). -
Lung problems. KISQALI may cause severe or life-threatening inflammation of the lungs during treatment that may lead to death. Tell your healthcare provider right away if you have any new or worsening symptoms, including:
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
Kisqali®Femara®CO-PACK
(ribociclib and letrozole) tabletsNDC: 0078-0909-61
Rx only
28-Day supply
200 mg daily dose
(one 200 mg tablet)
21 Film-coated tablets
2.5 mg
per tablet
28 Film-coated tablets
Novartis
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
Kisqali®Femara®CO-PACK
(ribociclib and letrozole) tabletsNDC: 0078-0916-61
Rx only
28-Day supply
400 mg daily dose
(two 200 mg tablets)
42 Film-coated tablets
2.5 mg
per tablet
28 Film-coated tablets
Novartis
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
Kisqali®Femara®CO-PACK
(ribociclib and letrozole) tabletsNDC: 0078-0923-61
Rx only
28-Day supply
600 mg daily dose
(three 200 mg tablets)
63 Film-coated tablets
2.5 mg
per tablet
28 Film-coated tablets
Novartis
-
INGREDIENTS AND APPEARANCE
KISQALI FEMARA CO-PACK
letrozole and ribociclib kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0909 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0909-61 1 in 1 KIT 05/04/2017 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 BOTTLE, PLASTIC 28 Part 2 1 BLISTER PACK 21 Part 1 of 2 FEMARA
letrozole tablet, film coatedProduct Information Item Code (Source) NDC: 0078-0881 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LETROZOLE (UNII: 7LKK855W8I) (LETROZOLE - UNII:7LKK855W8I) LETROZOLE 2.5 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) FERRIC OXIDE RED (UNII: 1K09F3G675) HYPROMELLOSES (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) STARCH, CORN (UNII: O8232NY3SJ) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POLYETHYLENE GLYCOL (UNII: 3WJQ0SDW1A) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color YELLOW (dark yellow) Score no score Shape ROUND (slightly biconvex with beveled edges) Size 7mm Flavor Imprint Code FV;CG Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0881-50 28 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020726 07/31/1997 Part 2 of 2 KISQALI
ribociclib tabletProduct Information Item Code (Source) NDC: 0078-0888 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RIBOCICLIB (UNII: TK8ERE8P56) (RIBOCICLIB - UNII:TK8ERE8P56) RIBOCICLIB 200 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (UNII: 2S7830E561) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FERRIC OXIDE RED (UNII: 1K09F3G675) LECITHIN, SOYBEAN (UNII: 1DI56QDM62) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) XANTHAN GUM (UNII: TTV12P4NEE) Product Characteristics Color PURPLE (light greyish violet) Score no score Shape ROUND Size 14mm Flavor Imprint Code RIC;NVR Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0888-21 21 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209092 03/13/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209935 05/04/2017 KISQALI FEMARA CO-PACK
letrozole and ribociclib kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0916 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0916-61 1 in 1 KIT 05/04/2017 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 BOTTLE, PLASTIC 28 Part 2 3 BLISTER PACK 42 Part 1 of 2 FEMARA
letrozole tablet, film coatedProduct Information Item Code (Source) NDC: 0078-0881 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LETROZOLE (UNII: 7LKK855W8I) (LETROZOLE - UNII:7LKK855W8I) LETROZOLE 2.5 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) FERRIC OXIDE RED (UNII: 1K09F3G675) HYPROMELLOSES (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) STARCH, CORN (UNII: O8232NY3SJ) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POLYETHYLENE GLYCOL (UNII: 3WJQ0SDW1A) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color YELLOW (dark yellow) Score no score Shape ROUND (slightly biconvex with beveled edges) Size 7mm Flavor Imprint Code FV;CG Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0881-50 28 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020726 07/31/1997 Part 2 of 2 KISQALI
ribociclib tabletProduct Information Item Code (Source) NDC: 0078-0895 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RIBOCICLIB (UNII: TK8ERE8P56) (RIBOCICLIB - UNII:TK8ERE8P56) RIBOCICLIB 200 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (UNII: 2S7830E561) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FERRIC OXIDE RED (UNII: 1K09F3G675) LECITHIN, SOYBEAN (UNII: 1DI56QDM62) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) XANTHAN GUM (UNII: TTV12P4NEE) Product Characteristics Color PURPLE (light greyish violet) Score no score Shape ROUND Size 11mm Flavor Imprint Code RIC;NVR Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0895-14 14 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209092 03/13/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209935 05/04/2017 KISQALI FEMARA CO-PACK
letrozole and ribociclib kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0923 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0923-61 1 in 1 KIT 05/04/2017 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 BOTTLE, PLASTIC 28 Part 2 3 BLISTER PACK 63 Part 1 of 2 FEMARA
letrozole tablet, film coatedProduct Information Item Code (Source) NDC: 0078-0881 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LETROZOLE (UNII: 7LKK855W8I) (LETROZOLE - UNII:7LKK855W8I) LETROZOLE 2.5 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) FERRIC OXIDE RED (UNII: 1K09F3G675) HYPROMELLOSES (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) STARCH, CORN (UNII: O8232NY3SJ) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POLYETHYLENE GLYCOL (UNII: 3WJQ0SDW1A) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color YELLOW (dark yellow) Score no score Shape ROUND (slightly biconvex with beveled edges) Size 7mm Flavor Imprint Code FV;CG Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0881-50 28 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020726 07/31/1997 Part 2 of 2 KISQALI
ribociclib tabletProduct Information Item Code (Source) NDC: 0078-0902 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RIBOCICLIB (UNII: TK8ERE8P56) (RIBOCICLIB - UNII:TK8ERE8P56) RIBOCICLIB 200 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (UNII: 2S7830E561) HYDROXYPROPYL CELLULOSE, UNSPECIFIED (UNII: 9XZ8H6N6OH) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FERRIC OXIDE RED (UNII: 1K09F3G675) LECITHIN, SOYBEAN (UNII: 1DI56QDM62) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) XANTHAN GUM (UNII: TTV12P4NEE) Product Characteristics Color PURPLE (light greyish violet) Score no score Shape ROUND Size 11mm Flavor Imprint Code RIC;NVR Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0902-21 21 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209092 03/13/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209935 05/04/2017 Labeler - Novartis Pharmaceuticals Corporation (002147023)
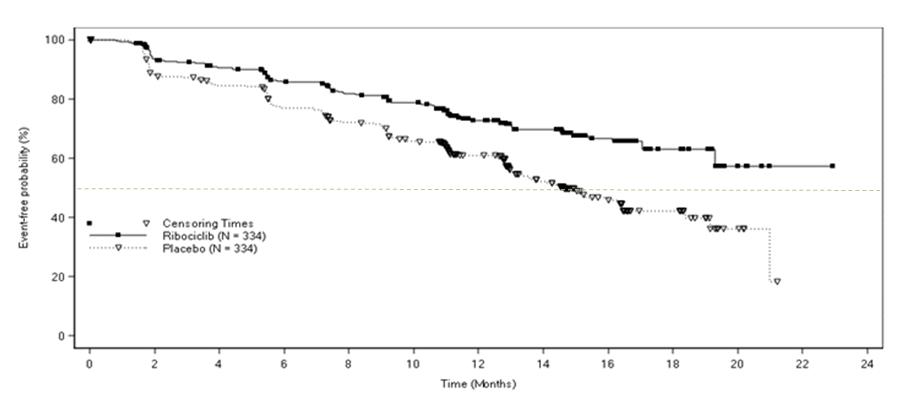
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.