AURYXIA- ferric citrate tablet, coated
Auryxia by
Drug Labeling and Warnings
Auryxia by is a Prescription medication manufactured, distributed, or labeled by Akebia Therapeutics, Inc., Keryx Biopharmaceuticals, Inc., Patheon Inc., Patheon Manufacturing Services LLC, Siegfried USA, LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use AURYXIA® safely and effectively. See full prescribing information for AURYXIA.
AURYXIA (ferric citrate) tablets, for oral use
Initial U.S. Approval: 2014INDICATIONS AND USAGE
Auryxia is a phosphate binder indicated for the control of serum phosphorus levels in adult patients with chronic kidney disease on dialysis (1)
Auryxia is an iron replacement product indicated for the treatment of iron deficiency anemia in adult patients with chronic kidney disease not on dialysis (1)
DOSAGE AND ADMINISTRATION
- Hyperphosphatemia in Chronic Kidney Disease on Dialysis:
- Iron Deficiency Anemia in Chronic Kidney Disease Not on Dialysis:
DOSAGE FORMS AND STRENGTHS
- Tablets: 210 mg ferric iron, equivalent to 1 g ferric citrate (3)
CONTRAINDICATIONS
- Iron overload syndromes (e.g., hemochromatosis) (4)
WARNINGS AND PRECAUTIONS
- Iron overload: Monitor ferritin and TSAT. Patients may require a reduction in dose or discontinuation of intravenous iron. (5.1)
- Accidental overdose of iron-containing products is a leading cause of fatal poisoning in children under 6 years of age. Keep this product out of reach of children. In case of accidental overdose, call a doctor or poison control center immediately. (5.2)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥5%) are discolored feces, diarrhea, constipation, nausea, vomiting, cough, abdominal pain, and hyperkalemia (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Keryx Biopharmaceuticals at 1-844-445-3799 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
When clinically significant drug interactions are expected, consider separation of the timing of administration. Consider monitoring clinical responses or blood levels of the concomitant medication (7)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Hyperphosphatemia in Chronic Kidney Disease on Dialysis
1.2 Iron Deficiency Anemia in Chronic Kidney Disease Not on Dialysis
2 DOSAGE AND ADMINISTRATION
2.1 Dosage for Hyperphosphatemia in Chronic Kidney Disease on Dialysis
2.2 Dosage for Iron Deficiency Anemia in Chronic Kidney Disease Not on Dialysis
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Iron Overload
5.2 Risk of Overdosage in Children Due to Accidental Ingestion
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Hyperphosphatemia in Chronic Kidney Disease on Dialysis
14.2 Iron Deficiency Anemia in Chronic Kidney Disease Not On Dialysis
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage for Hyperphosphatemia in Chronic Kidney Disease on Dialysis
The recommended starting dose is 2 tablets orally 3 times per day with meals. Monitor serum phosphorus levels and titrate the Auryxia dose in decrements or increments of 1 to 2 tablets per day as needed to maintain serum phosphorus at target levels, up to a maximum dose of 12 tablets daily. Dose can be titrated at 1-week or longer intervals.
In a clinical trial, patients required an average of 8 to 9 tablets a day to control serum phosphorus levels.
2.2 Dosage for Iron Deficiency Anemia in Chronic Kidney Disease Not on Dialysis
The recommended starting dose is 1 tablet orally 3 times per day with meals. Titrate the dose of Auryxia as needed to achieve and maintain hemoglobin at target levels, up to a maximum dose of 12 tablets daily.
In a clinical trial in patients with chronic kidney disease not on dialysis (CKD-NDD), patients required an average of 5 tablets per day to increase hemoglobin levels.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Auryxia is contraindicated in patients with iron overload syndromes (e.g., hemochromatosis) [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Iron Overload
Iron absorption from Auryxia may lead to excessive elevations in iron stores. Increases in serum ferritin and transferrin saturation (TSAT) levels were observed in clinical trials. In a 56-week safety and efficacy trial evaluating the control of serum phosphate levels in patients with chronic kidney disease on dialysis in which concomitant use of intravenous iron was permitted, 55 (19%) of patients treated with Auryxia had a ferritin level >1500 ng/mL as compared with 13 (9%) of patients treated with active control.
Assess iron parameters (e.g., serum ferritin and TSAT) prior to initiating Auryxia and monitor iron parameters while on therapy [see Contraindications (4), Overdosage (10) and Clinical Pharmacology (12.2)]. Patients receiving intravenous iron may require a reduction in dose or discontinuation of intravenous iron therapy.
5.2 Risk of Overdosage in Children Due to Accidental Ingestion
Accidental ingestion and resulting overdose of iron-containing products is a leading cause of fatal poisoning in children under 6 years of age [see Overdosage (10)]. Advise patients of the risks to children and to keep Auryxia out of the reach of children.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to adverse reaction rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Hyperphosphatemia in Chronic Kidney Disease on Dialysis
A total of 289 patients were treated with Auryxia and 149 patients were treated with active control (sevelamer carbonate and/or calcium acetate) during the 52-week, randomized, open-label, active control phase of a trial in patients on dialysis. A total of 322 patients were treated with Auryxia for up to 28 days in three short-term trials. Across these trials, 557 unique patients were treated with Auryxia; dosage regimens in these trials ranged from 210 mg to 2,520 mg of ferric iron per day, equivalent to 1 to 12 tablets of Auryxia.
Adverse reactions reported in more than 5% of patients treated with Auryxia in these trials included diarrhea (21%), discolored feces (19%), nausea (11%), constipation (8%), vomiting (7%), and cough (6%).
During the 52-week, active-control period, 61 patients (21%) on Auryxia discontinued study drug because of an adverse reaction, as compared to 21 patients (14%) in the active control arm. Patients who were previously intolerant to any of the active control treatments (calcium acetate and sevelamer carbonate) were not eligible to enroll in the study. Gastrointestinal adverse reactions were the most common reason for discontinuing Auryxia (14%).
Iron Deficiency Anemia in Chronic Kidney Disease Not on Dialysis
Across two trials, 190 patients with CKD-NDD were treated with Auryxia. This included a study of 117 patients treated with Auryxia and 116 patients treated with placebo in a 16-week, randomized, double-blind period and a study of 75 patients treated with Auryxia and 73 treated with placebo in a 12-week randomized double-blind period. Dosage regimens in these trials ranged from 210 mg to 2,520 mg of ferric iron per day, equivalent to 1 to 12 tablets of Auryxia.
Adverse reactions reported in at least 5% of patients treated with Auryxia in these trials are listed in Table 1.
Table 1: Adverse Events Reported in Two Clinical Trials in at least 5% of patients receiving Auryxia Body System
Adverse ReactionAuryxia %
(N=190)Placebo %
(N=188)Any Adverse Reaction 75 62 Metabolism and Nutrition Disorders Hyperkalemia 5 3 Gastrointestinal Disorders Discolored feces 22 0 Diarrhea 21 12 Constipation 18 10 Nausea 10 4 Abdominal Pain 5 2 During the 16-week, placebo-control trial, 12 patients (10%) on Auryxia discontinued study drug because of an adverse reaction, as compared to 10 patients (9%) in the placebo control arm. Diarrhea was the most common adverse reaction leading to discontinuation of Auryxia (2.6%).
-
7 DRUG INTERACTIONS
Table 2:
Oral drugs that can be administered concomitantly with AuryxiaAmlodipine
Aspirin
Atorvastatin
Calcitriol
Clopidogrel
Digoxin
Diltiazem
Doxercalciferol
Enalapril
Fluvastatin
Glimepiride
Levofloxacin
Losartan
Metoprolol
Pravastatin
Propranolol
Sitagliptin
Warfarin
Oral drugs that have to be separated from Auryxia and meals Dosing Recommendations Doxycycline Take at least 1 hour before Auryxia Ciprofloxacin Take at least 2 hours before or after Auryxia Oral medications not listed in Table 2
There are no empirical data on avoiding drug interactions between Auryxia and most concomitant oral drugs. For oral medications where a reduction in the bioavailability of that medication would have a clinically significant effect on its safety or efficacy, consider separation of the timing of the administration of the two drugs. The duration of separation depends upon the absorption characteristics of the medication concomitantly administered, such as the time to reach peak systemic levels and whether the drug is an immediate release or an extended release product. Consider monitoring clinical responses or blood levels of concomitant medications that have a narrow therapeutic range.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on Auryxia use in pregnant women to inform a drug-associated risk of major birth defects and miscarriage. Animal reproduction studies have not been conducted using Auryxia. Skeletal and encephalic malformation was observed in neonatal mice when ferric gluconate was administered intraperitoneally to gravid dams on gestation days 7-9. However, oral administration of other ferric or ferrous compounds to gravid CD1-mice and Wistar-rats caused no fetal malformation.
An overdose of iron in pregnant women may carry a risk for spontaneous abortion, gestational diabetes and fetal malformation.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies are 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
The effect of Auryxia on the absorption of vitamins and other nutrients has not been studied in pregnant women. Requirements for vitamins and other nutrients are increased in pregnancy.
8.2 Lactation
Risk Summary
There are no human data regarding the effect of Auryxia in human milk, the effects on the breastfed child, or the effects on milk production. Data from rat studies have shown the transfer of iron into milk by divalent metal transporter-1 (DMT-1) and ferroportin-1 (FPN-1). Hence, there is a possibility of infant exposure when Auryxia is administered to a nursing woman. The development and health benefits of breastfeeding should be considered along with the mother’s clinical need for Auryxia and any potential adverse effects on the breastfed child from Auryxia or from the underlying maternal condition.
8.4 Pediatric Use
The safety and efficacy of Auryxia have not been established in pediatric patients.
8.5 Geriatric Use
Clinical studies of Auryxia included 292 subjects aged 65 years and older (104 subjects aged 75 years and older). Overall, the clinical study experience has not identified any obvious differences in responses between the elderly and younger patients in the tolerability or efficacy of Auryxia.
-
10 OVERDOSAGE
No data are available regarding overdose of Auryxia in patients. In patients with chronic kidney disease, the maximum dose studied was 2,520 mg ferric iron (12 tablets of Auryxia) per day. Iron absorption from Auryxia may lead to excessive elevations in iron stores, especially when concomitant intravenous iron is used [see Warnings and Precautions (5.1)].
In clinical trials, one case of elevated iron in the liver as confirmed by biopsy was reported in a patient on dialysis administered intravenous iron and Auryxia.
-
11 DESCRIPTION
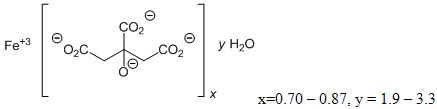
Auryxia (ferric citrate), a phosphate binder and iron replacement product, is known chemically as iron (+3), x (1, 2, 3-propanetricarboxylic acid, 2 hydroxy-), y (H2O)
Auryxia 210 mg ferric iron tablets for oral administration, equivalent to 1g ferric citrate, are film-coated, peach-colored, and oval-shaped tablets debossed with “KX52”. The inactive ingredients are pregelatinized starch and calcium stearate. In addition, the film-coating contains the following inactive ingredients; hypromellose, titanium dioxide, triacetin, and FD&C Yellow #6/Sunset Yellow FCF Aluminum Lake, FD&C Red #40/Allura Red AC Aluminum Lake, and FD&C Blue #2/Indigo Carmine Aluminum Lake.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Hyperphosphatemia in Chronic Kidney Disease on Dialysis
Ferric iron binds dietary phosphate in the GI tract and precipitates as ferric phosphate. This compound is insoluble and is excreted in the stool. By binding phosphate in the GI tract and decreasing absorption, ferric citrate lowers the phosphate concentration in the serum.
Iron Deficiency Anemia in Chronic Kidney Disease Not On Dialysis
Ferric iron is reduced from the ferric to the ferrous form by ferric reductase in the GI tract. After transport through the enterocytes into the blood, oxidized ferric iron circulates bound to the plasma protein transferrin, and can be incorporated into hemoglobin.
12.2 Pharmacodynamics
Hyperphosphatemia in Chronic Kidney Disease on Dialysis
Auryxia reduces serum phosphorus levels and has also been shown to increase serum iron parameters, including ferritin, iron and TSAT. In dialysis patients treated with Auryxia for hyperphosphatemia in a 52-week study in which intravenous iron could also be administered, mean (SD) ferritin levels rose from 593 (293) ng/mL to 895 (482) ng/mL, mean (SD) TSAT levels rose from 31% (11) to 39% (17) and mean (SD) iron levels rose from 73 (29) mcg/dL to 88 (42) mcg/dL. In contrast, in patients treated with active control, these parameters remained relatively constant [see Contraindications (4) and Warnings and Precautions (5.1)].
Iron Deficiency Anemia in Chronic Kidney Disease Not On Dialysis
Auryxia may increase hemoglobin levels and has also been shown to reduce serum phosphorus levels. In chronic kidney disease patients not on dialysis treated with Auryxia for iron deficiency anemia in a 16-week placebo-controlled study, mean (SD) phosphorus levels decreased from 4.23 (0.91) mg/dL at baseline to 3.72 (0.60) mg/dL. In comparison, in patients treated with placebo control, mean (SD) phosphorus levels decreased from 4.12 (0.68) mg/dL at baseline to 3.87 (0.68) mg/dL.
12.3 Pharmacokinetics
Absorption and Distribution
Formal pharmacokinetic studies have not been performed with Auryxia. Examination of serum iron parameters has shown that there is systemic absorption of iron from Auryxia [see Contraindications (4), Warnings and Precautions (5.1) and Clinical Pharmacology (12.2)].
Drug Interaction Studies
In vitro
Of the drugs screened for an interaction with ferric citrate in vitro, only doxycycline showed the potential for interaction with at least 70% decrease in its concentration. This interaction can be avoided by spacing the administration of doxycycline and ferric citrate [see Drug Interactions (7)].
In vivo
Six drug interaction studies (N=26-60/study) were conducted to establish the effects of Auryxia (administered as 3 x 2 g/day with meals) on the disposition of concomitantly orally administered clopidogrel, ciprofloxacin, digoxin, diltiazem, glimepiride and losartan in healthy subjects. With the exception of ciprofloxacin, Auryxia did not alter the systemic exposure of the tested drugs, as measured by the area under the curve (AUC) and Cmax of the tested drugs when either co-administered with Auryxia or given 2 hours later. Auryxia decreased the relative bioavailability of concomitantly administered ciprofloxacin by approximately 45%. However, there was no interaction when Auryxia and ciprofloxacin were taken 2 hours apart. Consequently, ciprofloxacin should be taken at least 2 hours before or after Auryxia is dosed [see Drug Interactions (7)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Data from carcinogenesis studies have shown that ferric citrate is not carcinogenic in mice and rats when administered intramuscularly or subcutaneously. Ferric citrate was neither mutagenic in the bacterial reverse mutation assay (Ames test) nor clastogenic in the chromosomal aberration test in Chinese hamster fibroblasts.
The potential for ferric citrate to impair reproductive performance or to cause fetal malformation has not been evaluated.
-
14 CLINICAL STUDIES
14.1 Hyperphosphatemia in Chronic Kidney Disease on Dialysis
The ability of Auryxia to lower serum phosphorus in patients with CKD on dialysis was demonstrated in randomized clinical trials: one 56-week, safety and efficacy trial, consisting of a 52-week active-controlled phase and a 4-week, placebo-controlled, randomized withdrawal period, and one 4-week open-label trial of different fixed doses of Auryxia. Both trials excluded subjects who had an absolute requirement for aluminum containing drugs with meals.
Study KRX-0502-304 (NCT 01191255)
Study KRX-0502-304 was a long-term, randomized, controlled, safety and efficacy trial. After the 2-week washout period during which phosphate binders were held, patients with a mean serum phosphorus of 7.5 mg/dL during washout were randomized 2:1 to Auryxia (N=292) or active control (calcium acetate and/or sevelamer carbonate; N=149). The majority (>96%) of subjects were on hemodialysis. The starting dose of Auryxia was 6 tablets/day, divided with meals. The starting dose of active control was the patient’s dose prior to the washout period. The dose of phosphate binder was increased or decreased as needed to maintain serum phosphorus levels between 3.5 and 5.5 mg/dL, to a maximum of 12 tablets/day.
As shown in the figure below, serum phosphorus levels declined following initiation of therapy. The phosphorus lowering effect was maintained over 52 weeks of treatment.
Figure 1: Serum Phosphorus Control over 52 Weeks
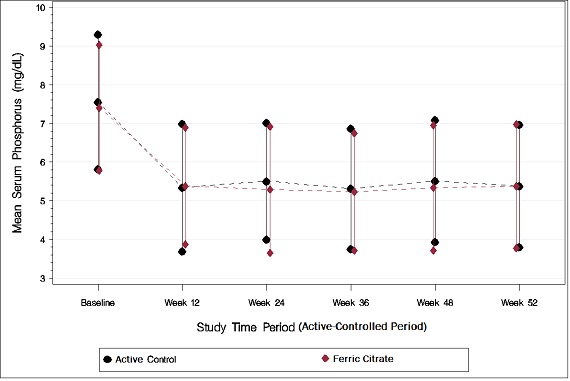
Following completion of the 52-week active-controlled phase, Auryxia-treated patients were eligible to enter a 4-week placebo-controlled randomized withdrawal phase, in which patients were re-randomized in a 1:1 ratio to receive Auryxia (N=96) or placebo (N=96). During the placebo-controlled period, the serum phosphorus concentration rose by 2.2 mg/dL on placebo relative to patients who remained on Auryxia.
Table 3: Effect of Auryxia on serum phosphorus during randomized withdrawal a The LS mean treatment difference and p-value for the change in mean were created via an ANCOVA model with treatment as the fixed effect and Week-52 baseline (phosphorus) as the covariate. Between-treatment differences were calculated as the LS mean (Auryxia) - LS mean (placebo or active control).
Note: Analyses using ANCOVA with last observation carried forward. ANCOVA=analysis of covariance; CI=confidence interval.Primary Endpoint (Week 56) Auryxia Placebo Treatment Difference
(95% CI)p-value Serum phosphorus (mg/dL) Mean baseline (Week 52) 5.12 5.44 Mean change from baseline (Week 56) -0.24 1.79 −2.18 (−2.59, −1.77) <0.0001a Study KRX-0502-305 (NCT 01074125)
Following a 1- to 2-week washout from all phosphate-binding agents, 154 patients with hyperphosphatemia (mean serum phosphorus of 7.5 mg/dL) and CKD on dialysis were randomized in a 1:1:1 ratio to 1, 6, or 8 tablets/day of Auryxia for 4 weeks. Auryxia was administered with meals; subjects receiving 1 tablet/day were instructed to take it with their largest meal of the day, and subjects on 6 or 8 tablets/day took divided doses in any distribution with meals. Dose-dependent decreases in serum phosphorus were observed by Day 7 and remained relatively stable for the duration of treatment. The demonstrated reductions from baseline to Week 4 in mean serum phosphorus were significantly greater with 6 and 8 tablets/day than with 1 tablet/day (p<0.0001). Mean reduction in serum phosphorus at Week 4 was 0.1 mg/dL with 1 tablet/day, 1.9 mg/dL with 6 tablets/day, and 2.1 mg/dL with 8 tablets/day.
14.2 Iron Deficiency Anemia in Chronic Kidney Disease Not On Dialysis
Study KRX-0502-306 (NCT 02268994)
The efficacy of Auryxia for the treatment of iron deficiency anemia in adult patients with CKD not on dialysis was demonstrated in a 24-week study consisting of a 16-week, randomized, double-blind, placebo-controlled, efficacy period followed by an 8-week open-label safety extension period in which all patients remaining in the study, including the placebo group, received Auryxia. Patients with eGFR <60 mL/min/1.73m2, who were intolerant of or have had an inadequate therapeutic response to oral iron supplements, with Hgb ≥9.0 g/dL and ≤11.5 g/dL, serum ferritin ≤200 ng/mL and TSAT ≤25% were enrolled. Patients were randomized to treatment with either Auryxia (n=117) or placebo (n= 117). Dosing with Auryxia or placebo was initiated at 3 tablets/day with meals. Dose titration could occur at Weeks 4, 8 and 12 during Randomized Period, and at Weeks 18 and 20 during Safety Extension Period based on Hgb response. Use of oral or intravenous iron, erythropoiesis stimulating agents (ESAs) was not permitted at any time during the study.
The mean age of the patients was 65 years (range 26 to 93); 63% were female, 69% Caucasian, 30% were African American and <2% were other races.
The main efficacy outcome measure was the proportion of subjects achieving an increase in Hgb of ≥1.0 g/dL at any time point between baseline and the end of the 16-week Randomized Period.
Table 4: Efficacy of Auryxia in Iron Deficiency Anemia in Chronic Kidney Disease (Not on Dialysis) Auryxia
(N=117)Placebo
(N=115)p-value Proportion of patients achieving an increase in hemoglobin of ≥ 1.0 g/dL at any time point during the 16 week randomized period 52% 19% <0.001 During the 16-week randomized period 49% of subjects in the Auryxia arm and 15% of subjects in the placebo arm (p <0.001) had a mean change in hemoglobin from baseline ≥0.75 g/dL over any 4-week time period provided that an increase of at least 1.0 g/dL had occurred during that 4-week period. Increases in mean hemoglobin (0.75 ± 0.09 g/dL), serum ferritin (163 ± 9 ng/mL) and transferrin saturation (18 ± 1%) were observed from baseline during the 16-week randomized period in the Auryxia arm.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Tablets: Auryxia 210 mg ferric iron tablets equivalent to 1 g of ferric citrate are supplied as 200 tablets in 400-cc high-density polyethylene bottles. The 210 mg ferric iron tablets are film-coated, peach-colored, and oval-shaped tablets debossed with “KX52.”
1 Bottle of 200-count 210 mg ferric iron tablets (NDC: 59922-631-01)
-
17 PATIENT COUNSELING INFORMATION
Dosing Recommendations
Instruct patients to take Auryxia as directed with meals and adhere to their prescribed diets. Instruct patients on concomitant medications that should be dosed apart from Auryxia [see Dosage and Administration (2)]. Advise patients not to chew or crush Auryxia because tablets may cause discoloration of mouth and teeth.
Adverse Reactions
Advise patients that Auryxia may cause discolored (dark) stools, but this staining of the stool is considered normal with oral medications containing iron.
Auryxia may cause diarrhea, nausea, constipation, vomiting, hyperkalemia, abdominal pain, and cough. Advise patients to report severe or persistent gastrointestinal symptoms to their physician [see Adverse Reactions (6.1)].
Accidental Ingestion
Advise patients to keep this product out of the reach of children and to seek immediate medical attention in case of accidental ingestion by a child.
Manufactured for and Distributed by:
Keryx Biopharmaceuticals, Inc.
245 First Street
Suite 1400
Cambridge, MA 02142Issued 12/2019 Rev 5.0
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
AURYXIA
ferric citrate tablet, coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59922-631 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TETRAFERRIC TRICITRATE DECAHYDRATE (UNII: Q91187K011) (FERRIC CATION - UNII:91O4LML611) FERRIC CATION 210 mg Product Characteristics Color PINK Score no score Shape OVAL Size 19mm Flavor Imprint Code KX52 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59922-631-01 200 in 1 BOTTLE; Type 0: Not a Combination Product 09/17/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205874 09/17/2014 Labeler - Keryx Biopharmaceuticals, Inc. (073504263)

Trademark Results [Auryxia]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 AURYXIA 86390566 4956680 Live/Registered |
Keryx Biopharmaceuticals, Inc. 2014-09-10 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.